

Abstract
Understanding the mechanisms underlying resistance is critical to improving therapeutic outcomes in patients with metastatic castration-resistant prostate cancer (mCRPC). Previous work showed dynamic interconversions between epithelial-mesenchymal transition (EMT) to mesenchymal-epithelial transition (MET) defines the phenotypic landscape of prostate tumors, as a potential driver of emergence of therapeutic resistance. In this study, we use in vitro and in vivo preclinical MDA PCa PDX models of resistant human prostate cancer to determine molecular mechanisms of cross-resistance between anti-androgen therapy and taxane chemotherapy, underlying the therapeutically resistant phenotype. Transcriptomic profiling revealed that resistant and sensitive prostate cancer C4-2B cells have a unique differential gene signature response to cabazitaxel. Gene pathway analysis showed that sensitive cells exhibit increase in DNA damage, while resistant cells express genes associated with protein regulation in response to cabazitaxel. These PDX specimens are from patients who have metastatic lethal CRPC, treated with androgen-deprivation therapy (ADT), antiandrogens and chemotherapy including 2nd line taxane chemotherapy, cabazitaxel. Immunohistochemistry revealed high expression of E-cadherin and low expression of vimentin resulting in re-differentiation toward an epithelial phenotype. Furthermore, the mitotic kinesin-related protein (HSET) involved in microtubule binding and the SLCO1B3 transporter (implicated in cabazitaxel intracellular transport), associated with resistance in these prostate tumors. Combinational targeting of kinesins (ispinesib) with cabazitaxel was more effective than single monotherapies in inducing cell death in resistant prostate tumors.
Keywords: Cell plasticity, tumor recurrence, anti-androgens, taxane chemotherapy, lethal prostate cancer
Introduction
Prostate cancer, the most common malignancy among men, has favorable survival rates in local disease and early detection (1). Lethality of prostate cancer arises from progression to metastatic disease, resulting in 1 in 33 American men with prostate cancer to succumb to the disease. Those with metastatic prostate cancer have a 70% chance of mortality within 5 years, largely due to the emergence of treatment resistance and tumor recurrence (1). Second-generation anti-androgens enzalutamide and abiraterone acetate target androgen signaling by preventing the binding of 5α-dihydrotestosterone (DHT) to the androgen receptor (AR) and subsequent translocation to the nucleus and inhibiting the biosynthesis of DHT respectively (2, 3). Androgen deprivation therapy (ADT) and AR signaling inhibitors (ARSI), offers survival benefits in mCRPC patients. The majority of prostate cancer patients treated with ADT eventually develop CRPC that ultimately recurs and progresses to lethal disease. These therapies have survival benefits for patients with CRPC, however there is often progression to resistant disease, no longer responsive to anti-androgen therapy (4, 5). At this stage, the only therapeutic strategies that confer improvement in patient survival are 1st line taxane chemotherapy and 2nd line taxane chemotherapy (docetaxel and cabazitaxel), and the recently FDA approved PARP inhibitors (olaparib) (6–8).
The primary mechanism of antitumor action by taxane chemotherapy proceeds via induction of G2-M cell cycle arrest and apoptosis in prostate cancer through stabilization of microtubules by binding to β-tubulin and preventing de-polymerization of microtubules, consequentially blocking mitosis (7). Further evidence (from our group) first showed that taxane chemotherapy prevents the translocation of AR to the nucleus, impairing AR signaling and transcriptional activity (9). Cabazitaxel was also shown to have a differential effect as a result of targeting expression of HSET (KIFC1), a kinesin protein involved in microtubule binding, assembly of the mitotic spindle and potentially associated with AR (10, 11). This is of major clinical significance, as HSET expression has been linked to therapeutic resistance and poor clinical outcomes in prostate cancer (12). There is potential therapeutic value in targeting HSET to overcome resistance, as in vitro studies have shown the re-sensitization of docetaxel resistant prostate cancer cells and apoptosis when treated with an HSET inhibitor CW069 (13). Our In vivo studies have shown effective targeting of HSET by treating mice with cabazitaxel therapy following ADT, showing the potential value for sequencing strategies to improve patient clinical response (10).
Prostate tumors acquire a more invasive and stem-like phenotype through the process of epithelial to mesenchymal transition (EMT) leading to the development of metastases and therapeutic resistance (14). This process is characterized by the loss of epithelial markers such as E-cadherin, insulin-like growth factor binding protein 3 (IGFBP-3), and their tight junctions, and the upregulation of mesenchymal markers including vimentin (15–17). Previous studies from our group showed that cabazitaxel treatment contributes to the phenotypic reprogramming of prostate cancer cells within the tumor microenvironment (TME), by reversing EMT to mesenchymal-epithelial-transition (MET) (11). The cell plasticity that drives interconversion between EMT and MET not only defines the phenotypic landscape of prostate tumors, but has also been linked to the emergence of therapeutic resistance and recurrent tumors, yet it provides a potential therapeutic vulnerability window where cells are primed for therapy (17–19). In this study, we profiled the effectors involved in phenotypic re-differentiation in models of therapeutic resistance in prostate cancer, towards the development of a gene signature that underpins the molecular landscape of resistance for potential leads to overcome tumor recurrence and lethal disease.
Materials and Methods
Cell Lines.
Human prostate cancer cell lines, 22RV1 (CRPC), LNCaP (androgen sensitive), and PC3 (androgen independent, AR-negative) were obtained from American Type Culture Collection (CRL-2505, CRL-1740, CRL-1435 respectively. ATCC, Manassas, VA). Therapeutically resistant human prostate cancer cell lines were generated according to and generously provided by Dr. Allen Gao (University of California, Davis) as previously described, C4-2BER (enzalutamide resistant), C4-2BAR (abiraterone resistant), C4-2BDR (docetaxel resistant) and C4-2B (parental) (20, 21). Cells were maintained using Roswell Park Memorial Institute (RPMI) 1640 (Corning, New York, NY) supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, Waltham, MA) and 1% penicillin-streptomycin (Thermo Fisher Scientific, Waltham, MA) in a 37°C incubator with 5% CO2. Experiments using DHT supplemented media with charcoal stripped FBS (Thermo Fisher Scientific, Waltham, MA). PC3-CR (cabazitaxel resistant) cell lines were generated through incremental increase in exposure to cabazitaxel, starting with 0.5nM, over a 2-month period. Cells were tested for mycoplasma contamination using the ATCC Universal Mycoplasma Detection Kit (30–1012K, ATCC, Manassas, VA).
Drugs:
Antiandrogen drugs enzalutamide and abiraterone obtained from Selleck chemicals and Sigma Aldrich respectively. Both drugs were reconstituted to 10mM stocks in DMSO and stored at −80°C. Docetaxel obtained from Sigma Aldrich (St Louis, MO) was dissolved in DMSO to 1mM stocks. Cabazitaxel (from Sigma Aldrich), dissolved in 100% ethanol and stored at −20°C in 1mM stocks. Kinesin inhibitors CW069 and ispinesib were purchased from Selleck Chemicals and Sigma Aldrich respectively.
Cell Viability.
Cell viability was assessed using the Thiazolyl Blue Tetrazolium bromide (MTT) assay. Cells (5×104 cells/mL) at 60–70% confluence in 96-well plates were exposed to the respective drugs, enzalutamide (10μM), abiraterone (10μM), or docetaxel (50nM) for 24 and 48hrs. Experiments investigating the effects of cabazitaxel (1–50nM) or Ispinesib (1–50nM) on cell viability were exposed to treatment for 24, 48, 72 and 96 hours. Cells were subsequently treated with 1mg/mL of MTT (Thermo Fisher Scientific, Waltham, MA) for 1hr at 37°C and formazan crystals were solubilized with dimethyl sulfoxide (DMSO). Absorbance was measured at 570nM using SpectraMaxx M5 spectrophotometer (Molecular Devices, San Jose, CA). All treatments were given in triplicate, and results presented as the average of three independent experiments.
Cell Migration.
Cell lines were seeded in 6-well plates in triplicate at a density of 2×105 cells/well, grown to 60% density. The cell monolayer was wounded with 10μL pipette tip. Following wounding, cells were exposed to enzalutamide (10μM), abiraterone (10μM), or docetaxel (50nM). At 24 and 48hrs, images were captured (10x magnification) and the number of cells migrating into the wound were counted in three fields per well. This experiment was replicated three times.
Cell Invasion.
The invasive potential of therapeutically resistant cell lines was investigated using the matrigel invasion assay. Cells were seeded into the upper chamber of the Biocoat Matrigel Transwell Chamber (Beckon Dickinson, Franklin Lakes, NJ) at a density of 50,000 cells/well. Non-invasive cells were removed using medium soaked cotton swabs after 24 and 48 hours and cell were fixed and stained using the Diff-Quick staining solutions (IMEB inc., San Marcos, CA). Images were captured at 10x magnification, and the number of invading cells were counted in 4 fields.
Western blot analysis.
Total cellular protein was isolated from cell lysates using RIPA buffer (Thermo Fisher Scientific, Waltham, MA) with protease inhibitor (Thermo Fisher Scientific, Waltham, MA). Protein concentration of cell lysates was determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA). Proteins were subjected to SDS-polyacrylamide gel electrophoresis using 10–12% SDS-polyacrylamide gels (Bio-Rad, Hercules, CA) and transferred to polyvinylidene fluoride (PVDF) membranes (Bio-Rad, Hercules, CA). Membranes were probed with primary antibodies overnight at 4°C followed by exposure to species-specific HRP-labelled secondary antibodies (Cell Signalling, Danvers, MA). Probes were detected with WesternBright ECL spray (Advansta, San Jose, CA) and visualized using GE ImageQuant chemiluminescence. GAPDH was used as a loading control.
In vivo xenograft experiments
All animal experiments were performed according to the Institutional Animal Care and Use Committee (IACUC) Office of Animal Care Use and Welfare (OACUW) at the Icahn School of Medicine at Mount Sinai. Six to 8 week old male NOD-scid gamma mice (The Jackson Laboratory) were injected subcutaneously into right flank with C4-2B or C4-2BDR cells (5 million in 100μL media) mixed with 100μL Matrigel (Corning). After 4 weeks, tumors reached palpable size and were treated with cabazitaxel at 3mg/kg every 3 days for 2 weeks, the vehicle control groups were administered 1:1:18 vol/vol of ethanol: polysorbate 80: 5% wt/vol glucose in sterile water. Treatments were administered via intraperitoneal injection. Tumors were measured every 3 days and tumor volume was calculated by length × width × 0.5236. 3 days after the final treatment, mice were euthanized using CO2 according to approved guidlines, and tumors were exised for analysis. One mouse was excluded from analysis due to significant toxicity from cabazitaxel.
Co-immunoprecipitation (Co-IP)
Cells were lysed using the Pierce IP Lysis buffer (Thermo Fisher scientific, Waltham, MA). Co-immunoprecipitation was performed using the Dynabeads Protein G Kit according to manufacturer’s instructions with cross-linking with the Pierce BS3 Crosslinker (Thermo Fisher Scientific, Waltham, MA). Anti-KIFC1 and Anti-Bcl2 Antibodies were used at 5μg/sample. Antibodies used are summarized in Supplementary Table 1.
RNA sequencing.
RNA-seq sequencing of PC3 and PC3-CR cells was done using the Human Clariom-S array analysis with the assistance of the University of Kentucky Microarray Core Facility. Total RNA was extracted from prostate cancer cells using TRIzol reagent (Invitrogen/Life Technologies) and recommended protocol for RNA-isolation. RNA sequencing of C4-2B samples was performed at the Genetic and Genomics Core Facilities at Icahn School of Medicine at Mount Sinai. We used 50bp single-end and 30 million reads per sample for a total of 18 samples. Illumina Stranded mRNA library prep kit was used to prepare the RNA material for sequencing. NextSeq 500/550 High output (75 Cycles) was utilized to amplify the mRNAs. Briefly, the resulting reads were quality controlled using FASTQC, Multi-QC and SamTools. The reads were aligned to Human Genomic Reference HG38 with splicing sensitive aligner STAR 2.7. Gene counts were quantified using FeatureCounts. Downstream differential expression analysis and pathway enrichment were performed using R packages Dream, mle4, VariancePartition, enrichR, GSVA and figures were generated with ggplot2, tidyverse complexheatmap and pheatmap packages. Data is available upon request and under SRA folder XXXXXXXXXXX.
Quantitative RT-PCR analysis.
Total RNA was extracted using PureLink® RNA Mini Kit (Invitrogen, Waltham, MA) according to manufacturer’s instructions. 1μg of RNA sample was subjected to reverse-transcription using the iScript™ cDNA synthesis kit (Bio-rad, Hercules CA). Quantitative real-time RT-PCR was performed in the CFX96 Real Time Detection System (Bio-Rad, Hercules CA) with Sybr green based detection using specific primers for CBLN2, NECTIN3, LRG1, ELF5 TRIM2, KIF5C, MARCKS, WLS, Snail, Slug, Zeb1 and Twist1 (Thermo Fisher Scientific, Waltham, MA). Each PCR reaction included three technical replicates, data represents the average of three repeated independent experiments, normalized to Actin expression (ΔΔCT) and expressed relative to untreated controls.
Transporter siRNA Silencing.
For gene silencing in prostate cancer cells, the ShRNA vector was obtained from Open Biosystems (Huntsville, AL). After transfection ShRNA SLCO1BP3 transporter gene in LNCaP cells were selected using puromycin (a resistance marker). Polyclonal populations were pooled under antibiotic selection media and after several passages, stable cell lines were characterized as previously described (22).
Immunohistochemical analysis of PDX Specimens.
Formalin-fixed paraffin embedded tissue specimens from patient derived xenograft models (PDX) sections (5μm) of male patients with advanced CRPC were provided by Dr. Nora Navone (MD Anderson Cancer Center) as part of the MDA PCa PDX series as previously described (23). Sections were subjected to immunohistochemical analysis using antibodies against E-cadherin, vimentin, cytokeratin 18, HSET, B-cell lymphoma-2 (Bcl-2), and solute carrier organic anion transporter family 1B3 (SLCO1B3) (Antibodies used are summarized in Supplementary table 1). Slides were de-paraffinized, rehydrated and heat-induced antigen retrieval was performed using the Dako antigen retrieval solution at 100°C. Sections were exposed to primary antibodies overnight at 4°C, followed by detection with IHC-Select species-specific biotinylated secondary antibodies and horseradish peroxidase (HRP)-streptavidin (Millipore, Burlington, MA). Visualized was achieved using 3’3-diaminobenzidine chromogen (DAB) substrate kit (Dako, Santa Clara, CA). Images were captured using Nanozoomer digital scanner (Hamamatsu photonics, Japan). The positively stained HSET (nuclear) was quantified at 40x magnification by two independent observers each in three random fields.
Statistical analysis.
Numerical assay data was analyzed by one way or two-way ANOVA to test for statistical significance between cell lines and treatments using GraphPad Prism 9. Analysis of PC3 data was analyzed using students t-test. Data is expressed as mean ± standard error of the mean. Statistical significance was inferred when P<0.05. RNA sequencing data was normalized and modeled using mixed effect models for precise modeling of time points and the 2 cellular conditions (resistant and sensitive). RNAseq quality control using count distribution, library size and gene detection of at least 5 counts between samples was performed to remove potential sequencing artifacts. Differential expression tests part of mixed effect models used moderate t statistics. Hierarchical clustering was performed using the R package HMISC implementing “ward.D2” method (24). Pathway enrichment analysis was done using local enrichR to survey all available gene set databases with focus on gene ontology (GO) and MsigBD. Pathways were ranked based on number of genes per pathway, odds ratio and false discovery rate adjusted p-values. All p-values were adjusted using multiple testing adjustment (25).
Data availability.
Data processing and analysis scripts are deposited at https://github.com/eegk and available upon request. All other relevant data that support the conclusions of the study are within the supplementary material or available from the authors upon request.
Results
Characterization of Cross-resistance to Anti-androgens and 1st Line Taxane Chemotherapy
We determined the response of human prostate cancer cell lines that are resistant to enzalutamide (C4-2BER), abiraterone (C4-2BAR) and docetaxel (C4-2BDR), to anti-androgens and docetaxel chemotherapy, based on cell viability assessment in response enzalutamide (10μM), abiraterone (10μM) and docetaxel (50nM). Docetaxel induced significant cell death, as identified by the significant reduction in cell viability in response to the drug at 24 and 48hrs of treatment (P<0.05), with the exception of the docetaxel resistant C4-2BDR (Fig. 1A and B). At 24hrs, 22RV1 cells (CRPC), exhibited partial sensitivity in response to docetaxel, and the antiandrogen abiraterone by 48hrs of treatment (Fig. 1A and B). The C4-2B cells showed significant loss of cell viability in response to both anti-androgens and docetaxel. The enzalutamide resistant C4-2BER and abiraterone resistant C4-2BAR failed to show a cell death induction after treatment (24hrs) to either anti-androgen. However, by 48hrs, C4-2BER showed sensitivity to abiraterone, and C4-2BAR exhibited differential response to all drugs (Fig. 1A and B). The taxane resistant C4-2BDR cells failed to respond to any of the drugs at 24hrs (Fig. 1A and B). We also examined the effect of anti-androgen on prostate cancer cell migration (Supplementary fig. 1A, Fig. 1G and H). 22RV1 cells showed minimal migration, while C4-2B parental cells showed the highest migration potential at 24 and 48hrs. Treatment with enzalutamide or abiraterone had no effect on the migration of 22RV1, C4-2B, C4-2BER and C4-2BAR. Only in response to abiraterone there was a significant reduction on cell migration of C4-2BDR cells (P<0.05) (Fig. 1G and H).
Figure 1. Differential response to anti-androgens and taxane chemotherapy in prostate cancer resistant cell lines.
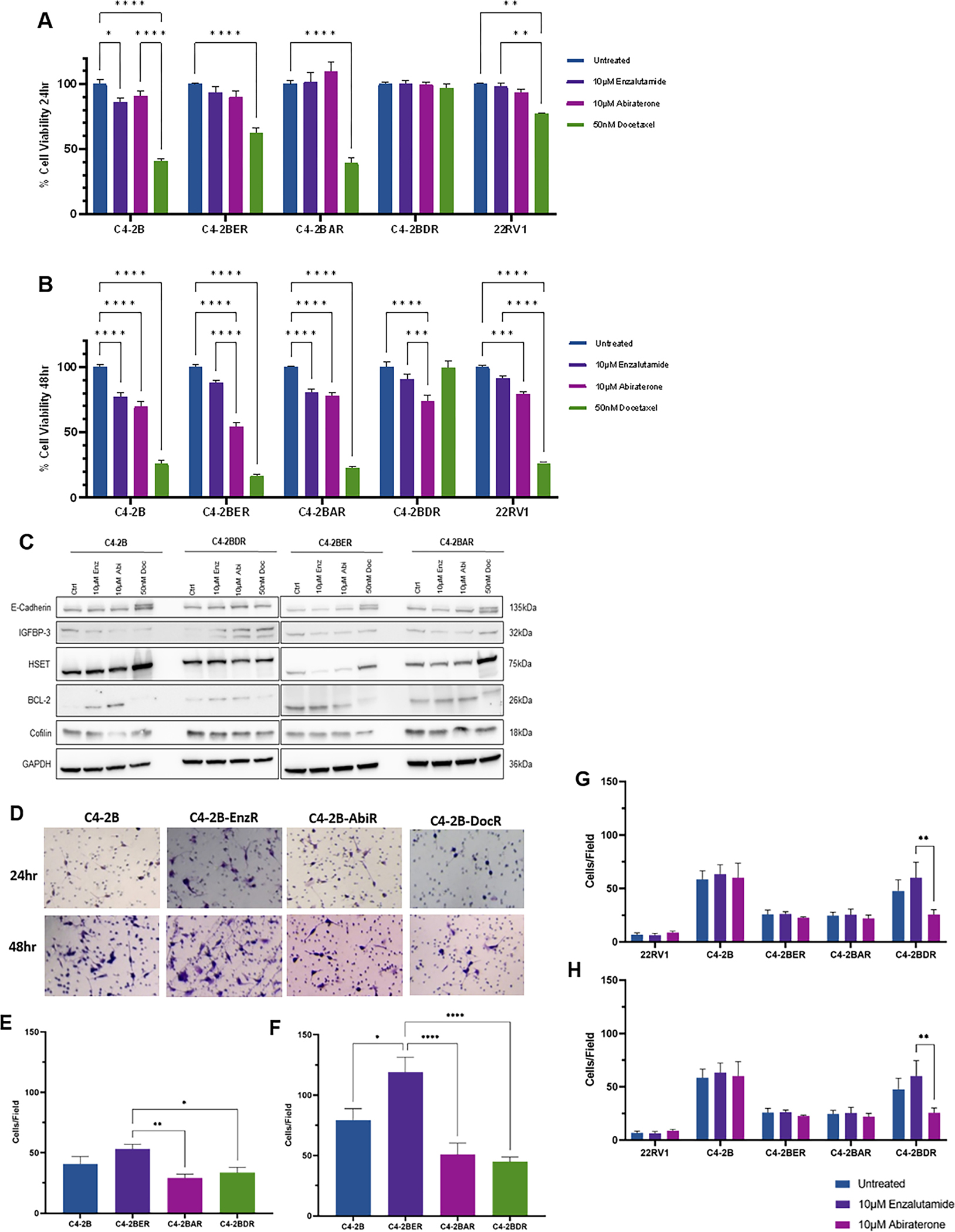
Panels A and B, show the results of cell viability of C4-2B, C4-2BER, C4-2BAR and C4-2BDR in response to enzalutamide (10μM), 10μM abiraterone or docetaxel (50nM) at 24 (panel A) and 48hrs (panel B) measured by the MTT assay. Panel C, Western blot analysis of E-cadherin, IGFBP-3, HSET, BCL-2 and cofilin protein levels in response to enzalutamide (10μM), abiraterone (10μM) or docetaxel (50nM) (for 24hrs), GAPDH was used as a loading control. Panel D shows representative images of matrigel invasion assay, cells were counted in 4 fields of view from three independent experiments at 24 (panel E) and 48 (panel F) hours. Following wound scratch assay, images were taken in 3 fields of view and quantified at 24 (panel G) and 48 (panel H) hrs. Data represents mean of three independent experiments in triplicate ±SEM, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 as determined by two-way ANOVA.
The invasive potential of resistant cell lines was investigated using matrigel invasion assays. All cell lines showed invading cells at 24 and 48hrs (Fig. 1D–F). At 24hrs, the resistant cell lines showed no difference in the number of invading cells compared to the C4-2B cells (Fig. 1D and E). At 48hrs, the enzalutamide resistant C4-2BER had significantly greater number of invading cells compared to C4-2B and C4-BDR (Fig. 1D and F).
Phenotypic profiling of the effectors of cross-resistance by western blot revealed that IGFBP-3 is upregulated in C4-2BDR cells compared to C4-2B cells (24hrs). Both anti-androgen resistant cell lines exhibited similar protein expression profiles, while Bcl-2 expression was downregulated in response to docetaxel in both cell lines. HSET expression is induced by docetaxel in parental and anti-androgen resistant cells, and not targeted as observed with cabazitaxel (Fig. 1C) (11).
Phenotypic interconversions Drive Cross-resistance to Cabazitaxel
A time course and dose response to cabazitaxel were investigated in C4-2B cell models; our findings showed that docetaxel resistant C4-2BDR cells exhibit significant cross-resistance to cabazitaxel (Fig. 2A–D). At 48hrs, C4-2BDR have significantly greater viability when exposed to 1–35nM of cabazitaxel compared to parental cells (Fig. 2B). In concentrations up to 10nM, C4-2BDR cells maintain greater than 50% cell viability through 96hrs. At higher doses (50nM), there is no difference in response to cabazitaxel (Fig. 2A–D). For subsequent experiments a dose of 20nM was used for 48hrs treatment period. At concentrations of cabazitaxel up to 20nM, C4-2BDR cells showed reduced sensitivity to the drug compared to all other cell lines (Supplementary Fig. 2A–D). Anti-androgen resistant cell lines C4-2BER and C4-2BAR, and 22RV1 CRPC cells showed similar viability profile to the parental C4-2B cells. This response was consistent at higher doses of cabazitaxel, (35–50nM, Fig. 2A–D, Supplementary fig. 2 and 3). Early results from in vivo xenograft C4-2B and C4-2BDR tumors suggests cabazitaxel is not effective in reducing tumor growth in C4-2BDR tumors compared to C4-2B tumors and vehicle controls (Supplementary fig. 3H–I).
Figure 2. Phenotypic profiling of cross-resistance in docetaxel resistant prostate cancer cells to cabazitaxel.
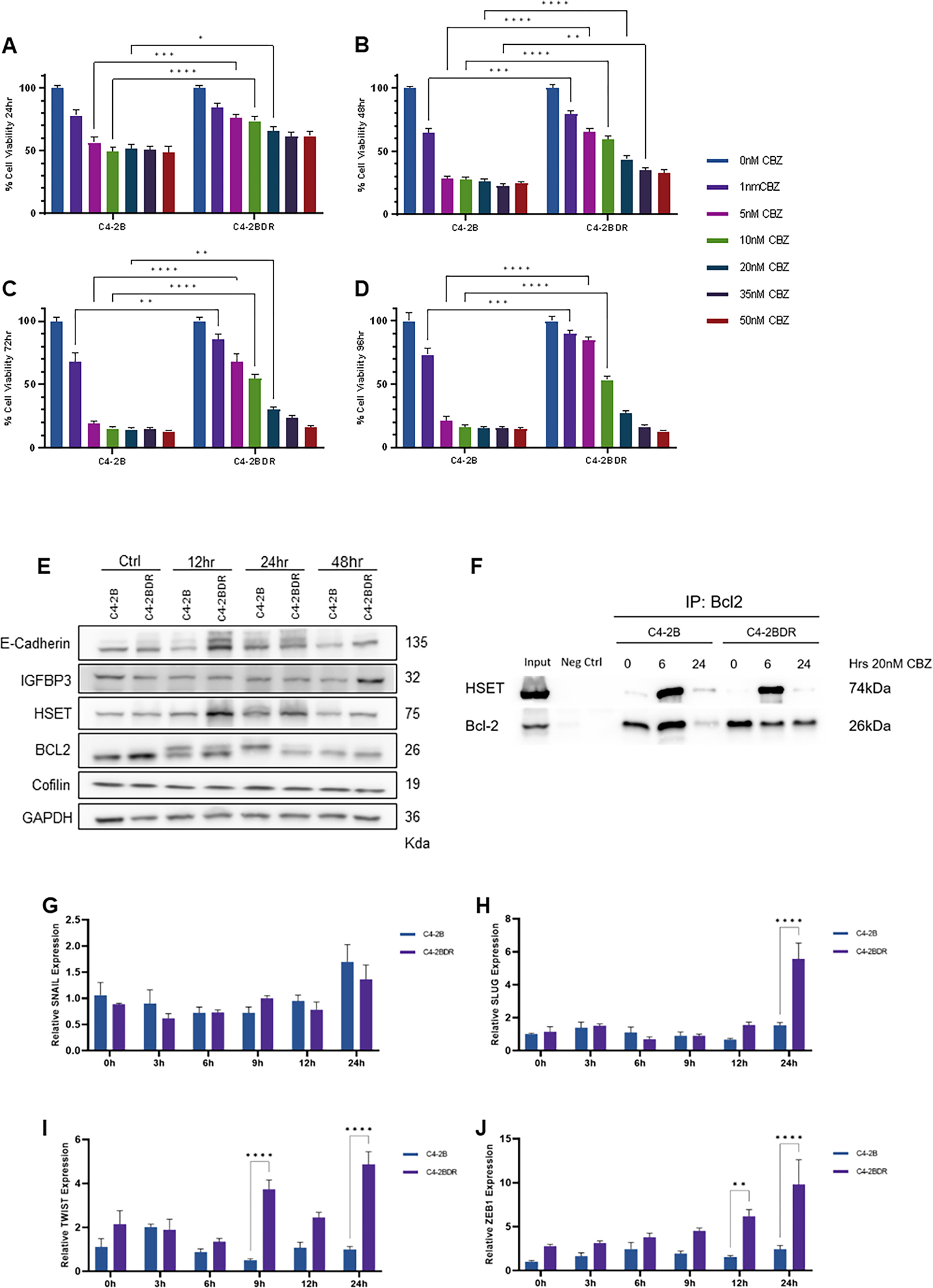
Panels A-D, reveal the dose response of C4-2B and C4-2BDR cells to increasing concentrations (0–50nM) of cabazitaxel at 24 (panel A), 48 (panel B), 72 (panel C) and 96hrs (panel D). The results represent the mean of three independent MTT assay experiments in triplicate as percentage of untreated controls. Panel E, Western blot analysis of E-cadherin, IGFBP-3, HSET, BCL-2 and cofilin protein levels in response to 20nM cabazitaxel in C4-2B and C4-2BDR cells (12 to 48hrs). GAPDH was used as a loading control; the data is representative of three experiments. Panel F, Bcl-2 co-immunoprecipitation of C4-2B and C4-2BDR cells treated with cabazitaxel (12 and 24hrs). Panels G-J, C4-2B and C4-2BDR cells were treated with cabazitaxel (20nM) as indicated and mRNA was analyzed by RT-PCR for transcriptional regulators of EMT were measured by qRT-PCR; panel G-SNAIL, panel H-SLUG, panel I-TWIST, panel J-ZEB1. Data presented as average relative expression to untreated C4-2B cells from three independent experiments performed in triplicate (mean±SEM, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 by ANOVA).
The phenotypic profile of this cross-resistance to cabazitaxel was analyzed by western blot (20nM, 0–48hrs). E-cadherin was consistently upregulated when exposed to cabazitaxel for 12–48 hours, and IGFBP-3 was increased in C4-2BDR cells resistant cells. This suggests that cabazitaxel is driving an interconversion between EMT and MET in resistant cells. No significant changes were detected in HSET levels in taxane resistant cells. Bcl-2 expression decreases in response to cabazitaxel treatment (Fig. 2E). By co-immunoprecipitation, an interaction between Bcl-2 and HSET was observed, and was enriched when exposed to cabazitaxel for 6 hours (Fig. 2F). Expression of genes involved in regulation of EMT were comparatively analyzed by qRT-PCR in the two cell lines. Expression of Slug, Twist and Zeb1 were upregulated in C42BDR cells in response to cabazitaxel after 9–24hrs (Fig. 2H–J), but no differences in Snail mRNA (Fig. 2G).
Tracing Genes Involved in Phenotypic Reprogramming and Therapeutic Resistance
To gain mechanistic insights into the phenotypic landscape that underpins therapeutic resistance, we performed an RNAseq on C4-2B and C4-2BDR cells, to determine their differential response to cabazitaxel (20nM, 6–12hrs). Principal component analysis (PCA) reveals that C42B and C4-2BDR cells have distinct gene profiles during treatment (Fig. 3A, Supplementary fig. 4 and 5). The heat maps depict the significant (FDR<0.05) gene signature responses of C4-2B (Fig. 3B), and C4-2BDR (Fig. 3C) to cabazitaxel over time and show the 30 most affected genes (LogFC>2, FDR<0.01), though C4-2B cells exhibited a greater number of gene changes in response to cabazitaxel. Differential expression analysis reveals the largest differences in mRNA expression in resistant cells, and response to cabazitaxel (Fig. 3D and E). Resistant C4-2BDR cells exhibited high expression of CBLN2, ELF5, ELOVL5, FAM155B, GNAQ and GCH1, downregulation of LRG1 and B4GALNT compared to sensitive C4-2B cells, and expression is maintained following cabazitaxel treatment (Fig. 3D). Treatment with cabazitaxel increased expression of WLS and NECTIN3, while expression of SLC16A3, COLEC12, EPHA3 and PDIA2 were reduced compared to C4-2B cells (Fig. 3E). In C4-2B cells, treatment with cabazitaxel, led to enhancement/activation of gene pathways involved in autophagy, cell cycle stress and apoptosis. In the resistant C4-2BDR cells, transcriptomic profiling revealed enrichment of DNA replication and repair pathways in response to taxane. Pathways engaged in stemness, and unfolded protein response were enriched in both sensitive and resistant prostate cancer cells after treatment; however this differential activation was significantly higher in the C4-2BDR resistant cells (Fig. 3F).
Figure 3. RNAseq analysis of differential response of cabazitaxel sensitive and resistant prostate cancer cells.
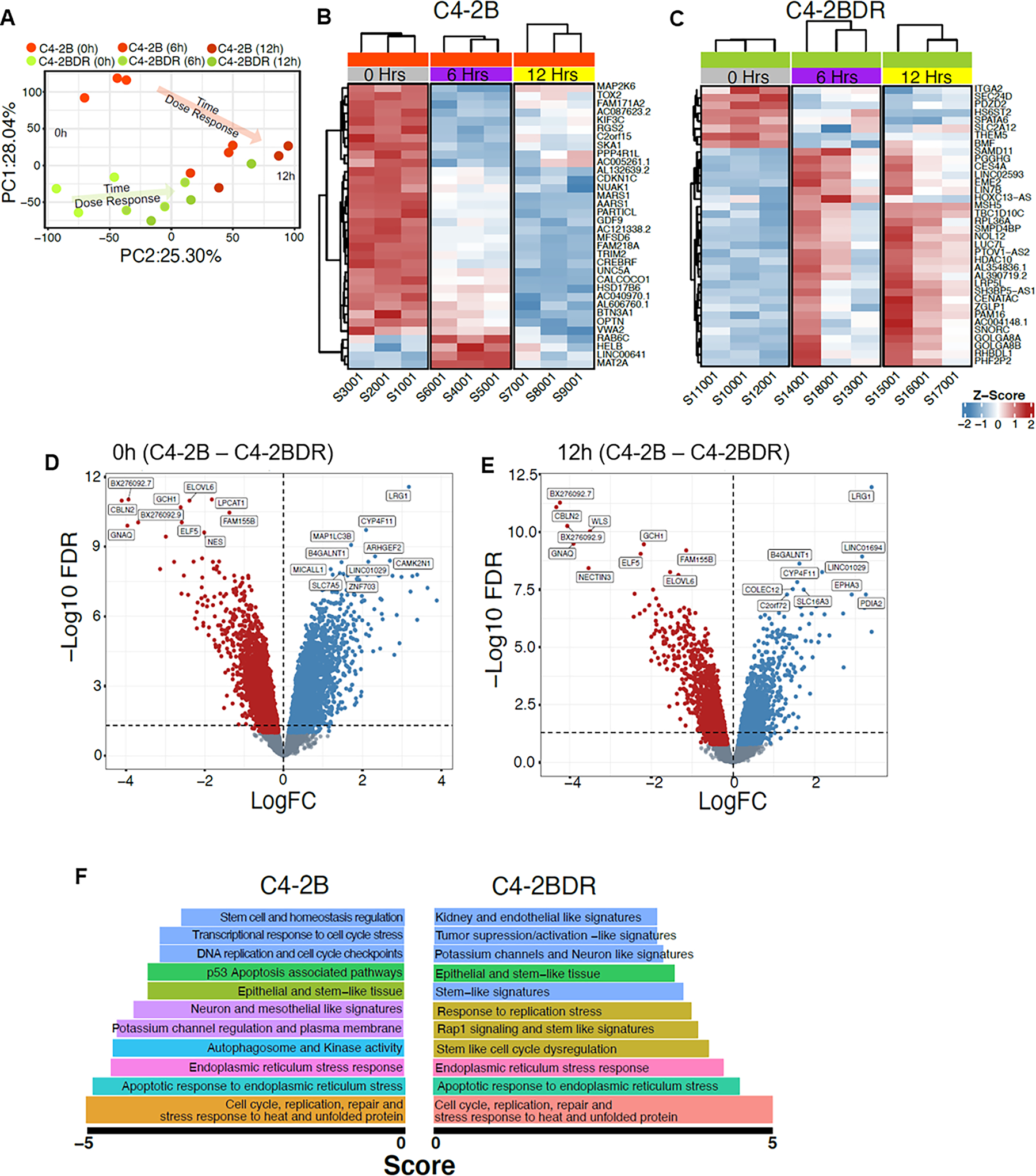
Panel A, PCA analysis between C4-2B and C4-2BDR in response to cabazitaxel (20nM). Panels B-C, heat-map of the unique gene signatures after cabazitaxel treatment of C4-2B and C4-2BDR, respectively. Volcano plot between C4-2B and C4-2BDR at 0hrs (panel D) and 12hrs (panel E) cabazitaxel treatment. Top enriched gene pathways in C4-2B and C4-2BDR cells in response to cabazitaxel (panel F).
To validate the results of RNAseq, qRT-PCR was completed on C4-2B and C4-2BDR cells after treatment with cabazitaxel (24hrs). Consistent with RNAseq, the mRNA expression of CBLN2, NECTIN3 and ELF5 were upregulated in untreated C4-2BDR cells, and this continued through exposure to cabazitaxel (Supplementary fig. 6A, B and F). Expression of MARCKS and TRIM2 was induced by cabazitaxel in C4-2BDR resistant cells lines. (Supplementary fig. 6E and G). LRG1 and KIF5C exhibited reduced expression in C4-2BDR resistant cells compared to C4-2B parental cells, which continued with cabazitaxel treatment (Supplementary fig. 6C and D). This was consistent with RNAseq analysis, however expression of WLS was variable (Supplementary fig. 6H).
Functional loss of SLCO1B3 to underpin therapeutic resistance
Transcriptomic analysis revealed alterations in solute carrier (SLC) family transport proteins associated with therapeutic resistance. Combined expression of SLC family transporters in C4-2B RNAseq analysis revealed a global downregulation of these transporters in resistant C4-2BDR cells, and reduced expression of transporters with treatment with cabazitaxel in both cell lines (Fig. 4A and B). To further investigate the role of SLC transporters in prostate cancer cells, androgen-independent PC3 cells were continually exposed to increasing doses of cabazitaxel to develop PC3-CR cells, which exhibit increase cell viability in response to cabazitaxel compared to PC3 cells (Fig. 4C). RNAseq analysis of these cells revealed downregulation of the SLC family membrane transporter SLCO1B3 in the cabazitaxel-resistant prostate cancer cells compared to the PC3 control cells (Figure 4D). This result was validated using qRT-PCR, and loss of SCLO1B3 in PC3-CR cells at a protein level was demonstrated via Western blot (Fig. 4E and F). To examine the functional contribution of SLCO1B3 to cabazitaxel resistance in PC3 cells, we silenced SLC01B3 expression by shRNA knockdown (a shCONTROL vector and three shSLCO1B3 constructs were selected). Dose response treatment for a period of 96hrs, demonstrated that shSLCO1B3 Clones C and D had greater cell viability in response to increasing doses of cabazitaxel, compared to control and shSLCO1B3-A cells (Fig. 4G). In publicly available clinical prostate cancer database TCGA, SCLO1B3 alterations, which were most commonly shallow deletions or deep deletions, were associated with a decrease in progression free survival, and overall survival (Fig. 4H, Supplementary fig. 8B). Immunohistochemical analysis for SLCO1B3 in MDA PCa PDX specimens from patients with advanced CRPC showed very little staining, except in PDX derived from Patient 4, derived from circulating tumor cells (Supplementary fig. 8A).
Figure 4. Loss of SLCO1B3 as a mediator of cabazitaxel resistance.
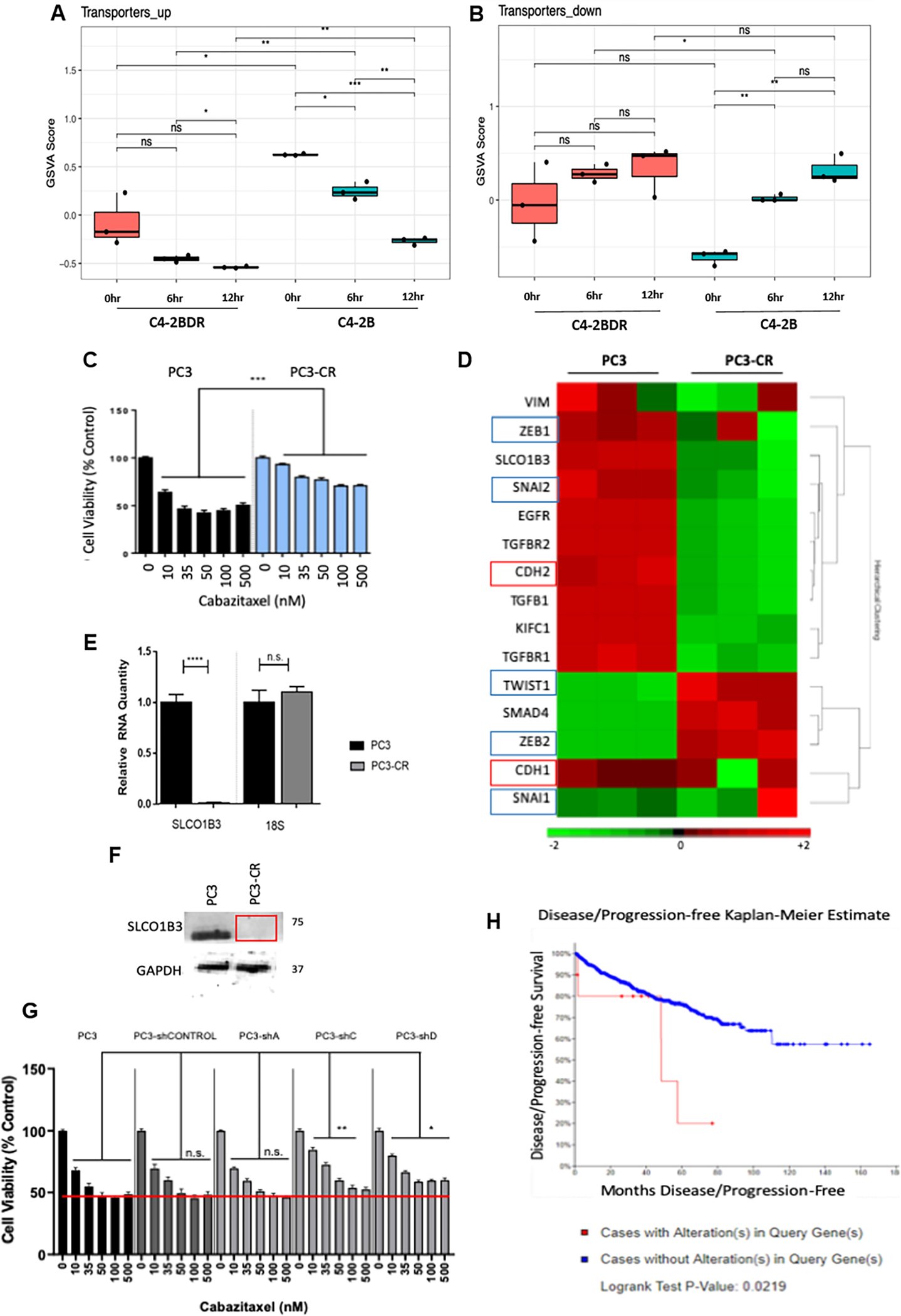
Panels A-B, RNAseq expression signatures of SLC family transporter genes in C4-2B (sensitive) and C4-2BDR (resistant) cells after 6 and 12hrs cabazitaxel treatment. Panel C, Dose response of parental PC3 and cabazitaxel resistant PC3-CR cells to cabazitaxel for 96hrs. Panel D, RNAseq analysis of PC3 and PC3-CR cells. Panels E-F, expression of SLCO1B3 in PC3 and PC3-CR cells via RT-PCR normalized to 18s (panel E), and western blot (panel F, GAPDH as loading control). Panel G, Cell viability analysis of PC3-shSLCO1B3 stable knockdown in dose response to cabazitaxel. Panel H, Kaplan-Meier plot depicts progression free survival of patients with alterations in the SLCO1B3 gene (red) and no alterations (blue) as determined by TCGA database. Statistical significance indicated by *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, students t-test.
Phenotypic Signatures of Lethal Prostate Cancer MDA PCa PDX Models
To investigate the clinical relevance of phenotypic signatures of therapeutic resistance, PDX tumors derived from patients with advanced, treatment resistant prostate cancer were utilized (MDA PCa PDX). These tumors were all derived from patients with advanced metastatic CRPC that had undergone different treatment sequences and regimes. All patients had received cabazitaxel, with the exception of Patient 1 (donor of MDA PCa 183-A), who is treatment naïve. Eight samples were studied, including four samples (MDA PCa 342-A+B, 350-A+B, 355–9, 355–15) derived from one patient longitudinally (Patient 5a, 5b, 5c, 5d). The clinical characteristics of these tumors are summarized in Supplementary Table 2. The tumors were phenotypically profiled for EMT-MET interconversion, based on E-cadherin and vimentin immunohistochemistry. The PDX from Patient sample 1, (treatment naïve), maintained strong expression of the epithelial marker E-cadherin, with minimal vimentin expression. Interestingly, the PDX from Patient 4, who had undergone cabazitaxel treatment following ADT and abiraterone treatment, exhibited strong expression of E-cadherin and weak vimentin staining. This suggests that treatment reverts to an epithelial phenotype (via MET). Conversely, PDX from Patient 2 and all longitudinal samples derived from Patient 5 (5a-d) appeared mesenchymal in phenotype (based on vimentin and E-cadherin immunostaining, Supplementary fig. 9).
Since we previously demonstrated sequencing cabazitaxel treatment after ADT in vivo reduces the expression of HSET in CSPC xenografts (10), we subsequently investigated the expression of HSET in PDX models of therapeutically resistant prostate cancer and the correlation with the sequencing regimen in the patients (with advanced disease) they were derived from. HSET protein expression was detected in all PDX samples with strong nuclear immunoreactivity (Fig. 5A). Models derived from Patients 2 and 3 had the highest level of immunoreactivity (approximately 33%). For PDX from Patient 5, there was variability in HSET expression levels ranging from 15–25% of cells expressing HSET. PDX derived from treatment naïve Patient 1 had low expression of HSET, with 10% positively stained cells. PDX from Patient 4 exhibited the lowest positivity for HSET, only 8% of cells were positively stained (Fig. 5A and B). This may be a result of treatment sequencing, targeting of HSET by cabazitaxel following anti-androgen therapy. However, RNAseq analysis of these tumors revealed variable expression of HSET (KIFC1) at an mRNA level, with no clear correlation of mRNA level with protein expression (Supplementary fig. 10D). In publically available TCGA data sets, alterations (most commonly amplification) was associated with decreased disease free survival and mRNA expression of HSET is higher in prostate cancer tissues compared to benign disease (Fig. 5C, Supplementary fig. 10E). Concurrently, immunohistochemistry of anti-apoptotic Bcl-2 was performed in these specimens. The highest Bcl-2 immunoreactivity was observed in PDX from Patient 3, which was derived from lymph node. Patient 4 and all Patient 5 PDX samples displayed Bcl-2 positivity, while PDX from treatment naive Patient 1, and Patient 2 failed to show any immunoreactivity (Fig. 5A).
Figure 5. Profiling of kinesin and survival protein in lethal prostate cancer MDA PCa PDX models.
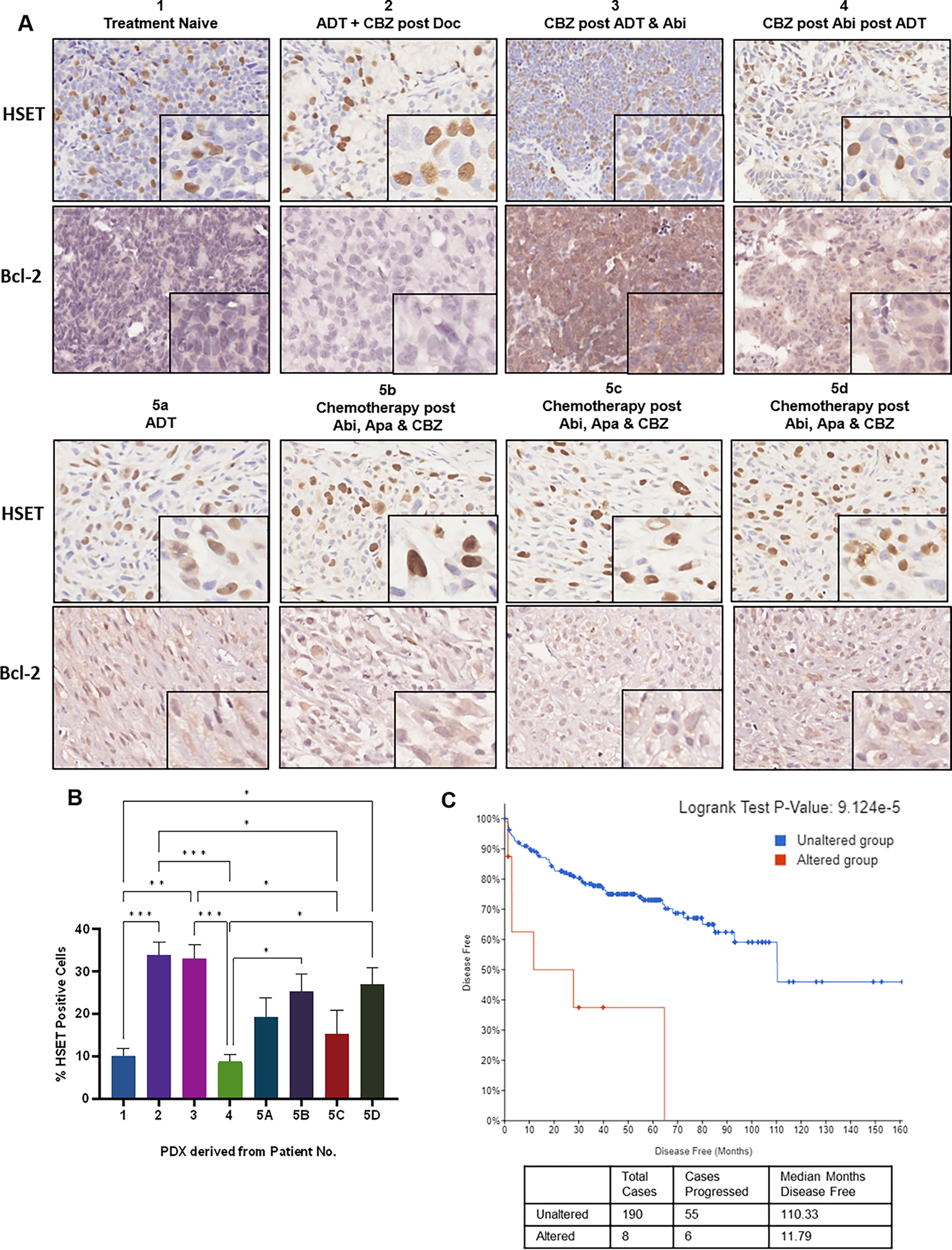
Panel A, PDX prostate cancer specimens were subjected to HSET and Bcl-2 immunohistochemistry, shown by representative images at 20x and 40x magnification. Panel B, Quantification analysis of HSET immunohistochemistry represented as the mean percentage of positive cells (from 6 fields of view), analyzed by two independent observers. Statistical significance indicated by *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, students t-test. Panel C, Kaplan-Meier plot depicts progression free survival of patients with alterations in the HSET gene (red) and no alterations (blue) as determined by TCGA database.
The AR expression status was also assessed by immunohistochemical analysis in serial sections. PDX tumors from two patients exhibited AR positivity, treatment naïve derived tumor specimen exhibited strong AR positivity (80% of cell population), and tumor derived from Patient 4 (PDX developed from circulating tumor cells) had 90% AR positivity. Expression of AR mRNA was assessed in these specimens using RNAseq, PDX from patients 1 and 4 showed expression of AR consistent with the immunohistochemical profile (Supplementary fig. 10A–C).
Targeting Kinesins to overcome resistance
The effect of kinesin inhibition to overcome taxane resistance was interrogated by targeting kinesin proteins. As shown on Figure 6 (A–D), a dose response of C4-2B and C4-2BDR cell lines to kinesin inhibitor ispinesib revealed that during treatment period of 24–96 hrs, the C4-2BDR cells showed resistance to ispinesib in a dose-dependent pattern (5–35nM). Exposure to higher dose resulted in 40% loss of cell viability (vs the untreated controls). The C4-2B cells exhibited high sensitivity to ispinesib with a 60% cell death when treated with ispinesib (5–50nM, 24–96hrs) (Fig. 6A–D). Similar response patterns were observed for other cell lines resistant to enzalutamide or abiraterone (Supplementary fig. 11). Optimal dose of CW069 was determined using dose response for 24–48hrs (Supplementary fig. 12). 5nM of ispinesib was used for subsequent experiments. To investigate the combined targeting of kinesins with cabazitaxel and kinesin inhibition, we used C4-2B, C4-2BDR and 22RV1 prostate cancer cells as cell models, treated with cabazitaxel as monotherapy or combination with kinesin or specific HSET inhibitors ispinesib or CW069 for 24–48hrs. As shown on Figure 6 (panels E and F) the combination of ispinesib or CW069 with cabazitaxel caused a significant induction of cell death than any of the drugs given alone (40% after 48hrs). Similarly, there was significant cell death in resistant C4-2BDR prostate cancer cells upon combination treatment of cabazitaxel with CW069 or ispinesib, compared to cabazitaxel alone (Fig. 6E and F).
Figure 6. Combinational targeting of kinesins to overcome therapeutic resistance in prostate cancer cells.
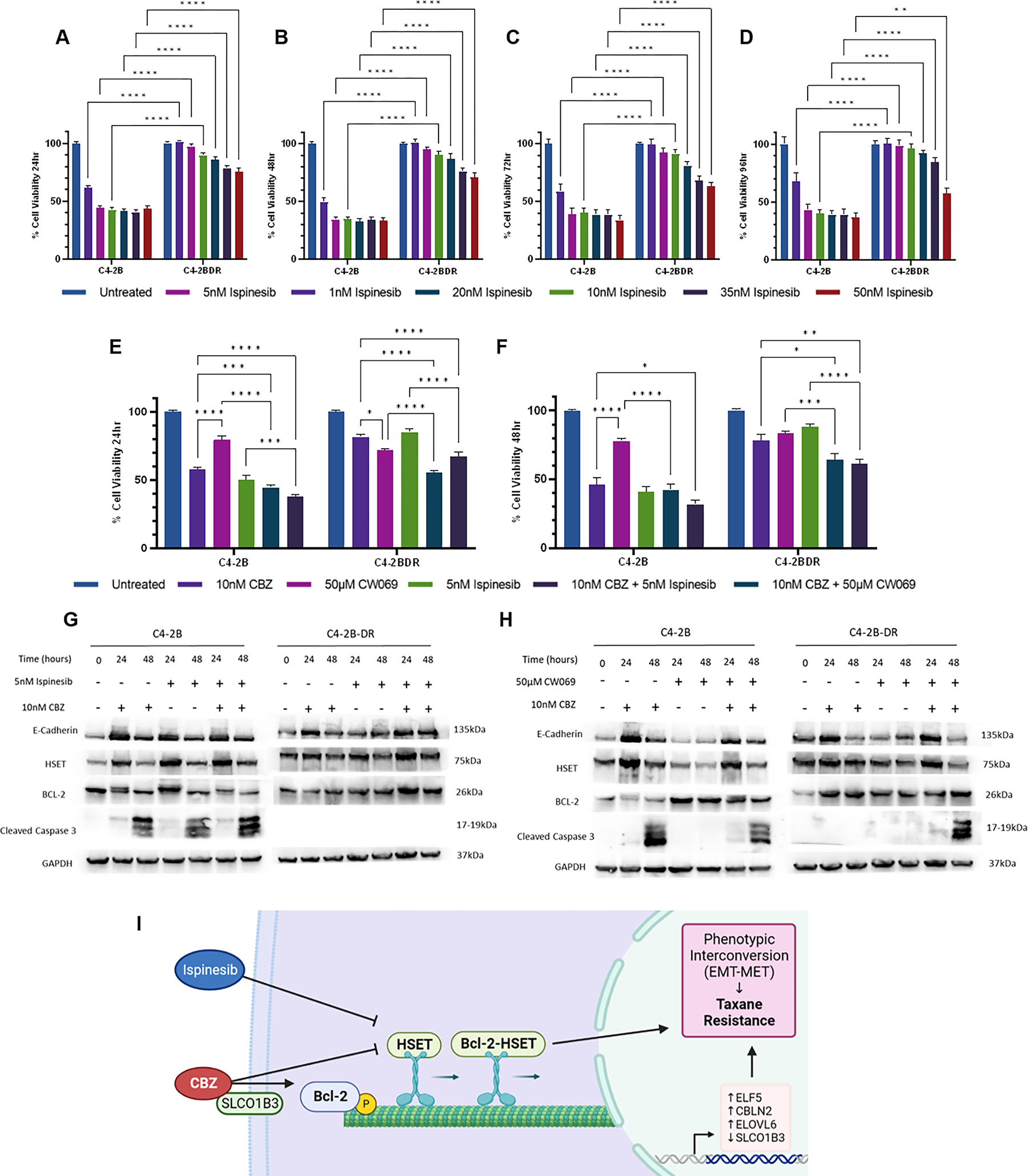
Dose response of cell viability of C4-2B and C4-2BDR prostate cancer cells to kinesin inhibitor ispinesib at 24, 48, 72 and 96hrs, respectively shown on panels A, B, C, and D;. (0–50nM). C4–2b and C4-2BDR cells were treated with kinesin inhibitors CW069 (50μM) or ispinesib (5nM) alone or in combination with cabazitaxel (10nM) for 24 and 48hrs. Viability was determined by MTT assay after 24 (panel E), and 48hrs (panel F). Data presented as mean ± SEM of three independent experiments in triplicate, expressed as percentage of untreated controls. Western blot analysis of EMT, HSET, and apoptotic proteins in C4-2B and C4-2BDR cells following treatment with combinations of CW069 (panel G) or Ispinesib (panel H) and cabazitaxel. GAPDH was used as a loading control. Statistical significance indicated by *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 as determined by two-way ANOVA. Panel I, Schematic illustration revealing that in taxane resistant prostate cancer cells there is upregulation of HSET and Bcl-2 proteins, loss of SLCO1B3 transporter, and increased gene expression of ELF5, CBLN2 and ELOVL6. These key players contribute to the interconversion dynamic between epithelial and mesenchymal phenotypes in response to cabazitaxel. Inhibition of mitotic kinesins (HSET) and survival (Bcl-2) proteins, along with microtubule targeting cabazitaxel provides a potentially powerful platform to overcome therapeutic resistance in CRPC.
Protein profiling of phenotypic effectors (by Western blot) in C4-2B and C4-2BDR cells revealed an increase in expression of E-cadherin in response to cabazitaxel in combination with ispinesib in sensitive cells. HSET expression peaks at 24hrs treatment in C4-2B cells after each treatment; however, in C4-2BDR cells expression remains high. Similarly, expression of anti-apoptotic protein Bcl-2 is high in C4-2BDR cells. In response to ispinesib and cabazitaxel combination treatment, we detected downregulation of Bcl-2 in sensitive, but not in resistant cells (C4-2B-DR) (Fig. 6G, Supplementary fig. 13). C4-2B cells exhibited reduced HSET expression when treated with CW069 alone, confirming specificity of drug targeting. E-cadherin followed a similar pattern in these cells, expression was low with treatment with CW069 but was increased when cabazitaxel was added, this result was also reflected in resistant C4-2BDR cells. HSET expression remained high in C4-2BDR cells, and increased at 24hrs with CW069 in combination with cabazitaxel before reducing at 48hrs. Bcl-2 levels remained consistently high in C4-2BDR cells as well as in sensitive C4-2B cells, in response to CW069 treatment. Caspase activation (cleaved caspase 3) was detected in both cell lines after 48hrs of CW069 in combination with cabazitaxel (Fig. 6H, Supplementary Fig. 14).
We subsequently examined the phenotypic traits of the resistant prostate cancer C4-2BDR cells in response to kinesin targeting by ispinesib and CW069 by qRT-PCR. Expression of EMT and HSET genes were assessed after combination treatment (with cabazitaxel) (6–24hrs). A compensatory increase effect of CW069 is observed in KIFC1 expression in both C4-2BDR and parental cells when combined with cabazitaxel. Similarly, KIFC1 expression is increased in response to ispinesib either as a monotherapy and in combination with cabazitaxel by 24hrs in C4-2B cells, and 12hrs in C4-2BDR. However, this reduced back to baseline by 24hrs (Supplementary fig. 15A and B). An increase in E-cadherin levels was detected in C4-2B cells at 24hrs of treatment with all drug combinations; however, in the resistant cells E-cadherin increase was detected in response to combination of CW069 with cabazitaxel (Supplementary fig. 15C and D). No significant changes were observed in the transient expression of Snail or Slug in response to different treatments in either cell line, and variable expression of RNAseq genes was observed in response to kinesin targeting. (Supplementary fig. 15 and 16).
Targeting HSET-AR Interactions Triggers Phenotypic Reprogramming
We recently showed that cabazitaxel targets the association between the kinesin HSET and the AR, which affects the EMT phenotype (10). To gain mechanistic insights into this interaction, the effect inhibiting HSET on transcriptional regulation of EMT was assessed in LNCaP cells. The results shown on Figure 10, indicate the cells treated with CW069 with and without androgens (DHT) in CSS for 6, 12 and 24hrs had PSA expression induced by DHT, that was significantly downregulated by HSET inhibition by CW069 (Fig. 7A). Expression of KIFC1 was downregulated by CW069, but restored by DHT administration. However, by 24hours expression is reduced in both conditions treated with CW069 (Fig. 7B). E-cadherin expression was markedly increased by CW069 (Fig. 7C). This was associated with expression of Snail, a transcriptional repressor of E-cadherin, that was significantly decreased by HSET inhibitor (CW069), regardless of androgens (Fig. 7D). At a protein level (by western blot), HSET expression was dramatically decreased by CW069. Concurrently, PSA was decreased by HSET inhibition by CW069 regardless of the presence of androgens (DHT, Figure 7E).
Figure 7. Transcriptional regulation of EMT by kinesins and androgens.
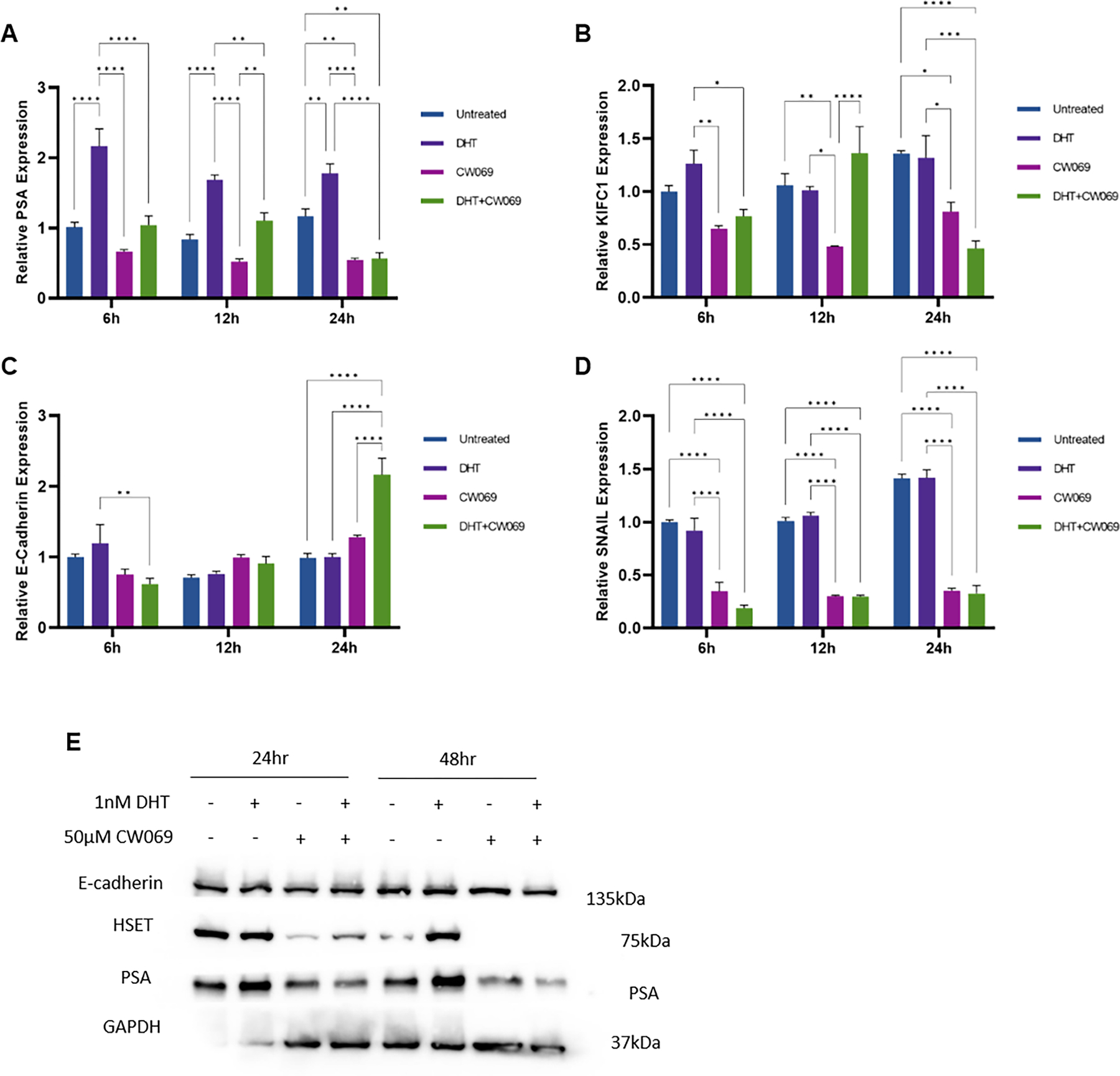
Human prostate cancer cells LNCaP, were treated with 50μM CW069 (HSET inhibitor) in the presence or absence of DHT (1nM) for 6, 12 and 24hrs in CSS. RT-PCR was subsequently performed to evaluate the mRNA levels PSA (panel A), KIFC1 (panel B), E-cadherin (panel C), and SNAIL (panel D) Data presented as average relative expression to untreated controls from three independent experiments performed in triplicate (mean±SEM, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001).
Discussion
The plasticity of prostate tumors contributes to the heterogeneity and acquisition of therapeutic resistance in advanced prostate cancer (26). Previous work from our lab demonstrated that EMT is induced by transforming growth factor beta (TGF-β) and/or androgens, with a threshold AR level determining the phenotypic outcome and invasive properties(27, 28). Moreover, there is clinical evidence that a switch from E- to N-cadherin expression predicts prostate tumor progression, recurrence, and mortality (29), and therapeutic targeting of N-cadherin in CRPC has emerged as a strategy for blocking metastasis (30, 31).
Previous studies by our group established an interaction between the mitotic spindle kinesin protein HSET and AR that that was targetable by sequencing of anti-androgens and cabazitaxel in pre-clinical models of castration sensitive prostate cancer (CSPC), but not CRPC (10). In the present study we provide the first evidence that pharmacologic targeting of kinesins [via HSET specific inhibitor (CW069) and kinesin inhibitor ispinesib] can potentially overcome (therapeutic) taxane resistance to cabazitaxel in cell models of advanced prostate cancer. Evidence on the re-sensitization of docetaxel resistant prostate cancer through combinations of CW069 and docetaxel (13), supports our observations. Significantly enough, limited efficacy has been demonstrated in the Southwest Oncology Group phase II clinical trials of kinesin-targeting ispinesib in the treatment of CRPC with docetaxel resistance despite HSET overexpression associated with resistance (12, 32). Further, our observations in in androgen sensitive human prostate cancer LNCaP cells demonstrate reduced PSA expression following HSET inhibition by CW069 (regardless of the presence of androgens). This evidence supports the use of combinational strategies before taxane resistance emerges to reduce HSET expression, priming prostate cancer cells for vulnerability to cabazitaxel treatment. Sequencing of cabazitaxel after ADT, effectively targets HSET levels and EMT to MET reversion in PDX models (MDA PCa PDX) of lethal prostate cancer (Fig. 5A, Supplementary fig. 8). Ongoing studies investigate the synergistic combination, as well as temporal sequencing strategies of kinesin inhibition to overcome cabazitaxel resistance using pre-clinical in vivo models of advanced prostate cancer.
The transcriptomic profiling of response to cabazitaxel in taxane resistant prostate cancer cells revealed a “global” loss of SLC family transport proteins and cabazitaxel resistant cells exhibited loss of SLC protein SLCO1B3. Functional studies further revealed that silencing SLCO1B3 induced resistance to cabazitaxel. SLCO1B3 plays a role in the influx of taxane chemotherapy into the cells, the loss of SLCO1B3 may lead to less internalization of these drugs, preventing their action on microtubules and thereby mediating tumor recurrence post taxane treatment. Direct support for our findings stems from recent studies (by other investigators) using docetaxel resistant PDX models (33, 34), that also demonstrated increased therapeutic vulnerability to taxane treatment upon SLCO1B3 upregulation (34). The SLCO1B3 transporter is functionally engaged in the transport of other taxanes, docetaxel, into cells (35–37), evidence of clinical significance as in clinical prostate cancer data from the TCGA dataset indicate that deletion/loss of expression of SLCO1B3 is associated with reduced progression-free and overall survival. Mechanistically the SLC family proteins may mediate resistance and progression to CRPC through drug transport and reprogramming of cellular metabolic pathways (38).
Bcl-2 as a determinant of apoptosis evasion is involved in emergence of CRPC (39). Topologically Bcl-2 is associated with microtubules (the primary target for taxane chemotherapy), and becomes functionally inactivated through phosphorylation, resulting in apoptosis (40–42). (Fig. 6I). The sustained high expression of Bcl-2 in C4-2BDR cells may suggest driving role for Bcl-2 in taxane resistance, further an association of Bcl-2 with HSET that is transiently enriched with cabazitaxel treatment was identified here (Fig. 6I). Inhibition of Bcl-2 has demonstrated some clinical success in re-sensitizing tumors to platinum-based therapies and Bcl-2 inhibition by Venetoclax in combination with anti-androgen enzalutamide for treatment of prostate cancer is currently being trialed (42, 43). However, the function of Bcl-2 and kinesins outside of apoptotic suppression as determinants of phenotypic reprogramming, through the association with microtubules and kinesins warrants further mechanistic pursuit.
Transcriptomic profiling of the response to cabazitaxel the docetaxel resistant C4-2B prostate cancer cells revealed that CBLN2 was highly expressed in taxane resistant cells before and after cabazitaxel treatment. CBLN2 is primarily expressed in the cerebellum, and is involved in synaptic cell-adhesion maintenance (44). A role for CBLN2 in cancer development is currently unknown; however, increased CBLN2 expression has been detected in cabazitaxel resistant 22Rv1 xenograft tumors and CRPC clinical samples (45, 46). CBLN2 may be contributing to the increased plasticity pathways in resistant cells, as it has been shown to drive transition of endothelial cells to mesenchymal phenotype through activation of Twist1 through the NF-κB/HIF-1α (47). In addition, our results revealed an upregulation of ELOVL6 associated with taxane resistance in prostate cancer cells. ELOVL family genes play a vital role in fatty acid lipid metabolism, through fatty acid chain elongation, a notable event in cancer cells to meet the metabolic demands of uncontrolled proliferation (48, 49). ELOVL6 is overexpressed in several cancers including breast cancer, lung cancer and hepatocellular cancer (50–52). While the contribution of ELOVL6 to prostate cancer progression has not yet been fully elucidated, other ELOVL family members (particularly ELOVL5 and 7) have been implicated in prostate cancer growth and metastasis, (53–55). Alterations in fatty acid metabolism by EMT regulator TGF-β suggests a relationship between lipid metabolism and phenotypic reprogramming with targeting potential (56, 57). E74-like transcription factor (ELF5) is consistently upregulated in resistant cell lines and retains high expression following cabazitaxel treatment (Fig. 6I). ELF5 is a multifunctional transcription factor that binds AR, expressed by epithelial cells with roles in lineage plasticity determination and EMT (58). In contrast to our findings, there are reports that ELF5 is a repressor of EMT through inhibition of TGF-β signaling by phosphorylation of SMAD3 (59). In this mechanistic context however one must also consider the temporal nature of the interconversion between EMT-MET phenotypes. Further, loss of ELF5 expression is associated with prostate cancer progression and enzalutamide resistance, providing a novel functional pursuit of ELF5 in preventing progression to therapeutic resistance (58). There were no changes in genes involved in lineage transitions between basal, luminal and neuroendocrine phenotypes (TP53/RB1, JAK-STAT and SOX2) (60).
Epigenetic reprogramming is responsible for silencing tumor-suppressor genes, activating oncogenic drivers and reprogramming of the cistrome of critical transcription factors in prostate cancer as the AR (61, 62). The present study identified kinesins and the SLC family transporters, as new actionable targets that call for further validation. Interrogating the chromatin landscape using ATAC-seq in a cohort of clinical prostate specimens (pre and post ADT) will provide an informative platform to study the contribution of epigenetic reprogramming to the lethal phenotype and guide clinical-decision making regarding therapeutic intervention and survival outcomes in patients with advanced disease. This work supports the use of combinational strategies of cabazitaxel with targeted inhibitors to mitotic kinesins (HSET) to overcome therapeutic resistance via phenotypic reprogramming within the TME in advanced prostate cancer.
Supplementary Material
Implications:
Our findings are of translational significance in identifying kinesin as a novel target of cross-resistance, towards enhancing therapeutic vulnerability and improved clinical outcomes in patients with advanced prostate cancer.
Acknowledgements:
We would like to acknowledge Yao Shen (Tisch Cancer Institute) for technical assistance with the immunohistochemical analysis and mouse xenograft studies, and Dr. Goutam Chakraborty (Department of Urology, Mount Sinai) for scientific discussions. Figure 6, panel I, was created using Biorender.com
Funding:
This work was supported by grant from the National Institutes of Health/NCI, R01 CA232574 (NK), R21 AG078848 (ND), P20 CA264076 (ND). Prostate Cancer Foundation, NCI Cancer Center Support Grant (P30CA16672), Cancer Center Prostate Cancer SPORE (NIH/NCI P50 CA140388), David H. Koch Center for Applied Research in Genitourinary Cancers at MD Anderson (Houston, TX), and NIH/NCI U01 CA224044 (NN).
Abbreviations
- ADT
androgen-deprivation therapy
- AR
androgen receptor
- ARSI
androgen receptor signaling inhibitor
- Bcl-2
B cell lymphoma-2
- CBZ
cabazitaxel
- CRPC
castration resistant prostate cancer
- CSPC
castration sensitive prostate cancer
- CSS
charcoal stripped serum
- DAB
3’3-diaminobenzidine
- DHT
Dihydrotestosterone
- DDR
DNA damage response
- DMSO
dimethyl sulfoxide
- EMT
epithelial-mesenchymal transition
- IGFBP-3
Insulin-like growth factor binding protein 3
- MET
mesenchymal-epithelial transition
- Enz
enzalutamide
- FBS
fetal bovine serum
- HSET
kinesin family member C1 (KIFC1)
- mCRPC
metastatic castration-resistant prostate cancer
- MET
mesenchymal-epithelial transition
- MTT
Thiazolyl Blue Tetrazolium bromide
- PCa
prostate cancer
- PDX
patient derived xenografts
- PSA
Prostate specific antigen
- SDS
sodium dodecyl sulphate
- PVDF
polyvinylidene fluoride
- RPMI
Roswell park memorial institute
- TGF-β
Transforming growth factor beta
- TME
Tumor microenvironment
- SLCO1B3
Solute carrier organic anion transporter family member 1B3
Footnotes
Conflict of Interest: The authors declare no potential conflicts of interest
References
- 1.Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. 2023;73(1):17–48. [DOI] [PubMed] [Google Scholar]
- 2.O’Donnell A, Judson I, Dowsett M, Raynaud F, Dearnaley D, Mason M, et al. Hormonal impact of the 17alpha-hydroxylase/C(17,20)-lyase inhibitor abiraterone acetate (CB7630) in patients with prostate cancer. British journal of cancer. 2004;90(12):2317–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science (New York, NY). 2009;324(5928):787–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. The New England journal of medicine. 2012;367(13):1187–97. [DOI] [PubMed] [Google Scholar]
- 5.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. The New England journal of medicine. 2011;364(21):1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. The New England journal of medicine. 2020;382(22):2091–102. [DOI] [PubMed] [Google Scholar]
- 7.Huizing MT, Misser VH, Pieters RC, ten Bokkel Huinink WW, Veenhof CH, Vermorken JB, et al. Taxanes: a new class of antitumor agents. Cancer investigation. 1995;13(4):381–404. [DOI] [PubMed] [Google Scholar]
- 8.de Bono JS, Oudard S, Ozguroglu M, Hansen S, Machiels JP, Kocak I, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet (London, England). 2010;376(9747):1147–54. [DOI] [PubMed] [Google Scholar]
- 9.Zhu ML, Horbinski CM, Garzotto M, Qian DZ, Beer TM, Kyprianou N. Tubulin-targeting chemotherapy impairs androgen receptor activity in prostate cancer. Cancer research. 2010;70(20):7992–8002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Begemann D, Wang Y, Yang W, Kyprianou N. Androgens modify therapeutic response to cabazitaxel in models of advanced prostate cancer. The Prostate. 2020;80(12):926–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin SK, Pu H, Penticuff JC, Cao Z, Horbinski C, Kyprianou N. Multinucleation and Mesenchymal-to-Epithelial Transition Alleviate Resistance to Combined Cabazitaxel and Antiandrogen Therapy in Advanced Prostate Cancer. Cancer research. 2016;76(4):912–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sekino Y, Oue N, Shigematsu Y, Ishikawa A, Sakamoto N, Sentani K, et al. KIFC1 induces resistance to docetaxel and is associated with survival of patients with prostate cancer. Urologic oncology. 2017;35(1):31.e13–31.e20. [DOI] [PubMed] [Google Scholar]
- 13.Sekino Y, Oue N, Koike Y, Shigematsu Y, Sakamoto N, Sentani K, et al. KIFC1 Inhibitor CW069 Induces Apoptosis and Reverses Resistance to Docetaxel in Prostate Cancer. Journal of clinical medicine. 2019;8(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nanda JS, Koganti P, Perri G, Ellis L. Phenotypic Plasticity - Alternate Transcriptional Programs Driving Treatment Resistant Prostate Cancer. Critical reviews in oncogenesis. 2022;27(1):45–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Current opinion in cell biology. 2005;17(5):548–58. [DOI] [PubMed] [Google Scholar]
- 16.Natsuizaka M, Ohashi S, Wong GS, Ahmadi A, Kalman RA, Budo D, et al. Insulin-like growth factor-binding protein-3 promotes transforming growth factor-{beta}1-mediated epithelial-to-mesenchymal transition and motility in transformed human esophageal cells. Carcinogenesis. 2010;31(8):1344–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stylianou N, Lehman ML, Wang C, Fard AT, Rockstroh A, Fazli L, et al. A molecular portrait of epithelial–mesenchymal plasticity in prostate cancer associated with clinical outcome. Oncogene. 2019;38(7):913–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dudas J, Ladanyi A, Ingruber J, Steinbichler TB, Riechelmann H. Epithelial to Mesenchymal Transition: A Mechanism that Fuels Cancer Radio/Chemoresistance. Cells. 2020;9(2):428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moreno-Bueno G, Portillo F, Cano A. Transcriptional regulation of cell polarity in EMT and cancer. Oncogene. 2008;27(55):6958–69. [DOI] [PubMed] [Google Scholar]
- 20.Liu C, Lou W, Zhu Y, Nadiminty N, Schwartz CT, Evans CP, et al. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20(12):3198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu Y, Liu C, Nadiminty N, Lou W, Tummala R, Evans CP, et al. Inhibition of ABCB1 expression overcomes acquired docetaxel resistance in prostate cancer. Molecular cancer therapeutics. 2013;12(9):1829–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sakamoto S, McCann RO, Dhir R, Kyprianou N. Talin1 promotes tumor invasion and metastasis via focal adhesion signaling and anoikis resistance. Cancer research. 2010;70(5):1885–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palanisamy N, Yang J, Shepherd PDA, Li-Ning-Tapia EM, Labanca E, Manyam GC, et al. The MD Anderson Prostate Cancer Patient-derived Xenograft Series (MDA PCa PDX) Captures the Molecular Landscape of Prostate Cancer and Facilitates Marker-driven Therapy Development. Clinical cancer research : an official journal of the American Association for Cancer Research. 2020;26(18):4933–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murtagh F, Legendre P. Ward’s Hierarchical Agglomerative Clustering Method: Which Algorithms Implement Ward’s Criterion? Journal of Classification. 2014;31(3):274–95. [Google Scholar]
- 25.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. 1995;57(1):289–300. [Google Scholar]
- 26.Livas L, Hasani S, Kyprianou N. Integrated Therapeutic Targeting of the Prostate Tumor Microenvironment. Advances in experimental medicine and biology. 2020;1296:183–98. [DOI] [PubMed] [Google Scholar]
- 27.Zhu M, and Kyprianou N. Role of androgens and the androgen receptor in epithelial-mesenchymal transition and invasion of prostate cancer cells. FASEB J. 2010;24(3):769–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh A, & Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gravdal K, Halvosten OJ., Haukaas SA., & Akslen LA. A switch from E-cadherin to N-cadherin expression indicates epithelial to mesenchymal transition and is of strong and independent importance for the progression of prostate cancer. Clin Cancer Res. 2007;13:7003–11. [DOI] [PubMed] [Google Scholar]
- 30.Tanaka H, Kono E., Tran CP., Miyazaki H., Yamashiro J., Shimomura T., et al. Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nat Med. 2010;16:1414–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jennbacken K, Tesan T., Wang W., Gustavsson H., Damber JE., Welen K. N-cadherin increases after androgen deprivation and is associated with metastasis in prostate cancer. Endocr Relat Cancer. 2010;17:469–79. [DOI] [PubMed] [Google Scholar]
- 32.Beer TM, Goldman B, Synold TW, Ryan CW, Vasist LS, Van Veldhuizen PJ, Jr., et al. Southwest Oncology Group phase II study of ispinesib in androgen-independent prostate cancer previously treated with taxanes. Clinical genitourinary cancer. 2008;6(2):103–9. [DOI] [PubMed] [Google Scholar]
- 33.de Morrée E, van Soest R, Aghai A, de Ridder C, de Bruijn P, Ghobadi Moghaddam-Helmantel I, et al. Understanding taxanes in prostate cancer; importance of intratumoral drug accumulation. The Prostate. 2016;76(10):927–36. [DOI] [PubMed] [Google Scholar]
- 34.de Morrée ES, Böttcher R, van Soest RJ, Aghai A, de Ridder CM, Gibson AA, et al. Loss of SLCO1B3 drives taxane resistance in prostate cancer. British journal of cancer. 2016;115(6):674–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ellen Morree RvS, Ashraf Aghai, Corrina de Ridder, Peter de Bruijn, Inge Ghobadi, Herman Burger, Ron Mathijssen, Erik Weimer, Ronald de Wit, Wytske van Weerden. Understanding Taxanes in prostate Cancer; Importance of Intratumoral Drug Accumulation. The Prostate. 2016;76:927–36. [DOI] [PubMed] [Google Scholar]
- 36.Mout L, Wit Rd, Sturrman D, Verhoef E, Mathijssen R, Ridder Cd, et al. Testosterone Diminishes Cabazitaxel Efficacy and Intratumoral Accumulation in a Prostate Cancer Xenograft Model. EBioMedicine. 2018;27:182–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Soest RJ, van Royen ME, de Morrée ES, Moll JM, Teubel W, Wiemer EAC, et al. Cross-resistance between taxanes and new hormonal agents abiraterone and enzalutamide may affect drug sequence choices in metastatic castration-resistant prostate cancer. European Journal of Cancer. 2013;49(18):3821–30. [DOI] [PubMed] [Google Scholar]
- 38.Kushwaha PP, Verma SS, Shankar E, Lin S, Gupta S. Role of solute carrier transporters SLC25A17 and SLC27A6 in acquired resistance to enzalutamide in castration-resistant prostate cancer. 2022;61(4):397–407. [DOI] [PubMed] [Google Scholar]
- 39.McDonnell TJ, Troncoso P, Brisbay SM, Logothetis C, Chung LW, Hsieh JT, et al. Expression of the protooncogene bcl-2 in the prostate and its association with emergence of androgen-independent prostate cancer. Cancer research. 1992;52(24):6940–4. [PubMed] [Google Scholar]
- 40.Haldar S, Basu A, Croce CM. Bcl2 is the guardian of microtubule integrity. Cancer research. 1997;57(2):229–33. [PubMed] [Google Scholar]
- 41.Scatena CD, Stewart ZA, Mays D, Tang LJ, Keefer CJ, Leach SD, et al. Mitotic phosphorylation of Bcl-2 during normal cell cycle progression and Taxol-induced growth arrest. The Journal of biological chemistry. 1998;273(46):30777–84. [DOI] [PubMed] [Google Scholar]
- 42.Ruiz de Porras V, Wang XC, Palomero L, Marin-Aguilera M, Solé-Blanch C, Indacochea A, et al. Taxane-induced Attenuation of the CXCR2/BCL-2 Axis Sensitizes Prostate Cancer to Platinum-based Treatment. European urology. 2021;79(6):722–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pilling AB, Hwang C. Targeting prosurvival BCL2 signaling through Akt blockade sensitizes castration-resistant prostate cancer cells to enzalutamide. The Prostate. 2019;79(11):1347–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seigneur E, Wang J, Dai J, Polepalli J, Südhof TC. Cerebellin-2 regulates a serotonergic dorsal raphe circuit that controls compulsive behaviors. Molecular Psychiatry. 2021;26(12):7509–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yun SJ, Kim SK, Kim J, Cha EJ, Kim JS, Kim SJ, et al. Transcriptomic features of primary prostate cancer and their prognostic relevance to castration-resistant prostate cancer. Oncotarget. 2017;8(70):114845–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ylitalo EB, Thysell E, Thellenberg-Karlsson C, Lundholm M, Widmark A, Bergh A, et al. Marked response to cabazitaxel in prostate cancer xenografts expressing androgen receptor variant 7 and reversion of acquired resistance by anti-androgens. 2020;80(2):214–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang EL, Zhang J-J, Luo F-M, Fu M-Y, Li D, Peng J, et al. Cerebellin-2 promotes endothelial-mesenchymal transition in hypoxic pulmonary hypertension rats by activating NF-κB/HIF-1α/Twist1 pathway. Life Sciences. 2023;328:121879. [DOI] [PubMed] [Google Scholar]
- 48.Nguyen PL, Ma J, Chavarro JE, Freedman ML, Lis R, Fedele G, et al. Fatty acid synthase polymorphisms, tumor expression, body mass index, prostate cancer risk, and survival. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28(25):3958–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hanahan D Hallmarks of Cancer: New Dimensions. Cancer Discovery. 2022;12(1):31–46. [DOI] [PubMed] [Google Scholar]
- 50.Su Y-C, Feng Y-H, Wu H-T, Huang Y-S, Tung C-L, Wu P, et al. Elovl6 is a negative clinical predictor for liver cancer and knockdown of Elovl6 reduces murine liver cancer progression. Scientific Reports. 2018;8(1):6586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Feng YH, Chen WY, Kuo YH, Tung CL, Tsao CJ, Shiau AL, et al. Elovl6 is a poor prognostic predictor in breast cancer. Oncology letters. 2016;12(1):207–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marien E, Meister M, Muley T, Gomez Del Pulgar T, Derua R, Spraggins JM, et al. Phospholipid profiling identifies acyl chain elongation as a ubiquitous trait and potential target for the treatment of lung squamous cell carcinoma. Oncotarget. 2016;7(11):12582–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Centenera MM, Scott JS, Machiels J, Nassar ZD, Miller DC, Zinonos I, et al. ELOVL5 Is a Critical and Targetable Fatty Acid Elongase in Prostate Cancer. Cancer research. 2021;81(7):1704–18. [DOI] [PubMed] [Google Scholar]
- 54.Tolkach Y, Merseburger A, Herrmann T, Kuczyk M, Serth J, Imkamp F. Signatures of Adverse Pathological Features, Androgen Insensitivity and Metastatic Potential in Prostate Cancer. Anticancer research. 2015;35(10):5443–51. [PubMed] [Google Scholar]
- 55.Tamura K, Makino A, Hullin-Matsuda F, Kobayashi T, Furihata M, Chung S, et al. Novel lipogenic enzyme ELOVL7 is involved in prostate cancer growth through saturated long-chain fatty acid metabolism. Cancer research. 2009;69(20):8133–40. [DOI] [PubMed] [Google Scholar]
- 56.Jiang L, Xiao L, Sugiura H, Huang X, Ali A, Kuro-o M, et al. Metabolic reprogramming during TGFβ1-induced epithelial-to-mesenchymal transition. Oncogene. 2015;34(30):3908–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kang H, Kim H, Lee S, Youn H, Youn B. Role of Metabolic Reprogramming in Epithelial⁻Mesenchymal Transition (EMT). International journal of molecular sciences. 2019;20(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li K, Guo Y, Yang X, Zhang Z, Zhang C, Xu Y. ELF5-Mediated AR Activation Regulates Prostate Cancer Progression. Sci Rep. 2017;7:42759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yao B, Zhao J, Li Y, Li H, Hu Z, Pan P, et al. Elf5 inhibits TGF-β-driven epithelial-mesenchymal transition in prostate cancer by repressing SMAD3 activation. 2015;75(8):872–82. [DOI] [PubMed] [Google Scholar]
- 60.Deng S, Wang C, Wang Y, Xu Y, Li X, Johnson NA, et al. Ectopic JAK-STAT activation enables the transition to a stem-like and multilineage state conferring AR-targeted therapy resistance. Nature cancer. 2022;3(9):1071–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pomerantz MM, Qiu X, Zhu Y, Takeda DY, Pan W, Baca SC, et al. Prostate cancer reactivates developmental epigenomic programs during metastatic progression. Nature genetics. 2020;52(8):790–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Davies A, Nouruzi S, Ganguli D, Namekawa T, Thaper D, Linder S, et al. An androgen receptor switch underlies lineage infidelity in treatment-resistant prostate cancer. Nature cell biology. 2021;23(9):1023–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data processing and analysis scripts are deposited at https://github.com/eegk and available upon request. All other relevant data that support the conclusions of the study are within the supplementary material or available from the authors upon request.