

This randomized clinical trial investigates the effects of solanezumab and gantenerumab on downstream biomarkers in individuals with dominantly inherited Alzheimer disease.
Key Points
Question
How do antiamyloid agents affect downstream biomarkers of Alzheimer-related pathophysiology regarding their target engagement with either soluble (solanezumab) or fibrillar (gantenerumab) amyloid?
Findings
This phase 2/3 double-blind, placebo-controlled, randomized clinical trial including 142 participants investigated gantenerumab and solanezumab in individuals with gene variants for dominantly inherited Alzheimer disease. Gantenerumab decreased cerebrospinal fluid (CSF) neurogranin and plasma glial fibrillary acidic protein levels while increasing CSF levels of soluble triggering receptor expressed on myeloid cells 2; in contrast, solanezumab treatment was associated with increased CSF neurofilament light protein levels.
Meaning
Antiamyloid agents removing fibrillar amyloid plaques demonstrated effects on glial and postsynaptic fluid biomarkers downstream of initial amyloid deposition, whereas binding soluble amyloid-β was associated with increased measures of neurodegeneration.
Abstract
Importance
Effects of antiamyloid agents, targeting either fibrillar or soluble monomeric amyloid peptides, on downstream biomarkers in cerebrospinal fluid (CSF) and plasma are largely unknown in dominantly inherited Alzheimer disease (DIAD).
Objective
To investigate longitudinal biomarker changes of synaptic dysfunction, neuroinflammation, and neurodegeneration in individuals with DIAD who are receiving antiamyloid treatment.
Design, Setting, and Participants
From 2012 to 2019, the Dominantly Inherited Alzheimer Network Trial Unit (DIAN-TU-001) study, a double-blind, placebo-controlled, randomized clinical trial, investigated gantenerumab and solanezumab in DIAD. Carriers of gene variants were assigned 3:1 to either drug or placebo. The present analysis was conducted from April to June 2023. DIAN-TU-001 spans 25 study sites in 7 countries. Biofluids and neuroimaging from carriers of DIAD gene variants in the gantenerumab, solanezumab, and placebo groups were analyzed.
Interventions
In 2016, initial dosing of gantenerumab, 225 mg (subcutaneously every 4 weeks) was increased every 8 weeks up to 1200 mg. In 2017, initial dosing of solanezumab, 400 mg (intravenously every 4 weeks) was increased up to 1600 mg every 4 weeks.
Main Outcomes and Measures
Longitudinal changes in CSF levels of neurogranin, soluble triggering receptor expressed on myeloid cells 2 (sTREM2), chitinase 3–like 1 protein (YKL-40), glial fibrillary acidic protein (GFAP), neurofilament light protein (NfL), and plasma levels of GFAP and NfL.
Results
Of 236 eligible participants screened, 43 were excluded. A total of 142 participants (mean [SD] age, 44 [10] years; 72 female [51%]) were included in the study (gantenerumab, 52 [37%]; solanezumab, 50 [35%]; placebo, 40 [28%]). Relative to placebo, gantenerumab significantly reduced CSF neurogranin level at year 4 (mean [SD] β = −242.43 [48.04] pg/mL; P < .001); reduced plasma GFAP level at year 1 (mean [SD] β = −0.02 [0.01] ng/mL; P = .02), year 2 (mean [SD] β = −0.03 [0.01] ng/mL; P = .002), and year 4 (mean [SD] β = −0.06 [0.02] ng/mL; P < .001); and increased CSF sTREM2 level at year 2 (mean [SD] β = 1.12 [0.43] ng/mL; P = .01) and year 4 (mean [SD] β = 1.06 [0.52] ng/mL; P = .04). Solanezumab significantly increased CSF NfL (log) at year 4 (mean [SD] β = 0.14 [0.06]; P = .02). Correlation analysis for rates of change found stronger correlations between CSF markers and fluid markers with Pittsburgh compound B positron emission tomography for solanezumab and placebo.
Conclusions and Relevance
This randomized clinical trial supports the importance of fibrillar amyloid reduction in multiple AD-related processes of neuroinflammation and neurodegeneration in CSF and plasma in DIAD. Additional studies of antiaggregated amyloid therapies in sporadic AD and DIAD are needed to determine the utility of nonamyloid biomarkers in determining disease modification.
Trial Registration
ClinicalTrials.gov Identifier: NCT04623242
Introduction
Alzheimer disease (AD) is characterized by progressive neuropathological changes years before clinical symptoms emerge. Pathophysiological hallmarks are the accumulation and aggregation of extracellular amyloid-β (Aβ), intracellular neurofibrillary tangles composed of hyperphosphorylated tau, neuroinflammation, synaptic toxicity, and neuronal death.1,2,3 Dominantly inherited AD (DIAD) is caused by variants in APP, PSEN1, or PSEN2 genes, with carriers developing cognitive impairment at a predictable, young age.4
The phase 2/3 placebo-controlled, double-blind, randomized clinical trial, the Dominantly Inherited Alzheimer Network Trial Unit (DIAN-TU-001), investigated 2 monoclonal immunoglobulin G1 antibodies against amyloid: gantenerumab targets Aβ fibrils, initiating plaque removal via fragment crystallizable (Fc) γ-receptor–mediated activation of microglial phagocytosis,5 and solanezumab binds to soluble forms of Aβ, thereby potentially ameliorating their synaptic toxicity.6,7,8 Although clear clinical benefits were not identified, target engagement was successful, showing a dose-dependent reduction in amyloid positron emission tomography (PET) burden with gantenerumab and significant increases of cerebrospinal fluid (CSF) Aβ42 for solanezumab.9
However, the effect on emerging markers of AD-related pathophysiology has not been sufficiently investigated. Neurogranin is a postsynaptic protein and considered a soluble marker of synaptic integrity due to its involvement in memory function and synaptic plasticity, both showing early impairment in AD.10 Glial fibrillary acidic protein (GFAP), chitinase 3–like protein 1 (YKL-40), and soluble triggering receptor expressed on myeloid cells 2 (sTREM2) are further biomarkers of interest reflecting neuroinflammatory processes of astrocytes and microglia,11,12,13,14 whereas neurofilament light protein (NfL) is a nonspecific marker of axonal degeneration in AD.15
Although the magnitude of clinical benefit when targeting Aβ in symptomatic AD is debated,16,17 recent trials have demonstrated a slowing of clinical decline in sporadic AD (sAD) with antiamyloid treatment, leading to traditional regulatory approval of lecanemab18 (US Food and Drug Administration news release July 2023). Considering the successful target engagement for both interventions in the DIAN-TU-001 trial, we investigated the effect of each drug on markers of AD-related pathology, in the context of their distinct mechanisms of action on respective forms of amyloid, and stage of disease by exploring longitudinal effects of gantenerumab and solanezumab on CSF and plasma levels of neurogranin, sTREM2, YKL-40, GFAP, and NfL.
Methods
Trial Design and Participants
The DIAN-TU-001 study ran as a double-blind, placebo-controlled, phase 3 randomized clinical trial from December 2012 until November 2019, spanning 25 sites in 7 countries (Supplement 1 and Supplement 2). It was approved by the Washington University Human Research Protection Office and local institutional review boards at each participating site. Eligible participants, after providing written informed consent, were tested for the presence of a DIAD gene variant via polymerase chain reaction–based amplification and subsequent Sanger sequencing. Baseline clinical status was determined using the Clinical Dementia Rating (CDR [Knight ADRC]) dementia staging instrument,19 grouping participants into cognitively unimpaired (CDR 0), very mild dementia (CDR 0.5), or mild dementia (CDR 1). Drug administration spanned 4 years, allocating participants 3:1 to either drug or placebo, with a midtrial increase to a maximal dosage of 1200 mg for gantenerumab and 1600 mg for solanezumab.9 Further details can be found in the original publication.20 Race and ethnicity information was collected from the participants through self-report; categories included Asian, Black, multiracial/other, and White. This study followed the Consolidated Standards of Reporting Trials (CONSORT) reporting guidelines.
Sample Collection and Fluid Biomarker Analysis
CSF samples were collected and processed as previously described,20 undergoing 2 freeze-thaw cycles before analysis. With limited availability of samples, analysis was restricted to relevant downstream biomarkers of AD-related pathology. When CSF and plasma were available, both were measured. Plasma samples were collected at baseline, along with CSF, and at years 1, 2, and 4. EDTA tubes were centrifuged at 3000g for 10 minutes at 4 °C and subsequently flash frozen in 1-mL aliquots for storage at −80 °C. CSF and plasma biomarkers were measured by the Roche NeuroToolKit (NTK), a portfolio of robust prototype assays, running on the fully automated Elecsys platform (Roche Diagnostics).21 Immunoassays for neurogranin, GFAP, sTREM2, YKL-40, and NfL were performed on the cobas e411 and e601 platforms (Roche Diagnostics) by individuals blinded to mutation and treatment status. Of note, these analyses were distinct from immunoassays previously reported.20
Neuroimaging
Study participants underwent carbon 11 Pittsburgh compound B (PiB) PET for amyloid imaging, magnetic resonance imaging (MRI) for structural and safety measures, and [18F]-fluorodeoxyglucose (FDG) PET for metabolic imaging at each time point of CSF collection. Neuroimaging protocols are detailed in the original publication.20
Statistical Analysis
Treatment effects in each outcome were assessed in the modified intention-to-treat (mITT) population, including all randomized participants who received at least 1 treatment dose and had baseline and postbaseline assessments of the primary efficacy measurements. Within the mITT population, subgroups were created based on baseline CDR Global scores: asymptomatic (CDR = 0) and symptomatic (CDR >0) populations. However, the original trial was not powered for subgroup analyses nor for post hoc biomarker analyses, with no formal sample size calculations conducted. Mixed models for repeated measures (MMRM) estimated treatment effects for each outcome within the entire mITT population and the asymptomatic and symptomatic subpopulation.
For the whole mITT population, MMRM analyses included fixed effects of baseline value, treatment, visit, and the interaction between treatment and visit. For asymptomatic and symptomatic subpopulations, MMRM analysis included additional fixed effects: baseline value, baseline status (asymptomatic vs symptomatic), treatment, visit, and various interactions involving these variables in order to estimate the change over time for each subpopulation (including baseline value × baseline status, treatment × visit, baseline status × treatment, and baseline status × treatment × visit). The model estimated least-squares mean changes from baseline to each postbaseline visit, their differences, and 95% CIs.
To examine correlations for rates of change in each outcome, individual rates of change were calculated using the least-squares mean method, and pairwise Spearman correlations were reported. Plasma and CSF NfL levels were log transformed following a previous convention, and a sensitivity analysis was conducted to exclude 1 extreme value (above 3 SD) identified in the gantenerumab arm.
All analyses were conducted with SAS, version 9.4 (SAS Institute). As post hoc analyses, these results are primarily descriptive, and their interpretation should focus on clinical relevance. With this and due to small sample sizes, no multiple comparison adjustments were made, and only nominal P values are presented from 2-sided t tests with type I error of .05 and 95% CI. P values <.05 were considered statistically significant.
Results
Baseline Demographics
Baseline characteristics are displayed in Table 1. Of 236 eligible participants screened, 43 were excluded. A total of 142 participants (mean [SD] age, 44 [10] years; 72 female [51%]; 70 male [49%]) were included in the study (gantenerumab, 52 [37%]; solanezumab, 50 [35%]; placebo, 40 [28%]). Participants self-identified with the following race and ethnicity categories: 3 Asian (2%), 1 Black (0.7%), 19 multiracial/other (6%), and 129 White (91%). Participants included in this analysis showed similar distributions for age, baseline estimated years to symptom onset, sex, clinical status, biomarker levels, APOE4 status, and gene variant type.
Table 1. Baseline Demographics and Mean Biomarker Levels of Participants in the Dominantly Inherited Alzheimer Network Trial Unit (DIAN-TU-001) Trial Included in the Analysis.
| Characteristic | Gantenerumab (n = 52) | Solanezumab (n = 50) | Placebo (n = 40) |
|---|---|---|---|
| Age, median (IQR), y | 44.00 (39.00 to 53.25) | 41.00 (36.00 to 50.00) | 44.00 (37.75 to 51.00) |
| Baseline EYO, median (IQR), y | −1.94 (−9.32 to 3.12) | −2.58 (−6.79 to 2.66) | −1.89 (−6.44 to 3.41) |
| Sex, No. (%) | |||
| Female | 21 (40) | 29 (58) | 22 (55) |
| Male | 31 (60) | 21 (42) | 18 (45) |
| APOE4 (≥1 ε4 allele), No. (%) | 16 (30.8) | 14 (28.0) | 13 (32.5) |
| Variant type, No. (%) | |||
| APP | 6 (11.5) | 8 (16.0) | 5 (12.5) |
| PSEN1 | 43 (82.7) | 40 (80.0) | 29 (80.0) |
| PSEN2 | 3 (5.8) | 2 (4.0) | 2 (7.5) |
| CDR 0, No. (%) | 31 (59.6) | 30 (60.0) | 22 (55.0) |
| CSF neurogranin, median (IQR), pg/mL | 1310.00 (973.00 to 1608.00) | 1236.00 (958.35 to 1611.50) | 1179.00 (956.20 to 1683.00) |
| CSF sTREM2, median (IQR), ng/mL | 9.14 (7.27 to 12.00) | 9.77 (7.15 to 11.55) | 9.05 (7.56 to 11.02) |
| CSF YKL-40, median (IQR), ng/mL | 140.20 (108.10 to 166.50) | 140.00 (107.20 to 164.20) | 121.40 (99.48 to 183.58) |
| CSF GFAP, median (IQR), ng/mL | 7.11 (4.84 to 10.12) | 5.62 (4.85 to 8.88) | 5.89 (4.06 to 9.71) |
| CSF NfL (log), median (IQR) | 4.65 (4.39 to 5.12) | 4.65 (4.26 to 5.11) | 4.73 (4.32 to 5.19) |
| Plasma GFAP, median (IQR), ng/mL | 0.10 (0.06 to 0.16) | 0.12 (0.06 to 0.17) | 0.13 (0.07 to 0.18) |
| Plasma NfL (log), median (IQR) | 0.45 (0.05 to 0.90) | 0.64 (0.06 to 1.17) | 0.46 (0.13 to 0.80) |
| PiB-PET composite (SUVR), median (IQR) | 2.45 (1.59 to 3.51) | 2.40 (1.70 to 3.52) | 2.40 (1.73 to 3.63) |
| FDG-PET precuneus (SUVR), median (IQR) | 1.83 (1.65 to 1.92) | 1.80 (1.68 to 1.96) | 1.80 (1.68 to 1.90) |
| MRI cortical thickness precuneus, median (IQR), mm | 2.26 (2.15 to 2.36) | 2.22 (2.11 to 2.35) | 2.25 (2.09 to 2.42) |
Abbreviations: APOE, apolipoprotein E; APP, amyloid-precursor protein; CDR, Clinical Dementia Rating; CSF, cerebrospinal fluid; EYO, estimated years to symptom onset; FDG, fluorodeoxyglucose; GFAP, glial fibrillary acidic protein; MRI, magnetic resonance imaging; NfL, neurofilament light protein; PET, positron emission tomography; PiB, Pittsburgh compound B; PSEN1, presenilin 1; PSEN2, presenilin 2; sTREM2, soluble triggering receptor expressed on myeloid cells 2; SUVR, standardized uptake value ratio; YKL-40, chitinase 3–like protein 1.
CSF and Plasma Measures
We investigated the impact of gantenerumab or solanezumab treatment on downstream CSF and plasma biomarkers (Figure 1, Figure 2, and Table 2) and further characterized both drugs with exploratory analyses within the presymptomatic and symptomatic subgroup (eFigures 1 and 2 and eTables 1 and 2 in Supplement 3).
Figure 1. Estimated Mean Change From Baseline for Gantenerumab, Solanezumab, and Placebo for Cerebrospinal Fluid (CSF) Markers.
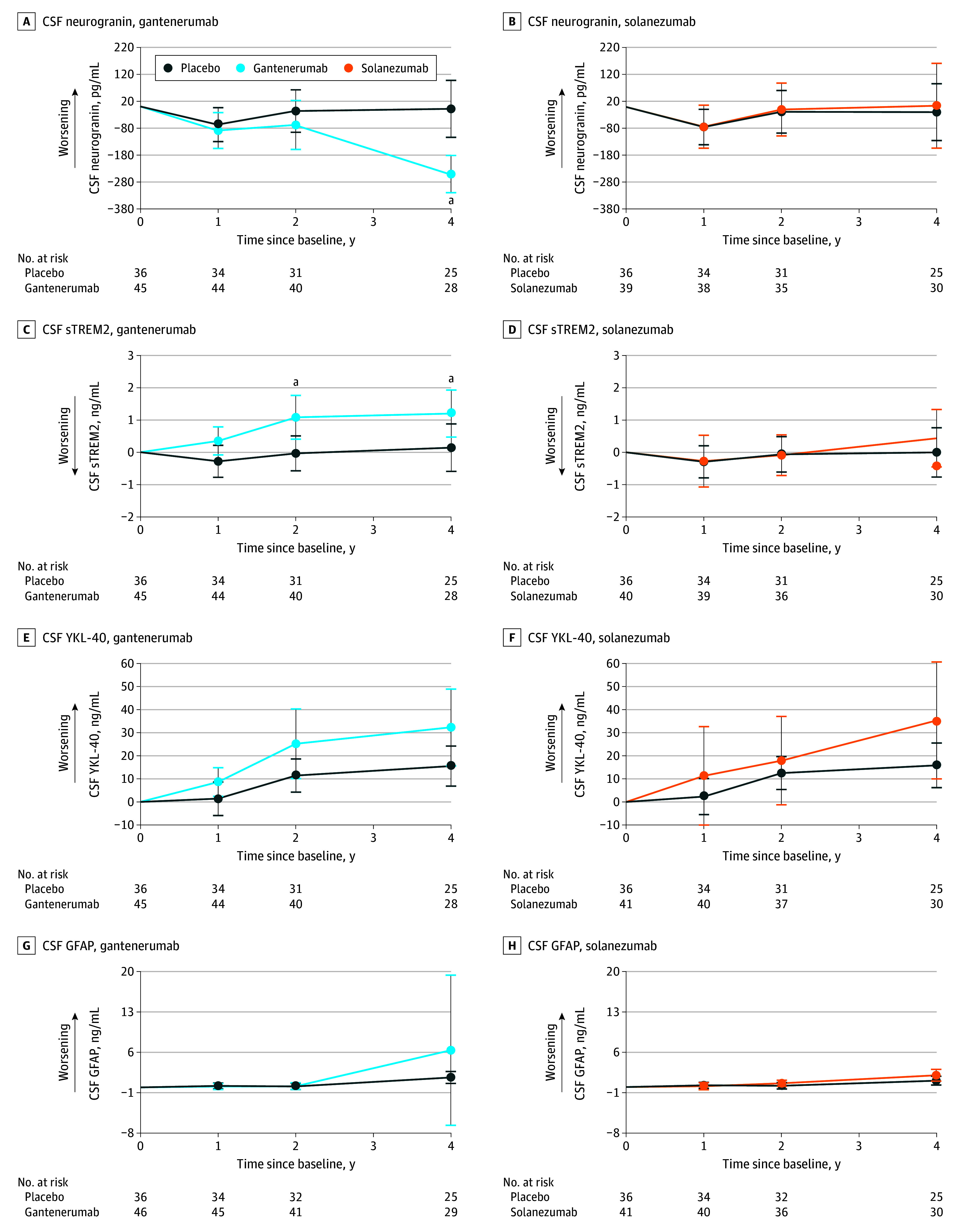
Assessment of CSF markers was done for both gantenerumab and solanezumab, respectively, in neurogranin (A and B), soluble triggering receptor expressed on myeloid cells 2 (sTREM2; C and D), chitinase 3–like protein 1 (YKL-40; E and F), and glial fibrillary acidic protein (GFAP; G and H),
Figure 2. Estimated Mean Change From Baseline for Gantenerumab, Solanezumab, and Placebo for Cerebrospinal Fluid (CSF) and Plasma Markers.
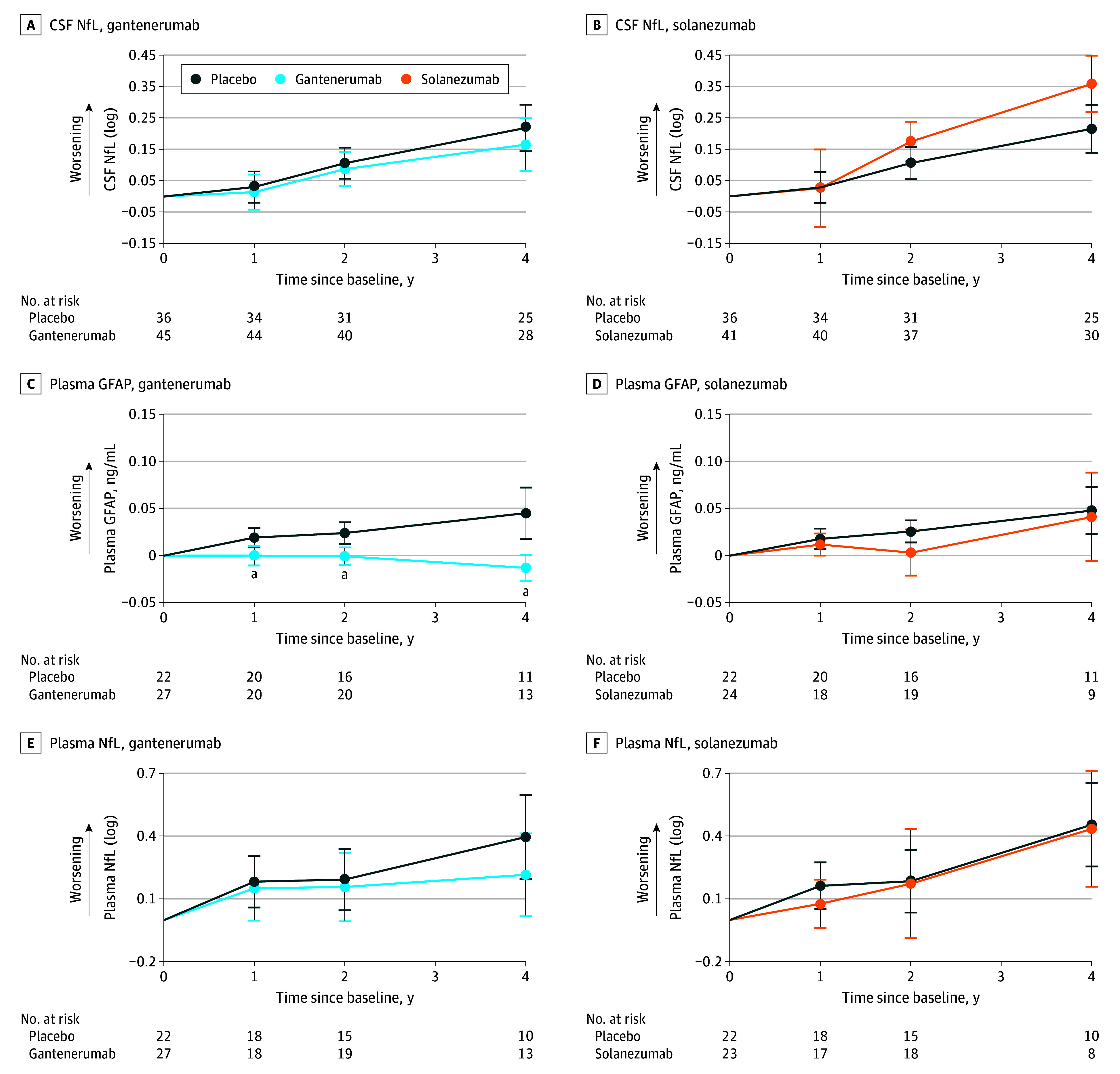
Assessment of CSF markers was done for both gantenerumab and solanezumab, respectively, in neurofilament light protein (NfL; A and B) and of plasma markers in glial fibrillary acidic protein (GFAP; C and D) and NfL (E and F). All estimations are shown with 95% CI error bars.
aResembles a significance of a P value <.05 or lower (Table 2).
Table 2. Results of the Model Analysis in the Whole Cohort Investigating the Longitudinal Changes of the Respective Biomarkers in Cerebrospinal Fluid (CSF) and Plasma for Each Drug.
| Yeara | Sample size | Estimated least-squares mean change from baseline | SE (95% CI) | P value |
|---|---|---|---|---|
| CSF neurogranin, pg/mL | ||||
| Gantenerumab | ||||
| 1 | 44 | −21.863 | 48.04 (−117.44 to 73.71) | .65 |
| 2 | 40 | −51.785 | 60.72 (−172.60 to 69.03) | .40 |
| 4 | 28 | −242.430 | 63.68 (−369.12 to −115.73) | <.001 |
| Solanezumab | ||||
| 1 | 38 | 1.182 | 53.87 (−106.16 to 108.53) | .98 |
| 2 | 35 | 8.507 | 63.98 (−118.98 to 135.99) | .90 |
| 4 | 30 | 23.712 | 96.44 (−168.44 to 215.87) | .81 |
| CSF sTREM2, ng/mL | ||||
| Gantenerumab | ||||
| 1 | 44 | 0.636 | 0.33 (−0.02 to 1.29) | .06 |
| 2 | 40 | 1.123 | 0.43 (0.26 to 1.99) | .01 |
| 4 | 28 | 1.063 | 0.52 (0.03 to 2.09) | .04 |
| Solanezumab | ||||
| 1 | 39 | 0.021 | 0.47 (−0.91 to 0.96) | .97 |
| 2 | 36 | −0.026 | 0.42 (−0.86 to 0.81) | .95 |
| 4 | 30 | 0.436 | 0.59 (−0.73 to 1.61) | .46 |
| CSF YKL-40, ng/mL | ||||
| Gantenerumab | ||||
| 1 | 44 | 7.196 | 4.86 (−2.47 to 16.86) | .14 |
| 2 | 40 | 13.795 | 8.38 (−2.88 to 30.47) | .10 |
| 4 | 28 | 16.822 | 9.39 (−1.86 to 35.50) | .08 |
| Solanezumab | ||||
| 1 | 40 | 8.994 | 10.70 (−12.32 to 30.31) | .40 |
| 2 | 37 | 5.394 | 9.83 (−14.19 to 24.98) | .59 |
| 4 | 30 | 19.511 | 13.28 (−6.93 to 45.96) | .15 |
| CSF GFAP, ng/mL | ||||
| Gantenerumab | ||||
| 1 | 45 | −0.105 | 0.36 (−0.82 to 0.61) | .77 |
| 2 | 41 | 0.024 | 0.40 (−0.78 to 0.83) | .95 |
| 4 | 29 | 4.713 | 6.55 (−8.32 to 17.74) | .47 |
| Solanezumab | ||||
| 1 | 40 | −0.156 | 0.40 (−0.95 to 0.64) | .70 |
| 2 | 36 | 0.434 | 0.39 (−0.34 to 1.21) | .27 |
| 4 | 30 | 0.941 | 0.64 (−0.34 to 2.22) | .15 |
| CSF NfL (log) | ||||
| Gantenerumab | ||||
| 1 | 44 | −0.016 | 0.04 (−0.09 to 0.06) | .66 |
| 2 | 40 | −0.019 | 0.04 (−0.09 to 0.05) | .61 |
| 4 | 28 | −0.053 | 0.06 (−0.17 to 0.06) | .35 |
| Solanezumab | ||||
| 1 | 40 | −0.002 | 0.07 (−0.14 to 0.13) | .97 |
| 2 | 37 | 0.068 | 0.04 (−0.01 to 0.15) | .10 |
| 4 | 30 | 0.143 | 0.06 (0.03 to 0.26) | .02 |
| Plasma GFAP, ng/mL | ||||
| Gantenerumab | ||||
| 1 | 20 | −0.019 | 0.01 (−0.03 to 0) | .02 |
| 2 | 20 | −0.025 | 0.01 (−0.04 to −0.01) | .002 |
| 4 | 13 | −0.058 | 0.02 (−0.09 to −0.03) | <.001 |
| Solanezumab | ||||
| 1 | 18 | −0.006 | 0.01 (−0.02 to 0.01) | .47 |
| 2 | 19 | −0.023 | 0.01 (−0.05 to 0) | .10 |
| 4 | 9 | −0.007 | 0.03 (−0.06 to 0.05) | .80 |
| Plasma NfL (log) | ||||
| Gantenerumab | ||||
| 1 | 18 | −0.032 | 0.09 (−0.21 to 0.15) | .72 |
| 2 | 19 | −0.035 | 0.11 (−0.25 to 0.18) | .74 |
| 4 | 13 | −0.180 | 0.13 (−0.45 to 0.09) | .19 |
| Solanezumab | ||||
| 1 | 17 | −0.085 | 0.08 (−0.24 to 0.07) | .29 |
| 2 | 18 | −0.012 | 0.15 (−0.31 to 0.29) | .94 |
| 4 | 8 | −0.020 | 0.17 (−0.36 to 0.32) | .91 |
Abbreviations: GFAP, glial fibrillary acidic protein; NfL, neurofilament light protein; sTREM2, soluble triggering receptor expressed on myeloid cells 2; YKL-40, chitinase 3–like protein 1.
Each year represents the time duration of drug administration since the initial biomarker assessment at baseline.
Gantenerumab treatment significantly decreased CSF neurogranin levels at year 4 compared with placebo (mean [SD] β = −242.43 [48.04] pg/mL; P < .001) (Figure 1A), whereas solanezumab exhibited no effect on CSF neurogranin (Figure 1B).
CSF sTREM2 levels (Figure 1C and D) increased steadily with gantenerumab compared with placebo (year 2: mean [SD] β = 1.12 [0.43] ng/mL; P = .01; year 4: mean [SD] β = 1.06 [0.52] ng/mL; P = .04). Solanezumab revealed no effect on sTREM2 level by year 4. CSF YKL-40 level (Figure 1E and F) was not significantly increased with gantenerumab or solanezumab. Further, no effect on CSF GFAP levels was seen with gantenerumab or solanezumab at any time point. Plasma GFAP levels (Figure 2C and D), however, stabilized under gantenerumab compared with placebo (year 1: mean [SD] β = −0.02 [0.01] ng/mL; P = .02; year 2: mean [SD] β = −0.03 [0.01] ng/mL; P = .002; year 4: mean [SD] β = −0.06 [0.02] ng/mL; P < .001) but were not affected by solanezumab.
Lastly, we found no difference in CSF NfL levels (Figure 2A and B) for gantenerumab. However, with solanezumab, CSF NfL (log) was significantly increased compared with placebo at year 4 (mean [SD] β = 0.14 [0.06]; P = .02). For plasma NfL (Figure 2E and F), solanezumab had no significant effect, and gantenerumab revealed a nonsignificant difference at year 4.
For the exploratory subgroup analysis (eTables 1 and 2 and eFigures 1 and 2 in Supplement 3), we found neurogranin and sTREM2 levels significantly decreased and increased, respectively, in presymptomatic participants receiving gantenerumab. CSF GFAP and NfL (log) levels showed significant increases with solanezumab and plasma NfL (log) level was significantly lowered in symptomatic carriers receiving gantenerumab, whereas plasma GFAP level significantly decreased in both groups for gantenerumab and increased in presymptomatics with solanezumab.
Correlation Analysis
Correlation analyses between the individually calculated rates of change of fluid and imaging biomarkers were conducted for gantenerumab or solanezumab separately (Figure 3 and eFigure 3 and eTable 3 in Supplement 3). Further details are also presented in eTables 4 and 5 in Supplement 3.
Figure 3. Correlations for Individual Rate of Change of Cerebrospinal Fluid (CSF) and Plasma Markers and Tests .

CSF biomarkers included neurogranin, soluble triggering receptor expressed on myeloid cells 2 (sTREM2), glial fibrillary acidic protein (GFAP), neurofilament light protein (NfL), and plasma markers included GFAP and NfL. Tests included Pittsburgh compound B (PiB) positron emission tomography (PET), [18F]-fluorodeoxyglucose (FDG) PET for precuneus, magnetic resonance imaging (MRI) precuneus thickness. Clinical status was assessed by Clinical Dementia Rating (CDR) sums of boxes (CDRS). The heatmap for the placebo group can be found in eFigure 3 in Supplement 3. NS indicates not significant. YKL-40 indicates chitinase 3–like 1 protein.
aP <.05.
bP <.01.
cP <.001.
Both interventions revealed similar patterns of positive correlations between all CSF biomarkers, with the solanezumab arm showing a tendency of higher correlation coefficients. Correlations of biomarkers with imaging for solanezumab found that CSF markers of sTREM2 (Spearman ρ = −0.36; P = .03), YKL-40 (Spearman ρ = −0.35; P = .03), GFAP (Spearman ρ = −0.38; P = .02), and NfL (log; Spearman ρ = −0.42; P = .01) were negatively correlated with PiB PET, whereas no relationship was detected for gantenerumab. However, participants receiving gantenerumab showed a correlation of lower GFAP (plasma Spearman ρ = −0.54; P = .008; CSF Spearman ρ = −0.36; P = .02) and NfL (log; plasma Spearman ρ = −0.49; P = .02; CSF Spearman ρ = −0.38; P = .01) levels in CSF and plasma with increased glucose metabolism in the precuneus, and solanezumab revealed a negative correlation with FDG precuneus for CSF NfL (log) level only (Spearman ρ = −0.47; P = .01). For CDR SB, there was moderate correlation with CSF NfL (log) and GFAP level only in both drugs arms (solanezumab: NfL [log] Spearman ρ = 0.38; P = .02; GFAP Spearman ρ = 0.33; P = .02; gantenerumab: NfL [log] Spearman ρ = 0.44; P = .002; GFAP Spearman ρ = 0.31; P = .03).
Discussion
We leveraged the Roche NeuroToolKit to assess multiple CSF and plasma markers of AD-related processes in the DIAN-TU-001 trial. As solanezumab and gantenerumab differ in target engagement, we aimed to elucidate the impact of each drug on biofluid markers of inflammation, synaptic loss, and neurodegeneration. We found that treatment with gantenerumab significantly decreased levels of CSF neurogranin and plasma GFAP levels while increasing CSF sTREM2 level. Meanwhile, solanezumab did not show beneficial changes in these biomarkers but significantly increased CSF NfL levels, which were previously demonstrated using a different immunoassay. With gantenerumab, lower levels of CSF YKL-40, GFAP, NfL (log), and plasma GFAP and NfL (log) significantly correlated with higher precuneus FDG-PET signals, and correlations between all CSF markers revealed slightly higher correlations for solanezumab relative to gantenerumab.
Early synaptic loss in AD is hypothesized to be induced by soluble forms of amyloid,22 rendering antiamyloid agents targeting soluble Aβ promising candidates against initial synapse loss. Neurogranin-level increases in the CSF in mild cognitive impairment and AD predict conversion from mild cognitive impairment to AD23,24 and correlate with hippocampal atrophy and cognitive decline.8,24,25 Increased neurogranin level also correlates with CSF phosphorylated tau (p-tau) 181 and total tau (t-tau)—but not Aβ42—in sAD10,24,26 and DIAD.27 We found that gantenerumab—but not solanezumab—decreased CSF levels of neurogranin at highest dosage. This suggests that a reduction of the specific soluble amyloid peptides targeted by solanezumab is not sufficient to decrease neurogranin levels. However, the administration of an agent against fibrillar amyloid might alleviate synaptic degeneration and therefore decrease CSF neurogranin levels. This is in line with reports of neurogranin increasing only after the point of amyloid PET positivity28 and correlating with neuropathological amyloid plaques,29 as well as with observations from a Study to Confirm Safety and Efficacy of Lecanemab in Participants With Early Alzheimer Disease (Clarity AD), reporting a decrease in neurogranin levels compared with placebo after 12 and 18 months of lecanemab administration,18 a drug with a similar binding profile, primarily targeting protofibrils and diffuse fibrils of Aβ.30,31 Exploratory results from the Study of Gantenerumab in Participants With Prodromal Alzheimer Disease (Scarlet Road) also suggested a dose-dependent reduction of CSF neurogranin level with gantenerumab, although careful interpretation is warranted as it was stopped prematurely due to futility.32
We further assessed sTREM2, YKL-40, and GFAP levels as markers of neuroinflammation. In AD, CSF sTREM2 concentrations seem to change dynamically, peaking at the early symptomatic stage of sAD and DIAD.33 Although some studies report higher levels of sTREM2 to be associated with higher degrees of AD-related pathology,34,35,36 others have found it to correlate with lower cross-sectional tau PET burden as well as CSF t-tau and p-tau levels,34,37 and less longitudinal increase of amyloid PET burden in sAD.11 Similarly, steeper annual increases of sTREM2 level result in a reduced rate of increase in PiB-PET burden in symptomatic carriers of a DIAD gene variant and a diminished rate in CSF Aβ42 decrease in presymptomatic carriers of a DIAD gene variant.38
In participants receiving gantenerumab, we found that CSF sTREM2 level increased compared with placebo, whereas solanezumab treatment remained without effect. Considering that decreased PiB PET levels were observed with gantenerumab, sTREM2 elevation might reflect an increase of microglia activity attributable to their receptor-mediated engagement with the drug, prompting increased glial activity with augmented plaque removal. Accordingly, a study reported that the dose-dependent effect of an agent against fibrillar amyloid on microglia was predominantly TREM2 mediated, with TREM2-depleted microglia exhibiting diminished ability to engulf Aβ and remove plaques, despite elevated levels of Fc receptors expected to compensate for deficits in phagocytic activity.39 Some investigations further suggest independent effects of sTREM2 on microglia by protecting them from apoptosis, promoting proinflammatory states40 and modulating Aβ clearance abilities.41
Elevated CSF levels of YKL-40 have been found in sAD and DIAD42,43 and seem to correlate with t-tau, p-tau, and increased cortical thinning in patients with reduced Aβ42 levels.44 Gantenerumab and solanezumab had no effect on YKL-40 compared with placebo. Although increased YKL-40 level has been proposed to precede amyloid plaques,45 studies in sAD and DIAD have found no correlation with CSF Aβ42,27,46 ultimately leaving the treatment-related changes in YKL-40 levels a subject of future research.
Although dynamics of CSF GFAP have been somewhat inconsistent in AD,47,48,49,50 recent studies show plasma GFAP levels to reliably increase in early stage sAD and DIAD,51,52 predict PiB-PET positivity49,53 and correlate with longitudinal amyloid PET54 and cognitive decline.49 Plasma GFAP levels in carriers of DIAD gene variants seem to diverge from noncarriers around 16 years before expected symptom onset, corroborating findings of early changes in sAD.55 Interestingly, we found no relevant treatment-related differences in CSF GFAP levels for either drug. GFAP plasma levels, however, revealed a significant decrease in participants receiving gantenerumab, with levels continuously rising in placebo, mirroring previous results with lecanemab18 and donanemab,56 where both trials reported a longitudinal decrease of plasma GFAP relative to baseline. Given that in AD, activated astrocytes colocalize more readily with fibrillar amyloid plaques57 and increased GFAP expression has been found to correlate predominantly with the presence of solid Aβ plaques,14 these results could hint at an indirect amelioration of astrocytic reactivity by gantenerumab due to successful cerebral plaque removal and explain why the engagement of solanezumab with soluble amyloid remained without effect on GFAP. The discrepancy between CSF and plasma hereby further underlines the theory that plasma levels might be more closely related to amyloid status due to an amyloid-dependent, direct secretion of GFAP into the bloodstream by astrocytic end feet, whereas CSF GFAP might respond to events in later disease stages, eg, neuroinflammation.49
Finally, we assessed NfL (log), which increases with age in CSF and blood and was found to correlate with progressive cognitive dysfunction in sAD and DIAD.58,59 CSF NfL levels increased with solanezumab, as it was reported in the main publication,20 but not gantenerumab, whereas significant correlations with imaging and CSF markers were seen for both drugs. These results differ from the original publication reporting significant decreases in CSF NfL level at year 1 and 4 for gantenerumab.20 However, only a subset of the original samples was included here, and original results were obtained using Simoa (Quanterix) instead of the NTK. Comparatively, effect sizes 3 times higher for NFL with Simoa (eTable 6 in Supplement 3) could be attributed to differences in assay standardization. Seeing no difference in CSF NfL level is, however, in line with our findings for plasma NfL, with no difference for either intervention compared with placebo. In sAD, donanemab and lecanemab did not affect plasma NfL (log) levels56 or NfL levels in the CSF and plasma,18 respectively. The increase in CSF NfL level with solanezumab treatment is, however, directionally consistent with cognitive worsening reported in the DIAN-TU-001 study20 and with the numerically greater cognitive decline observed in A4 in preclinical sAD.60 The reasons for increases of NfL level and cognitive decline are unclear, as a meta-analysis of the Trial of Solanezumab for Mild Dementia Due to Alzheimer’s Disease (EXPEDITION) 2 and 3 trials has found modest cognitive improvement in mildly symptomatic AD with solanezumab.61 Differences in the stage of disease could be one possible explanation.
Similar to prior biomarker findings in the DIAN-TU-001 trial,20 only gantenerumab significantly modulated markers of synaptic injury and neuroinflammation in a beneficial way. Though only exploratory, we found these differences predominantly within the presymptomatic group. In contrast, solanezumab did not show beneficial effects on biofluid markers or neuroimaging, in line with previous publications in DIAD and sAD,20,60 suggesting little impact of soluble Aβ42 or Aβ40 peptides on downstream pathophysiology. These discrepancies underscore the importance of targeting specific amyloid forms in AD treatment. Although results for gantenerumab imply a potential impact on early-stage AD-related pathology, the limited influence of solanezumab on the biomarkers calls for further investigation into its role in disease modification, especially in the context of its hypothesized neuroprotective effects against soluble amyloid-induced synaptic toxicity. These findings highlight the nuanced and complex nature of AD therapeutics, where the specific molecular targets of treatments can lead to varying outcomes in disease progression and biomarker profiles.
Correlation analysis revealed generally higher coefficients for solanezumab between fluid biomarkers and PiB PET that were similar to those receiving placebo. Considering that solanezumab had no significant effect on amyloid burden in PiB PET in this cohort, these findings suggest, in contrast to gantenerumab, where a lack of correlation for PiB PET and fluid markers hints at a decoupling due to significant target engagement and that solanezumab has little impact on biomarker progression in AD. With clinical progression, CDR-SB correlated moderately and to a similar degree with CSF NfL and GFAP for gantenerumab and solanezumab, suggesting that the pathophysiological modulations seen in this analysis do not translate to beneficial cognitive effects, similar to findings in the main publication.20
Limitations
Our analysis has limitations. The DIAN-TU-001 study was not intended to provide sufficient power to identify statistically significant differences for subgroups; results should be interpreted accordingly. Further, due to midtrial dose escalation,9 not all participants received the highest dose for the same time span, which might have implications for downstream biomarker levels. Moreover, a lack of racial and ethnic diversity limits generalizability of the presented results. Finally, although our findings offer valuable insights into changes of AD pathophysiology under antiamyloid treatment, the assessed biomarkers remain a tool of research with need for further standardization of assays, investigation of diagnostic and predictive value concerning clinical status and clinical function, as well as assessment of pathophysiological context. It is possible that with larger study cohorts or longer treatment durations, a novel magnitude of treatment effect might be found. As of now, results need to be interpreted with caution.
Conclusions
In summary, in DIAN-TU-001 randomized clinical trial, we report the beneficial impact of fibrillar amyloid reduction on fluid markers of synaptic dysfunction and neuroinflammation in DIAD, whereas the reduction of soluble Aβ42 or Aβ40 peptides did not show a positive effect on any of those markers. Results from further studies administering antiamyloid therapies in both sAD and DIAD are crucial to corroborate the utility of nonamyloid biomarkers in evaluating disease modification.
Trial Protocol.
Statistical Analysis Plan.
eTable 1. MMRM Estimated Differences in the Mean Change Between Treatments and Placebo From Baseline at Each Postbaseline Visit in Asymptomatic Participants (CDR0)
eTable 2. MMRM Estimated Differences in the Mean Change Between Treatments and Placebo From Baseline at Each Postbaseline Visit in Symptomatic Participants (CDR>0)
eTable 3. Spearman Correlations of Fluid Biomarkers and Imaging Measures in Participants Receiving Gantenerumab, Solanezumab, or Placebo
eTable 4. MRMM Analysis of the Longitudinal Changes Compared to Placebo in CSF and Plasma Biomarkers During Treatment With Gantenerumab or Solanezumab, Excluding a Visually Identified Outlier in the Gantenerumab Arm
eTable 5. MRMM Analysis of the Longitudinal Changes Compared to Placebo in CSF and plasma Biomarkers During Treatment With Gantenerumab or Solanezumab in Symptomatic Participants (CDR>0), Excluding a Visually Identified Outlier in the Gantenerumab Arm
eTable 6. Comparison of Simoa and Elecsys Assay for CSF NfL (log)
eFigure 1. Mean Change From Baseline and 95% Confident Intervals for the Treatment With Gantenerumab, or Placebo Based on Exploratory MMRM Analyses in the Separate CDR Subgroups
eFigure 2. Mean Change From Baseline and 95% Confident Intervals for the Treatment With Solanezumab, or Placebo Based on Exploratory MMRM Analyses in the Separate CDR Subgroups
eFigure 3. Heatmap Displaying Correlation Coefficients Derived From the Individual Rate of Change for CSF Levels of Neurogranin, sTREM2, GFAP, NfL and Plasma Levels of GFAP and NfL as Well as PiB PET Composite, FDG PET Tracer Binding in the Precuneus, and MRI Precuneus Thickness in Participants Receiving Placebo
Nonauthor Collaborators. Dominantly Inherited Alzheimer Network–Trial Unit
Data Sharing Statement.
References
- 1.Jack CR Jr, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12(2):207-216. doi: 10.1016/S1474-4422(12)70291-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bateman RJ, Xiong C, Benzinger TLS, et al. ; Dominantly Inherited Alzheimer Network . Clinical and biomarker changes in dominantly inherited Alzheimer disease. N Engl J Med. 2012;367(9):795-804. doi: 10.1056/NEJMoa1202753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McDade E, Wang G, Gordon BA, et al. ; Dominantly Inherited Alzheimer Network . Longitudinal cognitive and biomarker changes in dominantly inherited Alzheimer disease. Neurology. 2018;91(14):e1295-e1306. doi: 10.1212/WNL.0000000000006277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ryman DC, Acosta-Baena N, Aisen PS, et al. ; Dominantly Inherited Alzheimer Network . Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology. 2014;83(3):253-260. doi: 10.1212/WNL.0000000000000596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bohrmann B, Baumann K, Benz J, et al. Gantenerumab: a novel human anti-Aβ antibody demonstrates sustained cerebral amyloid-β binding and elicits cell-mediated removal of human amyloid-β. J Alzheimers Dis. 2012;28(1):49-69. doi: 10.3233/JAD-2011-110977 [DOI] [PubMed] [Google Scholar]
- 6.Samadi H, Sultzer D. Solanezumab for Alzheimer’s disease. Expert Opin Biol Ther. 2011;11(6):787-798. doi: 10.1517/14712598.2011.578573 [DOI] [PubMed] [Google Scholar]
- 7.Tu S, Okamoto S, Lipton SA, Xu H. Oligomeric Aβ-induced synaptic dysfunction in Alzheimer disease. Mol Neurodegener. 2014;9(1):48. doi: 10.1186/1750-1326-9-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mattsson N, Insel PS, Palmqvist S, et al. ; Alzheimer’s Disease Neuroimaging Initiative . Cerebrospinal fluid tau, neurogranin, and neurofilament light in Alzheimer disease. EMBO Mol Med. 2016;8(10):1184-1196. doi: 10.15252/emmm.201606540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang G, Li Y, Xiong C, et al. ; DIAN-TU Study Team . Evaluation of dose-dependent treatment effects after mid-trial dose escalation in biomarker, clinical, and cognitive outcomes for gantenerumab or solanezumab in dominantly inherited Alzheimer disease. Alzheimers Dement (Amst). 2022;14(1):e12367. doi: 10.1002/dad2.12367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tarawneh R, D’Angelo G, Crimmins D, et al. Diagnostic and prognostic utility of the synaptic marker neurogranin in Alzheimer disease. JAMA Neurol. 2016;73(5):561-571. doi: 10.1001/jamaneurol.2016.0086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ewers M, Biechele G, Suárez-Calvet M, et al. ; Alzheimer’s Disease Neuroimaging Initiative (ADNI) . Higher CSF sTREM2 and microglia activation are associated with slower rates of β-amyloid accumulation. EMBO Mol Med. 2020;12(9):e12308. doi: 10.15252/emmm.202012308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guerreiro R, Wojtas A, Bras J, et al. ; Alzheimer Genetic Analysis Group . TREM2 variants in Alzheimer disease. N Engl J Med. 2013;368(2):117-127. doi: 10.1056/NEJMoa1211851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Querol-Vilaseca M, Colom-Cadena M, Pegueroles J, et al. YKL-40 (Chitinase 3-like I) is expressed in a subset of astrocytes in Alzheimer disease and other tauopathies. J Neuroinflammation. 2017;14(1):118. doi: 10.1186/s12974-017-0893-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wharton SB, O’Callaghan JP, Savva GM, et al. ; MRC Cognitive Function and Ageing Neuropathology Study Group . Population variation in glial fibrillary acidic protein levels in brain ageing: relationship to Alzheimer-type pathology and dementia. Dement Geriatr Cogn Disord. 2009;27(5):465-473. doi: 10.1159/000217729 [DOI] [PubMed] [Google Scholar]
- 15.Gafson AR, Barthélemy NR, Bomont P, et al. Neurofilaments: neurobiological foundations for biomarker applications. Brain. 2020;143(7):1975-1998. doi: 10.1093/brain/awaa098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jicha GA. Is passive immunization for Alzheimer disease “alive and well” or “dead and buried”? Expert Opin Biol Ther. 2009;9(4):481-491. doi: 10.1517/14712590902828285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tolar M, Abushakra S, Hey JA, Porsteinsson A, Sabbagh M. Aducanumab, gantenerumab, BAN2401, and ALZ-801-the first wave of amyloid-targeting drugs for Alzheimer disease with potential for near term approval. Alzheimers Res Ther. 2020;12(1):95. doi: 10.1186/s13195-020-00663-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Dyck CH, Swanson CJ, Aisen P, et al. Lecanemab in early Alzheimer disease. N Engl J Med. 2023;388(1):9-21. doi: 10.1056/NEJMoa2212948 [DOI] [PubMed] [Google Scholar]
- 19.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43(11):2412-2414. doi: 10.1212/WNL.43.11.2412-a [DOI] [PubMed] [Google Scholar]
- 20.Salloway S, Farlow M, McDade E, et al. ; Dominantly Inherited Alzheimer Network–Trials Unit . A trial of gantenerumab or solanezumab in dominantly inherited Alzheimer disease. Nat Med. 2021;27(7):1187-1196. doi: 10.1038/s41591-021-01369-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson SC, Suárez-Calvet M, Suridjan I, et al. Identifying clinically useful biomarkers in neurodegenerative disease through a collaborative approach: the NeuroToolKit. Alzheimers Res Ther. 2023;15(1):25. doi: 10.1186/s13195-023-01168-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Selkoe DJ. Alzheimer disease is a synaptic failure. Science. 2002;298(5594):789-791. doi: 10.1126/science.1074069 [DOI] [PubMed] [Google Scholar]
- 23.Kester MI, Teunissen CE, Crimmins DL, et al. Neurogranin as a cerebrospinal fluid biomarker for synaptic loss in symptomatic Alzheimer disease. JAMA Neurol. 2015;72(11):1275-1280. doi: 10.1001/jamaneurol.2015.1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kvartsberg H, Duits FH, Ingelsson M, et al. Cerebrospinal fluid levels of the synaptic protein neurogranin correlates with cognitive decline in prodromal Alzheimer disease. Alzheimers Dement. 2015;11(10):1180-1190. doi: 10.1016/j.jalz.2014.10.009 [DOI] [PubMed] [Google Scholar]
- 25.Yoong SQ, Lu J, Xing H, Gyanwali B, Tan YQ, Wu XV. The prognostic utility of CSF neurogranin in predicting future cognitive decline in the Alzheimer disease continuum: a systematic review and meta-analysis with narrative synthesis. Ageing Res Rev. 2021;72:101491. doi: 10.1016/j.arr.2021.101491 [DOI] [PubMed] [Google Scholar]
- 26.Wellington H, Paterson RW, Portelius E, et al. Increased CSF neurogranin concentration is specific to Alzheimer disease. Neurology. 2016;86(9):829-835. doi: 10.1212/WNL.0000000000002423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thordardottir S, Almkvist O, Johansson C, Zetterberg H, Blennow K, Graff C. Cerebrospinal fluid YKL-40 and neurogranin in familial Alzheimer disease: a pilot study. J Alzheimers Dis. 2020;76(3):941-953. doi: 10.3233/JAD-191261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palmqvist S, Insel PS, Stomrud E, et al. Cerebrospinal fluid and plasma biomarker trajectories with increasing amyloid deposition in Alzheimer disease. EMBO Mol Med. 2019;11(12):e11170. doi: 10.15252/emmm.201911170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Portelius E, Olsson B, Höglund K, et al. Cerebrospinal fluid neurogranin concentration in neurodegeneration: relation to clinical phenotypes and neuropathology. Acta Neuropathol. 2018;136(3):363-376. doi: 10.1007/s00401-018-1851-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Söderberg L, Johannesson M, Nygren P, et al. Lecanemab, aducanumab, and gantenerumab—binding profiles to different forms of amyloid-β might explain efficacy and side effects in clinical trials for Alzheimer disease. Neurotherapeutics. 2023;20(1):195-206. doi: 10.1007/s13311-022-01308-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stern AM, Yang Y, Jin S, et al. Abundant Aβ fibrils in ultracentrifugal supernatants of aqueous extracts from Alzheimer disease brains. Neuron. 2023;111(13):2012-2020. doi: 10.1016/j.neuron.2023.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ostrowitzki S, Lasser RA, Dorflinger E, et al. ; SCarlet RoAD Investigators . A phase III randomized trial of gantenerumab in prodromal Alzheimer disease. Alzheimers Res Ther. 2017;9(1):95. doi: 10.1186/s13195-017-0318-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suárez-Calvet M, Kleinberger G, Araque Caballero MÁ, et al. sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer disease and associate with neuronal injury markers. EMBO Mol Med. 2016;8(5):466-476. doi: 10.15252/emmm.201506123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suárez-Calvet M, Morenas-Rodríguez E, Kleinberger G, et al. ; Alzheimer’s Disease Neuroimaging Initiative . Early increase of CSF sTREM2 in Alzheimer’s disease is associated with tau related-neurodegeneration but not with amyloid-β pathology. Mol Neurodegener. 2019;14(1):1. doi: 10.1186/s13024-018-0301-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vogels T, Murgoci AN, Hromádka T. Intersection of pathological tau and microglia at the synapse. Acta Neuropathol Commun. 2019;7(1):109. doi: 10.1186/s40478-019-0754-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Biel D, Suárez-Calvet M, Hager P, et al. ; Alzheimer’s Disease Neuroimaging Initiative (ADNI) . sTREM2 is associated with amyloid-related p-tau increases and glucose hypermetabolism in Alzheimer disease. EMBO Mol Med. 2023;15(2):e16987. doi: 10.15252/emmm.202216987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heslegrave A, Heywood W, Paterson R, et al. Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer disease. Mol Neurodegener. 2016;11(1):3. doi: 10.1186/s13024-016-0071-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morenas-Rodríguez E, Li Y, Nuscher B, et al. ; Dominantly Inherited Alzheimer Network . Soluble TREM2 in CSF and its association with other biomarkers and cognition in autosomal-dominant Alzheimer disease: a longitudinal observational study. Lancet Neurol. 2022;21(4):329-341. doi: 10.1016/S1474-4422(22)00027-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xiang X, Werner G, Bohrmann B, et al. TREM2 deficiency reduces the efficacy of immunotherapeutic amyloid clearance. EMBO Mol Med. 2016;8(9):992-1004. doi: 10.15252/emmm.201606370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhong L, Chen XF, Wang T, et al. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J Exp Med. 2017;214(3):597-607. doi: 10.1084/jem.20160844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhong L, Xu Y, Zhuo R, et al. Soluble TREM2 ameliorates pathological phenotypes by modulating microglial functions in an Alzheimer disease model. Nat Commun. 2019;10(1):1365. doi: 10.1038/s41467-019-09118-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hampel H, Toschi N, Baldacci F, et al. ; Alzheimer Precision Medicine Initiative (APMI) . Alzheimer disease biomarker-guided diagnostic workflow using the added value of six combined cerebrospinal fluid candidates: Aβ1-42, total-tau, phosphorylated-tau, NFL, neurogranin, and YKL-40. Alzheimers Dement. 2018;14(4):492-501. doi: 10.1016/j.jalz.2017.11.015 [DOI] [PubMed] [Google Scholar]
- 43.Schindler SE, Li Y, Todd KW, et al. ; Dominantly Inherited Alzheimer Network . Emerging cerebrospinal fluid biomarkers in autosomal dominant Alzheimer disease. Alzheimers Dement. 2019;15(5):655-665. doi: 10.1016/j.jalz.2018.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alcolea D, Vilaplana E, Pegueroles J, et al. Relationship between cortical thickness and cerebrospinal fluid YKL-40 in predementia stages of Alzheimer disease. Neurobiol Aging. 2015;36(6):2018-2023. doi: 10.1016/j.neurobiolaging.2015.03.001 [DOI] [PubMed] [Google Scholar]
- 45.Connolly K, Lehoux M, O’Rourke R, et al. Potential role of chitinase-3-like protein 1 (CHI3L1/YKL-40) in neurodegeneration and Alzheimer disease. Alzheimers Dement. 2023;19(1):9-24. doi: 10.1002/alz.12612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Craig-Schapiro R, Perrin RJ, Roe CM, et al. YKL-40: a novel prognostic fluid biomarker for preclinical Alzheimer disease. Biol Psychiatry. 2010;68(10):903-912. doi: 10.1016/j.biopsych.2010.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Olsson B, Lautner R, Andreasson U, et al. CSF and blood biomarkers for the diagnosis of Alzheimer disease: a systematic review and meta-analysis. Lancet Neurol. 2016;15(7):673-684. doi: 10.1016/S1474-4422(16)00070-3 [DOI] [PubMed] [Google Scholar]
- 48.Milà-Alomà M, Salvadó G, Gispert JD, et al. ; ALFA study . Amyloid β, tau, synaptic, neurodegeneration, and glial biomarkers in the preclinical stage of the Alzheimer continuum. Alzheimers Dement. 2020;16(10):1358-1371. doi: 10.1002/alz.12131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pereira JB, Janelidze S, Smith R, et al. Plasma GFAP is an early marker of amyloid-β but not tau pathology in Alzheimer disease. Brain. 2021;144(11):3505-3516. doi: 10.1093/brain/awab223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ishiki A, Kamada M, Kawamura Y, et al. Glial fibrillar acidic protein in the cerebrospinal fluid of Alzheimer disease, dementia with Lewy bodies, and frontotemporal lobar degeneration. J Neurochem. 2016;136(2):258-261. doi: 10.1111/jnc.13399 [DOI] [PubMed] [Google Scholar]
- 51.Benedet AL, Milà-Alomà M, Vrillon A, et al. ; Translational Biomarkers in Aging and Dementia (TRIAD) study, Alzheimer’s and Families (ALFA) study, and BioCogBank Paris Lariboisière cohort . Differences between plasma and cerebrospinal fluid glial fibrillary acidic protein levels across the Alzheimer disease continuum. JAMA Neurol. 2021;78(12):1471-1483. doi: 10.1001/jamaneurol.2021.3671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chatterjee P, Vermunt L, Gordon BA, et al. ; Dominantly Inherited Alzheimer Network . Plasma glial fibrillary acidic protein in autosomal dominant Alzheimer disease: associations with Aβ-PET, neurodegeneration, and cognition. Alzheimers Dement. 2023;19(7):2790-2804. doi: 10.1002/alz.12879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Verberk IMW, Thijssen E, Koelewijn J, et al. Combination of plasma amyloid β(1-42/1-40) and glial fibrillary acidic protein strongly associates with cerebral amyloid pathology. Alzheimers Res Ther. 2020;12(1):118. doi: 10.1186/s13195-020-00682-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Asken BM, Elahi FM, La Joie R, et al. Plasma glial fibrillary acidic protein levels differ along the spectra of amyloid burden and clinical disease stage. J Alzheimers Dis. 2021;80(1):471-474. doi: 10.3233/JAD-219001 [DOI] [PubMed] [Google Scholar]
- 55.O’Connor A, Abel E, Benedet AL, et al. Plasma GFAP in presymptomatic and symptomatic familial Alzheimer disease: a longitudinal cohort study. J Neurol Neurosurg Psychiatry. 2023;94(1):90-92. doi: 10.1136/jnnp-2022-329663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pontecorvo MJ, Lu M, Burnham SC, et al. Association of donanemab treatment with exploratory plasma biomarkers in early symptomatic Alzheimer disease: a secondary analysis of the TRAILBLAZER-ALZ randomized clinical trial. JAMA Neurol. 2022;79(12):1250-1259. doi: 10.1001/jamaneurol.2022.3392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Medeiros R, LaFerla FM. Astrocytes: conductors of the Alzheimer disease neuroinflammatory symphony. Exp Neurol. 2013;239:133-138. doi: 10.1016/j.expneurol.2012.10.007 [DOI] [PubMed] [Google Scholar]
- 58.Olsson B, Portelius E, Cullen NC, et al. Association of cerebrospinal fluid neurofilament light protein levels with cognition in patients with dementia, motor neuron disease, and movement disorders. JAMA Neurol. 2019;76(3):318-325. doi: 10.1001/jamaneurol.2018.3746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Preische O, Schultz SA, Apel A, et al. ; Dominantly Inherited Alzheimer Network . Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer disease. Nat Med. 2019;25(2):277-283. doi: 10.1038/s41591-018-0304-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sperling RA, Donohue MC, Raman R, et al. ; A4 Study Team . Trial of solanezumab in preclinical Alzheimer disease. N Engl J Med. 2023;389(12):1096-1107. doi: 10.1056/NEJMoa2305032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Holdridge KC, Yaari R, Hoban DB, Andersen S, Sims JR. Targeting amyloid β in Alzheimer disease: meta-analysis of low-dose solanezumab in Alzheimer disease with mild dementia studies. Alzheimers Dement. 2023;19(10):4619-4628. doi: 10.1002/alz.13031 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Trial Protocol.
Statistical Analysis Plan.
eTable 1. MMRM Estimated Differences in the Mean Change Between Treatments and Placebo From Baseline at Each Postbaseline Visit in Asymptomatic Participants (CDR0)
eTable 2. MMRM Estimated Differences in the Mean Change Between Treatments and Placebo From Baseline at Each Postbaseline Visit in Symptomatic Participants (CDR>0)
eTable 3. Spearman Correlations of Fluid Biomarkers and Imaging Measures in Participants Receiving Gantenerumab, Solanezumab, or Placebo
eTable 4. MRMM Analysis of the Longitudinal Changes Compared to Placebo in CSF and Plasma Biomarkers During Treatment With Gantenerumab or Solanezumab, Excluding a Visually Identified Outlier in the Gantenerumab Arm
eTable 5. MRMM Analysis of the Longitudinal Changes Compared to Placebo in CSF and plasma Biomarkers During Treatment With Gantenerumab or Solanezumab in Symptomatic Participants (CDR>0), Excluding a Visually Identified Outlier in the Gantenerumab Arm
eTable 6. Comparison of Simoa and Elecsys Assay for CSF NfL (log)
eFigure 1. Mean Change From Baseline and 95% Confident Intervals for the Treatment With Gantenerumab, or Placebo Based on Exploratory MMRM Analyses in the Separate CDR Subgroups
eFigure 2. Mean Change From Baseline and 95% Confident Intervals for the Treatment With Solanezumab, or Placebo Based on Exploratory MMRM Analyses in the Separate CDR Subgroups
eFigure 3. Heatmap Displaying Correlation Coefficients Derived From the Individual Rate of Change for CSF Levels of Neurogranin, sTREM2, GFAP, NfL and Plasma Levels of GFAP and NfL as Well as PiB PET Composite, FDG PET Tracer Binding in the Precuneus, and MRI Precuneus Thickness in Participants Receiving Placebo
Nonauthor Collaborators. Dominantly Inherited Alzheimer Network–Trial Unit
Data Sharing Statement.