

Conspectus
Chemists have long been inspired by biological photosynthesis, wherein a series of excited-state electron transfer (ET) events facilitate the conversion of low energy starting materials such as H2O and CO2 into higher energy products in the form of carbohydrates and O2. While this model for utilizing light-driven charge transfer to drive catalytic reactions thermodynamically “uphill” has been extensively adapted for small molecule activation, molecular machines, photoswitches, and solar fuel chemistry, its application in organic synthesis has been less systematically developed. However, the potential benefits of these approaches are significant, both in enabling transformations that cannot be readily achieved using conventional thermal chemistry and in accessing distinct selectivity regimes that are uniquely enabled by excited-state mechanisms. In this Account, we present work from our group that highlights the ability of visible light photoredox catalysis to drive useful organic transformations away from their equilibrium positions, addressing a number of long-standing synthetic challenges.
We first discuss how excited-state ET enabled the first general methods for the catalytic anti-Markovnikov hydroamination of unactivated alkenes with alkyl amines. In these reactions, an excited-state iridium(III) photocatalyst reversibly oxidizes secondary amine substrates to their corresponding aminium radical cations (ARCs). These electrophilic N-centered radicals can then react with olefins to furnish valuable tertiary amine products with complete anti-Markovnikov regioselectivity. Notably, some of these products are less thermodynamically stable than their corresponding amine and alkene starting materials. We next present a light-driven method for C–C bond cleavage in various aliphatic alcohols. In these reactions, alcohol O–H bonds are homolytically activated via excited-state proton-coupled electron transfer (PCET). The resulting alkoxy radical intermediates then undergo C–C β-scission to ultimately provide isomeric linear carbonyl products that are often higher in energy than their cyclic alcohol precursors. Applications of this chemistry for the light-driven depolymerization of lignin biomass, commercial phenoxy resin, hydroxylated polyolefin derivatives, and thermoset polymers are presented as well. We then describe a method for the contrathermodynamic positional isomerization of highly substituted olefins by means of cooperative photoredox and chromium(II) catalysis. In this work, generation of an allylchromium(III) species that can undergo highly regioselective in situ protodemetalation enables access to a less substituted and thermodynamically less stable positional isomer. Product selectivity in this reaction is determined by the large differential in oxidation potentials between differently substituted olefin isomers. Lastly, we discuss a light-driven deracemization reaction developed in collaboration with the Miller group, wherein a racemic urea substrate undergoes spontaneous optical enrichment upon visible light irradiation in the presence of an iridium(III) chromophore, a chiral Brønsted base, and a chiral peptide thiol. Excellent levels of enantioselectivity are achieved via sequential and synergistic proton transfer (PT) and H-atom transfer (HAT) steps. Taken together, these examples highlight the ability of excited-state ET events to enable access to non-equilibrium product distributions across a wide range of catalytic, redox-neutral transformations in which photons are the only stoichiometric reagents.
Graphical Abstract

Introduction
The majority of synthetically useful organic transformations are thermal processes that occur along a single potential energy surface. However, due to microscopic reversibility, thermal reactions are limited to transforming higher-energy starting materials into lower-energy products. In these scenarios, the favorable change in free energy provides directionality to the process.6 While generally taken for granted in reaction design, this fundamental aspect of thermal chemistry also represents a significant limitation with respect to its applications in synthesis. In many instances, it would be advantageous to convert a low-energy set of starting materials into higher-energy (and often higher-value) products, identify conditions that selectively drive known transformations in the reverse direction, or enable normally irreversible reactions to become dynamic. While there are significant synthetic opportunities in being able to drive organic reactions in opposition to a thermodynamic bias, doing so requires a means to decouple the reaction direction from the ground-state thermochemistry of the starting materials and products.
This long-standing challenge in organic chemistry has inspired numerous successful solutions, each with its own benefits and limitations. Common strategies involve offsetting the unfavorable energetics associated with a reaction of interest by coupling it to a more thermodynamically favorable process, often by utilizing stoichiometric reagents or modified substrates to elevate the energy of the starting materials. While highly effective, such strategies suffer from the generation of stoichiometric waste and the potential need for substrate prefunctionalization. Another popular approach is electrochemistry, where electrode potentials can be precisely controlled and modulated to drive even the most challenging charge transfer events.7 Although electrochemical transformations are often more environmentally benign than reagent-driven methods that rely on stoichiometric oxidants or reductants, they are most effective for processes that are either net-oxidative or net-reductive and can be challenging to apply to redox-neutral transformations.
An alternative strategy to drive organic reactions “uphill” in opposition to a thermodynamic bias is photoredox catalysis.8 Inspired by the excited-state charge transfer events that enable biological photosynthesis, these approaches harness the enhanced redox properties of electronic excited states to drive charge transfer events that would otherwise be disfavored.9,10 In addition to the exogenous driving force provided by photon absorption, photoredox manifolds exhibit unique kinetic benefits as a function of proceeding across multiple potential energy surfaces (e.g. ground and excited electronic states). These advantages can be illustrated in a simplified model reaction that converts lower energy starting material A into higher energy product B (Figure 1). For a thermal process, it is not possible to quantitatively convert A to B since microscopic reversibility necessitates a lower energy barrier for the conversion of B back to A. This establishes a thermal equilibrium that is dominated by the lower energy state A. However, in a photochemical transformation, irradiation of the chromophore generates an electronic excited state with augmented redox properties, enabling favorable ET with A to form a key charge-separated intermediate, CS. CS, comprised of the oxidized photocatalyst and the reduced form of A, is significantly higher in energy than the starting materials, indicating that some portion of the incident photon’s energy has been converted into chemical potential in the form of separated charge. CS can then either be consumed productively to form desired product B, or unproductively via back-electron transfer (BET) to return the ground-state chromophore and starting material A. Critically, the partitioning between the two pathways is kinetically controlled and is a function of the differential activation barriers for each reaction pathway that consumes CS. Since these selectivity-determining steps occur on the excited-state energy surface, the selectivity of the reaction can be decoupled from the ground-state thermochemistry of A and B, thereby providing a means to selectively populate the high-energy state. Similar considerations are applicable with regards to excited-state energy transfer mechanisms as well.
Figure 1.

A) Product equilibria in thermal reactions. B) Potential product equilibria in a photochemical reaction.
These ideas have been investigated in numerous chemical contexts for decades, particularly for solar fuel and small molecule-activation chemistry. Their specific adoption to address challenges in synthetic organic chemistry have only recently become the focus of research efforts, with seminal contributions from Bach,11 Gilmour,12 Wendlandt,13 MacMillan,14 Sorensen,15 Lei,16 Weaver,17 and others.9,10 In this Account, we present efforts from our lab to apply excited-state ET-based approaches to enable a variety of organic transformations that proceed in opposition to a thermodynamic bias and are challenging to catalyze using conventional thermal approaches. We illustrate how these excited-state ET schemes can facilitate a diverse array of redox-neutral transformations, including addition, elimination, and isomerization reactions that efficiently and selectively generate products that are higher in energy than the starting materials they are derived from. Moreover, we highlight that excited-state processes exhibit selectivity features that are distinct from those of conventional thermal reactions. These distinctions create exciting prospects for synthesis where chemists can, in principle, use molecular catalysts to kinetically select for any accessible product state in a dynamic system irrespective of its ground-state thermochemical preferences.
Intermolecular Anti-Markovnikov Hydroamination of Unactivated Olefins
Alkyl amines are ubiquitous in natural products, pharmaceuticals, and other bioactive small molecules. Consequently, strategies enabling selective C(sp3)–N bond construction are highly sought after, with catalytic intermolecular olefin hydroamination being an attractive approach that couples two classes of structurally diverse and accessible starting materials in a redox-neutral and atom-economical manner.18,19 While catalytic hydroamination technologies have significantly advanced, the intermolecular anti-Markovnikov hydroamination of electronically unactivated olefins with alkyl amines was a long-recognized challenge with no general solution. Established transition metal- and acid-catalyzed variants generally proceed with high levels of Markovnikov regioselectivity unless electronically biased olefins are employed.20 More fundamentally, the bimolecular addition of alkyl amine N–H bonds to substituted aliphatic alkenes generally lacks a significant thermodynamic driving force.21 For example, the anti-Markovnikov hydroamination of 2-methyl-2-butene with diethylamine is approximately thermoneutral (ΔG° = −0.1 kcal/mol at 298 K, CBS-QB3).1 Increasing the steric profile of the olefin, as demonstrated by the coupling of 2,3,-dimethyl-2-butene with diethylamine, renders the process endergonic by +4.9 kcal/mol (Figure 2A).
Figure 2.

A) Challenging thermodynamics for intermolecular hydroamination of unactivated olefins. B) Proposed catalytic cycle for intermolecular hydroamination. C) Kinetics of ARC olefin addition.
We hypothesized that both key issues in anti-Markovnikov olefin hydroamination could be overcome through radical-based approaches that are enabled by excited-state ET. Specifically, we envisioned a prospective catalytic cycle (Figure 2B) initiated by blue light irradiation of an iridium(III) polypyridyl photocatalyst to generate an excited-state oxidant capable of selectively oxidizing a secondary alkyl amine to its corresponding aminium radical cation (ARC).22 Pioneering studies by Newcomb and Lusztyk have shown that these highly electrophilic N-centered radicals undergo rapid and regioselective anti-Markovnikov addition to alkyl olefins at room temperature (k > 108 M−1s−1) (Figure 2C).23,24 After C–N bond formation, the nascent alkyl radical is reduced by an aryl thiol co-catalyst via HAT, resulting in formation of the closed-shell ammonium product and a thiyl radical. Reduction of the thiyl radical by the reduced photocatalyst furnishes a thiolate, which can deprotonate the ammonium to provide a tertiary amine and regenerate the thiol co-catalyst. Overall, this process would deliver the desired anti-Markovnikov hydroamination product in a redox-neutral fashion wherein the excitation event provides the exogenous driving force necessary for efficient product formation.
We ultimately discovered that blue light irradiation of toluene solutions containing 2 mol% [Ir(dF(Me)ppy)2(dtbbpy)]PF6 photocatalyst and 50 mol% 2,4,6-triisopropylbenzenethiol (TRIP thiol) HAT co-catalyst enabled effective anti-Markovnikov hydroamination of a wide variety of cyclic and acyclic secondary amines with a wide range of olefins, including terminal, 1,1- and 1,2-disubstituted, trisubstituted, and even tetrasubstituted olefins (Figure 3A).1 Enol ethers, allylic alcohols, and protected allyl amines were tolerated as well, all with complete anti-Markovnikov regioselectivity.
Figure 3.

A) Selected scope for photocatalytic intermolecular hydroamination of unactivated alkenes with secondary alkyl amines. B) Selected examples of intermolecular hydroamination with primary alkyl amines and heteroaryl amines.
Stern-Volmer studies showed that piperidine (Ep/2 = +0.56 V vs Fc+/Fc in MeCN) efficiently quenches the luminescence of the excited-state [Ir(dF(Me)ppy)2(dtbbpy)]PF6 photocatalyst (E1/2(*Ir(III)/Ir(II)) = +0.59 V vs Fc+/Fc in MeCN), consistent with ET to generate the ARC.1 Quenching was not observed for any of the aliphatic alkene coupling partners, indicating that this method does not proceed via alkene radical cation intermediates in contrast to seminal work by Nicewicz and coworkers.25 Through judicious photocatalyst selection, we have subsequently been able to expand this chemistry to realize the catalytic intermolecular anti-Markovnikov hydroamination reactions of unactivated alkenes with primary alkyl amines and more recently, heteroaryl amines (Figure 3B).26,27 Notably, these reactions selectively form secondary amines with minimal levels of overalkylation products. A detailed mechanistic study of the primary alkyl amine protocol by the Nocera lab resulted in both improved reaction conditions and the development of a scalable flow protocol.28
Photocatalytic Activation of Alcohol O–H Bonds for C–C Bond Cleavage
Over the last decade, our group has worked to develop PCET as a general catalytic platform for the homolytic activation of strong heteroatom–H bonds to access reactive heteroatom-centered radicals for organic synthesis.29 Of particular interest has been the use of PCET to directly generate alkoxy radicals from unfunctionalized alcohol starting materials. The protic character and high bond dissociation free energies (BDFEs) of alcohol O–H bonds (~105 kcal/mol) make them challenging to activate with conventional HAT reagents. Alternatively, the abstraction thermochemistry in PCET reactions is jointly determined by the pKa of the conjugate acid of the base and the reduction potential of the oxidant.30 Since these key properties can be independently modulated, the driving force for PCET can be elevated into ranges that enable favorable O–H cleavage. Accordingly, alkoxy radicals are versatile species that readily participate in hydrogen atom abstraction, olefin addition, and C–C bond β-scission reactions.31,32 The latter reactivity is particularly attractive in a catalytic context, as it would enable the functionalization of C–C bonds adjacent to common alcohol starting materials without requiring prefunctionalization of the hydroxyl group.
To this end, we set out to develop a redox-neutral isomerization of cyclic alcohols to linear carbonyl compounds via PCET-enabled C–C bond β-scission—an underdeveloped transformation that we hypothesized might have considerable utility in synthesis (Figure 4). Our initial efforts were inspired by the spectroscopic studies of Baciocchi and Steenken on alkoxy radical generation via radical relay in benzylic alcohol substrates.33 These reactions are initiated by single-electron oxidation of the substrate arene ring by an excited-state iridium(III) photocatalyst to form a transient arene radical cation. This arene radical cation then acts as an intramolecular oxidant for a bimolecular PCET event with the Brønsted base co-catalyst, activating the proximal O–H bond to furnish the key alkoxy radical intermediate. The alkoxy radical then undergoes C–C bond β-scission to open the carbocyclic ring, generating an aryl ketone and a tethered carbon-centered radical. This alkyl radical is subsequently reduced by a thiol H-atom donor to furnish a closed-shell linear aryl ketone product. The resulting thiyl can subsequently undergo ET with the reduced-state photocatalyst and PT with the protonated Brønsted base to close the catalytic cycle. While successful as both a method and a demonstration of PCET-based alcohol activation, the reaction scope of this first-generation process was restricted to cyclic alcohols bearing oxidizable aryl groups due to the redox-relay mechanism.34
Figure 4.

Proposed catalytic cycle for catalytic ring-opening isomerization of cyclic alcohols.
In 2019, we identified improved conditions that overcame this limitation, directly activating O–H bonds via PCET and circumventing the need for arene radical cation formation.2 In particular, we found that the identity of the Brønsted base was a critical factor governing reaction efficacy, as replacing a neutral collidine base with tetrabutylphosphonium dimethyl phosphate base resulted in dramatically improved reaction yields. The anionic phosphate is thought to promote formation of a key hydrogen-bonding complex between the alcohol substrate and base in the O–H PCET event, while the tetrabutylphosphonium cation is proposed to improve solubility in the nonpolar toluene solution. These new conditions greatly expanded the scope of the isomerization reaction and successfully facilitated the ring-opening isomerization of various cyclic aliphatic alcohols and hemiacetals, as well as the fragmentation of several acyclic tertiary alcohols (Figure 5).
Figure 5.

Selected scope and computed free energy change for ring-opening isomerization and β-fragmentation of aliphatic alcohols. aThermochemistry for isomerization of N-CO2Me substrate.
Notably, thermochemical calculations revealed that the linear carbonyl products were generally higher in energy than their corresponding cyclic alcohol starting materials, indicating that excited-state PCET processes are able to drive these redox-neutral isomerization reactions against a ground-state thermodynamic bias. To rationalize this light-driven contrathermodynamic reactivity, we considered the elementary steps necessary to isomerize cycloalkanol 1a to aldehyde 1b (Figure 6). After excluding mechanisms based on direct excitation or triplet sensitization of 2a, we postulated that an excited-state iridium(III) complex and phosphate base pair first engage in PCET with the O–H bond of 1a, forming the key alkoxy radical intermediate. This species can either undergo BET to return the closed-shell starting material 1a or undergo productive β-scission to form an alkyl radical. Subsequent HAT from the aryl thiol to the alkyl radical forms 1b, while ET/PT between the resultant thiyl radical, iridium(II), and protonated base returns the active forms of all three catalysts. Notably, the HAT and ET/PT steps in this mechanism are highly exergonic in the forward direction, effectively rendering these pathways irreversible. Even if the thiyl radical can be regenerated via excited-state S–H PCET, HAT between the thiyl radical and 1a to reform the requisite alkyl radical will be too slow to kinetically compete with charge recombination that drives the system back toward the products. This kinetic asymmetry in the forward and reverse directions is enforced by the charge recombination steps, which gate the lifetimes of key intermediates and thus enable selective formation of the higher-energy products. These processes are reminiscent of related ratchet-type mechanisms commonly invoked in supramolecular chemistry to drive unidirectional behavior in the reactions of molecular machines and motors.35,36
Figure 6.

Proposed free energy profile for the ring-opening isomerization of 1a to 1b, with the various molecular ensembles denoted as states A–G. Adapted from ref. 2. Copyright 2019 American Chemical Society.
We later harnessed this photocatalytic alkoxy radical-mediated C–C bond β-scission chemistry for the depolymerization of hydroxylated polymers. The depolymerization of polymers lacking hydrolyzable functionality along their backbone (e.g. polyesters and polyamides) typically requires iterative cleavage of strong aliphatic C–C bonds and is therefore thermodynamically challenging. While this issue can be addressed by subjecting the polymer to harsh conditions and featuring stoichiometric reagents, there is considerable interest in developing more selective and sustainable catalytic strategies for depolymerization that would allow polymer waste to become a source of chemical feedstock. For example, the depolymerization of lignin biomass, an abundant renewable resource, is considered a promising alternative to petroleum refining for producing arene feedstocks. However, current methods for lignin depolymerization often result in inseparable mixtures of poorly defined products. In our previous O–H PCET work, we were able to demonstrate that β-fragmentation of a representative lignin dimer into its constituent monomers could be achieved in good yields (Figure 7A).2 Encouraged by this result, we set out to establish an analogous system that could break down larger lignin polymers. Consequently, we developed a ternary catalyst system consisting of an iridium(III) photocatalyst, a phosphate base, and an aryl thiol H-atom donor that could selectively cleave up 42% of the C–C β-O-4 linkages from various native lignin samples to yield three well-defined monomer products (Figure 7B). Although the precise thermochemistry for the depolymerization of heterogeneous materials is difficult to calculate, we were able to determine that C–C bond β-scission of the aforementioned lignin dimer is approximately +1.2 kcal/mol uphill (M06–2X/6–311++G(d,p)//M06–2X/6–31G(d)).37 In collaboration with the Fors group, we also exploited this reactivity to perform photocatalytic depolymerizations of a commercial phenoxy resin and various high molecular weight hydroxylated polyolefin derivatives (Figure 8A).38 More recently, we adapted this chemistry to put forth a protocol for the selective depolymerization of highly cross-linked epoxy thermosets derived from bisphenol A (Figure 8B).5 Notably, this depolymerization results in the formation of four discrete monomers that can collectively be O-dealkylated using BBr3 to return bisphenol A, enabling a potential route for the chemical recycling of thiol epoxy thermosets.
Figure 7.

A) Computed thermochemistry for lignin dimer fragmentation. B) Photocatalytic depolymerization of native lignin biomass.
Figure 8.

A) Photocatalytic depolymerization of a commercial phenoxy resin. B) Photocatalytic depolymerization of a thiol epoxy thermoset.
Contrathermodynamic Olefin Isomerization
Positional alkene isomerization enables the interconversion between alkene regioisomers through double bond transposition along a carbon chain. While a great number of thermal transition metal-catalyzed isomerization protocols have been developed, the product distributions of such methods are dictated by the relative thermodynamic stabilities of the starting and product alkenes.39 As a result, such strategies can only transform less stable olefin substrates into their more stable positional isomers (Figure 9A). To achieve contrathermodynamic reactivity in which a more stable olefin is selectively isomerized into a less stable position, two-step methods involving sequential chain-walking hydrofunctionalization and elimination have been developed.40,41 In these methods, favorable consumption of stoichiometric reagents provides the driving force needed to compensate for the otherwise endergonic transformation. Alternatively, photochemical methods may be utilized to drive these reactions in opposition to a thermodynamic bias. Building on seminal works by Jorgenson,42 Pete,43 Arnold,44 and Gilmour45 in this area, we recently reported a photocatalytic method for the redox-neutral contrathermodynamic isomerization of various olefin classes through cooperative photoredox and chromium catalysis.4 Notably, an elegant light-driven method was published contemporaneously by the Wendlandt group that achieves similar types of contrathermodynamic isomerizations using a distinct set of catalysts.46
Figure 9.

A) Challenges associated with catalytic contrathermodynamic alkene transposition. B) Examples of chromium-catalyzed stereoselective allylation and regioselective reduction. C) Contrathermodynamic olefin isomerization enabled by dual photoredox and chromium catalysis.
Our approach was inspired by reports from Kanai and Glorius on the light-driven allylation of aldehydes with alkenes under the action of a photocatalyst and a chromium(II) salt (Figure 9B).47,48 In these reactions, single-electron oxidation of an electron-rich alkene forms a radical cation with significantly acidified allylic C–H bonds. Deprotonation subsequently affords an allylic radical that is captured by a chromium(II) catalyst to yield an allylchromium(III) complex. These classical organometallic nucleophiles can then add to aldehyde electrophiles to furnish homoallylic alcohol products with high levels of anti-diastereoselectivity and branched regioselectivity. The outcomes can be readily rationalized through cyclic six-membered transition-state models in which the Lewis acidic chromium(III) center is concomitantly bound to the less substituted terminus of the allyl fragment and coordinated to the oxygen of the aldehyde. We reasoned that protic electrophiles might function similarly, coordinating to chromium(III) and delivering a proton to the distal carbon of the allyl nucleophile to furnish a transposed alkene product. We found support for this proposal in stoichiometric studies by Castro and Kato, who reported that protodemetalation of allylchromium(III) complexes with alcohol proton donors was remarkably regioselective, favoring the less substituted alkene products.49,50 Together, these precedents suggested that a light-driven method could enable in situ generation of an organometallic nucleophile that would readily undergo regioselective protodemetalation to achieve alkene transposition (Figure 9C). As such, we proposed a catalytic mechanism for the contrathermodynamic positional isomerization of alkenes jointly mediated by a photocatalyst, a Brønsted base co-catalyst, a chromium(II) co-catalyst, and an alcohol proton donor (Figure 10). Importantly, we recognized that more substituted olefins often have oxidation potentials that are several hundred millivolts lower than those of less substituted olefins.51 Thus, with careful selection of the photocatalyst, we could ensure selective oxidation of the more substituted alkene starting material while rendering the isomerized products inert to oxidation and reentry into the catalytic cycle.
Figure 10.

Proposed catalytic cycle and key design principles for contrathermodynamic olefin isomerization.
After extensive optimization, we found that blue light irradiation of model silyl enol ether 2a (Ep/2 = +1.26 V vs Fc+/Fc in MeCN) in the presence of 4 mol% [Ir(dF(CF3)ppy)2(5,5’-d(CF3)bpy)]PF6 (Eo(*Ir(III)/Ir(II)) = 1.30 V Fc+/Fc in MeCN), 10 mol% CrCl2, 15 mol% dtbbpy, and 5 equiv. methanol in a solvent mixture of MeCN and PhCF3 (4:1 v/v) resulted in the formation of the desired isomerized allylic ether product 3b in nearly quantitative yield. Various silyl, benzyl, and aryl enol ethers bearing trisubstituted and tetrasubstituted internal olefins could also be successfully isomerized to give less stable terminal allylic ethers. However, we observed that the optimal conditions facilitating contrathermodynamic enol ether isomerization were less efficient for isomerizing other types of alkenes due to differences in their redox potentials. Further investigations revealed that by using 10 mol% CrCl3, dichloromethane as solvent, and various photocatalysts with appropriately matched redox potentials, a number of other alkene classes were amenable to our protocol. Specifically, enamides could be isomerized with [Ir(dF(CF3)ppy)2(bpy)]PF6 (Eo(*Ir(III)/Ir(II)) = 0.94 V Fc+/Fc in MeCN), styrenes and tetrasubstituted alkenes with [N-Ph Mes-t-Bu2-Acr]BF4 (Eo(*PC/PC−) = 1.72 V Fc+/Fc in MeCN), and 1,3-diene substrates with 4CzIPN (Eo(*PC/PC−) = 1.05 V Fc+/Fc in MeCN) (Figure 11).52 Computational evaluation (CBS-QB3) of the isomerization thermochemistry further confirmed the contrathermodynamic nature of this transformation.
Figure 11.
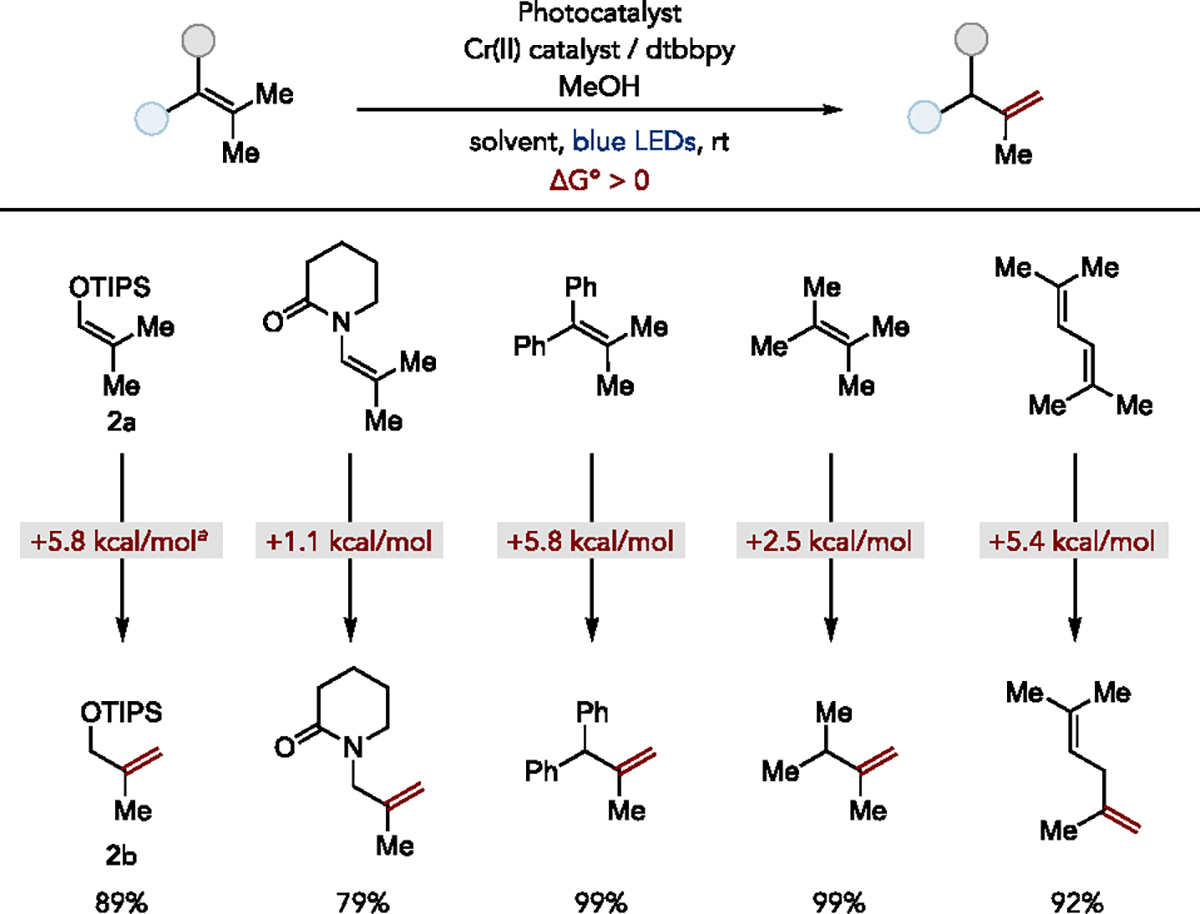
Selected scope and computed free energy change for olefin isomerization. aThermochemistry for isomerization of TMS substrate.
Light-Driven Deracemization of Cyclic Ureas
Excited-state ET events can also enable novel mechanisms for achieving selectivity that are distinct from those found in thermal processes. This is perhaps most clearly demonstrated in the recent development of photocatalytic deracemizations (Figure 12).53 Conventional approaches for accessing enantiopure materials either involve asymmetric synthesis—where an achiral reactant is converted to a chiral product—or kinetic resolution—where one stereoisomer of a racemic mixture is selectively converted into enantioenriched product. In both cases, the stereochemistry is set through either the bond forming or bond breaking reaction that lead to formation of the chiral product. By contrast, in a deracemization reaction, a racemic mixture of a given compound is wholly converted to a single enantiomer of the same compound. As such, the steps involved in the synthesis of the racemic substrate are distinct from the steps subsequently used to control stereoselectivity.
Figure 12.

Challenges associated with catalytic deracemization.
Despite its conceptual simplicity and potential practical benefits, examples of catalytic deracemization remain limited. This dearth of progress is largely attributed to two distinct challenges related to the energetic degeneracy of enantiomers. Although the reaction is enthalpically thermoneutral, conversion of a racemic mixture of enantiomers into a single enantiomer is entropically unfavorable (ΔG° = −TΔS° = RTln(2) = +0.42 kcal/mol at 298 K) and therefore requires an exogenous input of energy to proceed. The second and more formidable challenge stems from kinetic constraints related to microscopic reversibility. When considering individual particles on a potential energy surface rather than ensembles, the (R) and (S) enantiomers of a given chiral compound are, by definition, equal in energy. This ensures that any series of elementary steps that transform (R) into (S) will be equally accessible in converting (S) back to (R), necessarily resulting in a racemic mixture of enantiomers at equilibrium. To achieve deracemization, an energetic bias must be introduced for the interconversion of enantiomers such that all the elementary steps that convert one enantiomer into the other must be thermodynamically favorable and irreversible. As a consequence, the two steps that create and destroy stereochemistry in a deracemization reaction cannot be the microscopic reverse of one another and must proceed by distinct mechanisms.
These dual limitations are challenging to address and relatively few thermal catalytic deracemization reactions have been reported to date. Most of these protocols make use of chemically compatible or phase-separated combinations of oxidants and reductants that can drive the reversible formation and consumption of a common achiral intermediate.54 While effective, the scope and generality of these methods are often limited by the compatibility of these oxidant and reductant pairs. Since 2018, a number of highly enantioselective light-driven catalytic deracemizations have been reported that offer novel approaches to overcoming these constraints, including important contributions from the groups of Bach,55 Meggers,56 Luo,57 Gilmour,58 Jiang,59 and Zuo.60 Remarkably, the substrates in these reactions undergo spontaneous optical enrichment in the presence of an appropriate chromophore and chiral catalyst(s) simply upon irradiation with visible light! As these photochemical reactions occur across multiple potential energy surfaces, the individual steps responsible for creating and destroying stereochemistry can be made to be mechanistically distinct, while photon absorption provides the energy input necessary to drive these reactions forward.
In collaboration with the Miller group in 2019, we reported a light-driven method for the deracemization of cyclic ureas in up to 99% yield and 96:4 er using a three-component catalytic system comprised of an iridium(III) chromophore, a BINOL-derived chiral phosphate base, and a peptide-derived chiral thiol H-atom donor (Figure 13A).3 On the basis of experimental observations made during our optimization studies, we proposed a mechanism in which visible light irradiation of the iridium(III) photocatalyst generates an excited-state oxidant which reversibly oxidizes the urea substrate to form a mixture of transient radical cations with substantially acidified α-amino C–H bonds. These stereoisomeric radical cations are then kinetically resolved through selective C–H deprotonation by the chiral phosphate base, with the faster-reacting (R)-enantiomer undergoing PT to form a neutral α-amino radical while the slower-reacting (S)-enantiomer undergoes charge recombination with the reduced-state iridium(II) photocatalyst to return the original urea substrate with retention of configuration. The achiral α-amino radical intermediates then undergo enantioselective HAT with the chiral peptide thiol catalyst to preferentially reform the (S)-enantiomer of urea. Subsequent ET/PT between the reduced-state iridium(II), peptide thiyl radical, and protonated phosphate base regenerates all three active catalysts. Repeated iteratively, the reaction mixture becomes enriched in the slower-reacting (S)-enantiomer until a photostationary state is achieved in which the rate of formation of each enantiomer is equal to its rate of consumption.
Figure 13.

A) Photocatalytic deracemization of cyclic ureas enabled by excited-state redox events and the proposed mechanistic scheme. B) Kinetic scheme and selectivity expression for catalytic deracemization.
This mechanism is unusual in that both the C–H cleavage and C–H reformation steps proceed sequentially within the catalytic cycle and are mediated by two distinct chiral catalysts, raising the question of how each stereoselective step impacts the overall level of stereoselectivity observed. From the mechanistic proposal above, a kinetic expression can be derived describing the enantiomeric ratio at the photostationary state using the steady-state approximation. The expression is dependent on three terms: the enantioselectivity of the PT step between the enantiomeric radical cations and the chiral phosphate, the enantioselectivity of the HAT step between the prochiral α-amino radical and the chiral thiol, and the kinetic competition between PT and the BET of the reduced photocatalyst and the substrate radical cation (Figure 13B). In the limiting case where BET is much slower than PT (k-1 << k2, k2’), the ET step is rendered irreversible. In this scenario, both radical cation enantiomers are deprotonated by the phosphate base, negating any enantioselectivity in the PT step. In this regime the selectivity of the reaction depends solely on the enantioselectivity of the HAT step. Conversely, if BET is significantly faster than PT (k-1 >> k2, k2’), the ET step is fast and reversible relative to radical cation deprotonation. Consequently, one enantiomer is preferentially deprotonated while the other is converted back to the closed-shell urea. In this case, the overall selectivity depends equally on the stereoselectivity of both the PT and HAT steps, with the observed enantioselectivity being the product of the enantiomeric ratios for each elementary step (erobs= erPT × erHAT). Control experiments employing each chiral catalyst with its corresponding achiral co-catalyst showed that the individual PT and HAT steps were only modestly selective (erPT = 86:14 and erHAT = 79:21). However, when the stereochemically matched combination of thiol and base were employed together, the two modestly stereoselective steps operate in synergy, resulting in a very high enantiomeric ratio (erobs= 96:4) that was consistent with model prediction. This indicates that BET is faster than PT in these reactions—a unique outcome that is specifically enabled by the excited-state mechanism, as the lifetimes of the key radical cation intermediates are kinetically controlled by charge recombination in a stereochemically-dependent fashion.
Conclusion
This Account highlights the ability of excited-state redox events to drive a variety of redox-neutral transformations in opposition to a thermodynamic bias. This framework has the potential to be adapted for many reaction types, providing new opportunities to achieve reactions that are inaccessible under conventional thermal methods, drive known transformations in reverse, or introduce dynamic behavior into otherwise static systems. As such, we believe these strategies will lead to novel possibilities, efficiencies, and selectivities in synthesis, making the future of work in this area particularly bright.
Acknowledgements
Knowles group members, past and present, who have contributed to development of these projects and ideas. Nick Shin is acknowledged for contributions to an early version of this manuscript.
Funding Sources
The projects described above were supported by either the National Institutes of Health (NIGMS R35 GM134893) or through BioLEC, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences under Award No. DE-SC0019370. A.L acknowledges the NSF for a graduate fellowship (Grant DGE-2039656).
Biographies
Angela Lin
Angela hails from New York City and received her B.S. in chemistry from Yale University in 2020 under the advisement of Prof. Scott Miller. She then joined the Knowles Lab at Princeton University as an NSF graduate fellow, where she currently conducts research on developing light-driven methods for organic synthesis.
Sumin Lee
Sumin Lee was born and raised in Seoul, South Korea and obtained his B.S. and M.S degrees at Korea University, conducting research with Prof. Jong-Seung Kim. After five years at SK Chemicals, Sumin decided to pursue a career in academia and received his PhD in 2021 from Columbia University under the guidance of Prof. Tomislav Rovis. He then joined the Knowles Lab at Princeton University for his postdoctoral studies. Sumin began his independent career at Konkuk University as an assistant professor in September 2023.
Robert Knowles
A native of Virginia’s Shenandoah Valley, Rob received a B.S in chemistry from the College of William and Mary where he conducted research with Robert Hinkle and David Kranbuehl. He earned his PhD at Caltech with David MacMillan before moving to Eric Jacobsen’s lab at Harvard as a NIH postdoctoral fellow. Rob has been a faculty member in the Department of Chemistry at Princeton University since 2011.
Footnotes
The authors declare no competing financial interest.
References
- (1). Musacchio AJ; Lainhart BC; Zhang X; Naguib SG; Sherwood TC; Knowles RR Catalytic intermolecular hydroaminations of unactivated olefins with secondary alkyl amines. Science. 2017, 355, 727–730. Reports a general method for the photocatalytic intermolecular anti-Markovnikov hydroamination of unactivated olefins with dialkyl amines via the intermediacy of aminium radical cations.
- (2). Ota E; Wang H; Frye NL; Knowles RR A Redox Strategy for Light-Driven, Out-of-Equilibrium Isomerizations and Application to Catalytic C–C Bond Cleavage Reactions. J. Am. Chem. Soc. 2019, 141, 1457–1462. Describes a protocol for the direct PCET activation of alcohol O–H bonds to generate alkoxy radical intermediates. The β-scission reactivity of these intermediates is exploited for the unimolecular isomerization of cyclic aliphatic alcohols into linear carbonyl compounds.
- (3). Shin NY; Ryss JM; Zhang X; Miller SJ; Knowles RR Light-driven deracemization enabled by excited-state electron transfer. Science. 2019, 366, 364–369. Presents a strategy for the photocatalytic deracemization of cyclic urea racemates enabled by excited-state redox events. The reaction is mediated by three distinct catalysts and its enantioselectivity is determined by the synergistic stereoselectivity of the chiral ET and PT steps.
- (4). Zhao K; Knowles RR Contra-Thermodynamic Positional Isomerization of Olefins. J. Am. Chem. Soc. 2022, 144, 137–144. Discloses a light-driven approach for the contra-thermodynamic positional isomerization of highly substituted olefins to their less substituted counterparts under dual photoredox/chromium(II) catalysis. The out-of-equilibrium product distributions are gated by the differential oxidation potentials of the olefin isomers.
- (5). Nguyen ST; Fries LR; Cox JH; Ma Y; Fors BP; Knowles RR Chemical Recycling of Thiol Epoxy Thermosets via Light-Driven C–C Bond Cleavage. J. Am. Chem. Soc. 2023, 145, 11151–11160. Demonstrates the application of PCET-driven, alkoxy radical-mediated C–C bond cleavage chemistry for the photocatalytic depolymerization of thiol epoxy thermoset materials into well-defined monomeric products.
- (6).Blackmond DG “If pigs could fly” chemistry: a tutorial on the principle of microscopic reversibility. Angew. Chem. Int. Ed. 2009, 48, 2648–2654. [DOI] [PubMed] [Google Scholar]
- (7).Liu J; Lu L; Wood D; Lin S New Redox Strategies in Organic Synthesis by Means of Electrochemistry and Photochemistry. ACS Cent. Sci. 2020, 6, 1317–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kathan M; Hecht S Photoswitchable molecules as key ingredients to drive systems away from the global thermodynamic minimum. Chem. Soc. Rev. 2017, 46, 5536–5550. [DOI] [PubMed] [Google Scholar]
- (9).Wang H; Tian YM; König B Energy- and atom-efficient chemical synthesis with endergonic photocatalysis. Nat. Rev. Chem. 2022, 6, 745–755. [DOI] [PubMed] [Google Scholar]
- (10).Wang PZ; Xiao WJ; Chen JR Light-empowered contra-thermodynamic stereochemical editing. Nat. Rev. Chem. 2023, 7, 35–50. [DOI] [PubMed] [Google Scholar]
- (11).Bauer A; Westkämper F; Grimme S; Bach T Catalytic enantioselective reactions driven by photoinduced electron transfer. Nature. 2005, 436, 1139–1140. [DOI] [PubMed] [Google Scholar]
- (12).Metternich JB; Gilmour R A Bio-Inspired, Catalytic E → Z Isomerization of Activated Olefins. J. Am. Chem. Soc. 2015, 137, 11254–11257. [DOI] [PubMed] [Google Scholar]
- (13).Wang Y; Carder HM; Wendlandt AE Synthesis of rare sugar isomers through site-selective epimerization. Nature. 2020, 578, 403–408. [DOI] [PubMed] [Google Scholar]
- (14).Oswood CJ; MacMillan DWC Selective Isomerization via Transient Thermodynamic Control: Dynamic Epimerization of trans to cis Diols. J. Am. Chem. Soc. 2022, 144, 93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).West JG; Huang D; Sorensen EJ Acceptorless dehydrogenation of small molecules through cooperative base metal catalysis. Nat. Commun. 2015, 6, 10093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wang H; Gao X; Lv Z; Abdelilah T; Lei A Recent Advances in Oxidative R(1)-H/R(2)-H Cross-Coupling with Hydrogen Evolution via Photo-/Electrochemistry. Chem. Rev. 2019, 119, 6769–6787. [DOI] [PubMed] [Google Scholar]
- (17).Singh K; Staig SJ; Weaver JD Facile synthesis of Z-alkenes via uphill catalysis. J. Am. Chem. Soc. 2014, 136, 5275–5278. [DOI] [PubMed] [Google Scholar]
- (18).Muller TE; Hultzsch KC; Yus M; Foubelo F; Tada M Hydroamination: direct addition of amines to alkenes and alkynes. Chem. Rev. 2008, 108, 3795–3892. [DOI] [PubMed] [Google Scholar]
- (19).Huang L; Arndt M; Goossen K; Heydt H; Goossen LJ Late transition metal-catalyzed hydroamination and hydroamidation. Chem. Rev. 2015, 115, 2596–2697. [DOI] [PubMed] [Google Scholar]
- (20).Escorihuela J; Lledos A; Ujaque G Anti-Markovnikov Intermolecular Hydroamination of Alkenes and Alkynes: A Mechanistic View. Chem. Rev. 2023, 123, 9139–9203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Johns AM; Sakai N; Ridder A; Hartwig JF Direct measurement of the thermodynamics of vinylarene hydroamination. J. Am. Chem. Soc. 2006, 128, 9306–9307. [DOI] [PubMed] [Google Scholar]
- (22).Musacchio AJ; Nguyen LQ; Beard GH; Knowles RR Catalytic olefin hydroamination with aminium radical cations: a photoredox method for direct C–N bond formation. J. Am. Chem. Soc. 2014, 136, 12217–12220. [DOI] [PubMed] [Google Scholar]
- (23).Horner JH; Martinez FN; Musa OM; Newcomb M; Shahin HE Kinetics of Dialkylaminium Cation-Radical Reactions - Radical Clocks, Solvent Effects, Acidity Constants, and Rate Constants for Reactions with Hydrogen-Atom Donors. J. Am. Chem. Soc. 1995, 117, 11124–11133. [Google Scholar]
- (24).Wagner BD; Ruel G; Lusztyk J Absolute kinetics of aminium radical reactions with olefins in acetonitrile solution. J. Am. Chem. Soc. 1996, 118, 13–19. [Google Scholar]
- (25).Nguyen TM; Nicewicz DA Anti-Markovnikov Hydroamination of Alkenes Catalyzed by an Organic Photoredox System. J. Am. Chem. Soc. 2013, 135, 9588–9591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Miller DC; Ganley JM; Musacchio AJ; Sherwood TC; Ewing WR; Knowles RR Anti-Markovnikov Hydroamination of Unactivated Alkenes with Primary Alkyl Amines. J. Am. Chem. Soc. 2019, 141, 16590–16594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Geunes EP; Meinhardt JM; Wu EJ; Knowles RR Photocatalytic Anti-Markovnikov Hydroamination of Alkenes with Primary Heteroaryl Amines. J. Am. Chem. Soc. 2023, 145, 21738–21744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Qin YZ; Zhu QL; Sun R; Ganley JM; Knowles RR; Nocera DG Mechanistic Investigation and Optimization of Photoredox Anti-Markovnikov Hydroamination. J. Am. Chem. Soc. 2021, 143, 10232–10242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Gentry EC; Knowles RR Synthetic Applications of Proton-Coupled Electron Transfer. Acc. Chem. Res. 2016, 49, 1546–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Agarwal RG; Coste SC; Groff BD; Heuer AM; Noh H; Parada GA; Wise CF; Nichols EM; Warren JJ; Mayer JM Free Energies of Proton-Coupled Electron Transfer Reagents and Their Applications. Chem. Rev. 2022, 122, 1–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Chang L; An Q; Duan L; Feng K; Zuo Z Alkoxy Radicals See the Light: New Paradigms of Photochemical Synthesis. Chem. Rev. 2022, 122, 2429–2486. [DOI] [PubMed] [Google Scholar]
- (32).Tsui E; Wang HJ; Knowles RR Catalytic generation of alkoxy radicals from unfunctionalized alcohols. Chem. Sci. 2020, 11, 11124–11141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Baciocchi E; Bietti M; Steenken S Base-catalyzed C–H deprotonation of 4-methoxybenzyl alcohol radical cations in water: Evidence for a carbon-to-oxygen 1,2-H-shift mechanism. J. Am. Chem. Soc. 1997, 119, 4078–4079. [Google Scholar]
- (34).Yayla HG; Wang H; Tarantino KT; Orbe HS; Knowles RR Catalytic Ring-Opening of Cyclic Alcohols Enabled by PCET Activation of Strong O–H Bonds. J. Am. Chem. Soc. 2016, 138, 10794–10797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Cheng C; McGonigal PR; Stoddart JF; Astumian RD Design and Synthesis of Nonequilibrium Systems. ACS Nano. 2015, 9, 8672–8688. [DOI] [PubMed] [Google Scholar]
- (36).Borsley S; Leigh D; Roberts BMW Molecular Ratchets and Kinetic Asymmetry: Giving Chemistry Direction. Angew. Chem. Int. Ed. 2024, e202400495. [DOI] [PubMed] [Google Scholar]
- (37).Nguyen ST; Murray PRD; Knowles RR Light-Driven Depolymerization of Native Lignin Enabled by Proton Coupled Electron Transfer. Acs Catal. 2020, 10, 800–805. [Google Scholar]
- (38).Nguyen ST; McLoughlin EA; Cox JH; Fors BP; Knowles RR Depolymerization of Hydroxylated Polymers via Light-Driven C–C Bond Cleavage. J. Am. Chem. Soc. 2021, 143, 12268–12277. [DOI] [PubMed] [Google Scholar]
- (39).Larionov E; Li HH; Mazet C Well-defined transition metal hydrides in catalytic isomerizations. Chem. Commun. 2014, 50, 9816–9826. [DOI] [PubMed] [Google Scholar]
- (40).Hanna S; Butcher TW; Hartwig JF Contra-thermodynamic Olefin Isomerization by Chain-Walking Hydrofunctionalization and Formal Retro-hydrofunctionalization. Org. Lett. 2019, 21, 7129–7133. [DOI] [PubMed] [Google Scholar]
- (41).Li J; Qu SL; Zhao WX Rhodium-Catalyzed Remote C(sp3)–H Borylation of Silyl Enol Ethers. Angew. Chem. Int. Ed. 2020, 59, 2360–2364. [DOI] [PubMed] [Google Scholar]
- (42).Yang NC; Jorgenson MJ Photochemical Isomerization of Simple α,β-Unsaturated Ketones. Tetrahedron Lett. 1964, 1203–1207. [Google Scholar]
- (43).Pete JP; Henin F; Mortezaei R; Muzart J; Piva O Enantioselective Photodeconjugation of Conjugated Esters and Lactones. Pure App. Chem. 1986, 58, 1257–1262. [Google Scholar]
- (44).Arnold DR; Mines SA Radical ions in photochemistry. 21. The Photosensitized (electron transfer) tautomerization of alkenes; the phenyl alkene system. Can. J. Chem. 1989, 67, 689–698. [Google Scholar]
- (45).Morack T; Onneken C; Nakakohara H; Mück-Lichtenfeld C; Gilmour R Enantiodivergent Prenylation via Deconjugative Isomerization. Acs Catal. 2021, 11, 11929–11937. [Google Scholar]
- (46).Occhialini G; Palani V; Wendlandt AE Catalytic, contra-Thermodynamic Positional Alkene Isomerization. J. Am. Chem. Soc. 2022, 144, 145–152. [DOI] [PubMed] [Google Scholar]
- (47).Schwarz JL; Schafers F; Tlahuext-Aca A; Luckemeier L; Glorius F Diastereoselective Allylation of Aldehydes by Dual Photoredox and Chromium Catalysis. J. Am. Chem. Soc. 2018, 140, 12705–12709. [DOI] [PubMed] [Google Scholar]
- (48).Mitsunuma H; Tanabe S; Fuse H; Ohkubo K; Kanai M Catalytic asymmetric allylation of aldehydes with alkenes through allylic C(sp3)-H functionalization mediated by organophotoredox and chiral chromium hybrid catalysis. Chem. Sci. 2019, 10, 3459–3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Castro CE; Kray WC Cleavage of Bonds by Low Valent Transition Metal Ions. Homogeneous Reduction of Alkyl Halides by Chromous Sulfate. J. Am. Chem. Soc. 1963, 85, 2768–2773. [Google Scholar]
- (50).Omoto M; Kato N; Sogon T; Mori A Revisit to the reduction of allylic chlorides to less substituted olefins by a low-valent chromium species in the presence of a proton source. Tetrahedron Lett. 2001, 42, 939–941. [Google Scholar]
- (51).Roth HG; Romero NA; Nicewicz DA Experimental and Calculated Electrochemical Potentials of Common Organic Molecules for Applications to Single-Electron Redox Chemistry. Synlett. 2016, 27, 714–723. [Google Scholar]
- (52).Murray PRD; Cox JH; Chiappini ND; Roos CB; McLoughlin EA; Hejna BG; Nguyen ST; Ripberger HH; Ganley JM; Tsui E; et al. Photochemical and Electrochemical Applications of Proton-Coupled Electron Transfer in Organic Synthesis. Chem. Rev. 2022, 122, 2017–2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Grosskopf J; Bach T Catalytic Photochemical Deracemization via Short-Lived Intermediates. Angew. Chem. Int. Ed. 2023, 62, e202308241. [DOI] [PubMed] [Google Scholar]
- (54).Huang M; Pan T; Jiang X; Luo S Catalytic Deracemization Reactions. J. Am. Chem. Soc. 2023, 145, 10917–10929. [DOI] [PubMed] [Google Scholar]
- (55).Hölzl-Hobmeier A; Bauer A; Silva AV; Huber SM; Bannwarth C; Bach T Catalytic deracemization of chiral allenes by sensitized excitation with visible light. Nature. 2018, 564, 240–243. [DOI] [PubMed] [Google Scholar]
- (56).Zhang CH; Gao AZ; Nie X; Ye CX; Ivlev SI; Chen SM; Meggers E Catalytic α-Deracemization of Ketones Enabled by Photoredox Deprotonation and Enantioselective Protonation. J. Am Chem. Soc. 2021, 143, 13393–13400. [DOI] [PubMed] [Google Scholar]
- (57).Huang MX; Zhang L; Pan TR; Luo SZ Deracemization through photochemical E/Z isomerization of enamines. Science. 2022, 375, 869–874. [DOI] [PubMed] [Google Scholar]
- (58).Onneken C; Morack T; Soika J; Sokolova O; Niemeyer N; Mück-Lichtenfeld C; Daniliuc CG; Neugebauer J; Gilmour R Light-enabled deracemization of cyclopropanes by Al-salen photocatalysis. Nature. 2023, 621, 753–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Gu Z; Zhang L; Li H; Cao S; Yin Y; Zhao X; Ban X; Jiang Z Deracemization through Sequential Photoredox-Neutral and Chiral Bronsted Acid Catalysis. Angew. Chem. Int. Ed. 2022, 61, e202211241. [DOI] [PubMed] [Google Scholar]
- (60).Wen L; Ding J; Duan LF; Wang S; An Q; Wang HX; Zuo ZW Multiplicative enhancement of stereoenrichment by a single catalyst for deracemization of alcohols. Science. 2023, 382, 458–464. [DOI] [PubMed] [Google Scholar]