

Abstract
Background:
Hemophilia A arises from dysfunctional or deficient coagulation factor VIII (FVIII) and leads to inefficient fibrin clot formation and uncontrolled bleeding events. The development of antibody inhibitors is a clinical complication in hemophilia A patients receiving FVIII replacement therapy. LE2E9 is an anti-C1 domain inhibitor previously isolated from a mild/moderate hemophilia A patient and disrupts FVIII interactions with VWF and FIXa, though the intermolecular contacts that underpin LE2E9-mediated FVIII neutralization are undefined.
Objective:
To determine the structure of the complex between FVIII and LE2E9 and characterize its mechanism of inhibition.
Methods:
FVIII was bound to the antigen binding fragment (Fab) of NB2E9, a recombinant construct of LE2E9, and its structure was determined by cryogenic electron microscopy (cryo-EM).
Results:
This report communicates the 3.46 Å structure of FVIII bound to NB2E9, with its epitope comprised of FVIII residues S2040-Y2043, K2065-W2070, and R2150-H2155. Structural analysis reveals that the LE2E9 epitope overlaps with portions of the epitope for 2A9, a murine-derived inhibitor, suggesting these residues represent a shared antigenic region on the C1 domain between FVIII−/− mice and hemophilia A patients. Furthermore, the FVIII:NB2E9 structure elucidates the orientation of the LE2E9 glycan, illustrating how the glycan sterically blocks interactions between the FVIII C1 domain and the VWF D’ domain. A putative model of the FVIIIa:FIXa complex suggests potential clashing between the NB2E9 glycan and FIXa light chain.
Conclusion:
These results describe an antigenic “hot-spot” on the FVIII C1 domain and provide a structural basis for engineering FVIII replacement therapeutics with reduced antigenicity.
Keywords: antibody inhibitor, hemophilia, blood coagulation, cryo-electron microscopy, factor VIII
Introduction
Coagulation factor VIII (FVIII) is secreted as a heterodimer consisting of a heavy chain (A1–A2) and light chain (A3-C1-C2) and circulates as a complex with von Willebrand factor (VWF) which extends the circulatory half-life of FVIII [1–3]. In the event of vascular rupture, thrombin cleaves FVIII yielding activated FVIII (FVIIIa) as a heterotrimer (A1/A2/A3-C1-C2) that dissociates from VWF and binds to platelet membranes; here, FVIIIa initiates localized assembly of the intrinsic tenase complex by binding to activated factor IX (FIXa), which activates factor X (FX) and triggers fibrin clot formation [4]. Congenital hemophilia A (HA) is an X-linked recessive bleeding disorder characterized by uncontrolled bleeding events due to dysfunctional FVIII. Hemostasis in HA patients is maintained through administration of FVIII replacement products. Frequent infusions of recombinant or plasma-derived FVIII products can induce the development of neutralizing antibodies that disrupt FVIII activity. These antibody inhibitors occur in approximately 30% of severe congenital HA patients receiving FVIII infusions [5]. Furthermore, acquired HA can present in healthy individuals whereby antibody inhibitors spontaneously arise and inhibit syngeneic FVIII [6].
Antibody inhibitors that target FVIII predominantly bind to the A2, C1 and C2 domains and disrupt FVIII procoagulant activity through distinct mechanisms [5]. Pathogenic anti-C1 domain inhibitors block FVIII interactions with VWF, phospholipid membranes, and/or FIXa. Recent studies demonstrated that anti-C1 domain inhibitors are categorized into two distinct groups, group A and group B, which bind to non-overlapping epitopes [7]. Group A inhibitors enhance FVIII clearance rates by disrupting interactions with VWF and display weak type II inhibitory kinetics under saturating concentrations of inhibitor [7]. In addition to blocking interactions with VWF, group B inhibitors also inhibit procoagulant FVIII activity by blocking FVIII binding to phospholipid membranes and formation of the intrinsic tenase complex. Previous studies also demonstrated that group B inhibitors disrupt endocytosis of FVIII by dendritic cells for processing and antigen presentation [7–9]. Structures of FVIII bound to group A and group B inhibitors have revealed how inhibitors target the C1 domain with exquisite specificity and disrupt FVIII procoagulant activity [10,11].
LE2E9 is a pathogenic antibody inhibitor previously isolated from an individual with mild/moderate hemophilia A carrying an R2150H missense mutation located on a solvent-exposed region of the C1 domain (Figure S1) [12]. The R2150H variant has been identified in other hemophilia A patients and is strongly associated with inhibitor development [13–18]. Binding measurements between FVIII and LE2E9 demonstrated strong affinity toward the wildtype C1 domain which blocked interactions with VWF (IC50 0.25 μg/mL) [19]. Point mutations to the C1 domain which disrupted LE2E9 binding suggested classification as a group A inhibitor [20]. Intriguingly, the peptide sequence of LE2E9 revealed an N-linked glycosylation site at the paratope [21]. Substituting the glycosylated asparagine with glutamine (LE2E9Q), thereby knocking out the glycosylation site, abolished the inhibition of VWF binding to FVIII and reduced inhibitory activity by 50% yet retained affinity toward FVIII similar to LE2E9, suggesting that the inhibitor potency of LE2E9 is predominantly dependent on the glycan moiety [21]. Because LE2E9Q demonstrated reduced FVIII inhibitory activity, it has been pursued as an antithrombotic under the name TB-402 [22–25].
Here, we report on the structure of FVIII bound to an antigen binding fragment (Fab) of NB2E9, a recombinant construct of LE2E9 with identical variable light and heavy domains, determined by cryogenic electron microscopy (cryo-EM). The LE2E9 epitope is consistent with group A anti-C1 domain inhibitors and demonstrates a shared antigenic role for the FVIII C1 domain between FVIII−/− mice and hemophilia A patients. Further analysis reveals that the LE2E9 glycan occludes FVIII binding sites for VWF and potentially FIXa, providing a structure-based rationale for the mechanism of inhibition by LE2E9.
Methods
Expression and purification of B domain-deleted ET3i factor VIII
ET3i, a bioengineered chimera of human and porcine FVIII, was expressed and purified as previously described and stored in 50 mM HEPES (pH 7.4), 5 mM CaCl2, and 350 mM NaCl at 0.8 mg/mL at −80 °C [26].
Expression and purification of NB2E9 monoclonal antibodies
NB2E9 monoclonal antibodies were expressed and purified as previously described [27]. Fab fragments of NB2E9 were generated by papain cleavage and purified by protein A affinity chromatography as previously described and stored in 20 mM HEPES (pH 7.4) and 150 mM NaCl [10]. Chromogenic and one-stage clotting assays were performed using ET3i as previously described [27].
Cryo-EM sample preparation
The ET3i:NB2E9 complex was prepared by combining ET3i (5 μM) and NB2E9 Fabs (9.5 μM) at a 1:1.2 stoichiometric molar ratio (ET3i:NB2E9) and diluting the sample in 20 mM HEPES (pH 7.4), 150 mM NaCl to a final concentration of 0.1 mg/mL based on the absorbance at 280 nm. After allowing the sample to incubate on ice for 10 minutes, 3 μL of sample were applied to plasma-cleaned lacey 400 mesh grids with a continuous carbon support (Ted Pella) under 95–100% humidity at 4 °C. Following a 5 second wait time, the grid was blotted for 3 seconds with a −10 blot force and plunge frozen in liquid ethane using a Vitrobot Mark IV vitrification instrument (Thermo Fisher). After confirming optimal particle monodispersity (Figures S2A,S3A), the grid was stored under liquid nitrogen prior to data collection.
Cryo-EM data collection and processing
Cryo-EM data collection was performed with a Titan Krios cryogenic transmission electron microscope operating at 300 kV, resulting in 7,793 dose fractionated movies at a raw pixel size of 0.394 Å/pixel and a total exposure dose of 50.0 e−/Å2. A complete list of data collection parameters is provided in Table 1.
Table 1.
Cryo-EM data collection parameters and refinement statistics
| Map | |
|---|---|
| Microscope | Titan Krios |
| Magnification | 29,000 |
| Voltage (kV) | 300 |
| Defocus range (μm) | −0.8 to −2.0 |
| Exposure time (s) | 1.498 |
| Dose (e−/Å2) | 50.0 |
| Pixel size (Å) | 0.788 (super res, 0.394) |
| Micrographs | 7,793 |
| Initial particle images | 5,364,884 |
| Final particle images | 94,935 |
| Symmetry imposed | C1 |
| Map-sharpening B factor (Å2) | −82.3 |
| Map resolution, unmasked (Å) | 4.40 |
| Map resolution, masked (Å) | 3.46 |
| FSC threshold | 0.143 |
| Model | |
| Refinement program | PHENIX, ISOLDE |
| Model composition | |
| Non-hydrogen atoms | 13,509 |
| Amino acids | 1,675 |
| Ligands | 12 |
| RMSD | |
| Bond lengths (Å) | 0.005 |
| Bond angles (°) | 0.857 |
| Ramachandran plot (%) | |
| Favored | 88.9 |
| Allowed | 10.6 |
| Disallowed | 0.5 |
| Validation | |
| Clashscore | 22.72 |
| MolProbity score | 2.43 |
| Rotamer outliers (%) | 0.68 |
All processing of cryo-EM data was performed through cryoSPARC (Figure S2B) [28,29]. Pre-processing was carried out using patch motion correction (binned 2x) and patch CTF estimation. Corrected movies were curated to remove micrograph images with suboptimal ice thickness and poor CTF fit resolution. Blob picking resulted in 5,364,884 particles which were extracted at 1.576 Å/pixel in a 150-pixel box. Two rounds of 2D classification and ab initio reconstruction identified 70,428 particles displaying unequivocal densities for ET3i and NB2E9 domains (Figure S3B). Subsequent non-uniform refinement yielded a 4.13 Å map which was low-pass-filtered to 20 Å and used for template picking, resulting in 943,360 particles. Particles were extracted (3.152 Å/pixel, 100-pixel box) and underwent several rounds of 2D classification and ab initio reconstruction. Selected particles were re-extracted (1.576 Å/pixel, 270-pixel box) and used for non-uniform refinement, resulting in a 3.96 Å map with discernable densities for glycans on ET3i and NB2E9. Following a second round of template picking and 2D classification, particles were pooled and duplicates were removed, resulting in 109,994 unique particles. Ab initio reconstruction, heterogeneous and non-uniform refinement yielded a 4.0 Å map. Re-extracting particles (0.788 Å/pixel, 540-pixel box) and performing a second round of heterogeneous and non-uniform refinement generated a final 3.46 Å sharpened map from 94,935 particles (Figures S2C–S2E).
Model building
The 3.2 Å crystal structure of ET3i (PDB: 6MF0) and a model of NB2E9, built through the SWISS-MODEL server using the 2.0 Å crystal structure of BO2C11 as a template (PDB: 1IQD) [30], were rigid body fit into the final sharpened map and iteratively refined by PHENIX [31] and ISOLDE [32]. Glycans were built using Coot. The final sharpened map was deposited into the Electron Microscopy Data Bank and the refined model was deposited into the Protein Data Bank under accession codes EMD-41710 and 8TY1, respectively.
N-glycan preparation
RapiFluor-MS (RFMS) labelled N-glycans were prepared using Waters Glycoworks RapiFluor-MS kit reagents and protocols (Waters, Milford, MA, USA). Samples were buffer exchanged into water and concentrated to 2 mg/mL (calculated using a conversion factor of 120 IU/mg for each product) using 10000 Da molecular weight cutoff (MWCO) spin concentrators (Millipore, Burlington, MA, USA). Volumes of 7.5 μL were deglycosylated using PNGase F and labelled with RFMS according to the manufacturers’ instructions. Labelled N-glycans were purified using a Waters hydrophilic interaction chromatography-mass spectrometry (HILIC) μElution plate (Waters, Milford, MA, USA), as per manufacturers’ instructions. Samples were eluted with three 30 μL of 200 mM ammonium acetate in 5% acetonitrile, and then directly analyzed via liquid chromatography-mass spectrometry with fluorescence detection (LC-FLR-MS). To enable larger injection volumes, 9 μl samples of purified, labelled N-glycans were diluted with 10 μL of dimethylformamide (Waters, Milford, MA, USA) and 21 μL of acetonitrile (Fisher Scientific, Hampton, NH, USA).
Liquid chromatography-fluorescence detection-mass spectrometry (LC-FLR-MS)
LC-FLR-MS analysis was performed using an Acquity H-Class Bio UPLC system equipped with a single quadrupole detector (SQD2) (Waters Corp., Milford, MA, USA). RFMS-labelled N-glycan separations were achieved by injection of 1 μL sample volumes onto a 2.1 × 150 mm, 1.7 μm, 130 Å column (ACQUITY UPLC BEH Amide, Waters, Milford, MA) maintained at 55 °C. Dilution of 9 μL N-glycans in 10 μL of dimethylformamide and 21 of μL acetonitrile enabled injection of 30 μL sample volumes for enhanced signal intensity. Gradient elution was performed using 50 mM ammonium formate pH 4.4 (A) and acetonitrile (B), with transition from 20:80 to 42:58 (A:B) over 39 minutes, at a flow rate of 0.4 mL/min. Fluorescence was monitored at 265/425 nm (excitation/emission). Mass spectrometry analysis of RFMS-labelled N-glycans was performed using the SQD2 instrument in positive mode electrospray (ESI+), with a capillary voltage of 3 kV, cone voltage of 40 V, source temperature of 150 °C, desolvation temperature of 350 °C and desolvation gas flow of 800 L/hour. Data was acquired, processed and analyzed using Empower 3.1 software.
Modeling of the intrinsic tenase complex
A model of the intrinsic tenase complex was built using the ROBETTA structure prediction server with the structure of the prothrombinase complex (PDB code: 7TPP) as a template and sequences for human FVIII and FIX.
Results
Cryo-EM structure of ET3i:NB2E9
The structure of ET3i, a bioengineered chimera of human and porcine FVIII, in-complex with a Fab fragment of NB2E9 was determined to a nominal resolution of 3.46 Å by single-particle cryo-EM using 94,935 particles (Figures 1A,B). The conformations of the ET3i domains (A1-A2/A3-C1-C2) are consistent with previously described FVIII structures [33–35]. Alignment with the crystal structure of isolated ET3i (PDB ID: 6MF0) revealed no significant conformational rearrangements to the ET3i molecule peptide backbone (RMSD of 1.12 Å across 969 Cα atoms) [35]. Throughout cryo-EM data processing, no structural heterogeneity was observed, specifically with respect to the C2 domain which retains the canonical conformation. The quality of the ET3i:NB2E9 cryo-EM map also allowed atomic modeling of the NB2E9 Fab variable (Fv) and constant (Fc) domains and complementarity determining region (CDR) loops (Figure S4).
Figure 1. ET3i:NB2E9 cryo-EM map and structure.

(A,B) Cryo-EM map (A) and model (B) of the ET3i:NB2E9 complex. A1 domain, dark blue; A2 domain, light blue; A3 domain, cyan; C1 domain, orange; C2 domain, yellow; NB2E9 light chain, light green; NB2E9 heavy chain, dark green. Glycans are colored white in (A) and represented as sticks in (B).
The NB2E9 epitope localizes to the ET3i C1 domain, which carries the complete sequence of the human FVIII C1 domain, and has a buried surface area of 746 Å2, consistent with previously reported inhibitor-bound FVIII structures [10,11,30,36,37]. NB2E9 recognizes a discontinuous epitope comprised predominantly of FVIII residues S2040-Y2043, K2065-W2070, and R2150-H2155. FVIII residue E2066 forms several hydrogen bonds with adjacent hydrophilic amino acids contributed by the NB2E9 L1 and L3 loops (Figure 2A). The NB2E9 CDR H1 and H3 loops, which are longer than the other NB2E9 CDR loops by approximately 10 amino acids, wrap around and form extensive contacts with the C1 domain. Interestingly, FVIII residue F2068 is coordinated by two histidine residues from the NB2E9 CDR H1 and H3 loops through a bidentate cation-π interaction on both sides of the phenyl ring and is further stabilized by adjacent hydrophobic interactions (Figure 2B). Additionally, the NB2E9 CDR H3 loop forms extensive hydrophobic contacts with ET3i, including bulky residues W2070 and W2112 (Figure 2C). While the ET3i:NB2E9 structure demonstrates no interactions between NB2E9 and the FVIII A3 domain, the cryo-EM map reveals density at lower contour levels (4–5σ) between NB2E9 residue D58 and the FVIII N1810 A3 domain glycan which is conserved between human and porcine FVIII (Figure 3).
Figure 2. The ET3i:NB2E9 interface.

Intermolecular contacts between ET3i C1 domain (orange) and (A) NB2E9 light chain (light green) and (B,C) heavy chain (dark green).
Figure 3. Structures of ET3i:NB2E9 glycans.
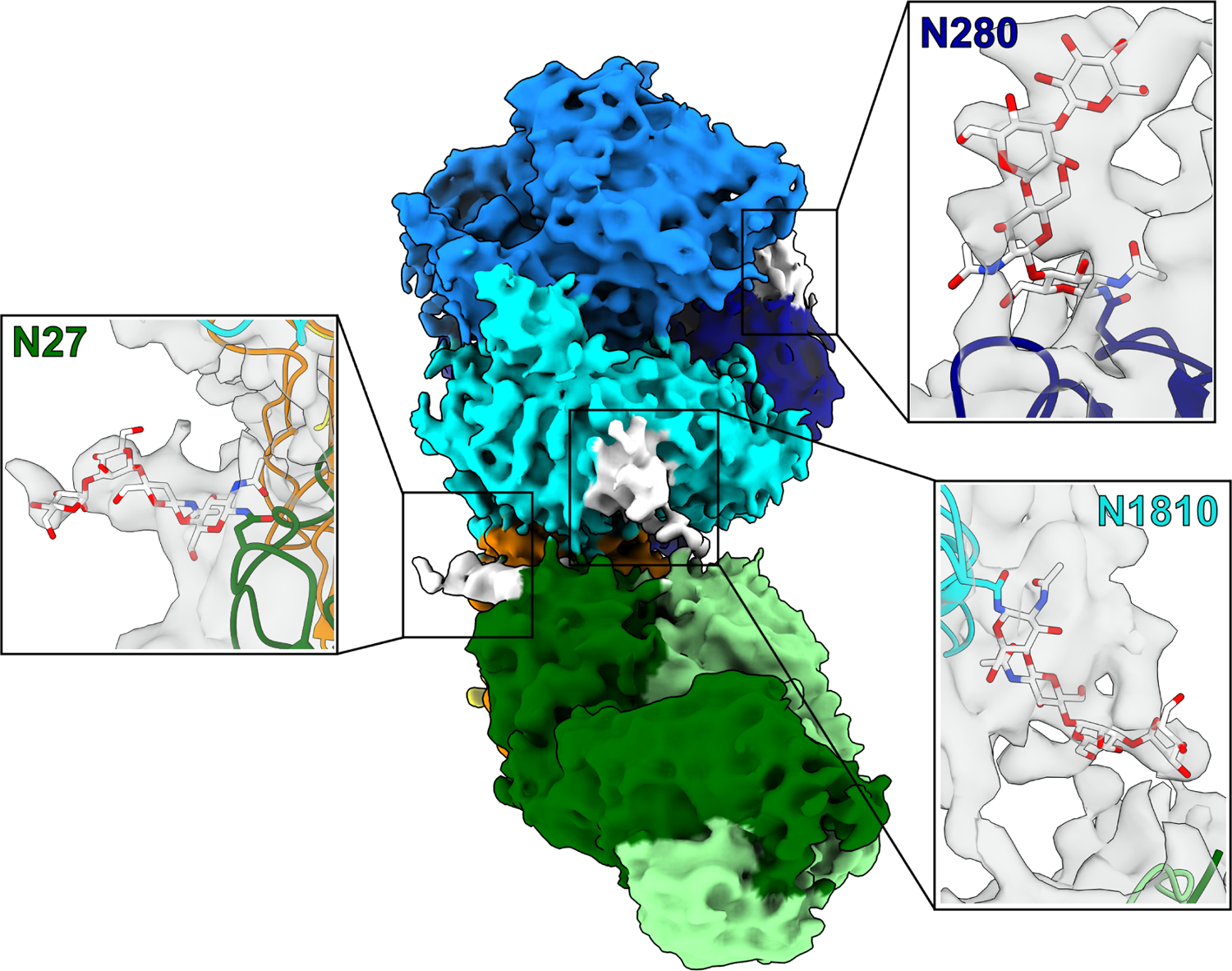
Cryo-EM map of the ET3i:NB2E9 complex. Insets depict glycans on ET3i residues N280 and N1810 and NB2E9 residue N27. A1 domain, dark blue; A2 domain, light blue; A3 domain, cyan; C1 domain, orange; C2 domain, yellow; NB2E9 light chain, light green; NB2E9 heavy chain, dark green. Glycans are colored white and represented as sticks.
Characterization of NB2E9 glycan
To confirm neutralization of ET3i activity by NB2E9, we performed several inhibition assays and compared the results with the inhibitory activity of NB33, a group AB anti-C1 domain inhibitor (Figures S5A,B) [11,27]. NB2E9 displayed IC50 values of 0.51 nM and 8.6 nM using chromogenic and one-stage clotting assays, respectively, which are comparable with inhibition of human FVIII by LE2E9 and NB2E9 [19,27].
Previous analysis of natively and recombinantly expressed LE2E9 using normal phase liquid chromatography revealed the presence of a mixture of N-glycans covalently attached to residue N27 on the CDR H1 loop [21]. Whilst native LE2E9 possessed a predominant biantennary, di-sialylated glycan with a bisecting N-acetylglucoasmine (NAG) (FA2BG2S2), the Chinese hamster ovary (CHO)-derived version predominantly contained the non-bisected mono- (FA2G2S1) and di-sialylated (FA2G2S2) equivalent glycans [21]. Our cryo-EM map allows visualization of several glycosylation sites on the ET3i:NB2E9 protein complex, including the NB2E9 glycan, showing sufficient density near residue N27 for a glycan core consisting of N-NAG2-MAN2 (Figure 3). This represents only a small portion of the structure of each N-glycan identified in NB2E9, with further N-glycan analysis using HILIC-MS identifying 20 N-glycans (Figures S5C,D), all of which are larger than the N-NAG2-MAN2 core identified by cryo-EM. Most prevalent were the mono- (FA1) and bi- (FA2) antennary structures at approximately 20% and 50% relative abundances, respectively. As the most abundant glycan moiety, the FA2 glycan had an estimated molecular weight of 1463 Da which is slightly smaller than previous calculations for LE2E9 when grown in CHO cells [21]. The remainder were a mixture of high-mannose (~6%) and complex asialylated bi- and tri- (~5%) antennary glycoforms. This differs from the N-glycosylation profiles previously observed for native and CHO-derived LE2E9 [21], in which larger di-sialylated structures were identified. Interestingly, NB33 displayed a simpler N-glycosylation profile, with approximately 85% composed of FA2 structures. The ET3i:NB2E9 structure suggests that no interactions occur between the NB2E9 glycan and the adjacent A3–C1 domains of FVIII and is instead oriented away from the ET3i:NB2E9 interface. Intramolecular interactions with the CDR H1 and H2 loops contact the glycan, including NB2E9 residue Y32 which stabilizes the glycan core and blocks interactions with the FVIII C1 domain (Figure S6).
Discussion
The C1 domain of FVIII is a key determinant for binding to VWF, docking onto lipid membranes, and formation of the intrinsic tenase complex [38–41]. In this study, we provide structural evidence for the mechanism of inhibition by LE2E9, an anti-C1 domain antibody inhibitor previously shown to disrupt FVIII interactions with VWF and formation of the intrinsic tenase complex through a novel N-linked glycan [21,42].
LE2E9 is a group A inhibitor that binds to a shared epitope amongst FVIII−/− mice and hemophilia A patients
Previous studies demonstrated strong similarities between LE2E9 and 2A9, a group A anti-C1 domain inhibitor purified from a hemophilia A murine model that disrupts FVIII binding to VWF and lipid membranes (Figure S7) [7,20]. We previously determined the 3.9 Å crystal structure of ET3i bound to 2A9, allowing for direct structural comparison between the LE2E9 and 2A9 epitopes [10]. Superposition of the ET3i:2A9 and ET3i:NB2E9 structures reveals multiple overlapping regions between the 2A9 and LE2E9 epitopes which center on FVIII residues P2067-W2070, R2150-Y2156, and portions of residues I2102-V2125 (Figure 4A), supporting classification of LE2E9 as a group A inhibitor. This region of FVIII has previously been described as highly antigenic in healthy donors and hemophilia A patients. Isolated peptides encoding portions of the C1 domain were used to probe the T cell response to FVIII, revealing residues I2098-W2112 induced the strongest immune response [43]. Two mutations to this peptide, M2104K and L2107T, significantly lowered the risk of inhibitor development. Furthermore, epitope mapping using hydrogen-deuterium exchange coupled with mass spectrometry (HDX-MS) demonstrated reduced deuterium uptake to FVIII residues S2063-I2071 and N2129-K2136 in the presence of several murine-derived anti-C1 domain inhibitors, including 2A9 [7]. Correlating these observations with our structural analysis suggests that this region of the C1 domain represents a potent antigenic region that is recognized by both FVIII−/− mice and hemophilia A patients. Future research will determine the immunogenicity of the FVIII C1 domain, specifically overlapping portions of the 2A9 and NB2E9 epitopes, in mice and humans and whether modification to this region of FVIII can reduce recognition by CD4+ T cells.
Figure 4. Structural comparison between LE2E9/2A9 epitopes and vWF binding site.

Surface representation of the A3–C1 domains from ET3i (PDB ID: 6MF0) (grey). (A) Blue and red colored surfaces highlight FVIII amino acids which exclusively bind inhibitors 2A9 and LE2E9, respectively. Purple colored surface illustrates FVIII amino acids which overlap between 2A9 and LE2E9 epitopes. (B) Orange and light purple surfaces highlight FVIII amino acids which exclusively bind vWF and LE2E9, respectively. Green colored surface illustrates FVIII amino acids which overlap between vWF and LE2E9 binding sites.
Comparison of the ET3i:2A9 and ET3i:NB2E9 structures reveals subtle, yet significant, differences between binding mechanisms. Previous studies elucidated FVIII variant E2066D abolished LE2E9 binding yet retained affinity toward 2A9 [20]. Comparison of the ET3i:2A9 and ET3i:NB2E9 structures reveals how the NB2E9 inhibitor docks closely to E2066 and forms several hydrogen bonds, while 2A9 is oriented away from residue E2066 and closer to the A3–C1 domain interface (Figures 2A and S7A,B). Conversely, FVIII F2068H abolished 2A9 affinity, yet had no impact LE2E9 binding. We previously described how the 2A9 paratope utilizes a cluster of hydrophobic residues to bind F2068 and proposed that these interactions would be destabilized by the F2068H variant [10]. The ET3i:NB2E9 structure reveals that NB2E9 binds F2068 through cation-π interactions formed by residues H37 and H122 at the CDR H1 and H3 loops (Figure 2B). These residues would presumably hydrogen bond with the FVIII F2068H variant and retain binding to FVIII. Lastly, the ET3i:2A9 crystal structure revealed how the FVIII C2 domain adopts a 20° swivel-like conformational change when bound to 2A9 [10]. The ET3i:NB2E9 cryo-EM structure demonstrated no rearrangement to the C2 domain, suggesting that the C2 domain conformational change observed in the ET3i:2A9 structure is unique to 2A9 binding to FVIII.
LE2E9 was originally isolated from a patient with mild/moderate hemophilia A carrying an R2150H missense mutation and demonstrated inhibition of allogeneic, but not syngeneic, FVIII [12]. Subsequent binding studies demonstrated that the R2150H variant has a lower affinity toward group A inhibitors, including 2A9 and LE2E9, as well as group B inhibitors, despite both groups binding to non-overlapping epitopes [19,20]. The ET3i:NB2E9 structure presented in this study illustrates that NB2E9 occludes FVIII residue R2150, but does not make direct non-covalent contacts with the guanidino functional group. Analysis of previously published FVIII structures reveals how residue R2150 is sandwiched between two solvent-exposed tryptophan residues, W2070 and W2112, forming a bidentate cation-π interaction [35,39]. Because both tryptophan residues form hydrophobic contacts with the NB2E9 H3 loop (Figure 2C), it is plausible that the R2150H variant disrupts intramolecular contacts with FVIII residues W2070 and W2112. The FVIII R2150H variant also disrupts binding to VWF, yet the recent cryo-EM structure of BIVV001, a bioengineered fusion of human FVIII and the D’D3 domains of VWF, revealed no direct interactions between residue R2150 and VWF [19,39]. Together, these observations suggest that the FVIII R2150H variant may induce a conformational change to the C1 domain which disrupts binding to VWF and anti-C1 domain inhibitors such as 2A9 and LE2E9, as previously hypothesized [19,20].
LE2E9 glycan sterically blocks binding to VWF
Glycosylation of antibody Fv domains occurs in approximately 20% of all naturally occurring IgG antibodies [44]. LE2E9 was previously shown to carry a glycan covalently attached to the CDR H1 loop [21]. LE2E9Q, a deglycosylated variant of LE2E9, exhibited 50% lower inhibitory activity in a chromogenic assay yet retains binding affinity toward FVIII similar to glycosylated LE2E9, suggesting that the glycan mediates LE2E9 pathogenicity without making direct contacts with FVIII [21]. The ET3i:NB2E9 structure supports these predictions, demonstrating that the NB2E9 glycan is oriented away from the C1:NB2E9 interface. Superposition of the ET3i:NB2E9 structure with the cryo-EM structure of BIVV001 highlights several overlapping regions on the FVIII C1 domain while illustrating how the NB2E9 glycan orientation blocks FVIII binding to VWF through steric hindrance (Figures 4B,5). Indeed, multiple hydrophobic residues on the C1 domain which directly bind to the VWF D’ domain are occluded by the NB2E9 glycan, including V2125, F2127, and V2130 [38,39]. This steric hindrance is relieved by deglycosylation of LE2E9, as previous studies demonstrated that LE2E9Q did not disrupt FVIII binding to VWF [21]. Structural comparison between the ET3i:2A9 and ET3i:NB2E9 structures demonstrates how 2A9 forms direct contacts with portions of the FVIII A3 domain, specifically FVIII residues D1740-Y1748, which participate in interactions with the VWF D’ domain (Figures 4A,B). FVIII residues D1740-Y1748 are adjacent to the NB2E9 glycan in the ET3i:NB2E9 structure and may be occluded by larger branched glycans identified in the HILIC-MS glycan profile (Figure S5D).
Figure 5. Superposition of ET3i:NB2E9 and BIVV001 structures.

Structures of BIVV001 (PDB ID: 7KWO) and ET3i:NB2E9 were aligned, demonstrating significant steric clashing between the NB2E9 glycan (white sticks) and VWF D’ domain (pink). C1 domain, orange; NB2E9 light chain, light green; NB2E9 heavy chain, dark green.
Our analysis of the glycan profile of NB2E9 differs slightly from the glycostructure previously described for native and CHO-derived LE2E9 [21], revealing a complex mixture of mono- and bi- antennary structures as the dominant glycoform and lower abundances of high-mannose and complex asialylated bi- and tri- antennary glycoforms. The density in the cryo-EM map for the NB2E9 glycan was only sufficient for a glycan core consisting of N-NAG2-MAN2 residues presumably due to glycan flexibility and microheterogeneity. While the NB2E9 glycan displays no direct interactions with the C1 domain, we cannot rule out intermolecular interactions with unmodeled branches of the NB2E9 glycan.
LE2E9 glycan possibly blocks FVIII binding to FIXa/FX
The inhibitory activity of LE2E9 has been previously investigated with respect to inhibition of the intrinsic tenase complex assembly and FX activation [42]. Indeed, LE2E9 and LE2E9Q lowered the apparent Vmax of the intrinsic tenase complex by 77% and 22%, respectively, indicative of the LE2E9 glycan as a possible determinant in inhibition. Conversely, LE2E9 had no effect on intrinsic tenase complex activity in the presence of Gla-domainless FX.
To further investigate the role of LE2E9 in disrupting intrinsic tenase complex activity, we generated a homology model of the FVIIIa:FIXa complex using the ROBETTA structure prediction server and the recent cryo-EM structure of the prothrombinase complex, formed by activated factors V and X which carry 41% and 45% sequence identity with FVIIIa and FIXa, respectively, as a template (Figure 6A) [45,46]. Our putative model of the FVIIIa:FIXa complex suggests that the FIXa light chain packs closely against the FVIIIa A3–C1 domains and the FIXa catalytic domain docks onto the FVIIIa A2 domain, consistent with previous studies [41,47–50]. Superposition with the ET3i:NB2E9 structure indicates minimal overlap between the LE2E9 epitope and putative binding site for the FIXa light chain on the C1 domain. However, there is potential for steric clashing between the LE2E9 glycan and the FIXa Gla-EGF1 domains (Figure 6B). Substituting wildtype FX with the Gla-domainless FX could allow FIXa to conformationally rearrange to overcome steric clashing with the LE2E9 glycan while maintaining optimal interactions with FVIIIa for FX activation. Because there is currently no high-resolution structure of the intrinsic tenase complex, it is plausible that FX binds to the FVIIIa A2/A3-C1 domains and clashes with the LE2E9 glycan, rather than FIXa. This would support previous results demonstrating inhibition of the intrinsic tenase complex by glycosylated LE2E9 only in the presence of full-length, but not Gla-domainless, FX [42].
Figure 6. Superposition of ET3i:NB2E9 structure and model of the intrinsic tenase complex.

(A) Model of the human intrinsic tenase complex was generated by the ROBETTA structure prediction server using the structure of the prothrombinase complex as a template (PDB ID: 7TPP). (B) The C1:NB2E9 structure was superimposed with the model of the intrinsic tenase complex, showing significant steric clashing between the NB2E9 glycan and FIXa light chain. A1 domain, dark blue; A2 domain, light blue; A3 domain, cyan; C1 domain, orange; C2 domain, yellow; FIXa heavy chain, light red; FIXa light chain, maroon; NB2E9 light chain, light green; NB2E9 heavy chain, dark green. Glycans are colored white and represented as sticks.
Because LE2E9Q retains inhibitory properties against the intrinsic tenase complex [42], NB2E9 may inhibit FVIII through other mechanisms independent of the glycan group. Indeed, the murine-derived inhibitor 2A9, which has strong epitope overlap with NB2E9 (Figure 4A) and lacks a glycosylation site at the Fv domain, partially blocked FVIII binding to lipid membranes (IC50 0.9 μg/mL) [7]. While the 2A9 and NB2E9 epitopes do not incorporate FVIII residues predicted to be involved directly in membrane binding, the observed disruption to FVIII binding to phospholipid surfaces in the presence of 2A9 most likely occurs through steric hindrance. LE2E9Q may disrupt FVIII binding to phospholipid membranes similarly. Alternatively, LE2E9Q may prevent the formation of optimal interfacial contacts between the FVIIIa A3–C1 domains and FIXa light chain necessary for FX activation. Determining a high-resolution structure of the intrinsic tenase complex will aid in elucidating the extent to which the glycan plays in mediating LE2E9 inhibition.
In summary, this study describes the structural basis for FVIII inhibition by NB2E9, a recombinant derivative of LE2E9. Our analysis of the LE2E9 epitope reveals strong overlap with 2A9, a murine-derived anti-C1 domain inhibitor, indicating the C1 domain plays a significant role in FVIII antigenicity that is shared between mice and humans. Further, the ET3i:NB2E9 structure reveals the structural basis for inhibition FVIII by the LE2E9 glycan, revealing how the glycan sterically blocks FVIII binding to VWF and FIXa. Future work will delineate how patients with the R2150H variant are at a higher risk for inhibitor development. Because LE2E9Q has been pursued as an antithrombotic in the treatment of pulmonary thromboembolism, our results also provide a new avenue in designing antithrombotic therapeutics targeting the intrinsic tenase complex.
Supplementary Material
Table 2.
List of non-covalent contacts between ET3i and NB2E9 (≤ 5 Å)
| ET3i amino acid | NB2E9 amino acid | Distance (Å) |
|---|---|---|
| S2040 | A30 (Light chain) | 4.4 |
| G2041 (NH) | S28 (CO) (Light chain) | 3.9 |
| Y2043 | Q27 (Light chain) | 4.1 |
| K2065 | S95 (Light chain) | 4.3 |
| E2066 | Q27 (Light chain) | 4.1 |
| Q91 (Light chain) | 3.4 | |
| T94 (Light chain) | 3.3 | |
| S95 (Light chain) | 4.2 | |
| P2067 | T94 (Light chain) | 3.8 |
| F2068 | H37 (Heavy chain) | 3.5 |
| F113 (Heavy chain) | 3.6 | |
| T120 (Heavy chain) | 3.3 | |
| H122 (Heavy chain) | 3.6 | |
| W2070 | I115 (Heavy chain) | 4.3 |
| V116 (Heavy chain) | 4.3 | |
| I2102 | H37 (Heavy chain) | 4.8 |
| M2104 | T117 (Heavy chain) | 3.7 |
| W2112 | T117 (Heavy chain) | 4.8 |
| V2125 | S35 (Heavy chain) | 5.0 |
| P2153 (CO) | H37 (Heavy chain) | 4.5 |
| T2154 | H37 (Heavy chain) | 3.8 |
| H2155 | S33 (Heavy chain) | 4.4 |
Funding
This work was supported by the National Bleeding Disorders Foundation Judith Graham Pool Postdoctoral Research Fellowship (K.C.C.) and the National Institutes of Health (NIH), National Heart, Lung, and Blood Institute (award numbers R15HL135658 and U54HL141981 [P.C.S.] and award numbers R44HL117511, R44HL110448, U54HL112309, and U54HL141981 [C.B.D., P.L.]). A portion of this research was supported by NIH grant U24GM129547 and performed at the PNCC at OHSU and accessed through EMSL (grid.436923.9), a DOE Office of Science User Facility sponsored by the Office of Biological and Environmental Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest Disclosure
P.L. is listed as an inventor on a patent application describing ET3i and on patents owned by Emory University claiming compositions of matter that include modified FVIII proteins with reduced reactivity with anti-FVIII antibodies. C.B.D. and P.L. are cofounders of Expression Therapeutics and own equity in the company. Expression Therapeutics owns the intellectual property associated with ET3i. The terms of this arrangement have been reviewed and approved by Emory University in accordance with its conflict-of-interest policies. The remaining authors declare no competing financial interests.
References
- 1.Fay PJ. Factor VIII structure and function. Int J Hematol 2006; 83: 103–8. [DOI] [PubMed] [Google Scholar]
- 2.Leyte A, Van Schijndel HB, Niehrs C, Huttner WB, Verbeet MP, Mertens K, Van Mourik JA. Sulfation of Tyr1680 of human blood coagulation Factor VIII is essential for the interaction of Factor VIII with von Willebrand factor. J Biol Chem 1991; 266: 740–6. [PubMed] [Google Scholar]
- 3.Wise RJ, Dorner AJ, Krane M, Pittman DD, Kaufman RJ. The role of von Willebrand factor multimers and propeptide cleavage in binding and stabilization of factor VIII. J Biol Chem 1991; 266: 21948–55. [PubMed] [Google Scholar]
- 4.Pittman DD, Kaufman RJ. Proteolytic requirements for thrombin activation of anti-hemophilic factor (factor VIII). Proc Natl Acad Sci U S A 1988; 85: 2429–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lollar P. Pathogenic antibodies to coagulation factors. Part one: Factor VIII and Factor IX. J Thromb Haemost 2004; 2: 1082–95. [DOI] [PubMed] [Google Scholar]
- 6.Tiede A, Collins P, Knoebl P, Teitel J, Kessler C, Shima M, Di Minno G, D’Oiron R, Salaj P, Jiménez-Yuste V, Huth-Kühne A, Giangrande P. International recommendations on the diagnosis hemophiliaand a treatment of acquired hemophilia A. Haematologica 2020; 105: 1791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Batsuli G, Deng W, Healey JF, Parker ET, Baldwin WH, Cox C, Nguyen B, Kahle J, Königs C, Li R, Lollar P, Meeks SL. High-affinity, noninhibitory pathogenic C1 domain antibodies are present in patients with hemophilia A and inhibitors. Blood 2016; 128: 2055–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meems H, Van Den Biggelaar M, Rondaij M, Van Der Zwaan C, Mertens K, Meijer AB. C1 domain residues Lys 2092 and Phe 2093 are of major importance for the endocytic uptake of coagulation factor VIII. Int J Biochem Cell Biol 2011; 43: 1114–21. [DOI] [PubMed] [Google Scholar]
- 9.Herczenik E, Van Haren SD, Wroblewska A, Kaijen P, Van Den Biggelaar M, Meijer AB, Martinez-Pomares L, Ten Brinke A, Voorberg J. Uptake of blood coagulation factor VIII by dendritic cells is mediated via its C1 domain. J Allergy Clin Immunol 2012; 129: 501–9. [DOI] [PubMed] [Google Scholar]
- 10.Gish JS, Jarvis L, Childers KC, Peters SC, Garrels CS, Smith IW, Spencer HT, Doering CB, Lollar P, Spiegel PC. Structure of blood coagulation factor VIII in complex with an anti–C1 domain pathogenic antibody inhibitor. Blood 2021; 137: 2981–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Childers KC, Avery NG, Estrada Alamo KA, Davulcu O, Haynes RM, Lollar P, Doering CB, Coxon CH, Spiegel PC. Structure of coagulation factor VIII bound to a patient-derived anti-C1 domain antibody inhibitor. Blood J 2023; 142: 197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peerlinck K, Jacquemin MG, Arnout J, Hoylaerts MF, Gilles JGG, Lavend’homme R, Johnson KM, Freson K, Scandella D, Saint-Remy J-MR, Vermylen J. Antifactor VIII Antibody Inhibiting Allogeneic but not Autologous Factor VIII in Patients With Mild Hemophilia A. Blood 1999; 93: 2267–73. [PubMed] [Google Scholar]
- 13.Santagostino E, Gringeri A, Tagliavacca L, Mannucci PM. Inhibitors to factor VIII in a family with mild hemophilia: Molecular characterization and response to factor VIII and desmopressin. Thromb Haemost 1995; 74: 619–21. [PubMed] [Google Scholar]
- 14.Tavassoli K, Eigel A, Wilke K, Pollmann H, Horst J. Molecular diagnostics of 15 hemophilia a patients: Characterization of eight novel mutations in the factor VIII gene, two of which result in exon skipping. Hum Mutat 1998; 12: 301–3. [DOI] [PubMed] [Google Scholar]
- 15.Liu ML, Nakaya S, Thompson AR. Non-inversion factor VIII mutations in 80 hemophilia A families including 24 with alloimmune responses. Thromb Haemost 2002; 87: 273–6. [PubMed] [Google Scholar]
- 16.Fernández-López O, García-Lozano JR, Núñez-Vázquez R, Pérez-Garrido R, Núñez-Roldán A. The spectrum of mutations in Southern Spanish patients with hemophilia A and identification of 28 novel mutations. Haematologica 2005; 90: 707–10. [PubMed] [Google Scholar]
- 17.Kempton CL, Allen G, Hord J, Kruse-Jarres R, Pruthi RK, Walsh C, Young G, Soucie JM. Eradication of factor viii inhibitors in patients with mild and moderate hemophilia A. Am J Hematol 2012; 87: 933–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwaab R, Pavlova A, Albert T, Caspers M, Oldenburg J. Significance of F8 missense mutations with respect to inhibitor formation. Thromb Haemost 2013; 109: 464–70. [DOI] [PubMed] [Google Scholar]
- 19.Jacquemin M, Benhida A, Peerlinck K, Desqueper B, Vander Elst L, Lavend’homme R, D’Oiron R, Schwaab R, Bakkus M, Thielemans K, Gilles J-G, Vermylen J, Saint-Remy J-M. A human antibody directed to the factor VIII C1 domain inhibits factor VIII cofactor activity and binding to von Willebrand factor. Blood 2000; 95: 156–63. [PubMed] [Google Scholar]
- 20.Kahle J, Orlowski A, Stichel D, Healey JF, Parker ET, Jacquemin M, Krause M, Tiede A, Königs C, Schwabe D, Lollar P. Frequency and epitope specificity of anti–factor VIII C1 domain antibodies in acquired and congenital hemophilia A. Blood 2017; 130: 808–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jacquemin M, Radcliffe CM, Lavend’Homme R, Wormald MR, Vanderels L, Wallays G, Dewaele J, Collen D, Vermylrn J, Dwek RA, Saint-Remy JM, Rudd PM, Dewerchin M. Variable region heavy chain glycosylation determines the anticoagulant activity of a factor VIII antibody. J Thromb Haemost 2006; 4: 1047–55. [DOI] [PubMed] [Google Scholar]
- 22.Singh I, Smith A, Vanzieleghem B, Collen D, Burnand K, Saint-Remy JM, Jacquemin M. Antithrombotic effects of controlled inhibition of factor VIII with a partially inhibitory human monoclonal antibody in a murine vena cava thrombosis model. Blood 2002; 99: 3235–40. [DOI] [PubMed] [Google Scholar]
- 23.Dewerchin M, van der Elst L, Singh I, Grailly S, Saint-Remy JM, Collen D, Jacquemin M. Inhibition of factor VIII with a partially inhibitory human recombinant monoclonal antibody prevents thrombotic events in a transgenic model of type II HBS antithrombin deficiency in mice. J Thromb Haemost Elsevier Masson SAS; 2004; 2: 77–84. [DOI] [PubMed] [Google Scholar]
- 24.Jacquemin M, Stassen JM, Saint-Remy JM, Verhamme P, Lavend’Homme R, Vanderelst L, Meiring M, Pieters H, Lamprecht S, Roodt J, Badenhorst P. A human monoclonal antibody inhibiting partially factor VIII activity reduces thrombus growth in baboons. J Thromb Haemost 2009; 7: 429–37. [DOI] [PubMed] [Google Scholar]
- 25.Emmerechts J, Vanassche T, Loyen S, Van Linthout I, Cludts K, Kauskot A, Long C, Jacquemin M, Hoylaerts MF, Verhamme P. Partial versus complete factor VIII inhibition in a mouse model of venous thrombosis. Thromb Res 2012; 129: 514–9. [DOI] [PubMed] [Google Scholar]
- 26.Doering CB, Denning G, Shields JE, Fine EJ, Parker ET, Srivastava A, Lollar P, Spencer HT. Preclinical Development of a Hematopoietic Stem and Progenitor Cell Bioengineered Factor VIII Lentiviral Vector Gene Therapy for Hemophilia A. Hum Gene Ther 2018; 29: 1183–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coxon CH, Yu X, Beavis J, Diaz‐Saez L, Riches‐Duit A, Ball C, Diamond SL, Raut S. Characterisation and application of recombinant FVIII‐neutralising antibodies from haemophilia A inhibitor patients. Br J Haematol 2021; 193: 976–87. [DOI] [PubMed] [Google Scholar]
- 28.Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA. CryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 2017; 14: 290–6. [DOI] [PubMed] [Google Scholar]
- 29.Punjani A, Zhang H, Fleet DJ. Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat Methods 2020; 17: 1214–21. [DOI] [PubMed] [Google Scholar]
- 30.Spiegel PC, Jacquemin M, Stoddard BL, Pratt KP. Structure of a factor VIII C2 domain–immunoglobulin G4k Fab complex: identification of an inhibitory antibody epitope on the surface of factor VIII. Blood 2001; 98: 13–20. [DOI] [PubMed] [Google Scholar]
- 31.Liebschner D, Afonine PV., Baker ML, Bunkoczi G, Chen VB, Croll TI, Hintze B, Hung LW, Jain S, McCoy AJ, Moriarty NW, Oeffner RD, Poon BK, Prisant MG, Read RJ, Richardson JS, Richardson DC, Sammito MD, Sobolev OV, Stockwell DH, et al. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr Sect D Struct Biol 2019; 75: 861–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Croll TI. ISOLDE: A physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr Sect D Struct Biol 2018; 74: 519–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen BW, Spiegel PC, Chang C-H, Huh J-W, Lee J-S, Kim J, Kim Y-H, Stoddard BL. The tertiary structure and domain organization of coagulation factor VIII. Blood 2007; 111: 1240–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ngo JCK, Huang M, Roth DA, Furie BC, Furie B. Crystal Structure of Human Factor VIII: Implications for the Formation of the Factor IXa-Factor VIIIa Complex. Structure 2008; 16: 597–606. [DOI] [PubMed] [Google Scholar]
- 35.Smith IW, D’Aquino AE, Coyle CW, Fedanov A, Parker ET, Denning G, Spencer HT, Lollar P, Doering CB, Spiegel PC. The 3.2 Å structure of a bioengineered variant of blood coagulation factor VIII indicates two conformations of the C2 domain. J Thromb Haemost 2020; 18: 57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walter JD, Werther RA, Brison CM, Cragerud RK, Healey JF, Meeks SL, Lollar P, Spiegel PC. Structure of the factor VIII C2 domain in a ternary complex with 2 inhibitor antibodies reveals classical and nonclassical epitopes. Blood 2013; 122: 4270–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ronayne EK, Peters SC, Gish JS, Wilson C, Spencer HT, Doering CB, Lollar P, Spiegel PC, Childers KC. Structure of Blood Coagulation Factor VIII in Complex With an Anti-C2 Domain Non-Classical, Pathogenic Antibody Inhibitor. Front Immunol 2021; 12: 2981–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Przeradzka MA, Freato N, Boon-Spijker M, van Galen J, van der Zwaan C, Mertens K, van den Biggelaar M, Meijer AB. Unique surface-exposed hydrophobic residues in the C1 domain of factor VIII contribute to cofactor function and von Willebrand factor binding. J Thromb Haemost 2020; 18: 364–72. [DOI] [PubMed] [Google Scholar]
- 39.Fuller JR, Knockenhauer KE, Leksa NC, Peters RT, Batchelor JD. Molecular determinants of the factor VIII/von Willebrand factor complex revealed by BIVV001 cryo-electron microscopy. Blood 2021; 137: 2970–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meems H, Meijer AB, Cullinan DB, Mertens K, Gilbert GE. Factor VIII C1 domain residues Lys 2092 and Phe 2093 contribute to membrane binding and cofactor activity. Blood 2009; 114: 3938–46. [DOI] [PubMed] [Google Scholar]
- 41.Wakabayashi H, Fay PJ. Replacing the factor VIII C1 domain with a second C2 domain reduces factor VIII stability and affinity for factor IXa. J Biol Chem 2013; 288: 31289–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gilbert GE, Bhimavarapu A, Price P, Jacquemin M. Antibody to C1 Domain of Factor VIII Alters Interaction of Factor Xase Complex with Factor X. Blood 2004; 104: 1738–1738. [Google Scholar]
- 43.Jones TD, Phillips WJ, Smith BJ, Bamford CA, Nayee PD, Baglin TP, Gaston JSH, Bakert MP. Identification and removal of a promiscuous CD4+ T cell epitope from the C1 domain of factor VIII. J Thromb Haemost 2005; 3: 991–1000. [DOI] [PubMed] [Google Scholar]
- 44.van de Bovenkamp FS, Hafkenscheid L, Rispens T, Rombouts Y. The Emerging Importance of IgG Fab Glycosylation in Immunity. J Immunol 2016; 196: 1435–41. [DOI] [PubMed] [Google Scholar]
- 45.Kim DE, Chivian D, Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res 2004; 32: 526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruben EA, Summers B, Rau MJ, Fitzpatrick JAJ, Di Cera E. Cryo-EM structure of the prothrombin-prothrombinase complex. Blood 2022; 139: 3463–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fay PJ, Koshibu K. The A2 subunit of factor VIIIa modulates the active site of factor IXa. J Biol Chem 1998; 273: 19049–54. [DOI] [PubMed] [Google Scholar]
- 48.Bajaj SP, Schmidt AE, Mathur A, Padmanabhan K, Zhong D, Mastri M, Fay PJ. Factor IXa:factor VIIIa interaction: Helix 330–338 of factor IXa interacts with residues 558–565 and spatially adjacent regions of the A2 subunit of factor VIIIa. J Biol Chem 2001; 276: 16302–9. [DOI] [PubMed] [Google Scholar]
- 49.Jenkins PV, Dill JL, Zhou Q, Fay PJ. Contribution of Factor VIIIa A2 and A3-C1-C2 Subunits to the Affinity for Factor IXa in Factor Xase. Biochemistry 2004; 43: 5094–101. [DOI] [PubMed] [Google Scholar]
- 50.Jagannathan I, Ichikawa HT, Kruger T, Fay PJ. Identification of Residues in the 558-Loop of Factor VIIIa A2 Subunit That Interact with Factor IXa. J Biol Chem 2009; 284: 32248–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.