

Abstract
Background
Ichthyosis is a common keratotic skin disease with high clinical, etiological and genetic heterogeneity. There are four types of non‐syndromic hereditary ichthyoses, among which autosomal recessive congenital ichthyosis (ARCI) is a heterogeneous group of recessive Mendelian disorders. ARCI present with different phenotypes and ABCA12 pathogenic variants have been shown to cause complex ARCI phenotypes, including harlequin ichthyosis (HI), lamellar ichthyosis (LI) and congenital ichthyosiform erythroderma (CIE).
Methods
A sporadic male patient, clinically diagnosed with CIE, was enrolled in this study. Exome sequencing was combined with Sanger sequencing to confirm the diagnosis and identify the pathogenic variants. In silico predictions were made using multiple software programs, and the identified variants were interpreted using the ACMG guidelines. A review of all literature reported ABCA12 variants was performed to explore genotype–phenotype correlations.
Results
Compound heterozygous ABCA12 variants [c.5381+1G>A and c.5485G>C (p.Asp1829His)] (NM_173076) were identified. The two variants were not detected in the public database. c.5381+1G>A is predicted to affect ABCA12 mRNA splicing and Asp1829 is highly conserved among various species. In silico analysis suggested that these two variants were responsible for the phenotype of the patient. Genotype–phenotype correlation analysis showed that biallelic truncation variants and/or exon/amino acid deletions in ABCA12 are the most common causes of HI. Biallelic missense variants are most common in LI and CIE.
Conclusions
The compound heterozygous ABCA12 variants caused the CIE phenotype observed in the patient. The spectrum of ABCA12 pathogenic variants were broaden. Genotype–phenotype correlation analysis provided detailed evidence which can be used in future prenatal diagnosis and can inform the need for genetic counselling for patients with ABCA12‐related ARCIs.
Keywords: ABCA12, autosomal recessive congenital ichthyosis (ARCI), compound heterozygous variants, congenital ichthyosiform erythroderma (CIE), genotype–phenotype correlations

1. INTRODUCTION
Ichthyosis is a common keratotic skin disorder characterised by dry, thick and scaly skin. Ichthyoses are a clinically, etiologically and genetically heterogeneous group of acquired and hereditary disorders. Acquired ichthyosis is often associated with malignancies, autoimmune/inflammatory diseases and nutritional deficiencies (Dar et al., 2011; Lee et al., 2006). Hereditary ichthyosis, which belongs to Mendelian disorders of cornification, is often associated with germline genetic variants (Oji et al., 2010). Hereditary ichthyosis is further divided into syndromic and non‐syndromic forms. Non‐syndromic ichthyosis is characterised by the phenotypic expression, that is lesions, of the disorder only on the skin. According to the results of the First Ichthyosis Consensus Conference in Sorèze 2009, non‐syndromic hereditary ichthyosis is divided into four categories: common ichthyoses, autosomal recessive congenital ichthyosis (ARCI), keratinopathic ichthyosis (KPI) and other rare forms (Oji et al., 2010).
ARCI describes a continuous symptom spectrum of ichthyoses consisting of three major clinical subtypes: harlequin ichthyosis (HI), lamellar ichthyosis (LI) and congenital ichthyosiform erythroderma (CIE). The overall prevalence of ARCI is approximately 1 in 100,000 (Hernandez‐Martin et al., 2012). ARCI, involving anomalies in intercellular lipid layers and cornified cell envelopes in the skin barrier of stratum corneum, results from pathogenic variants in at least 13 genes (ABCA12, ALOX12B, ALOXE3, CASP14, CERS3, CYP4F22, LIPN, NIPAL4, PNPLA1, SDR9C7, SULT2B1, ST14 and TGM1) (Chulpanova et al., 2022). Pathogenic variants within TGM1, account for the majority of ARCI cases (32%–68%) (Fischer, 2009; Israeli et al., 2013; Pigg et al., 2016).
Pathogenic variants in ABCA12 can cause complex ARCI phenotypes. These are HI, by far the most severe ichthyosis, as well as milder LI and CIE. Family history informs ichthyosis diagnosis, therefore, diagnosis is challenging when a patient, with no familial history of the disorder, presents with symptoms. However, exome‐sequencing enables rapid and reliable clinical diagnosis of a rare suspected Mendelian disorder. Herein, we report a Chinese male patient with CIE. Exome sequencing was combined with Sanger sequencing to identify the associated ABCA12 variants. We reviewed the genotype–phenotype relationship of the identified ABCA12 pathogenic variants. Overall, this case expands the spectrum of ABCA12‐reported disease‐causing variants.
2. MATERIALS AND METHODS
See Supplementary Materials and Methods.
3. RESULTS
3.1. Case presentation
A 27‐year‐old male presented with diffuse skin erythema and desquamation with itching (Figure 1). Symptoms were initiated at 9‐day‐old and have since gradually worsened. No bullous, mucosal involvement, hair or nail dystrophy was observed. The patient was delivered at full term via spontaneous vaginal birth. He was from a non‐consanguineous family. No similar skin defects were observed in any of the patient's family members. No signs of collodion membrane were reported at birth. The physical examination revealed erythroderma affecting the entire body, with slight scaling. No ectropion, eclabium or joint contracture was observed. The patient exhibited normal teeth, hair and nails. Both intellectual and physical development were generally within the normal range. The patient was prescribed Vaseline for external use and occasional oral cetirizine to manage itching.
FIGURE 1.
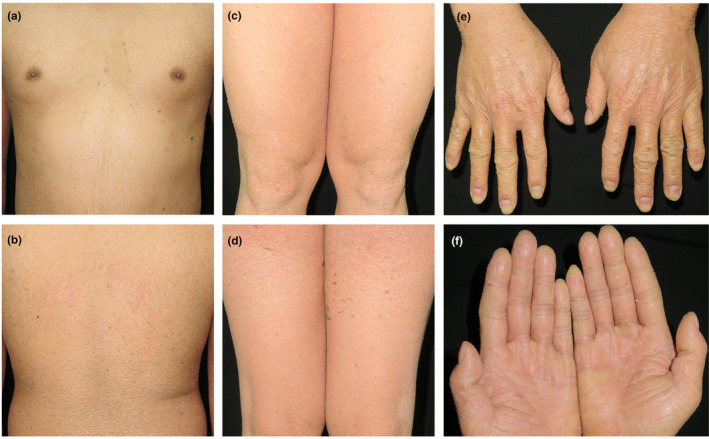
Clinical pictures of the patient. (a–e) depicting generalised erythematous desquamative skin rash affecting the entire body. (f) Clinical pictures of the patient's palms depicting keratosis and scaling.
3.2. Compound heterozygous ABCA12 variants were identified in the patient's genomic DNA
Exome‐sequencing results showed that the patient carried two ABCA12 variants (NM_173076), c.5381+1G>A and c.5485G>C (p.Asp1829His) (Figure 3b). Sanger sequencing confirmed that the splice site variant c.5381+1G>A was inherited from the paternal parent and the missense variant c.5485G>C (p.Asp1829His) was inherited from the maternal parent (Figure 2a–c).
FIGURE 3.
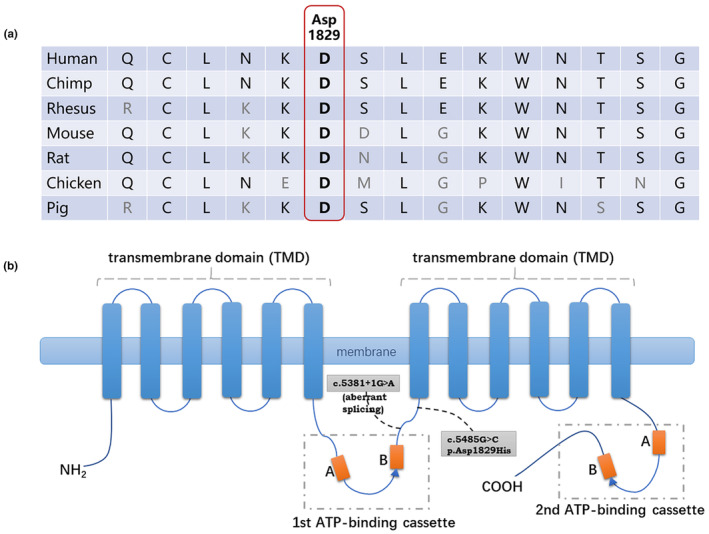
ABCA12 protein and partial protein conservation analysis. (a) Asp1829 of ABCA12 is highly conserved in multiple vertebrate species. (b) Schematic diagram of ABCA12 protein structure and two variants identified in the patient. Two variants that may affect the protein function detected in our patient are depicted as black dashed lines on the drawing. The orange box A and B represents Walker A Motif and Walker B Motif. The blue triangle represents the active transport signature.
FIGURE 2.
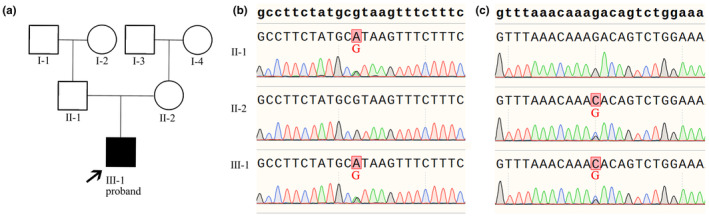
Pedigree of patient and Sanger‐sequencing results of ABCA12. (a) Patient pedigree. (b) Sanger‐sequencing highlighting the heterozygous ABCA12 variant c.5381+1G>A in the patient and the patient's father. (c) Sanger‐sequencing highlighting the heterozygous ABCA12 variant c.5485G>C (p.Asp1829His) in the patient and the patient's mother.
3.3. In silico analysis
The ABCA12 c.5485G>C (p.Asp1829His) variant was not detected in any of the public control databases, including the 1000 Genome Project, ESP6500, gnomAD and in‐house databases. Asp1829 is highly conserved among various species (human, chimp, rhesus, mouse, rat, chicken and pig) (sequences obtained from http://genome.ucsc.edu/). (Figure 3a). Several prediction programs predicted c.5485G>C (p.Asp1829His) to be a damaging variant (PolyPhen2, 0.453 probably damaging; SIFT, 0.029 damaging; CADD_Phred 22.8, damaging). The variant was classified as “likely pathogenic” (PM1+PM2+PM3), according to the ACMG/AMP 2015 guideline.
The ABCA12 c.5381+1G>A variant is a canonical +1 splice site variant located in intron 34. It is predicted to affect ABCA12 mRNA splicing: dbscSNV scores (ADA score:1.0000; RF score:0.938) and the Spidex method (−5.1122). The variant was not detected in the 1000 Genome Project, ESP6500, gnomAD or in the in‐house database. The classification of the variant is “likely pathogenic” (PVS1+PM2) according to the ACMG/AMP 2015 guideline.
4. DISCUSSION
Ichthyosis occurs due to impaired skin barrier function. The large majority of ichthyoses phenotypically manifest as abnormal epidermal granular and cuticle layers. The associated skin problems in affected individuals usually last a lifetime, and in severe cases, quality of life is drastically affected. Symptomatic treatments such as moisturisers, keratolytics and retinoids are often used to relieve dry and itchy skin. However, damaged protein function persists, and so does the skin damage. Gene therapy, is a promising targeted therapy for ichthyosis, however, precision therapy depends on accurate diagnosis (Chulpanova et al., 2022). Accurate ichthyosis diagnosis remains elusive since the disorder is highly heterogeneous, with more than 20 genes involved in non‐syndromic hereditary ichthyosis. In this study, exome sequencing was performed in an ARCI sporadic male patient with a clinical diagnosis of CIE. CIE can be caused by variants in eight known genes (ABCA12, ALOX12B, ALOXE3, CERS3, CYP4F22, NIPAL4, PNPLA1 and TGM1). Eventually, the above genes were initially checked and excluded, combined with DNA verification from the patient's parents, we identified compound heterozygous ABCA12 variants [c.5381+1G>A and c.5485G>C (p.Asp1829His)] in the patient.
ATP‐binding cassette (ABC) transporters are a large family of transmembrane proteins widely distributed in eukaryotic and prokaryotic cells (Lane et al., 2016). They transport diverse substrates, including amino acids, peptides, iron, sugars, lipids, drugs and toxins, by hydrolysing ATP and generating energy (Xiong et al., 2015). ABC transporters in eukaryotes are classified into seven main subfamilies (ABC‐A to ABC‐G) based on sequence homology and domain organisation (Xiong et al., 2015). The core topology of ABC transporters contains a highly hydrophobic transmembrane domain (TMD) and nucleotide‐binding domain (NBD). Different TMDs and NBDs arrangement and number combinations yield a variety of protein structures. Human ABC transporters participate in many key physiological processes and, once impaired, maybe the causative factors of several diseases (Theodoulou & Kerr, 2015). The ABC‐A subfamily contains 12 members and is of great significance for lipid transport and metabolism, which is usually a “full structure” transporter, that is, presented with TMD‐NBD‐TMD‐NBD tandem organisation from N‐terminal to C‐terminal (Kaminski et al., 2006).
ABCA12, cloned in 2002 and located on chromosome 2q35, encodes a 2595 amino acid protein (NM_173076) with a molecular mass of approximately 293 kDa (Annilo et al., 2002). ABCA12 is the second‐largest molecular weight transporter in the ABC‐A subfamily, with 53 exons in its coding region. ABCA12 typically consists of two TMDs and two NBDs (Annilo et al., 2002). NBDs, as the ATP‐binding cassette, contain three highly conserved motifs: the Walker A motif, the Walker B motif and the active transport signature (Figure 3b). With a strong expression in keratinocytes which indicates that ABCA12 play a significant role in skin barrier function (Lefevre et al., 2003). ABCA12 is abundantly localised in lamellar granules (LGs) of keratinocytes and functions as an epidermal keratinocyte lipid transporter (Akiyama et al., 2005). ABCA12 can bind and hydrolyse ATP to promote lipid transport from the Golgi apparatus to the LGs in granular layer cells (Sakai et al., 2007). Impaired ABCA12 function results in malformation of stratum corneum intercellular lipid layers (Akiyama et al., 2005).
Since 2003, ABCA12 homozygous missense variants have been identified and were shown to be associated with human LI, in nine consanguineous families (Lefevre et al., 2003). In 2005, two independent research groups showed that severely truncated ABCA12 proteins cause new‐born HI (Akiyama et al., 2005; Kelsell et al., 2005), which is an extremely severe and frequently lethal form of ARCI, with an incidence of 1 in 300,000 births (Ahmed & O'Toole, 2014). ABCA12 is the unique gene responsible for HI. Typical features of HI include thick, plate‐like scales with ectropion, eclabium and flattened ears (Kelsell et al., 2005). Respiratory distress, feeding problems and systemic infections which can be life‐threatening complications in new borns. Natsuga et al identified that a non‐bullous CIE phenotype is caused by homozygous ABCA12 missense variants (Natsuga et al., 2007). Patients with LI appear to be colloidal infants at birth, and the membrane then peels covered with large, dark, plate‐like epidermal scales with small erythema (Lefevre et al., 2003). Distinguished from LI, in CIE, scales are fine, white and on an erythematous background (Fischer, 2009). ARCI can be regarded as a whole spectrum of HI, LI and CIE, with clinical manifestations ranging from severe to mild.
It is estimated that ABCA12 variants have been detected in 252 ichthyosis families of diverse ethnicity (Table S1). It included 121 HI families, 21 LI families, 79 CIE families and 31 other ichthyosis families (non‐classified congenital ichthyosis, non‐classified ARCI, ichthyosis linearis circumflexa, mixed LI/HI phenotype, mixed LI/CIE phenotype, HI‐like phenotype, collodion baby, palmoplanar keratosis, erythema, translucent superficial scaling, ichthyosis congenita gravis, mild ichthyosis, arthrogryposis, self‐improving congenital ichthyosis, CIE with palmoplanar keratosis and LI and no HI). Phenotypic overlap occurs sometimes. It has been shown that ABCA12 variants correlate with phenotypic severity (Akiyama, 2010; Hotz et al., 2023; Loo et al., 2018; Montalvan‐Suarez et al., 2019; Umemoto et al., 2011). Current studies generally support that most biallelic truncation variants and/or exon/amino acid deletions lead to HI. In addition, most biallelic missense variants cause CIE and LI.
We summarised the combinations of variants to explore the genotype–phenotype relationship among ABCA12 variants (Tables 1 and S1). Biallelic truncation variants and/or exon/amino acid deletions were confirmed to be the most common causes of HI. Severe damage caused by some missense variants in highly conserved regions cannot be ignored. The variant c.3535G>A (p.Gly1179Arg), located in the first conserved TMD of ABCA12, and its single amino acid change may have drastic effects on the protein structure and function (Umemoto et al., 2011). For example, patient 16 harbouring homozygous c.3535G>A (p.Gly1179Arg) presented with the HI phenotype (Thomas et al., 2006), and patient 92 harbouring compound heterozygous c.3535G>A (p.Gly1179Arg) as well as a truncation variant also presented with the HI phenotype (Umemoto et al., 2011) (Table S1). To date, only 21 simple LI families have been reported. Fifteen of the 21 families harboured homozygous or compound heterozygous missense variants. In CIE, most of the pathogenic variants contain missense variants. Compound heterozygous missense and truncation variants are also important for CIE pathogenesis. The c.4139A>G (p.Asn1380Ser) variant appears to be a hotspot variant that occurs in many ethnicities, including Czech, Swedish, Moroccan, Algerian, Spanish, Pakistani, Turk, Chinese, German, Japanese, French, Scandinavian and Italian. Interestingly, patients with the same homozygous c.4139A>G (p.Asn1380Ser) showed clinical heterogeneity. Patients 25, 26, 28 and 34 presented with LI; however, patients 27, 32 and 33 presented with CIE. A total of 13 Chinese families and 31 Japanese families of ABCA12 variants have been reported in the East Asian population. There were no significant mutation hotspots.
TABLE 1.
Percentage distribution of reported ABCA12‐related ichthyosis probands numbers and variants forms.
| Biallelic variants resultant change combination | Reported ABCA12‐related ichthyosis probands numbers | ||||
|---|---|---|---|---|---|
| HI (%) | LI (%) | CIE (%) | Other ichthyosis (%) | Total (100%) | |
| Missense + missense | 8 (10.7%) | 15 (20%) | 36 (48%) | 16 (21.3%) | 75 |
| Missense +? | – | – | 4 (100%) | – | 4 |
| Missense + truncation | 16 (30.8%) | 5 (9.6%) | 22 (42.3%) | 9 (17.3%) | 52 |
| Missense + splicing | 2 (14.3%) | – | 9 (64.3%) | 3 (21.4%) | 14 |
| Missense + small deletion | 1 (100%) | – | – | – | 1 |
| Missense + initiation codon variant | – | – | – | 1 (100%) | 1 |
| Truncation + truncation | 60 (96.8%) | 1 (1.6%) | 1 (1.6%) | – | 62 |
| Truncation +? | 3 (60.0%) | – | 2 (40.0%) | – | 5 |
| Splicing + splicing | 8 (80%) | – | 2 (20%) | – | 10 |
| Splicing +? | 1 (100%) | – | – | – | 1 |
| Splicing + truncation | 14 (73.7%) | – | 3 (15.8%) | 2 (10.5%) | 19 |
| Truncation + small deletion | 2 (100%) | – | – | – | 2 |
| Splicing + gross deletion | 2 (100%) | – | – | – | 2 |
| Small deletion + small deletion | 1 (100%) | – | – | – | 1 |
| Gross deletion + gross deletion | 3 (100%) | – | – | – | 3 |
| Total probands numbers | 121 | 21 | 79 | 31 | 252 |
Note: Other ichthyosis includes non‐classified congenital ichthyosis, non‐classified autosomal recessive congenital ichthyosis, ichthyosis linearis circumflexa, mixed LI/HI phenotype, mixed LI/CIE phenotype, HI‐like phenotype, collodion baby, palmoplanar keratosis, erythema, translucent superficial scaling, ichthyosis congenita gravis, mild ichthyosis, arthrogryposis, CIE with palmoplanar keratosis and LI and no HI.
Abbreviations: CIE, congenital ichthyosiform erythroderma; HI, harlequin ichthyosis; LI, lamellar ichthyosis.
To our knowledge, our study is the 14th reported ABCA12‐related Chinese ichthyosis family and the 7th CIE family (Table S1). Our patient was clinically diagnosed with CIE and carried compound heterozygous ABCA12 variants c.5381+1G>A and c.5485G>C (p.Asp1829His). Neither variant exists in public control databases. c.5381+1G>A is a canonical splice site variant in intron 34 that most likely disrupts the 5′ splicing signals, according to the GU–AG rule. In silico analysis predicted that the ABCA12 mRNA aberrant splicing occurs. Multiple programs predicted that the c.5485G>C (p.Asp1829His) variant to be pathogenic, and Asp1829 was found to be highly conserved among species. The ACMG scores were ‘likely pathogenic’ and ‘likely pathogenic’, respectively of c.5381+1G>A and c.5485G>C (p.Asp1829His). We speculated that p.Asp1829His was not located in the significant ATP‐binding cassette or TMD, which might be one of the reasons for the milder phenotype.
The management of CIE presents a challenging task. Traditional therapeutic approaches primarily concentrate on external moisturisation and keratolytic agents to alleviate skin manifestations. In severe cases, oral retinoids such as acitretin or other vitamin A analogues are recommended, albeit their utility is constrained by adverse effects, including skin irritation, teratogenic potential, hypertriglyceridemia and hyperostosis. The discovery of ABCA12 as a pathogenic gene has provided insights into the treatment of CIE. Dysfunctional ABCA12 disrupts lipid transport into lamellar granules, subsequently impeding their release into the interstitial lipid domain among terminally differentiated keratinocytes, resulting in the deterioration of skin barrier integrity and ultimately leading to the development of CIE. Functional barrier deterioration was reported to be correlate with serum and tissue IL‐17 level (Paller, 2019). Secukinumab, an IL‐17A inhibitor, showed its efficiency and safety in the treatment of CIE (Yogarajah et al., 2021). Non‐viral gene transfer system as a gene therapy method for ichthyosis has been used for ABCA12 delivery to the patients' keratinocytes (Chulpanova et al., 2022).
In conclusion, this case expands the spectrum of ABCA12‐related inherited ichthyosis‐causing variants. Identifying specific causative genes and genetic modes will allow for the accurate diagnosis of ichthyosis. Once an accurate diagnosis is made, families can be referred to and may benefit from genetic counselling, which will cover carrier screening and disease prevention. Genotype–phenotype correlation analysis provides detailed evidence which in future can lead to a more precise pathway of diagnosis and therapy for ABCA12‐related ARCIs.
AUTHOR CONTRIBUTIONS
Wang R, Ma D and Zhang X contributed to the conception and design of the study. Liu J and Ma D were involved in the clinical evaluation of the patient and his family members. Guo K and Zhang R performed the experiments and analysed the data. Guo K wrote the manuscript, which was revised critically by all authors. All authors read and approved the final manuscript.
CONFLICT OF INTEREST STATEMENT
The author(s) declare that they have no conflict of interest.
ETHICS STATEMENT
Written informed consent was obtained from all participants. The present study was approved by the Ethics Committee of the Peking Union Medical College Hospital and the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences.
Supporting information
Data S1: Supporting Information.
Table S1.
ACKNOWLEDGMENTS
We are grateful to the patients and their family members for their participation. This work was financially supported by the National Natural Science Foundation of China (grant number 81788101), the National Key Research and Development Program of China (grant numbers 2022YFC2703900 and 2022YFC2703903), the CAMS Innovation Fund for Medical Sciences (grant numbers 2021‐I2M‐1‐018 and 2022‐I2M‐JB‐004) and National High Level Hospital Clinical Research Funding (grant number 2022‐PUMCH‐A‐161).
Liu, J.‐W. , Guo, K. , Zhang, R. , Wang, R. , Ma, D.‐L. , & Zhang, X. (2024). Compound heterozygous ABCA12 variants identified in a Chinese patient with congenital ichthyosiform erythroderma: Advancing genotype–phenotype correlations and literature review. Molecular Genetics & Genomic Medicine, 12, e2431. 10.1002/mgg3.2431
Jia‐Wei Liu and Kexin Guo contributed equally to this work.
Contributor Information
Rongrong Wang, Email: rongrongbwl@ibms.pumc.edu.cn.
Dong‐Lai Ma, Email: mdonglai@sohu.com.
DATA AVAILABILITY STATEMENT
The data that supports the findings of this study are available in the supplementary material of this article.
REFERENCES
- Ahmed, H. , & O'Toole, E. A. (2014). Recent advances in the genetics and management of harlequin ichthyosis. Pediatric Dermatology, 31(5), 539–546. 10.1111/pde.12383 [DOI] [PubMed] [Google Scholar]
- Akiyama, M. (2010). ABCA12 mutations and autosomal recessive congenital ichthyosis: A review of genotype/phenotype correlations and of pathogenetic concepts. Human Mutation, 31(10), 1090–1096. 10.1002/humu.21326 [DOI] [PubMed] [Google Scholar]
- Akiyama, M. , Sugiyama‐Nakagiri, Y. , Sakai, K. , McMillan, J. R. , Goto, M. , Arita, K. , Tsuji‐Abe, Y. , Tabata, N. , Matsuoka, K. , Sasaki, R. , Sawamura, D. , & Shimizu, H. (2005). Mutations in lipid transporter ABCA12 in harlequin ichthyosis and functional recovery by corrective gene transfer. The Journal of Clinical Investigation, 115(7), 1777–1784. 10.1172/JCI24834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annilo, T. , Shulenin, S. , Chen, Z. Q. , Arnould, I. , Prades, C. , Lemoine, C. , Maintoux‐Larois, C. , Devaud, C. , Dean, M. , Denefle, P. , & Rosier, M. (2002). Identification and characterisation of a novel ABCA subfamily member, ABCA12, located in the lamellar ichthyosis region on 2q34. Cytogenetic and Genome Research, 98(2–3), 169–176. 10.1159/000069811 [DOI] [PubMed] [Google Scholar]
- Chulpanova, D. S. , Shaimardanova, A. A. , Ponomarev, A. S. , Elsheikh, S. , Rizvanov, A. A. , & Solovyeva, V. V. (2022). Current strategies for the gene therapy of autosomal recessive congenital ichthyosis and other types of inherited ichthyosis. International Journal of Molecular Sciences, 23(5), 2506. 10.3390/ijms23052506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar, N. R. , Raza, N. , Khan, A. , & Amin, M. U. (2011). Paraneoplastic Addisonian pigmentation and acquired ichthyosis as presenting features of multiple myeloma. Journal of the College of Physicians and Surgeons–Pakistan, 21(1), 40–42. https://jcpsp.pk/archive/2011/Jan2011/12.pdf [PubMed] [Google Scholar]
- Fischer, J. (2009). Autosomal recessive congenital ichthyosis. The Journal of Investigative Dermatology, 129(6), 1319–1321. 10.1038/jid.2009.57 [DOI] [PubMed] [Google Scholar]
- Hernandez‐Martin, A. , Garcia‐Doval, I. , Aranegui, B. , de Unamuno, P. , Rodriguez‐Pazos, L. , Gonzalez‐Ensenat, M. A. , Vicente, A. , Martin‐Santiago, A. , Garcia‐Bravo, B. , Feito, M. , Baselga, E. , Ciria, S. , de Lucas, R. , Ginarte, M. , Gonzalez‐Sarmiento, R. , & Torrelo, A. (2012). Prevalence of autosomal recessive congenital ichthyosis: A population‐based study using the capture‐recapture method in Spain. Journal of the American Academy of Dermatology, 67(2), 240–244. 10.1016/j.jaad.2011.07.033 [DOI] [PubMed] [Google Scholar]
- Hotz, A. , Kopp, J. , Bourrat, E. , Oji, V. , Sussmuth, K. , Komlosi, K. , Bouadjar, B. , Tantcheva‐Poor, I. , Hellstrom Pigg, M. , Betz, R. C. , Giehl, K. , Schedel, F. , Weibel, L. , Schulz, S. , Stolzl, D. V. , Tadini, G. , Demiral, E. , Berggard, K. , Zimmer, A. D. , … Fischer, J. (2023). Mutational spectrum of the ABCA12 gene and genotype‐phenotype correlation in a cohort of 64 patients with autosomal recessive congenital ichthyosis. Genes (Basel), 14(3), 717. 10.3390/genes14030717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israeli, S. , Goldberg, I. , Fuchs‐Telem, D. , Bergman, R. , Indelman, M. , Bitterman‐Deutsch, O. , Harel, A. , Mashiach, Y. , Sarig, O. , & Sprecher, E. (2013). Non‐syndromic autosomal recessive congenital ichthyosis in the Israeli population. Clinical and Experimental Dermatology, 38(8), 911–916. 10.1111/ced.12148 [DOI] [PubMed] [Google Scholar]
- Kaminski, W. E. , Piehler, A. , & Wenzel, J. J. (2006). ABC A‐subfamily transporters: Structure, function and disease. Biochimica et Biophysica Acta, 1762(5), 510–524. 10.1016/j.bbadis.2006.01.011 [DOI] [PubMed] [Google Scholar]
- Kelsell, D. P. , Norgett, E. E. , Unsworth, H. , Teh, M. T. , Cullup, T. , Mein, C. A. , Dopping‐Hepenstal, P. J. , Dale, B. A. , Tadini, G. , Fleckman, P. , Stephens, K. G. , Sybert, V. P. , Mallory, S. B. , North, B. V. , Witt, D. R. , Sprecher, E. , Taylor, A. E. , Ilchyshyn, A. , Kennedy, C. T. , … O'Toole, E. A. (2005). Mutations in ABCA12 underlie the severe congenital skin disease harlequin ichthyosis. American Journal of Human Genetics, 76(5), 794–803. 10.1086/429844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane, T. S. , Rempe, C. S. , Davitt, J. , Staton, M. E. , Peng, Y. , Soltis, D. E. , Melkonian, M. , Deyholos, M. , Leebens‐Mack, J. H. , Chase, M. , Rothfels, C. J. , Stevenson, D. , Graham, S. W. , Yu, J. , Liu, T. , Pires, J. C. , Edger, P. P. , Zhang, Y. , Xie, Y. , … Stewart, C. N., Jr. (2016). Diversity of ABC transporter genes across the plant kingdom and their potential utility in biotechnology. BMC Biotechnology, 16(1), 47. 10.1186/s12896-016-0277-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, H. W. , Ahn, S. J. , Choi, J. C. , Chang, S. E. , Choi, J. H. , Moon, K. C. , & Koh, J. K. (2006). Acquired ichthyosis associated with an overlap syndrome of systemic sclerosis and systemic lupus erythematosus. The Journal of Dermatology, 33(1), 52–54. 10.1111/j.1346-8138.2006.00010.x [DOI] [PubMed] [Google Scholar]
- Lefevre, C. , Audebert, S. , Jobard, F. , Bouadjar, B. , Lakhdar, H. , Boughdene‐Stambouli, O. , Blanchet‐Bardon, C. , Heilig, R. , Foglio, M. , Weissenbach, J. , Lathrop, M. , Prud'homme, J. F. , & Fischer, J. (2003). Mutations in the transporter ABCA12 are associated with lamellar ichthyosis type 2. Human Molecular Genetics, 12(18), 2369–2378. 10.1093/hmg/ddg235 [DOI] [PubMed] [Google Scholar]
- Loo, B. K. G. , Batilando, M. J. , Tan, E. C. , & Koh, M. J. A. (2018). Compound heterozygous mutations with novel missense ABCA12 mutation in harlequin ichthyosis. BML Case Reports, 2018. 10.1136/bcr-2017-222025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montalvan‐Suarez, M. , Esperon‐Moldes, U. S. , Rodriguez‐Pazos, L. , Ordonez‐Ugalde, A. , Moscoso, F. , Ugalde‐Noritz, N. , Santome, L. , Fachal, L. , Tettamanti‐Miranda, D. , Ruiz, J. C. , Ginarte, M. , & Vega, A. (2019). A novel ABCA12 pathologic variant identified in an Ecuadorian harlequin ichthyosis patient: A step forward in genotype‐phenotype correlations. Molecular Genetics & Genomic Medicine, 7(5), e608. 10.1002/mgg3.608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsuga, K. , Akiyama, M. , Kato, N. , Sakai, K. , Sugiyama‐Nakagiri, Y. , Nishimura, M. , Hata, H. , Abe, M. , Arita, K. , Tsuji‐Abe, Y. , Onozuka, T. , Aoyagi, S. , Kodama, K. , Ujiie, H. , Tomita, Y. , & Shimizu, H. (2007). Novel ABCA12 mutations identified in two cases of non‐bullous congenital ichthyosiform erythroderma associated with multiple skin malignant neoplasia. The Journal of Investigative Dermatology, 127(11), 2669–2673. 10.1038/sj.jid.5700885 [DOI] [PubMed] [Google Scholar]
- Oji, V. , Tadini, G. , Akiyama, M. , Blanchet Bardon, C. , Bodemer, C. , Bourrat, E. , Coudiere, P. , DiGiovanna, J. J. , Elias, P. , Fischer, J. , Fleckman, P. , Gina, M. , Harper, J. , Hashimoto, T. , Hausser, I. , Hennies, H. C. , Hohl, D. , Hovnanian, A. , Ishida‐Yamamoto, A. , … Traupe, H. (2010). Revised nomenclature and classification of inherited ichthyoses: Results of the first ichthyosis consensus conference in Soreze 2009. Journal of the American Academy of Dermatology, 63(4), 607–641. 10.1016/j.jaad.2009.11.020 [DOI] [PubMed] [Google Scholar]
- Paller, A. S. (2019). Profiling immune expression to consider repurposing therapeutics for the ichthyoses. The Journal of Investigative Dermatology, 139(3), 535–540. 10.1016/j.jid.2018.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigg, M. H. , Bygum, A. , Ganemo, A. , Virtanen, M. , Brandrup, F. , Zimmer, A. D. , Hotz, A. , Vahlquist, A. , & Fischer, J. (2016). Spectrum of autosomal recessive congenital ichthyosis in Scandinavia: Clinical characteristics and novel and recurrent mutations in 132 patients. Acta Dermato‐Venereologica, 96(7), 932–937. 10.2340/00015555-2418 [DOI] [PubMed] [Google Scholar]
- Sakai, K. , Akiyama, M. , Sugiyama‐Nakagiri, Y. , McMillan, J. R. , Sawamura, D. , & Shimizu, H. (2007). Localization of ABCA12 from Golgi apparatus to lamellar granules in human upper epidermal keratinocytes. Experimental Dermatology, 16(11), 920–926. 10.1111/j.1600-0625.2007.00614.x [DOI] [PubMed] [Google Scholar]
- Theodoulou, F. L. , & Kerr, I. D. (2015). ABC transporter research: Going strong 40 years on. Biochemical Society Transactions, 43(5), 1033–1040. 10.1042/BST20150139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, A. C. , Cullup, T. , Norgett, E. E. , Hill, T. , Barton, S. , Dale, B. A. , Sprecher, E. , Sheridan, E. , Taylor, A. E. , Wilroy, R. S. , DeLozier, C. , Burrows, N. , Goodyear, H. , Fleckman, P. , Stephens, K. G. , Mehta, L. , Watson, R. M. , Graham, R. , Wolf, R. , … Kelsell, D. P. (2006). ABCA12 is the major harlequin ichthyosis gene. The Journal of Investigative Dermatology, 126(11), 2408–2413. 10.1038/sj.jid.5700455 [DOI] [PubMed] [Google Scholar]
- Umemoto, H. , Akiyama, M. , Yanagi, T. , Sakai, K. , Aoyama, Y. , Oizumi, A. , Suga, Y. , Kitagawa, Y. , & Shimizu, H. (2011). New insight into genotype/phenotype correlations in ABCA12 mutations in harlequin ichthyosis. Journal of Dermatological Science, 61(2), 136–139. 10.1016/j.jdermsci.2010.11.010 [DOI] [PubMed] [Google Scholar]
- Xiong, J. , Feng, J. , Yuan, D. , Zhou, J. , & Miao, W. (2015). Tracing the structural evolution of eukaryotic ATP binding cassette transporter superfamily. Scientific Reports, 5, 16724. 10.1038/srep16724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yogarajah, J. , Gouveia, C. , Iype, J. , Hafliger, S. , Schaller, A. , Nuoffer, J. M. , Fux, M. , & Gautschi, M. (2021). Efficacy and safety of secukinumab for the treatment of severe ABCA12 deficiency‐related ichthyosis in a child. Skin Health and Disease, 1(2), e25. 10.1002/ski2.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1: Supporting Information.
Table S1.
Data Availability Statement
The data that supports the findings of this study are available in the supplementary material of this article.