

Abstract
Cap-independent or eukaryotic initiation factor (eIF) 4E-independent, translation initiation in eukaryotes requires scaffolding protein eIF4G or its homolog, death-associated protein 5 (DAP5). eIF4G associates with the 40S ribosomal subunit, recruiting the ribosome to the RNA transcript. A subset of RNA transcripts, such as fibroblast growth factor 9 (FGF-9), contain 5′ untranslated regions (5′ UTRs) that directly bind DAP5 or eIF4GI. For viral mRNA, eIF recruitment usually utilizes RNA structure, such as a pseudoknot or stem–loops, and the RNA-helicase eIF4A is required for DAP5- or 4G-mediated translation, suggesting these 5′ UTRs are structured. However, for cellular IRES-like translation, no consensus RNA structures or sequences have yet been identified for eIF binding. However, the DAP5-binding site within the FGF-9 5′ UTR is unknown. Moreover, DAP5 binds to other, dissimilar 5′ UTRs, some of which require an unpaired, accessible 5′ end to stimulate cap-independent translation. Using SHAPE-seq, we modeled the 186 nt FGF-9 5′-UTR RNA's complex secondary structure in vitro. Further, DAP5 footprinting, toeprinting, and UV cross-linking experiments identify DAP5–RNA interactions. Modeling of FGF-9 5′-UTR tertiary structure aligns DAP5-interacting nucleotides on one face of the predicted structure. We propose that RNA structure involving tertiary folding, rather than a conserved sequence or secondary structure, acts as a DAP5-binding site. DAP5 appears to contact nucleotides near the start codon. Our findings offer a new perspective in the hunt for cap-independent translational enhancers. Structural, rather than sequence-specific, eIF-binding sites may act as attractive chemotherapeutic targets or as dosage tools for mRNA-based therapies.
Keywords: biophysics, cap-independent translation, DAP5, DAP5-binding site, FGF-9 RNA
INTRODUCTION
Under normoxic conditions, translation initiation occurs in a cap-dependent fashion. Eukaryotic initiation factor 4E (eIF4E) recognition of the RNA transcript's m7G cap is necessary for further eIF binding and 43S ribosomal recruitment to the transcript (Shatsky et al. 2018). eIF4G binds to the transcript via association with eIF4E and RNA-helicase eIF4A, forming the eIF4F complex. eIF4F then recruits the 40S ribosome to the transcript via 40S interaction with bound eIF4G (Leppek et al. 2018). However, when oxygen is scarce, 4E-binding protein (4E-BP) sequesters eIF4E and prevents recognition of the m7G cap (Braunstein et al. 2007). Any ribosomal recruitment to an RNA transcript under hypoxic conditions must therefore proceed in a 4E-independent, or cap-independent, manner: eIF4G and its homolog, death-associated protein 5 (DAP5), among other eIFs, can bind directly to the 5′ untranslated region (5′ UTR) of the RNA (Haizel et al. 2020). Eukaryotic viruses and proliferating cancer cells use cap-independent translation to survive and evade host defense mechanisms (Lacerda et al. 2017).
Cap-independent translation is poorly understood and occurs through a variety of mechanisms with varying eIF requirements. However, a subset of cellular mRNAs are believed to initiate via one of two pathways: a cap-independent translational enhancer (CITE)-like mechanism or a cellular internal ribosome entry site (IRES)-like mechanism (Haizel et al. 2020). As we define them here, a CITE-like mechanism, such as for HIF-1α-encoding mRNA, requires an unpaired 5′ end on the transcript for successful eIF binding, ribosomal recruitment and scanning, and cap-independent translation (Haizel et al. 2020). Under a CITE-like mechanism of translation initiation, scanning for the start codon by the recruited ribosome is likely. In contrast, an IRES-like mechanism recruits eIFs to the 5′ UTR, where RNA elements enhance eIF binding. An unpaired 5′ end was not necessary for a cellular IRES-like mechanism, in which ribosomal recruitment likely occurs at or near the AUG start codon (Haizel et al. 2020; Marques et al. 2022). The cellular IRES is still poorly defined and subject to rigorous debate, and no consensus sequence or structures have been identified (Akirtava et al. 2022; Smirnova et al. 2022). Additionally, different transcripts may interact with different mixes of eIFs, making a “true” cellular IRES mechanism difficult to distinguish from alternative cap-independent initiation mechanisms. Although cellular IRES are considered CITEs, not all CITEs are IRES-like. Interestingly, eIF4A is required for eIF4GI-binding specificity, suggesting these 5′ UTRs are highly structured and that structure may direct eIF4G binding (Gentry et al. 2023).
We have previously shown that cap-independent translational upregulation of fibroblast growth factor 9 (FGF-9) occurs in an IRES-like fashion (Haizel et al. 2020). FGF-9, an angiogenic growth factor, is translationally upregulated in breast and colorectal cancer cells, where it triggers mitogenesis (Chen et al. 2014). In normal lung tissue, FGF-9 expression is localized to smooth muscle around the airways, alveolar ducts, alveolar sacs, and blood vessels (Coffey et al. 2013). The 5′ UTR of FGF-9-encoding mRNA contains a regulatory uORF and a predicted IRES (Chen et al. 2014). Under normoxic conditions, ribosomes preferentially gather at the uORF start codon; translation of this uORF depresses FGF-9 expression (Chen et al. 2014). Under hypoxic conditions, ribosomal presence at the uORF decreases, and ribosomes instead gather at the FGF-9 start codon (Chen et al. 2014). The FGF-9 5′ UTR need not be located near an unpaired, accessible 5′ end to stimulate cap-independent translation, suggesting the ribosome is recruited away from the m7G cap during cellular stress (Haizel et al. 2020).
Lack of dependence on an unpaired 5′ end invites classification of the FGF-9's 5′ UTR as a traditional IRES. However, known viral IRES elements assume complex and functional tertiary structures (Balvay et al. 2009), and the secondary and tertiary structures of the FGF-9 5′ UTR are unknown. It has been previously speculated that cellular IRES may assume diverse secondary structures distinct from, yet less complex than, those of known viral IRES elements (Komar and Hatzoglou 2011; Leppek et al. 2018). A cellular IRES may instead require internal trans-acting factors (ITAFs) to fold the 5′ UTR into a functional structure (Leppek et al. 2018). Indeed, such a mechanism has already been suggested for the FGF-9 5′ UTR, although the exact ITAF that mediates such a change is as yet undiscovered (Chen et al. 2014), and the tertiary structure or binding site for such an ITAF has not been identified. Given that cap-independent translation of FGF-9 appears to rely on an IRES-like mechanism, it is therefore important to understand what structures, if any, result in successful ribosomal recruitment.
We have previously shown that DAP5 binds the FGF-9 5′ UTR directly; this binding initiates translation of a downstream reporter (Haizel et al. 2020). A highly conserved eIF4GI homolog, DAP5 is expressed in proliferating cells and is required for embryonic cellular differentiation (Virgili et al. 2013; Weingarten-Gabbay et al. 2014; Smirnova et al. 2022). For structured 5′ UTRs, DAP5 was found to require eIF4A for RNA unwinding, as does eIF4GI (Liberman et al. 2015; Gentry et al. 2023). Previous work by Lee and McCormick found DAP5 to make up 0.1%–0.3% of total proteins in ectopic breast carcinoma cells (Lee and McCormick 2006). The C-terminal two-thirds of DAP5 are similar to those of eIF4GI; however, the N-terminal region of DAP5 lacks eIF4E and PABP-binding sites (Virgili et al. 2013; Alard et al. 2019). Additionally, DAP5 possesses a longer half-life in the cell under hypoxic conditions as compared to eIF4GI and eIF4GII (Alard et al. 2019). DAP5, like eIF4GI, associates with key initiation factors to initiate cap-independent translation; its comparatively long half-life suggests it may substitute for eIF4GI when canonical translation initiation is hindered. Beyond eIF4A for structured 5′ UTRs, DAP5-mediated translation may require different initiation factors depending on the transcript (Smirnova et al. 2022). Although DAP5 does not unilaterally drive translation initiation when eIF4GI is readily available, it appears to play unique roles in cell survival and immune response, even under normoxic conditions. For example, DAP5 drives Treg cell differentiation in an mTOR/4E-independent, but not cap-independent, manner, via association with FOXP3 5′-UTR mRNA (Volta et al. 2021). DAP5 will bind type I viral IRES elements, but not type II or III viral IRES elements, suggesting an ability to discern between structural translational enhancers (Dave et al. 2019).
Indeed, DAP5 has been previously demonstrated to distinguish between cellular IRESs (Weingarten-Gabbay et al. 2014). A p53 isoform, Δ40p53, has a single IRES within its 5′ UTR and a second IRES within its coding sequence. DAP5 binds both IRES elements with similar affinity, but DAP5 preferentially binds the second IRES in vivo (Weingarten-Gabbay et al. 2014). How DAP5 distinguishes between these different translational enhancers is unknown, and a universal DAP5-binding site has not yet been identified. Moreover, we have previously shown that DAP5 binding does not always stimulate cap-independent translation in an IRES-like manner (Haizel et al. 2020). A more complete understanding of DAP5's role requires characterization of its binding site on one or more 5′ UTRs. Given DAP5's ability to mediate both cap-dependent and cap-independent translation efficiency, elucidating one or more potential DAP5-binding sites on these transcripts is of broad biological interest.
Computational prediction of cellular IRES-like elements has not yet produced a putative DAP5-binding site. Neither our group nor others have found significant sequence or secondary structural similarities among 5′ UTRs bound by DAP5 (de la Parra et al. 2018), further frustrating efforts to categorize and computationally predict CITEs. It has previously been suggested that the presence of an uORF, such as FGF-9's, distinguishes the “true” cellular IRES from other -9s (Leppek et al. 2018). However, not all DAP5-dependent transcripts contain uORFs (Shestakova et al. 2023). The DAP5-binding site within an IRES-like 5′ UTR is more complex than a single-nucleotide sequence or motif which can be identified computationally.
Given the importance of structure to the function of viral IRES elements, we therefore asked if a complex RNA structural element, rather than a conserved nucleotide sequence or secondary structure, binds DAP5. Cap-dependent, but DAP5-mediated, translation requires eIF4AI for successful ribosomal scanning (Liberman et al. 2015). eIF4AI dependence increases with 5′-UTR structural complexity, and mutating eIF4AI will decrease IRES-mediated translation in the presence of a truncated, DAP5-like eIF4GI (Svitkin et al. 2001). It is possible that a conserved tertiary structure within the FGF-9 5′ UTR is responsible for DAP5 recruitment, thereby necessitating eIF4AI for unwinding and ribosomal scanning.
Here, we present data identifying DAP5 protection on the FGF-9 5′ UTR and, using tertiary modeling, suggest that DAP5 binds a specific surface of the proposed tertiary structure of the FGF-9 5′-UTR mRNA. Using selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE), we found that the FGF-9 5′-UTR mRNA likely takes on a stable and highly structured conformation in vitro in the presence of 1 mM magnesium. DAP5 footprinting experiments reveal that the majority of protein–nucleotide contacts occur near nt 93. UV cross-linking experiments confirm a localized DAP5-binding site at a junction consisting of multiple stem–loops (SLs). Tertiary modeling aligned the DAP5-protected residues to a surface of the proposed RNA structure, further suggesting the importance of tertiary structure in binding.
RESULTS
The FGF-9 5′ UTR
We first sought to determine the FGF-9 5′-UTR secondary structure. We probed the secondary structure of a wt FGF-9 5′-UTR RNA construct using 1-methyl anhydride (1M7) treatment as previously outlined in Powell et al. (2022). Certain structural elements, although not directly involved in DAP5 or eIF binding, may still be necessary to form a CITE's catalytically active structure (Lozano et al. 2018). To ensure the full FGF-9 5′ UTR was well-represented, a short segment of the firefly luciferase reporter sequence downstream from the 5′ UTR, as well as a T7 promoter sequence upstream of the 5′ UTR, was PCR-amplified. This PCR product was then transcribed in vitro and folded in the presence of magnesium (Fig. 1). cDNA was reverse-transcribed from 1M7-treated RNA using a primer specific to the short firefly luciferase segment. Situating the wt FGF-9 5′ UTR in a cassette with the short luciferase sequence prevented 3′ end bias on our 5′-UTR sequencing data, as reverse transcriptase (RT) primer peaks on the 3′ end will always be higher than peaks closer to the 5′ end of the RNA (Vasa et al. 2008).
FIGURE 1.
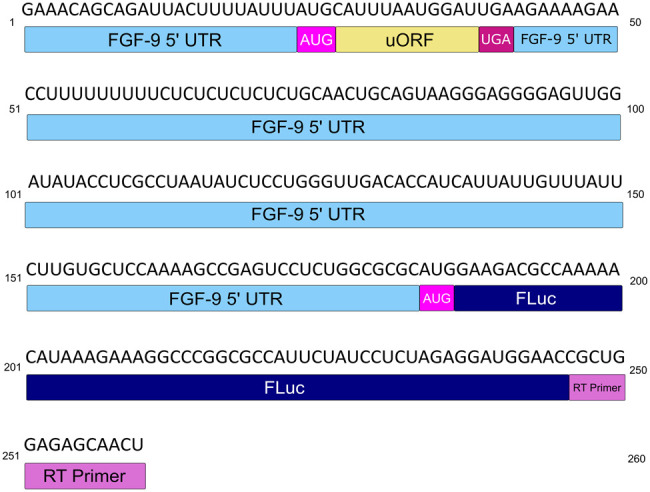
wt FGF-9 5′-UTR construct. The FGF-9 5′ UTR (sky blue) and partial firefly luciferase (FLuc) reporter sequence (navy) RNA used for this study. The FGF-9 uORF (nt [nucleotides] 27–38) is annotated in goldenrod, uAUG in magenta, and uUGA in maroon. A cy5-tagged reverse transcript primer (orchid), corresponding to part of the FLuc reporter, was used to create cDNA from treated RNA.
SHAPEFinder software (Vasa et al. 2008) calculated 1M7 reactivity per nucleotide as compared to a DMSO-treated negative control (Supplemental Fig. S1; Mortimer and Weeks 2007). 1M7 reactivity was used in RNAStructure software (Reuter and Mathews 2010) as a folding constraint at 37°C. The lowest-free-energy fold that considers 1M7 reactivity was chosen. Our predictive fold was generated using a 186 nt region containing the FGF-9 5′ UTR (177 nt) and ending with an AUG (nt 184–186) based on the minimal 5′ UTR studied in Chen et al. (2014) and in Haizel et al. (2020). Because these RNA structures are flexible, certain nucleotides predicted to be base-paired may still retain some 1M7 reactivity.
We found that the wt FGF-9 5′-UTR RNA takes on a stable, highly structured conformation (Fig. 2). The minimum-free-energy prediction (−73.1 kJ) at 37°C was chosen for analysis (Mortimer and Weeks 2007). Seven stem–loops (SL1–SL7) were shown to form, with SL1 forming the base of a “pinwheel”-like structure containing SL2–SL5. SL7 includes the AUG codon (nt 184–186) at its base. Nucleotides with high 1M7 reactivity were predicted to reside within single-stranded or bulge regions. The uORF start codon (nuceotides 24–26) was largely unreactive to 1M7.
FIGURE 2.

SHAPE-derived 2′ structure of the FGF-9 5′-UTR mRNA. Normalized SHAPE data from three SHAPE reactions were pooled and input into RNAStructure software as a binding constraint. The minimum-free-energy predicted structure (ΔG = −73.1 kJ) was selected. Nucleotides were annotated in RNA2Drawer on a scale from 0 (ivory, unreactive to 1M7) to 1.0 and above (medium slate blue, always reactive to 1M7, seven SL were predicted to form). The AUG start codon is visible at the base of SL7.
DAP5 binding
We next asked where the location of DAP5 binding occurs along this highly structured 5′ UTR. To achieve a “ballpark” estimate of where DAP5 binds, we designed a modified version of ribosome toeprinting experiments, nicknamed “DAP5 toeprinting.” Our previous fluorescence binding experiments revealed that DAP5-bound RNA with a 1:1 stoichiometry showing a single binding site (Haizel et al. 2020). The full-length DAP5 peptide was expressed from a plasmid vector and purified as previously described (Haizel et al. 2020). A total of 300 nM DAP5 was added to 20 pmol unmodified FGF-9 5′-UTR mRNA; this concentration is far above our previously calculated Kd (136 ± 10.0 nM) for the DAP5–FGF-9 5′-UTR mRNA interaction, thus ensuring all available RNA is bound (Tijerina et al. 2007; Haizel et al. 2020). As RT proceeds along the DAP5-bound transcript, it eventually collides with the 3′-most edge of DAP5. Thus, the resulting cDNA library should overwhelmingly terminate at the 3′-most edge of the DAP5-binding site. As a negative control, a cDNA library created from unbound, unmodified FGF-9 5′-UTR mRNA was sequenced simultaneously. To identify DAP5-mediated RT stops, stops from the “unbound” sample were directly compared to stops from the “DAP5-bound” toeprinting sample. For largely unknown reasons, RT is known to pause across unmodified transcripts (Weeks and Mauger 2011; Sexton et al. 2017). Therefore, all DAP5-mediated stops were identified relative to “natural” RT stops on unmodified FGF-9 5′-UTR RNA (Lozano et al. 2018).
We found clear evidence of RT stops almost exclusively at nt 93 of the FGF-9 5′ UTR (Fig. 3). This indicates that the bound DAP5 sits among the first four SLs of the transcript. Smaller RT stops were found 3′ of nt 93 between nt 120 and 140 (between SL1 and SL7); these may be weaker or more transient DAP5 contacts. A large number of cDNA within both the bound and control samples terminated at the 5′ end of the RNA, indicating that the full 5′ UTR was well-represented.
FIGURE 3.

FGF-9 5′ UTR–DAP5 toeprinting data. FGF-9 5′-UTR cDNA was reverse-transcribed from DAP5-bound mRNA. n = 4. Higher peak area corresponds to a higher proportion of RT stops in the presence of DAP5. Size standard peaks visible at 20, 40, 50, 60, 70, 80, 100, 120, 140, 160, 170, and 180 nt. Strong RT stop at nt 93 (black arrow). Start codon: nts184–186 (magenta arrow).
To better discern the exact nucleotides contacted by DAP5, SHAPE footprinting experiments were performed in the presence of DAP5. Unlike toeprinting, DAP5 footprinting involves SHAPE chemistry. DAP5-bound FGF-9 5′-UTR mRNA was modified with 1M7 at 37°C. To ensure all modified RNA was recovered after cleanup, and to prevent RT stops due to bound protein, DAP5 is digested off of the 1M7-modified RNA using proteinase K (Duncan and Weeks 2008). Nucleotides that experience a significant decrease in 1M7 reactivity (>0.3 units) in the presence of bound DAP5 are likely shielded from adduct formation by the protein. “Strong” shielding, in which previously accessible nucleotides become inaccessible to 1M7, indicates a high probability of protein–RNA interaction (Powell et al. 2022).
As expected, strong DAP5 contacts (a difference in 1M7 reactivity >1 unit) were found in the first two SL of the FGF-9 5′ UTR (Fig. 4A, in green). The DAP5 toeprinting site at nt 93 experienced a sharp decrease in 1M7 reactivity in the presence of DAP5. However, some shielding from 1M7 in the presence of DAP5 was found 3′ of nt 93. As mentioned with our toeprinting experiments, any DAP5 contact close to the 3′ end may be too weak to stop RT during toeprinting, explaining why nt 93 was a favored toeprinting site. Additionally, there may be single-nucleotide, DAP5-induced conformational changes 3′ of nt 93 that could alter 1M7 reactivity. Smaller decreases in 1M7 reactivity were visible at individual nucleotides throughout the FGF-9 5′-UTR transcript (Fig. 4A). These decreases appear distributed across our proposed secondary structure (Fig. 4B).
FIGURE 4.

FGF-9 5′ UTR–DAP5 footprinting data. 1M7 reactions were performed on DAP5-bound FGF-9 5′-UTR mRNA. n = 2. (A) Changes in 1M7 reactivity in the presence of DAP5. Normalized 1M7 reactivity in the presence of DAP5 was subtracted from normalized 1M7 reactivity in the absence of DAP5. For clarity, reactivity changes >1 (from a reactive site to a nonreactive site) are shown here. A decrease in 1M7 reactivity > 1 (green) indicates possible contact with DAP5. An increase in 1M7 reactivity (magenta) indicates increased flexibility in the presence of DAP5. Start codon: nt 184–186. Full length of mRNA = 209 nt. Size standard peaks (20, 40, 50, 60, 70, 80, 100, 120, 140, 160, 170, and 180 nt) are not shown here. (B) DAP5-induced protection from 1M7 annotated across the predicted secondary structure of the FGF-9 5′-UTR mRNA. Decreased 1M7 reactivity is annotated in green, with darker green = sharper decrease in reactivity. Toeprinting site (nt 93) from Figure 3 is annotated in aqua blue. AUG (nt 184–186) in magenta.
We also noticed a significant increase in 1M7 reactivity (magenta) for some nucleotides (Fig. 4A). Increased accessibility to 1M7 may indicate a change in secondary or tertiary structure: for example, a previously base-paired nucleotide may become single-stranded. In particular, the short luciferase reporter region after the start codon (nt 187+) became more accessible to 1M7 modification in the presence of DAP5. SL3 and SL4 (nt 40–90) were slightly more reactive to 1M7, suggesting increased flexibility.
UV cross-linking
To further clarify the location of DAP5 binding, and to further distinguish between bases contacted by DAP5 and those involved in conformational changes, we cross-linked DAP5 to the folded wt FGF-9 5′ UTR using 100 mJ/cm2 light. Ultraviolet light catalyzes an irreversible covalent bond between aromatic rings at short distances, thereby cross-linking participating aromatic amino acids to their bound nucleobases (Poria and Ray 2017; Urdaneta and Beckmann 2020). Proteinase K digestion leaves behind small peptide fragments on the RNA that will stall RT (Urdaneta and Beckmann 2020). To distinguish between RNA–RNA cross-linking products, we sequenced a second cDNA library made from unbound, UV-treated wt FGF-9 5′-UTR RNA. As with toeprinting and footprinting, DAP5 cross-linking sites were identified relative to an unbound, UV-treated RNA sample. To ensure full RT readthrough, a small amount of non-cross-linked FGF-9 RNA was added to the reverse transcription reaction.
Increased RT stops after cross-linking DAP5s were found largely near the start codon (nt 182–183) and at sites between nt 130 and 139 (Fig. 5A, orange). DAP5 contacts between nt 130 and 139 would explain the small RT stops found there during our toeprinting experiment. As expected, increased RT stops in the FGF-9-only sample (Fig. 5A, blue) were found 5′-most of these sites; without cross-linked DAP5, nothing hinders RT from reading through these nucleotides. Additional DAP5-mediated RT stops were found toward the 5′ end of the FGF-9 RNA (nt 4–7, 19, and 26–27). In particular, the RT stops at 26–27 are proximal to the FGF-9 uORF's start codon (nt 29–31). When these DAP5-mediated RT stops were mapped on our predicted secondary structure with our footprinting and toeprinting data, we found them in close proximity to sites protected from 1M7 (Fig. 5B). Importantly, DAP5 cross-linking contacts occurred on SL2, SL3, SL6, and SL7. Nt 19 and 131 were identified as both DAP5 cross-linking sites and DAP5 footprinting sites (Fig. 5B, ivory with red outline). Interestingly, SL3 and SL4 do not appear to either contact DAP5 or experience DAP5-induced changes in 1M7 reactivity.
FIGURE 5.

DAP5–FGF-9 5′-UTR UV cross-linking. DAP5 was cross-linked to the wt FGF-9 5′-UTR RNA construct using UV light. For clarity, DAP5 footprinting data are shown alongside UV cross-linking stops. (A) Difference in peak area between RT stops occurring with and without DAP5. (Orange) More frequent stops with DAP5. (Blue) More frequent stops found without DAP5. Size standard peaks (20, 40, 50, 60, 70, 80, 100, 120, 140, 160, 170, and 180 nt) are not shown here. (B) DAP5 footprinting, toeprinting, and UV cross-linking sites mapped on our putative 2D model. Ivory with red outline = RT stops shared between footprinting stops from Figure 4 (green) and UV cross-linking data (orange). Toeprinting site (nt 93) shown in aqua blue. Start codon (nt 184–186) shown in magenta.
Modeling the FGF-9 5′-UTR's tertiary structure
RNAStructure's predicted minimum-free-energy fold has been shown to be up to 91% accurate when constrained by 1M7 reactivity (Mortimer and Weeks 2007). However, RNAStructure cannot predict tertiary conformations. Additionally, our DAP5 toeprinting, footprinting, and cross-linking sites were scattered across our predicted secondary structure. We therefore decided to use a computational prediction of 3D RNA structure to help explain these results. Our 2D structure was fed into the CentroidFold algorithm using RNAComposer software (Popenda et al. 2012; Antczak et al. 2016). RNAComposer will generate a putative minimum-free-energy tertiary structure based on known RNA crystal structures (Popenda et al. 2012; Antczak et al. 2016). We then annotated 1M7 reactivity on our tertiary model using UCSF ChimeraX software (Goddard et al. 2018; Pettersen et al. 2020). Our model of the wt FGF-9 5′ UTR takes on a low-energy (−3060 kcal/mol) 3D conformation (Fig. 6). Despite its distance from the 5′ end in the 2D structure, SL6 associates closely with SL1–SL5 in a bouquet-like cluster. The 5′ end of the 5′ UTR is tucked within this cluster. A main face, consisting of the SL1 structure, part of the 2D “pinwheel” structure, and SL7, is most accessible to solvent. Interestingly, the FGF-9 start codon is tucked nearest SL6 and is largely inaccessible to 1M7. Using the IPKnot algorithm in lieu of CentroidFold did not predict pseudoknot formation (data not shown; Sato et al. 2011).
FIGURE 6.

Predicting the FGF-9 5′-UTR tertiary structure. The minimum-free-energy tertiary conformation was predicted by RNAComposer using Figure 2 as a folding constraint. USCF ChimeraX was used to annotate 1M7 reactivity. ΔG = −3060 kcal/mol. SL1–SL5 aggregate closely around the 5′ end of the mRNA, whereas SL6 and SL7 project away. The AUG start codon, tucked between SL5 and SL6, is visualized in an insert (bottom left). SL1 = nt 5−12 and 117–124, SL2 = 20–38, SL3 = 40–62, SL4 = 63–92, SL5 = 94–112, SL6 = 128–138, and SL7 = 140–186. Start codon: 184–186.
Interestingly, when viewed on our proposed tertiary structure, DAP5-induced shielding sites and DAP5 cross-linking sites coalesce along a single face (Fig. 7). Notably, DAP5 shielding and cross-linking are centered around the start codon (magenta) at the end of SL7, where it sits in a nest of bound nucleotides. Nt 93 is also near the start codon in our proposed tertiary structure. Given that our laboratory has proposed that DAP5 binding to the FGF-9 5′-UTR mRNA recruits the ribosome close to the start codon (Haizel et al. 2020), strong DAP5 contact near the start codon is to be expected.
FIGURE 7.

DAP5 contacts mapped on our tertiary model of the FGF-9 5′-UTR mRNA. Tertiary prediction from Figure 6 annotated with DAP5 toeprinting, footprinting, and cross-linking data. Ivory with red outline = RT stops shared between footprinting data (green) and UV cross-linking data (orange). Toeprinting site (nt 93) shown in aqua blue. Start codon (nt 184–186) shown in magenta. All other nucleotides are annotated in black.
DISCUSSION
The mechanism of DAP5-mediated translation initiation is still being determined. Previous literature has shown that, like other members of the eIF4G family, DAP5 requires eIF4A to initiate the translation of sequences downstream from structured 5′ UTRs; this interaction is also crucial for DAP5 binding to eIF2β (Liberman et al. 2015). Whether or not DAP5 itself also participates in RNA restructuring is unknown; however, this finding suggests that some degree of RNA unwinding is crucial for the initiation on structured transcripts. eIF3 may play a smaller role in DAP5-dependent initiation, possibly in reinitiation on the same transcript (Smirnova et al. 2022). A unified DAP5-binding site on these transcripts, however, has not been identified.
In an effort to clarify the mechanism of DAP5-mediated, cap-independent translation initiation, we sought to characterize the FGF-9 5′ UTR and identify its DAP5-binding site. Our SHAPE-derived model of the FGF-9 5′-UTR mRNA predicts a complex secondary structure. Integrating 1M7 reactivity into our computational predictions allowed us to characterize this IRES-like element. To understand the disperse DAP5 interactions with FGF-9 5′ UTR, we utilized a predicted tertiary structure model. When DAP5 cross-linking, toeprinting, and footprinting contacts were mapped onto our predicted tertiary structure, DAP5 cross-linking contacts surround sites of 1M7 protection (Fig. 7). In our model, SL3 and SL4 extend in opposite directions from each other, with SL1 and SL5 bisecting them in a cross-like formation. DAP5 contacts favor this junction, with the toeprinting site (Fig. 7, aqua) in the center. The start codon (magenta) is in the center of DAP5 cross-linking contacts, as are nt 19 and 131 (ivory with red outline). Further, DAP5 appears to wrap around our predicted tertiary structure, making contacts near the start codon while avoiding two SLs entirely. Although DAP5 contact appears scattered across our secondary structure model, these localized contacts coalesce into a “binding surface” across our tertiary model. We therefore propose that the DAP5-binding site is not a localized sequence or single nucleotide, but a preference for one face of the 5′ UTR.
Although FGF-9 has long been speculated to be a cellular IRES (Chen et al. 2014), no distinct conserved secondary or tertiary structures for the cellular IRES have been identified. Our model of the FGF-9 5′-UTR's tertiary structure does not contain obvious similarities to existing type I viral IRES (Martinez-Salas et al. 2018). Although our toeprinting site occurs in a guanine-rich region, no other guanine-rich sequence is available for G-quadruplex formation. We likewise did not find evidence of pseudoknot formation using IPknot (data not shown; Sato et al. 2011). The pinwheel structure formed by SL1–SL5 is reminiscent of SL K of the EMCV IRES, where eIF4GI binds (Balvay et al. 2009). SL K of the EMCV IRES, which juts from SL J, is surrounded by two nucleotide bulges, each of which bore eIF4GI footprints: One bulge sits directly downstream from SL K, whereas the other bulge is further upstream on SL J (Balvay et al. 2009). Our DAP5 footprinting experiments on the FGF-9 5′ UTR showed contact within several single-stranded regions and bulges: Footprinting showed changes in 1M7 reactivity within the SL5 bulge and the single-stranded pinwheel downstream from SL1, whereas UV cross-linking showed contact with the SL1 bulge. The distance between eIF4GI footprinting sites on the EMCV IRES's secondary structure is ∼100 nt—a similar distance as between DAP5 cross-linking sites on the FGF-9 5′ UTR. However, EMCV is a type II IRES, and DAP5 does not bind EMCV: Successful infection does not require eIF4GI cleavage into DAP5 (Dave et al. 2019).
Nevertheless, the predicted structural complexity of the FGF-9 5′ UTR indicates that the structures of cellular IRESes are likely, as previously speculated, distinct from those of viral IRESes (Komar and Hatzoglou 2011). In the absence of a more suitable term, we will continue to refer to the FGF-9 5′ UTR as “IRES-like.”
Through DAP5 footprinting, toeprinting, and UV cross-linking, we found DAP5 contacts the region near the FGF-9 start codon. This agrees with previous findings showing ribosomal preference for the FGF-9 start codon under hypoxia (Chen et al. 2014). DAP5 also cross-linked to nucleotides upstream of the FGF-9 uORF, suggesting that DAP5 can recruit ribosomes directly to the uORF and repress FGF-9 translation. Our SHAPE experiments suggest increased nucleotide flexibility at single-nucleotide sites across the FGF-9 5′ UTR in the presence of DAP5. Some predicted DAP5 footprinting sites were near, but did not correspond to, DAP5 cross-linking sites, suggesting that a change in 1M7 reactivity may be due to DAP5 shielding, rather than peptide–nucleotide contact. Such changes have been seen during ribosomal binding on the FMDV viral IRES, where SHAPE accessibility changed due to ribosome and eIF binding (Lozano et al. 2018). Additionally, small structural changes in DAP5 amino acid residues may account for slight discrepancies between DAP5 footprinting and UV cross-linking sites.
However, small changes in RNA folding in the presence of DAP5 may also be possible. It has been previously suggested that, as with other cellular IRES, an unknown ITAF changes FGF-9's secondary structure into a CITE under hypoxia (Chen et al. 2014). DAP5 has already been named as an ITAF for other IRES-like translational enhancers, such as Δ40p53 (Marques et al. 2022).
Our experiments suggest DAP5 makes contact on both the 5′- and 3′-most ends of the SL1–SL5 pinwheel. Our proposed tertiary fold for the FGF-9 5′ UTR brings these contact sites close to one another. However, DAP5 does not appear to contact SL3 and SL4. This suggests an alternative function for these SLs beyond that of a DAP5-binding site. The most obvious explanation is that SL3 and SL4 must form to preserve the overall 5′-UTR structure. The FMDV IRES requires domains II and III to retain viral IRES activity, although these domains do not participate in ribosome binding (Lozano et al. 2018). A polypyrimidine tract does exist in the FGF-9 5′ UTR, suggesting the need for polypyrimidine-tract binding protein (PTB); in our predicted model, this tract spans SL3 and SL4. It follows that DAP5 would not interact with this region, leaving it open for potential PTB binding. However, additional experiments would be needed to confirm a requirement for PTB.
Overall, we find that our predicted model of DAP5 binding agrees with a proposed IRES-like model (Haizel et al. 2020). Here, we provide strong evidence that DAP5 likely favors one side of the FGF-9 5′ UTR, with direct contacts found near the uORF start codon and the reporter start codon. This model provides insight into recognition features of the RNA, which offer the potential for use in designing novel chemotherapeutic targets and may aid in identifying other complex, IRES-like RNA structural elements.
MATERIALS AND METHODS
Isolation of FGF-9 5′-UTR region
Plasmid reporter constructs containing a T7 promoter region and the 5′-UTR sequence of FGF-9 (177 nt; GenBank accession number AY682094.1) directly upstream of a firefly luciferase coding region were a generous gift from Dr. Solomon Haizel (see Fig. 1; Haizel et al. 2020). To simplify later SHAPE-seq analysis, the T7 promoter, FGF-9 5′-UTR sequence, and the first 75 nt of the firefly luciferase reporter were PCR-amplified using the Thermo Scientific Phusion High-Fidelity PCR Kit (Supplemental Table 1). The resulting PCR product was purified into nuclease-free water using the Zymo Oligo Clean & Concentrator Kit. The concentration was verified using a NanoDrop UV-Vis Spectrophotometer, and integrity was confirmed using a native 1.5% (w/v) agarose gel.
FGF-9 RNA transcription
RNA was in vitro transcribed overnight from the isolated FGF-9 5′-UTR region using the NEB HiScribe T7 High Yield RNA Synthesis Kit. To prevent RNA degradation, 1 µL of a 1.6 units/µL Thermo Scientific RiboLock RNase Inhibitor solution was included. The 75 nt of the firefly luciferase reporter and a short 20 nt region upstream of the FGF-9 5′ UTR were retained in the resulting transcript to ensure the entire FGF-9 5′ UTR was well-represented in later SHAPE data. After DNase digestion, RNA was purified into nuclease-free water using the Zymo Oligo Clean & Concentrator Kit. As with the PCR product, RNA concentration was verified using a NanoDrop UV-Vis Spectrophotometer, and RNA integrity was confirmed using a native 1.5% (w/v) agarose gel.
SHAPE-seq
In total, 50 mM 1-methyl-7-nitroisatoic anhydride (1M7) and pure DMSO stock solutions were preheated to 37°C prior to SHAPE reactions. Twenty picomoles of FGF-9 5′-UTR mRNA was suspended in 20 mM HEPES buffer (pH = 7.5) with 100 mM KCl. As described previously (Haizel et al. 2020), nascent RNA secondary and tertiary structure was disrupted by heating to 90°C over 2 min, followed by slow cooling at room temperature for 1 h. To refold RNA, MgCl2 was added to a final concentration of 1 mM Mg2+ before incubation on ice for 1 h (Haizel et al. 2020).
SHAPE-seq reactions were executed according to existing literature (Rice et al. 2014; Powell et al. 2022). For positive (+) SHAPE reactions, preheated 1M7 stock was added to the folded RNA sample to a final concentration of 5 mM. For negative control (−) reactions, an equivalent volume of preheated DMSO was added to the sample in lieu of 1M7. Both solutions were incubated at 37°C for 70 sec, equivalent to five 1M7 half-lives (Mortimer and Weeks 2007). Sample tubes were wrapped in tinfoil to protect 1M7-modified RNA from light. RNA was purified into 9 µL nuclease-free water using the Zymo Oligo Clean & Concentrator Kit.
A cyanine-5 (cy5)-labeled cDNA primer corresponding to part of the firefly luciferase reporter region preserved in the amplified DNA sequence was a generous gift from Dr. Paul Powell (Supplemental Table 1). Reverse transcription of (+) and (−) SHAPE samples was executed using the Invitrogen SuperScript III Kit, as previously described (Powell et al. 2022). Ten picomoles of cy5-labeled cDNA primer, 1 µL of RiboLock RNase Inhibitor, 2 µL of NEB10 µM dNTP mix, 1 µL of 0.1 mM DTT, 1 µL nuclease-free water, and 4 µL Invitrogen 5× first-strand buffer were added to SHAPE samples. CTP-seq samples were created using unreacted FGF-9 5′-UTR mRNA and 1 µL ddGTP in lieu of nuclease-free water. For UTP-seq samples, ddATP was used in lieu of ddGTP. To allow primer annealing, all samples were incubated at 65°C for 5 min before slow cooling to 55°C. Invitrogen SuperScript III reverse-transcriptase (RT) was added before incubation at 55°C for 40 min. Unmodified transcripts (negative [−] control reactions) show where RT pauses or stops on unmodified RNA transcripts (Weeks and Mauger 2011).
Remaining RNA was digested via the addition of 2 µL 2M NaOH and incubation at 95°C for 3 min, followed by immediate neutralization with 2 µL 2M HCl. cDNA was recovered via ethanol precipitation at 4°C overnight (Powell et al. 2022). cDNA was sequenced using capillary electrophoresis on the Beckman GenomeLab GeXP Genetic Analysis System using custom parameters (Mitra et al. 2008; Powell et al. 2022). All traces were evaluated using the Beckman-Coulter GenomeLab 400 bp Size Standard Kit, which contains fragments of lengths 20, 40, 50, 60, 70, 80, 100, 120, 140, 160, 170, 180, 200, 220, 240, 260, 280, 300, 320, 340, 360, 380, 400, and 420 nt.
5′-UTR structural prediction
Simultaneous capillary electrophoresis runs were first aligned according to their size standards using the MATLAB CEQ alignment suite of software developed by the Laederach Lab at UNC Chapel Hill (available at https://ribosnitch.bio.unc.edu/software/). .shape files were then created using one (+) trace, one (−) trace, one CTP-seq trace, and one UTP-seq trace. SHAPEFinder software, developed by the Giddings laboratory at UNC Chapel Hill (Vasa et al. 2008), was used to determine 1M7 reactivity per nucleotide. The difference in peak area between (+) capillary electrophoresis trace and the (−) capillary electrophoresis trace defined the degree of 1M7 SHAPE reactivity. A higher difference in peak area corresponds to a more frequent RT stop in the (+) sample and, therefore, a more reactive nucleotide.
Individual SHAPE runs were normalized against themselves using the “simple” normalization method described in Low and Weeks (2010). The highest 2% of reactivities across all nucleotides were first temporarily removed from the data pool. The following top 8% of reactivities were averaged against each other to create a normalization factor. The removed reactivity values were then re-added to the data pool, and all reactivities were divided by the calculated normalization factor. This resulted in a normalized 1M7 reactivity value for each nucleotide ranging between 0 (not reactive to 1M7) to >1.5 (extremely reactive to 1M7).
To determine the average 1M7 reactivity for each nucleotide, multiple SHAPE runs were normalized against each other using the “box plot” normalization method (Low and Weeks 2010; Rice et al. 2014). Normalized 1M7 reactivity values were created for each individual nucleotide across multiple runs. The 25th and 75th quartiles were calculated for each nucleotide's data pool, as well as the Q value. If a datum was >1.5Q, it was labeled as an outlier. However, if 1.5Q < 90th percentile, data greater than the 90th percentile were labeled as outliers instead. Chosen outliers were summarily removed from the data pool. Remaining data points were averaged and fed into RNAStructure software as a folding constraint for the first 185 nt of our construct at 37°C (Reuter and Mathews 2010). Nucleotides beyond nt 186 were not considered in our structural prediction. The RNAStructure prediction was annotated for SHAPE reactivity and base-pairing probabilities using RNA2Drawer software (Johnson et al. 2019).
RNAComposer software (Popenda et al. 2012; Antczak et al. 2016) was used to generate 3D structural predictions for normalized, average 1M7 reactivity. The MFE structure for nt 1–186, generated by RNAStructure, was fed into RNAComposer. The MFE conformation of these 186 nt at 37°C was then generated. Visualization and annotation of this 3D structure, as well as a “spin video” showing the structure throughout a 360° turn, were made using UCSF Chimera X software (Goddard et al. 2018; Pettersen et al. 2020).
DAP5 isolation
A plasmid construct, including the DAP5 coding sequence followed by an N-terminal 6× histidine tag, was a generous gift from Dr. Solomon Haizel (see Haizel et al. 2020). A TEV protease cut site preceding the 6× histidine tag was previously introduced using the Q5 Site-Directed Mutagenesis Kit from NEB and the GeneJET Plasmid Miniprep Kit from Thermo Fisher. DAP5 was expressed in Escherichia coli BL21-CodonPlus (DE3)-RIL cells from Agilent and purified as previously described (Haizel et al. 2020). DAP5 was first purified from bacterial cell lysate using a 5 mL His-Trap HP (Ni-NTA) column from GE Healthcare Life Sciences. To cleave the N-terminal 6× histidine tag, purified DAP5 was dialyzed overnight at 4°C against storage buffer (20 mM HEPES [pH 7.5], 200 mM KCl, 10% glycerol, 20 mM BME) in the presence of TEV protease. Final purification and concentration of the untagged DAP5 was performed using a 1 mL HiTrap heparin HP column from GE Healthcare Life Sciences. Elution fraction quality was verified using a 10% SDS-PAGE gel; chosen elution fractions (>95% purity) were pooled and distributed into uniform aliquots. Protein concentration was then determined using Thermo Scientific's Coomassie Blue protein assay reagent. Aliquots were stored at −80°C. To ensure protein integrity, used aliquots were discarded after three freeze–thaw cycles.
DAP5 toeprinting
Twenty picomoles of FGF-9 5′-UTR RNA was first folded in the presence of magnesium. To ensure the majority of RNA was bound by protein, purified DAP5 was introduced to a final concentration of 300 µM, well above our group's determined Kd for DAP5 binding to the FGF-9 5′ UTR (Tijerina et al. 2007; Haizel et al. 2020). To match SHAPE solutions, the final volume of these protein–RNA solutions was 9 µL. As a negative control, 9 µL of a 20 pmol FGF-9 5′-UTR RNA solution was run in parallel. Protein–RNA solutions were incubated at room temperature for 10 min. Ten picomoles of cy5-labeled cDNA primer, 1 µL of RiboLock RNase Inhibitor, 2 µL of NEB10 µM dNTP mix, 1 µL of 0.1 mM DTT, 1 µL nuclease-free water, and 4 µL Invitrogen 5× first-strand buffer were added directly to protein–RNA solutions. Parallel CTP-seq and UTP-seq reactions were run with unbound FGF-9 mRNA and ddNTPs. Reverse transcription, cDNA purification, and sequencing via capillary electrophoresis were performed as described for SHAPE samples (see SHAPE-seq). Peak height in the presence of DAP5 was determined using SHAPEFinder (Vasa et al. 2008). To determine the frequency of RT stops in the presence of DAP5, the peak area from an RNA-only trace was subtracted from the peak area of the DAP5 sample trace (Lozano et al. 2018). The “box plot” method was then used for data normalization (Low and Weeks 2010; Rice et al. 2014).
DAP5 footprinting
1M7 and DMSO were preheated to 37°C. As with toeprinting experiments, 300 µM purified DAP5 was bound to folded 20 pmol FGF-9 5′-UTR RNA at room temperature for 10 min; 5 mM 1M7 or an equivalent volume of DMSO was added to protein–RNA solutions, followed by incubation at 37°C for 70 sec. To digest DAP5 off of RNA and ensure all modified RNA was fully recovered, 60 µg proteinase K was added, followed by incubation at 37°C for 10 min (Duncan and Weeks 2008). Sample tubes were again wrapped in tinfoil to protect 1M7-modified RNA from light. 1M7-modified RNA was purified into 9 µL nuclease-free water using the Zymo Oligo Clean & Concentrator Kit. Reverse transcription, cDNA isolation, sequencing, SHAPEFinder analysis, and data normalization were performed as described with SHAPE samples. As with toeprinting samples, the cDNA trace from DAP5 footprinting samples was compared to cDNA traces from RNA-only samples (Lozano et al. 2018). To determine changes in 1M7 reactivity, normalized and averaged 1M7 reactivity in the presence of DAP5 was subtracted from normalized and averaged 1M7 reactivity in the absence of DAP5. Because size standard peaks were visible in normalized data, peaks at 20, 40, 50, 60, 70, 80, 100, 120, 140, 160, 170, and 180 nt are not presented here.
DAP5 UV cross-linking
Our UV cross-linking experiments were optimized to the wt FGF-9 5′ UTR based on existing literature (Poria and Ray 2017; Urdaneta and Beckmann 2020). A 96-well plate was left to chill at −20°C until the sample was prepared; 300 µM purified DAP5 was bound to folded 60 pmol FGF-9 5′-UTR RNA at room temperature for 10 min. To identify RNA-to-RNA cross-linking, an equivalent volume of folded 60 pmol FGF-9 5′-UTR RNA was prepared without DAP5 (Urdaneta and Beckmann 2020). Samples were then transferred to the chilled 96-well plate for cross-linking. The plate was irradiated with 100 mJ/cm2 light atop a frozen ice pack in a VWR UV Crosslinker. Cross-linked samples were transferred to fresh Eppendorf tubes. Non-cross-linked DAP5 residues were digested off of RNA, and cross-linked RNA was recovered in 9 µL nuclease-free water as described for DAP5 footprinting. Just prior to reverse transcription, 20 pmol of non-cross-linked wt FGF-9 5′-UTR RNA was added to the cross-linked sample in lieu of 1 µL nuclease-free water. cDNA made from cross-linked RNA overwhelmingly terminated at the first or second DAP5 cross-linking site, making it difficult to match peaks to sequencing traces. The addition of excess unreacted RNA at this stage ensured a complete transcript to align sequencing lanes. Reverse transcription, cDNA isolation, sequencing, SHAPEFinder analysis, and data normalization was then executed as described for DAP5 footprinting and toeprinting. As with DAP5 footprinting, size-standard peaks at 20, 40, 50, 60, 70, 80, 100, 120, 140, 160, 170, and 180 nt are not presented here.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
This project was funded by National Institutes of Health grant (NIH) RO1 GM 128239 and National Science Foundation grant (NSF) MCB 2317112. The authors wish to thank Dr. Usha Bhardwaj, Dr. Solomon Haizel, and Dr. Paul Powell for gifting their plasmid constructs, primers, and knowledge of SHAPE. Open-source software was crucial for this publication. The authors thank the Laederach Lab (UNC) for their MATLAB CEQ alignment suite. Additional thanks are in order for the Weeks Lab and Giddings Lab (both at UNC Chapel Hill) for providing us with SHAPEFinder software. Thank you to the Mathews Lab (URMC) for RNAStructure, used for our 2D structural predictions. 2D structures were annotated thanks to RNA2Composer, developed by the Simon Lab (UMD) in collaboration with the NCI. We are grateful to the RNAComposer team (Polish Academy of Sciences, Poznan University of Technology) for their 3D modeling software. 3D structures were annotated thanks to UCSF ChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080013.124.
MEET THE FIRST AUTHOR
Amanda Whittaker.

Meet the First Author is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Amanda Whittaker is the first author of this paper, “Modeling the structure and DAP5-binding site of the FGF-9 5′-UTR RNA utilized in cap-independent translation.” Amanda is a sixth-year PhD candidate in Biochemistry with The Graduate Center, CUNY, and Hunter College, CUNY. She is currently finishing her dissertation about the role of RNA structure in cap-independent translation initiation. She received her bachelor's degree in Biochemistry from Boston College in 2015. She spent a few years after college working in the biotech industry, most notably as a research associate at Siemens Healthineers in Norwood, Massachusetts from 2016 to 2018.
What are the major results described in your paper and how do they impact this branch of the field?
Several proteins, like FGF-9, are cap-independently upregulated during hypoxic stress, but the exact mechanism of that upregulation is still unknown. We know that transcript binding to an eIF4GI homolog (DAP5) plays a role. Our group and others haven't predicted any significant sequence or structural similarity between DAP5-bound transcripts—there's no easy answer as to what DAP5 could be recognizing! We used chemical probing to characterize the structure of the FGF-9 5′-UTR RNA, and then performed probing again in the presence of DAP5. It turns out DAP5 likely recognizes a face of the RNA, rather than a single sequence or secondary structural element. UV cross-linking of DAP5 to the FGF-9 5′-UTR transcript confirms this. We suggest that how these RNA sit in space, rather than a single shared motif, may be key for understanding transcriptional regulation in cancer cells.
What led you to study RNA or this aspect of RNA science?
I fell in love with genetics and bioinformatics in undergraduate, so I knew I wanted to do something with either the genome or the transcriptome. Learning about epigenetics for the first time absolutely blew my mind: We focus so much on sequence, but important information can be conferred with nucleic acid structure, orientation, and modification. The implications of that—that we may be sitting on a wealth of valuable information not immediately apparent from sequencing—haunted me. Seeing Moderna and other groups suggest that RNA can be used as a therapeutic tool made me sure that I wanted to work with RNA!
During the course of these experiments, were there any surprising results or particular difficulties that altered your thinking and subsequent focus?
We wanted to be 100% sure where DAP5 bound, so Professor Goss suggested UV cross-linking. It was more challenging than it looked at first glance. Everyone cross-links RNA to proteins in slightly different ways, with slightly different purification methods. I fiddled with existing methods with varying success. At first, I was practically frying the DAP5 and RNA with too much energy. There was at least a month where I was coming in daily, doing the experiment over and over with different parameters, purifying more DAP5 or transcribing more RNA as needed … I still remember the day when I began getting reproducible results. I'm so excited to finally share what worked and hopefully help others with their own RNA–protein cross-linking!
What are some of the landmark moments that provoked your interest in science or your development as a scientist?
I've always been deeply fascinated by how the world worked. I spent a large part of my childhood in Orlando, Florida, which is full of cool wildlife. I fell in love with science by observing the native reptiles and insects. My parents saw this fascination right away and encouraged it with educational books and toys. The shape and form of what I wanted to do in science changed over time, but becoming a scientist was always a lifelong goal.
I also have a strong liberal arts background—I focused on theater in high school and took a ton of creative writing, literature, and arts courses in undergrad. I thought I'd have to leave my love of the arts behind if I became a scientist, but that wasn't true. All scientists are trying to tell a story! I realized I could put those communication skills to work for my scientific career. I drew my own animations for a talk at CSH 2022, and everyone who spoke to me afterward knew me as “the girl with the animations.” Getting to integrate those two sides of my brain gave me so much joy.
If you were able to give one piece of advice to your younger self, what would that be?
“You belong here. Keep going.”
Are there specific individuals or groups who have influenced your philosophy or approach to science?
I owe so much to my former coworkers and supervisors. I miss every single one of them! Sitting passively in a university lecture hall is one thing, but research at the bench is a different animal entirely. Having supportive mentors gave me the push I needed to stay in this field. All three of my PhD recommendation letters were from scientists I worked closely with at the very beginning of my career (shout-out to Paul, Bob, and Wei!).
I am a big fan of popular science educators like Carl Sagan, Neil deGrasse Tyson, and Bill Nye. I have a small portrait of Carl Sagan on my desk in our lab. His humility and curiosity in particular made me feel the “human” part of what we do. I always want my work to be approachable, even for a general audience.
What are your subsequent near- or long-term career plans?
I am currently prepping for my PhD defense and am looking for work! I would love to either move into clinical research or return to R&D within the biotech and biomedical industry. I am especially excited about where RNA therapeutics will go next. Perhaps this is my bias talking, but I'd love to keep exploring RNA structure and its uses for drug and vaccine design. I can see myself studying it for the rest of my life. Beyond that, I am keeping my mind open. I love to learn and wouldn't mind diving into a completely different topic.
REFERENCES
- Akirtava C, May GE, McManus CJ. 2022. False-positive IRESes from Hoxa9 and other genes resulting from errors in mammalian 5′ UTR annotations. Proc Natl Acad Sci 119: e2122170119. 10.1073/pnas.2122170119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alard A, Marboeuf C, Fabre B, Jean C, Martineau Y, Lopez F, Vende P, Poncet D, Schneider RJ, Bousquet C, et al. 2019. Differential regulation of the three eukaryotic mRNA translation initiation factor (eIF) 4Gs by the proteasome. Front Genet 10: 254. 10.3389/fgene.2019.00254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antczak M, Popenda M, Zok T, Sarzynska J, Ratajczak T, Tomczyk K, Adamiak RW, Szachniuk M. 2016. New functionality of RNAComposer: application to shape the axis of miR160 precursor structure. Acta Biochim Pol 63: 737–744. 10.18388/abp.2016_1329 [DOI] [PubMed] [Google Scholar]
- Balvay L, Rifo RS, Ricci EP, Decimo D, Ohlmann T. 2009. Structural and functional diversity of viral IRESes. Biochim Biophys Acta 1789: 542–557. 10.1016/j.bbagrm.2009.07.005 [DOI] [PubMed] [Google Scholar]
- Braunstein S, Karpisheva K, Pola C, Goldberg J, Hochman T, Yee H, Cangiarella J, Arju R, Formenti SC, Schneider RJ. 2007. A hypoxia-controlled cap-dependent to cap-independent translation switch in breast cancer. Mol Cell 28: 501–512. 10.1016/j.molcel.2007.10.019 [DOI] [PubMed] [Google Scholar]
- Chen T-M, Shih Y-H, Tseng JT, Lai M-C, Wu C-H, Li Y-H, Tsai S-J, Sun HS. 2014. Overexpression of FGF9 in colon cancer cells is mediated by hypoxia-induced translational activation. Nucleic Acids Res 42: 2932–2944. 10.1093/nar/gkt1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey E, Newman DR, Sannes PL. 2013. Expression of fibroblast growth factor 9 in normal human lung and idiopathic pulmonary fibrosis. J Histochem Cytochem 61: 671–679. 10.1369/0022155413497366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave P, George B, Raheja H, Rani P, Behera P, Das S. 2019. The mammalian host protein DAP5 facilitates the initial round of translation of coxsackievirus B3 RNA. J Biol Chem 294: 15386–15394. 10.1074/jbc.RA119.009000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Parra C, Ernlund A, Alard A, Ruggles K, Ueberheide B, Schneider RJ. 2018. A widespread alternate form of cap-dependent mRNA translation initiation. Nat Commun 9: 3068. 10.1038/s41467-018-05539-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan CDS, Weeks KM. 2008. SHAPE analysis of long-range interactions reveals extensive and thermodynamically preferred misfolding in a fragile group I intron RNA. Biochemistry 47: 8504–8513. 10.1021/bi800207b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry RC, Ide NA, Comunale VM, Hartwick EW, Kinz-Thompson CD, Gonzalez RL. 2023. The mechanism of mRNA activation. bioRxiv 10.1101/2023.11.15.567265 [DOI]
- Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, Ferrin TE. 2018. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci 27: 14–25. 10.1002/pro.3235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haizel SA, Bhardwaj U, Gonzalez RL, Mitra S, Goss DJ. 2020. 5′-UTR recruitment of the translation initiation factors eIF4GI or DAP5 drives cap-independent translation of a subset of human mRNAs. J Biol Chem 295: 11693–11706. 10.1074/jbc.RA120.013678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson PZ, Kasprzak WK, Shapiro BA, Simon AE. 2019. RNA2Drawer: geometrically strict drawing of nucleic acid structures with graphical structure editing and highlighting of complementary subsequences. RNA Biol 16: 1667–1671. 10.1080/15476286.2019.1659081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komar AA, Hatzoglou M. 2011. Cellular IRES-mediated translation. Cell Cycle 10: 229–240. 10.4161/cc.10.2.14472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacerda R, Menezes J, Romão L. 2017. More than just scanning: the importance of cap-independent mRNA translation initiation for cellular stress response and cancer. Cell Mol Life Sci 74: 1659–1680. 10.1007/s00018-016-2428-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, McCormick F. 2006. P97/DAP5 is a ribosome-associated factor that facilitates protein synthesis and cell proliferation by modulating the synthesis of cell cycle proteins. EMBO J 25: 4008–4019. 10.1038/sj.emboj.7601268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppek K, Das R, Barna M. 2018. Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat Rev Mol Cell Biol 19: 158–174. 10.1038/nrm.2017.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberman N, Gandin V, Svitkin YV, David M, Virgili G, Jaramillo M, Holcik M, Nagar B, Kimchi A, Sonenberg N. 2015. DAP5 associates with eIF2β and eIF4AI to promote Internal Ribosome Entry Site driven translation. Nucleic Acids Res 43: 3764–3775. 10.1093/nar/gkv205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low JT, Weeks KM. 2010. SHAPE-directed RNA secondary structure prediction. Methods 52: 150–158. 10.1016/j.ymeth.2010.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano G, Francisco-Velilla R, Martinez-Salas E. 2018. Ribosome-dependent conformational flexibility changes and RNA dynamics of IRES domains revealed by differential SHAPE. Sci Rep 8: 5545. 10.1038/s41598-018-23845-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques R, Lacerda R, Romão L. 2022. Internal ribosome entry site (IRES)-mediated translation and its potential for novel mRNA-based therapy development. Biomedicines 10: 1865. 10.3390/biomedicines10081865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Salas E, Francisco-Velilla R, Fernandez-Chamorro J, Embarek AM. 2018. Insights into structural and mechanistic features of viral IRES elements. Front Microbiol 8: 2629. 10.3389/fmicb.2017.02629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra S, Shcherbakova IV, Altman RB, Brenowitz M, Laederach A. 2008. High-throughput single-nucleotide structural mapping by capillary automated footprinting analysis. Nucleic Acids Res 36: e63. 10.1093/nar/gkn267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimer SA, Weeks KM. 2007. A fast-acting reagent for accurate analysis of RNA secondary and tertiary structure by SHAPE chemistry. J Am Chem Soc 129: 4144–4145. 10.1021/ja0704028 [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris JH, Ferrin TE. 2020. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci 30: 70–82. 10.1002/pro.3943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popenda M, Szachniuk M, Antczak M, Purzycka KJ, Lukasiak P, Bartol N, Blazewicz J, Adamiak RW. 2012. Automated 3D structure composition for large RNAs. Nucleic Acids Res 40: e112. 10.1093/nar/gks339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poria D, Ray P. 2017. RNA-protein UV-crosslinking assay. Bio Protoc 7: e2193. 10.21769/BioProtoc.2193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell P, Bhardwaj U, Goss D. 2022. Eukaryotic initiation factor 4F promotes a reorientation of eukaryotic initiation factor 3 binding on the 5′ and the 3′ UTRs of barley yellow dwarf virus mRNA. Nucleic Acids Res 50: 4988–4999. 10.1093/nar/gkac284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter JS, Mathews DH. 2010. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics 11: 129. 10.1186/1471-2105-11-129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GM, Leonard CW, Weeks KM. 2014. RNA secondary structure modeling at consistent high accuracy using differential SHAPE. RNA 20: 846–854. 10.1261/rna.043323.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Kato Y, Hamada M, Akutsu T, Asai K. 2011. IPknot: fast and accurate prediction of RNA secondary structures with pseudoknots using integer programming. Bioinformatics 27: i85–i93. 10.1093/bioinformatics/btr215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexton AN, Wang PY, Rutenberg-Schoenberg M, Simon MD. 2017. Interpreting reverse transcriptase termination and mutation events for greater insight into the chemical probing of RNA. Biochemistry 56: 4713–4721. 10.1021/acs.biochem.7b00323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shatsky IN, Terenin IM, Smirnova VV, Andreev DE. 2018. Cap-independent translation: What's in a name? Trends Biochem Sci 43: 882–895. 10.1016/j.tibs.2018.04.011 [DOI] [PubMed] [Google Scholar]
- Shestakova ED, Smirnova VV, Shatsky IN, Terenin IM. 2023. Specific mechanisms of translation initiation in higher eukaryotes: the eIF4G2 story. RNA 29: 282–299. 10.1261/rna.079462.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova VV, Shestakova ED, Nogina DS, Mishchenko PA, Prikazchikova TA, Zatsepin TS, Kulakovskiy IV, Shatsky IN, Terenin IM. 2022. Ribosomal leaky scanning through a translated uORF requires eIF4G2. Nucleic Acids Res 50: 1111–1127. 10.1093/nar/gkab1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkin YV, Pause A, Haghighat A, Pyronnet S, Witherell G, Belsham GJ, Sonenberg N. 2001. The requirement for eukaryotic initiation factor 4A (elF4A) in translation is in direct proportion to the degree of mRNA 5′ secondary structure. RNA 7: 382–394. 10.1017/s135583820100108x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tijerina P, Mohr S, Russell R. 2007. DMS footprinting of structured RNAs and RNA–protein complexes. Nat Protoc 2: 2608–2623. 10.1038/nprot.2007.380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urdaneta EC, Beckmann BM. 2020. Fast and unbiased purification of RNA–protein complexes after UV cross-linking. Methods 178: 72–82. 10.1016/j.ymeth.2019.09.013 [DOI] [PubMed] [Google Scholar]
- Vasa SM, Guex N, Wilkinson KA, Weeks KM, Giddings MC. 2008. ShapeFinder: a software system for high-throughput quantitative analysis of nucleic acid reactivity information resolved by capillary electrophoresis. RNA 14: 1979–1990. 10.1261/rna.1166808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgili G, Frank F, Feoktistova K, Sawicki M, Sonenberg N, Fraser CS, Nagar B. 2013. Structural analysis of the DAP5 mIF4G domain and its interaction with eIF4A. Structure 21: 517–527. 10.1016/j.str.2013.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volta V, Pérez-Baos S, de la Parra C, Katsara O, Ernlund A, Dornbaum S, Schneider RJ. 2021. A DAP5/eIF3d alternate mRNA translation mechanism promotes differentiation and immune suppression by human regulatory T cells. Nat Commun 12: 6979. 10.1038/s41467-021-27087-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks KM, Mauger DM. 2011. Exploring RNA structural codes with SHAPE chemistry. Acc Chem Res 44: 1280–1291. 10.1021/ar200051h [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weingarten-Gabbay S, Khan D, Liberman N, Yoffe Y, Bialik S, Das S, Oren M, Kimchi A. 2014. The translation initiation factor DAP5 promotes IRES-driven translation of p53 mRNA. Oncogene 33: 611–618. 10.1038/onc.2012.626 [DOI] [PubMed] [Google Scholar]