

Abstract

Darunavir is a potent HIV protease inhibitor that has been established as an effective tool in the fight against the progression of HIV/AIDS in the global community. The successful application of this drug has spurred the development of derivatives wherein strategic regions (e.g., P1, P1’, P2, and P2’) of the darunavir framework have been structurally modified. An alternate route for the synthesis of darunavir and three related P1 and P1’ derivatives has been developed. This synthetic pathway involves the use of a Crimmins titanium tetrachloride-mediated oxazolidine-2-thione-guided asymmetric glycolate aldol addition reaction. The resultant aldol adduct introduces the P1 fragment of darunavir via an aldehyde. Transamidation with a selected amine (isobutylamine or 2-ethyl-1-butylamine) to cleave the auxiliary yields an amide wherein the P1’ component is introduced. From this stage, the amide is reduced to the corresponding β-amino alcohol and the substrate is then bis-nosylated to introduce the requisite p-nitrobenzenesulfonamide component and activate the secondary alcohol for nucleophilic substitution. Treatment with sodium azide yielded the desired azides, and the deprotection of the p-methoxyphenoxy group is achieved with the use of ceric ammonium nitrate. Finally, hydrogenation to reduce both the aniline and azide functionalities with concurrent acylation yields darunavir and its derivatives.
Introduction
Darunavir (1) is a highly effective HIV protease inhibitor1,2 that is used in combination antiretroviral therapies (cART) along with other antiretroviral medicinal agents for suppressing viral replication and ultimately reducing the viral load in patients.3 This drug was initially approved by the FDA in 2006 and the European Directorate for the Quality of Medicines and Health Care (The European Pharmacopoeia) in 2007 and fully approved in 2008 due to its high efficacy in combating HIV infection.4 The successful application of darunavir in combination therapies has inspired the pursuit of improved syntheses of darunavir5−9 and syntheses of derivatives with the potential for even greater potency (Figure 1).10−15 In this context, the seminal efforts of Ghosh and coworkers laid the foundation for structural aspects of darunavir and the domains where the structure could be modified. In 2023, Raines and coworkers modified the structure of darunavir by modifying the P2’ region with the use of a benzoborolone group (see 2a and 2b) and demonstrated the utility of installing such groups as pharmacophores.10 Schiffer, Ali, and coworkers modified the P2 domain by introducing a phenolic methylene diethylphosphonate group in conjunction with the introduction of modifications of the P1’ and P2’ domains to afford darunavir derivative 3.11 This modification resulted in sustained potency against highly resistant HIV-1 variants.11
Figure 1.

Darunavir (1) and related derivatives.
Reiser and coworkers changed the P2 domain of darunavir by the use of an ethereal bicyclic cyclopentyl system resulting in low nanomolar IC50 values.12 Ghosh and coworkers have been leaders in this field with the development of a number of darunavir derivatives wherein the P2 domain has been changed to enhance drug efficacy.13−15 In this context, darunavir derivative 5 was designed with modifications to the P1, P2, and P2’domains to enhance interactions with the active site of the HIV-1 protease. The combined modifications led to enzyme inhibition that was markedly potent and yielded more insight into the nature of the binding properties of a series of related derivatives with the protease.
The dominant synthetic pathway for the preparation of darunavir and many of its derivatives has involved the use of either β-epoxy azide 6(15) or commercially available β-epoxy carbamates 7a,b (a: R = -Boc;7,11 b: R = -CBz.10 An alternate pathway to the chiral structural framework of darunavir, which was developed by Funicello and coworkers,9 involves the preparation of a suitable vinyl ester (9) obtained from a ligand-free Suzuki-Miyaura coupling reaction (Figure 2). The Sharpless asymmetric dihydroxylation reaction is then used to create the requisite chiral centers necessary for the synthesis of darunavir.
Figure 2.
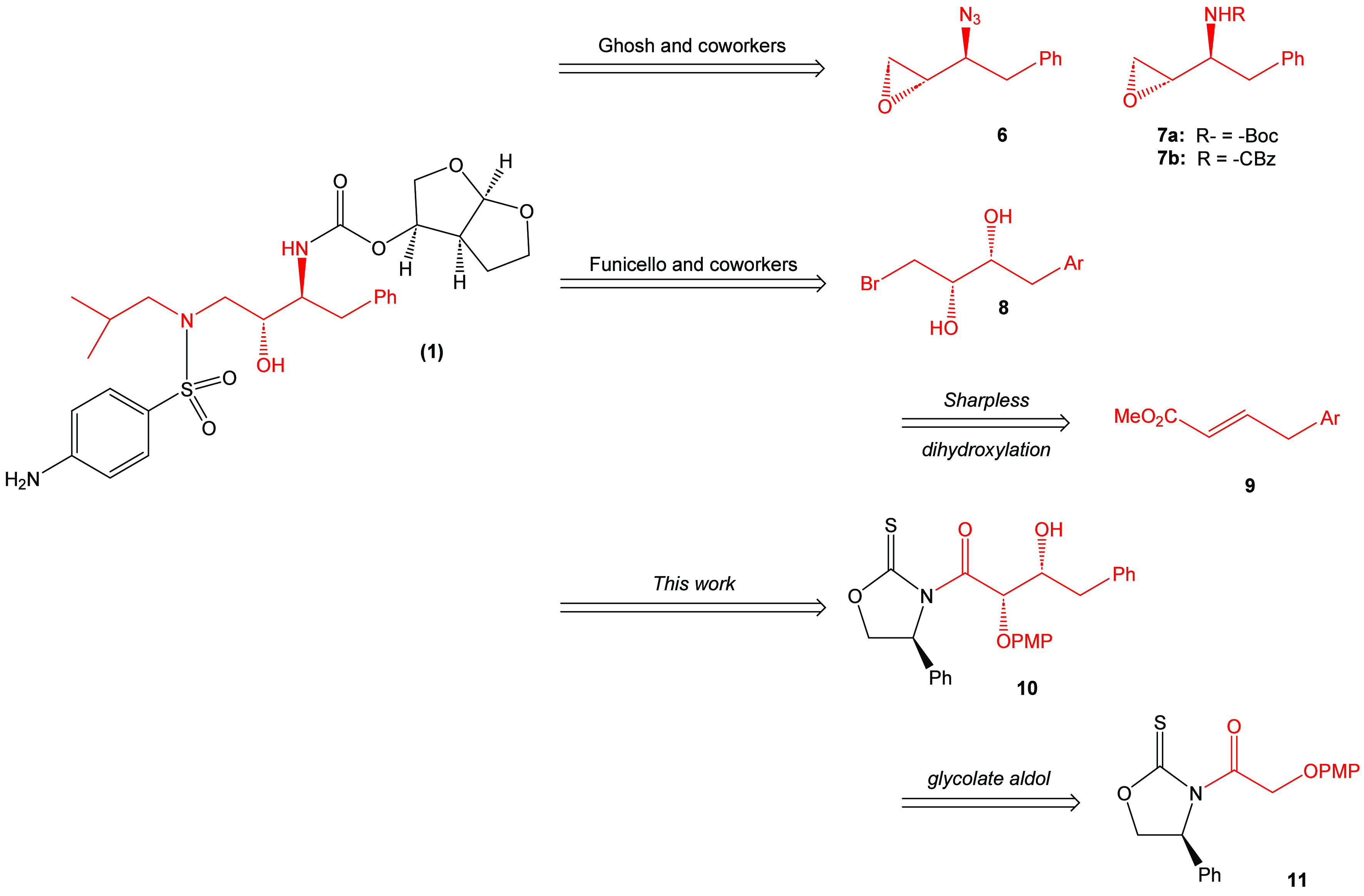
Synthetic pathways to darunavir (1).
There was an interest in determining the potential of using an asymmetric glycolate aldol addition pathway to achieve the synthesis of darunavir. Retrosynthetically, it is proposed that darunavir may be derived from the known intermediate β-azido alcohol 12 (Scheme 1). This material, in turn, may be derived from precursor 13 that possesses an alkoxy-protecting group. Intermediate 13 may be accessed by the bis-nosylation of intermediate 14. It is proposed that the bis-nosylation process will introduce more convergence into the overall synthesis of the target molecule. Amino alcohol 14 can be envisioned to come from the reduction of amide 15. This amide can be directly obtained by a process of transamidation with the aldol adduct 10 arising from the syn-stereoselective Crimmins glycolate aldol addition reaction with (S)-phenylglycinol-derived oxazolidine-2-thione 11.
Scheme 1. Retrosynthetic Analysis of Darunavir.

Results and Discussion
Oxazolidine-2-thione [(S)-16] was prepared in 77% yield using the method of Wu and workers16 and subsequently acylated with p-methoxyphenoxyacetic acid in the presence of EDC and DMAP in dichloromethane to yield the N-(p-methoxyphenoxyacetyl)oxazolidine-2-thione (S)-10 in 83% yield after recrystallization (Scheme 2). The reaction conditions for this process were optimized (slow addition of the carboxylic acid as the final reagent to the reaction mixture) to suppress the formation of the self-condensation byproduct that is proposed to arise from a putative Claisen condensation.17 The acylated thione (S)-11 was then reacted with one equivalent of titanium tetrachloride and two equivalents of triethylamine in an asymmetric glycolate aldol addition reaction18,19 with phenylacetaldehyde to generate the Evan syn-adduct (S,S,R)-10 in 86% yield and ≥95% diastereomeric purity after flash chromatography on silica gel.
Scheme 2. Synthesis of the Evans and Non-Evans syn-Glycolate Aldol Adducts 10.

The synthesis of the aldol adduct (S,S,R)-10 was successful but required chromatographic purification. In order to optimize this process, an alternate route for obtaining the key aldol adduct side chain was considered.20 Thus, (R)-phenylglycinol [(R)-16] was cyclized using the Wu conditions to afford thione (R)-17 in 78% yield after recrystallization. The thione was acylated as before with EDC and DMAP using the optimized conditions to yield (R)-11 in 81% yield as a crystalline solid. This material was then reacted with two equivalents of titanium tetrachloride at −78 °C and enolized by the addition of two equivalents of triethylamine. Treatment of the titanium enolate with phenylacetaldehyde yielded the desired non-Evans syn-aldol adduct (R,S,R)-10 in 87% yield (crude d.r. = 15:1) after recrystallization. The stereochemistry of the adduct was confirmed by single-crystal X-ray crystallography with the presence of the sulfur atom allowing clear assignment of absolute stereochemistry from the observed anomalous dispersion effects.21
With the synthesis of the key aldol reaction optimized, syn-aldol adduct (R,S,R)-10 was reacted with isobutylamine and imidazole to afford the transamidation product, β-hydroxyamide 15, in 82% after recrystallization (Scheme 3). In like fashion, 2-ethylbutylamine was employed in the transamidation process to yield amide 18 in 87% isolated yield. The introduction of this group introduces the 2-ethylbutylamine group as an eventual P1’ modification of darunavir. Subsequent reduction of amide 15 using the Meyers-Periasamy conditions (NaBH4/I2 in THF)22,23 yielded the β-aminoalcohol 14 in 89% yield after flash chromatography. Amide 18 was reduced using a commercially available borane-dimethylsulfide complex and yielded the β-amino alcohol 19 in 62% yield after chromatographic purification.
Scheme 3. Synthesis of the γ-Azido-N-sulfonamide.

At this stage, β-amino alcohols 14 and 19 were treated with two equivalents of p-nitrobenzenesulfonyl chloride, triethylamine, and DMAP. It was anticipated that the bis-p-nosylation process would streamline the overall reaction. However, the reaction proved to have challenges, apparently due to the stereoelectronic factors governing the initial N-sulfonylation vs the O-sulfonylation. Ultimately, the reaction required 1.1 equivalents of catalytic DMAP to reach the complete formation of the bis-p-nosylation product. The target O-p-nitrobenzenesulfonyl-N-p-nitrobenzenesulfonamides 13 and 20 were obtained in 52% and 56% yield, respectively, as crystalline solids after recrystallization. Treatment of sulfonamides 13 and 20 with sodium azide in DMSO afforded a near quantitative conversion to the corresponding azides 21 and 22 in 99% and 96% recovered yield, respectively. The azides were not purified as the analysis of the NMR spectra suggested that the reactions were clean transformations.
γ-Azidosulfonamides 21 and 22 were deprotected using ceric ammonium nitrate as a means of oxidatively removing the p-methoxyphenoxy group (Scheme 4).24,25 The isolated yield for the formation of β-azido alcohols 12 and 23 were 45% and 40%, respectively, after flash chromatography. Efforts to improve the reaction by adjusting the stoichiometry and changing the solvent and reaction temperature were not fruitful.26 The use of alternate deprotecting agent DDQ did not improve the yield of the reaction.
Scheme 4. Synthesis of Darunavir (1) and N-2-Ethylbutyl Darunavir (27).

This process was followed by the hydrogenolysis of the β-azido alcohols 12 and 23 using hydrogen gas (balloon) to form β-amino alcohol 16in situ. The intermediates 24 and 25 were not isolated but were reacted with the bis(tetrahydrofuranyl) N-hydroxysuccinimidyl carbonate (26) to afford the target darunavir (1) and the related N-2-ethylbutyl darunavir derivative (27) in 52% and 50%, respectively, for the one-pot, two-reaction pathway.
There was an interest in demonstrating that the overall synthetic pathway could be employed to create a P1 modification of the darunavir base structure (Scheme 5). In this regard, oxazolidine-2-thione (R)-11 was reacted with titanium tetrachloride and triethylamine to form the titanium enolate that was subsequently alkylated with isobutyraldehyde to yield the aldol adduct 28 in 74% isolated yield in greater than 95% d.e. This material was reacted with isopropylamine and imidazole in dichloromethane. The crude reaction mixture yielded the desired product in combination with byproducts that appeared to be ring-opening products. To circumvent this issue, an alternate approach was taken in which the acyl imidazole was formed in situ and then the isopropylamine was added.18b,27 This process yielded the targeted N-isopropylamide 30 in 96% yield after recrystallization. The amide was reduced using borane-dimethylsulfide in THF to afford β-amino alcohol 31 in 82%. The β-amino alcohol was subsequently treated with an excess of p-nitrobenzenesulfonyl chloride and DMAP to afford the desired bis(p-nitrobenzenesulfonylated) adduct 32 in 57% yield after chromatographic purification. This material was then reacted with sodium azide to yield the corresponding azide 33 in high yield. Based on the quality of the crude 1H NMR spectrum of this material, it was moved forward to the process of deprotection via the use of ceric ammonium nitrate. As with the synthesis of darunavir and its N-2-ethylbutyl derivative, the ceric ammonium nitrate only proved to have limited efficiency in removing the p-methoxyphenoxy protecting group. The deprotection yielded β-azido alcohol 34 in 45% yield after purification by flash chromatography. Treatment of 34 with hydrogen gas in the presence of palladium on carbon and bis(tetrahydrofuranyl) N-hydroxysuccinimidyl carbonate (26) yielded the desired P1-isopropyl target 35 in 73% yield after chromatographic purification for the two-step process.
Scheme 5. Synthesis of the P1-Isopropyl Derivative 35.
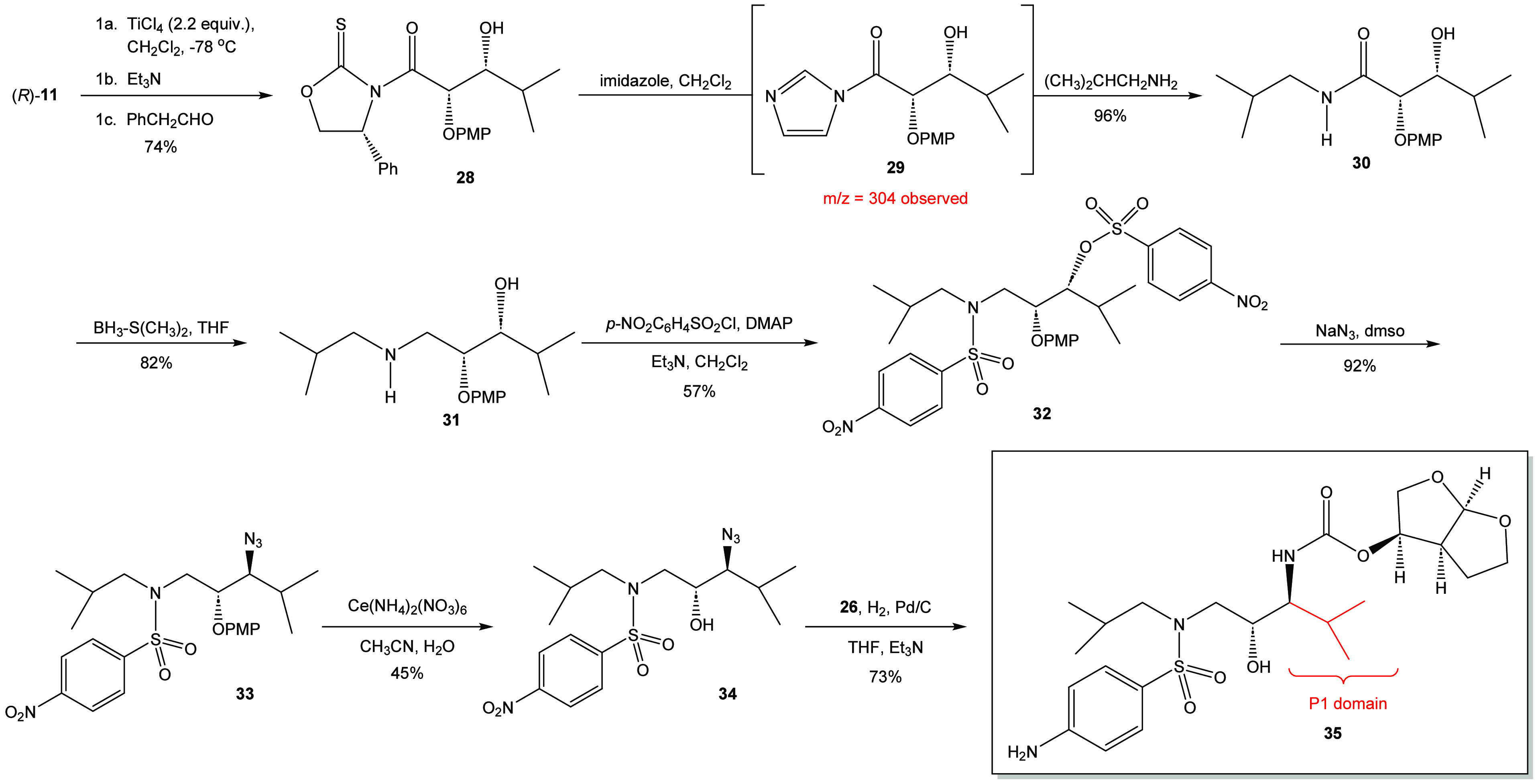
The synthesis of the P1-isopropyl derivative proved led to the pursuit of another P1-darunavir derivative based on the P1-adamantyl darunavir derivative prepared by Ghosh and coworkers (see Scheme 8 in ref (15)). It was originally proposed that the introduction of a hydrophobic and sterically demanding adamantyl group might provide further understanding of the protease inhibitor (PI)–protein interaction. It is proposed here that a contracted and truncated variant of the adamantyl system would also be a useful tool in probing the PI–protein interaction. To this end, the P1-adamantyl system was envisioned to be truncated down to the P1-neopentyl system illustrated in Scheme 6.
Scheme 6. Origin of the P1-Neopentyl Darunavir Design.

The pursuit of 38 was initiated with the titanium-mediated asymmetric aldol reaction of (R)-11 with 3,3-dimethylbutyraldehyde (Scheme 7). The reaction proved to be incomplete, perhaps due to the steric demand of the neopentyl-type structure of the aldehyde. As a recourse, oxazolidine-2-thione (S)-11 was employed in the asymmetric aldol addition reaction to afford aldol adduct 39 in 82% yield and greater than 95:5 d.r. This product was reacted with imidazole in the presence of dichloromethane, leading to the in situ formation of the acyl imidazole 40 that was tentatively identified by ESI-HRMS. Treatment of 40 with isobutylamine yielded amide 41 in 96% yield. As before, the amide was reduced to the corresponding β-amino alcohol 42 using a borane-dimethylsulfide complex. This process afforded a 93% yield after chromatographic purification. An attempt was made at the bis(sulfonylation) of β-amino alcohol 42 using two equivalents of the p-nitrobenzenesulfonyl chloride. Unfortunately, the product that was obtained in 50% yield was the monsulfonylated product 43. The failure of the product to undergo bis(sulfonylation) was attributed to the steric hindrance imparted by the proximal neopentyl fragment. Thus, sulfonamide 43 was treated with an excess of p-nitrobenzenesulfonyl chloride to afford the desired bis(sulfonylated) product 44 in 77% yield. The reaction of 44 with sodium azide in DMSO led to the isolation of the azido neopentyl darunavir intermediate 45. Deprotection of the p-methoxyphenoxy protecting group was accomplished by the use of a 4:1 acetonitrile/water solution of ceric ammonium nitrate. The product β-azido alcohol 46 was obtained in a 45% yield after chromatographic purification. Hydrogenation of β-azido alcohol 46 in the presence of Pd/C and carbonate 26 yielded neopentyl darunavir derivative 38 in 73% isolated yield after chromatography.
Scheme 7. Synthesis of the Azido Neopentyl Darunavir Derivative.

In conclusion, we have described an alternate preparative route to the HIV protease inhibitor darunavir using an asymmetric glycolate aldol addition reaction to form the key stereocenters. A strategy that involved the using enantiomeric oxazolidine-2-thione auxiliaries with opposing stereochemical outcomes in the asymmetric aldol addition reaction was successfully employed. A convergent strategy of introducing the key p-nitrobenzene sulfonamide and activating the chiral alcohol in the form of p-nitrobenzene sulfonate in the same chemical step was also employed. The overall synthesis of darunavir proved to be successful via the aldol addition pathway, but the potential for improving the current route remains open.
Experimental Section
Safety Considerations
It is noted that the formation of oxazolidine-2-thiones (R)-17 and (S)-17 involves an exothermic reduction/oxidation transformation that employs 30% hydrogen peroxide. It is recommended that a highly efficient water condenser is used for this reaction. The use of reagents such as titanium tetrachloride (1 M in CH2Cl2) and borane-dimethylsulfide complex should be handled with care using air-free techniques.
General Methods28,29
Unless otherwise noted, all chemical reagents and solvents were purchased and used without further purification. All reactions were conducted under a nitrogen atmosphere in glassware that was flame-dried. Unless otherwise indicated, chemical reagents that served as starting materials, chemical reactants, and solvents were used as received from commercial vendors (e.g., MilliporeSigma, ThermoFisher Scientific, and Combi-Blocks, Inc.) without further purification. Melting points were recorded on a Mel-Temp apparatus and were uncorrected. Unless otherwise noted, all 1H and proton-decoupled 13C NMR spectra were collected in deuterated chloroform (CDCl3) using a Bruker Ultrashield Avance III NMR spectrometer operating at either 500 or 400 MHz (1H NMR) and 125 or 100 MHz (13C{1H} NMR), respectively. Chemical shifts were reported in parts per million (δ scale), and coupling constants (J values) were reported in Hertz (Hz). Tetramethylsilane (TMS) was used as an internal standard (δ = 0 ppm). Signals appearing in the 1H NMR spectra were recorded in abbreviated form: s = singlet; d = doublet; t = triplet; q = quartet; p = pentet; m = multiplet, dd = doublet of doublets. 1H-decoupled 13C NMR [13C{1H}] spectral signals were referenced and reported relative to the central peak of deuterated chloroform (CDCl3) taken as 77.0 ppm. Optical rotation data were collected at ambient laboratory temperatures (22–25 °C) on a JASCO p-1010 digital polarimeter operating at 589 nm and using a cylindrical 10 × 100 mm quartz cell. Infrared spectra were recorded using NaCl plates. IR values were reported in reciprocal centimeters (cm–1) and were measured either as a nujol mull or as a neat liquid film from an evaporated chloroform solution. Mass spectral data were collected on a ThermoScientific Q-Exactive electrospray ionization high-resolution mass spectrometer (ESI HRMS) equipped with an Orbitrap mass analyzer. Samples were prepared in concentrations of 5–25 ppm in high-performance liquid chromatographygrade methanol/water/formic acid (1:1:0.01).
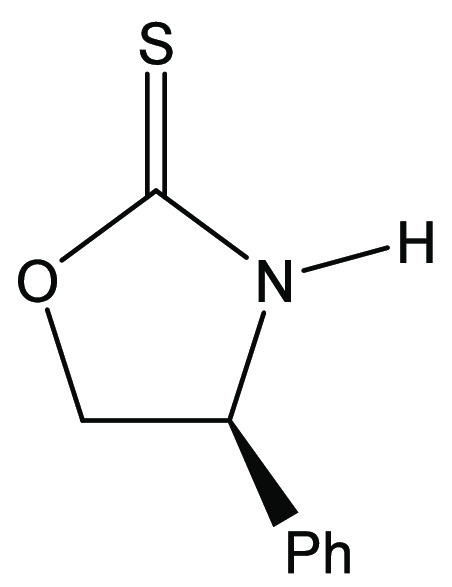
(4S)-4-Phenyl-1,3-oxazolidine-2-thione [(S)-17]28
To a flame-dried, nitrogen-purged 5 L round-bottom flask fitted with a Claisen adapter with a pressure-equalizing addition funnel and a tall condenser equipped with a stir bar were added l-phenylglycinol (13.7 g, 100 mmol, purchased from Combi-Blocks, Inc.), ethanol (100 mL), potassium carbonate (6.95 g, 50.0 mmol), and carbon disulfide (12 mL). The reaction was heated to 50 °C using a heating mantle controlled by a variable transformer, and a 30% (w/v) aqueous solution of hydrogen peroxide (17.0 mL, 167 mmol) was added dropwise. Upon complete addition of the hydrogen peroxide, the reaction was stirred for 15 min. The reaction is highly exothermic and must be carefully monitored during this time. It is important that the condenser is efficient and the system is open to the air. The reaction was cooled to ambient temperature and gravity-filtered into a 1 L flask to remove any solid material. The reaction flask was washed with ethyl acetate (3 × 30 mL), and the rinses were combined with the filtrate. The reaction solvent was then removed by rotary evaporation. The reaction was then reconstituted in ethyl acetate (200 mL). The organic layer was then washed with an aqueous solution of 1 M HCl (2 × 80 mL) and brine (80 mL). The organic layer was then dried (MgSO4) and filtered, and the solvents were removed by rotary evaporation. The crude solid product was then recrystallized with ethyl acetate and hexanes to afford the title compound as a light-yellow crystalline solid (13.7 g, 77 mmol, 77% yield). Melting point: 121–123 °C. [α]D = +71.6 (c = 1.00, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.71 (broad singlet, 1H), 7.46–7.32 (m, 5H), 5.12 (dd, J = 8.9, 7.0 Hz, 1H), 5.00 (apparent triplet, J = 8.9 Hz, 1H), 4.49 (dd, J = 8.9, 7.0 Hz, 1H) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ 189.7, 138.0, 129.2, 129.2, 126.3, 77.7, 60.2 ppm. This material was identical to that described by Wu and coworkers.16
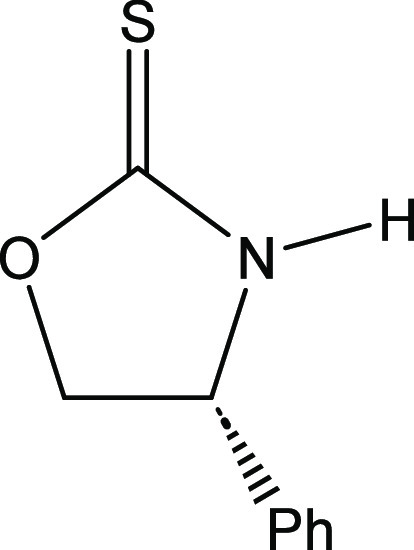
(4R)-4-Phenyl-1,3-oxazolidine-2-thione [(R)-17]28
d-Phenylglycinol (41.2 g, 300 mmol, purchased from Combi-Blocks, Inc.) was employed. The product was recrystallized and was recovered as a light-yellow crystalline solid (41.9 g, 234 mmol, 78% yield). Melting point: 121–122 °C. [α]D = −70.4 (c = 0.27, CHCl3). 1H NMR (400 MHz, CDCl3): δ 7.51 (broad singlet, 1H), 7.45–7.30 (m, 5H), 5.11 (dd, J = 8.9, 6.9 Hz, 1H), 5.00 (apparent triplet, J = 9.2 Hz, 1H), 4.49 (dd, J = 8.9, 6.9 Hz, 1H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 189.7, 138.0, 129.3, 129.1, 126.3, 77.7, 60.2 ppm. This material was identical to that described by Wu and coworkers.16

(4S)-3-[(p-Methoxyphenoxy)acetyl]-4-phenyl-1,3-oxazolidine-2-thione [(S)-11]28
To a flame-dried, nitrogen-purged 1000 mL round-bottom flask equipped with a stir bar were added oxazolidine-2-thione [(S)-17] (11.1 g, 61.4 mmol), dichloromethane (200 mL), EDC (12.9 g, 67.5 mmol), and DMAP (1.90 g, 15.4 mmol). Once these reagents had been added, p-methoxyphenoxy acetic acid (11.2 g, 61.4 mmol) was added portion-wise. The reaction mixture was then stirred overnight, and then, the contents of the round-bottom flask were transferred into a separatory funnel. The reaction mixture was then treated with an aqueous solution of 1 M HCl (60 mL). The organic layer was separated from the aqueous layer and then treated with an aqueous solution of 1 M NaOH (2 × 60 mL). The organic layer was separated and finally washed with brine (60 mL). The organic layer was collected, dried (MgSO4), and gravity-filtered. The solvent was removed by rotary evaporation, and the crude product was recrystallized from ethyl acetate and hexanes to afford compound (S)-11 as a white crystalline solid (17.5 g, 50.9 mmol, 83%). Mp: 108–109 °C. [α]D = +83.2 (c = 1.00, CHCl3,). 1H NMR (400 MHz, CDCl3): δ 7.40–7.30 (m, 5H), 6.82–6.76 (m, 4H), 5.73 (dd, J = 9.0, 3.3 Hz, 1H), 5.57 (d, J = 17.7 Hz, 1H), 5.45 (d, J = 17.7 Hz, 1H), 4.90 (apparent triplet, J = 9.0 Hz, 1H), 4.58 (dd, J = 9.0, 3.3 Hz, 1H), 3.74 (s, 3H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 185.0, 168.9, 154.5, 151.8, 138.2, 129.3, 129.0, 126.3, 115.9, 114.7, 75.4, 70.2, 62.0, 55.7 ppm. IR (CHCl3): 1724, 1211, 821, 780 cm–1. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C18H17NNaO4S366.0770; Found, 366.0777. The material has been previously prepared.18a

(4R)-3-[(p-Methoxyphenoxy)acetyl]-4-phenyl-1,3-oxazolidine-2-thione [(R)-11]28
Using the above procedure, oxazolidine-2-thione (R)−17 (17.9 g, 100 mmol) was employed as the substrate. The crude solid product was recrystallized using ethyl acetate and hexanes to afford the product as a crystalline solid (27.8 g, 81.0 mmol, 81%). Mp: 109–110 °C. [α]D = −73.9 (c = 1.04, CHCl3). 1H NMR (400 MHz, CDCl3): δ 7.41–7.30 (m, 5H), 6.82–6.76 (m, 4H), 5.73 (dd, J = 9.0, 3.2 Hz, 1H), 5.57 (d, J = 17.7 Hz, 1H), 5.45 (d, J = 17.7 Hz, 1H), 4.90 (apparent triplet, J = 9.0 Hz, 1H), 4.58 (dd, J = 9.0, 3.2 Hz, 1H), 3.74 (s, 3H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 185.0, 169.0, 154.5, 151.8, 138.2, 129.3, 129.1, 126.3, 115.9, 114.7, 75.4, 70.2, 62.1, 55.7 ppm. IR (CHCl3): 1721, 1206, 826, 786 cm–1. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C18H17NNaO4S 366.0770; Found, 366.0776.

(4S)-3-[(2S’,3R’)-3-Hydroxy-2-(p-methoxyphenoxy)-4-phenylbutanoyl]-4-phenyl-1,3-oxazolidine-2-thione[(S,S,R)-10]28
To a flame-dried, nitrogen-purged 5 L round-bottom flask equipped with a large stir bar were added acylated thione [(S)-11)] (10.0 g, 29.1 mmol) and anhydrous dichloromethane (1 L). The reaction vessel was chilled to −78 °C with a dry ice/ethanol bath, and titanium tetrachloride (1 M in CH2Cl2, 32.0 mL, 32.0 mmol) was added dropwise by syringe. This solution was stirred for 30 min, at which point triethylamine (8.90 mL, 64 mmol) was added by syringe. The color of the solution transitioned from an amber color to that of deep purple upon the addition of the triethylamine. This solution was stirred for 60 min at −78 °C, and freshly distilled phenylacetaldehyde (7.2 mL, 64 mmol) was added to the reaction vessel by syringe. The reaction mixture was stirred for an additional 4 h, after which time the dry ice bath was removed, and brine (200 mL) was added dropwise to quench the reaction. The reaction mixture was allowed to warm up to room temperature with vigorous stirring, transferred to a separatory funnel, and extracted with an aqueous solution of 1 M HCl (2 × 100 mL). The organic layer was separated and subsequently washed with brine (100 mL), dried (MgSO4), and filtered. The solvent was then removed by rotary evaporation to yield the crude reaction product as a viscous oil. The product was purified by flash chromatography on silica gel using a solvent gradient [100% hexanes; 85:15, hexanes:ethyl acetate; 60:40, hexanes:ethyl acetate] to achieve the isolation of the pure product (11.54 g, 24.89 mmol, 86% yield). 1H NMR (500 MHz, CDCl3): δ 7.30–7.14 (m, 10 H), 6.76 (d, J = 2.0 Hz, 1H), 6.69–6.63 (m, 4H), 5.58 (dd, J = 8.0, 2.3 Hz, 1H), 4.67 (dd, J = 9.0, 8.0 Hz), 4.55 (td, J = 7.5, 2.0 Hz, 1H), 4.36 (dd, J = 9.0, 2.3 Hz, 1H), 3.71 (s, 3H), 3.11–3.04 (m, 2H). 13C{1H} NMR (125 MHz, CDCl3): δ 185.2, 169.9, 154.6, 151.0, 138.3, 137.2, 129.6, 129.2, 128.8, 128.5, 126.8, 125.7, 116.1, 114.7, 77.3 (obscured by the CDCl3 signal), 74.7, 73.0, 62.7, 55.7, 40.6 ppm. The material has been previously prepared.18a

(4R)-3-[(2S’,3R’)-3-hydroxy-2-(p-methoxyphenoxy)-4-phenylbutanoyl]-4-phenyl-1,3-oxazolidine-2-thione[(R,S,R)-10]28
To a flame-dried, nitrogen-purged 5 L round-bottom flask equipped with a large stir bar were added acylated thione (R)-11 (10.0 g, 29.1 mmol) and anhydrous dichloromethane (1 L). The reaction vessel was chilled to −78 °C with a dry ice/ethanol bath, and titanium tetrachloride (1 M in CH2Cl2, 64 mL, 64 mmol) was added dropwise by syringe. This solution was stirred for 30 min, at which point triethylamine (8.90 mL, 64 mmol) was added by syringe. The color of the solution transitioned from an amber color to that of deep purple upon the addition of the triethylamine. This solution was stirred for 60 min at −78 °C, and freshly distilled phenylacetaldehyde (7.2 mL, 64 mmol) was added to the reaction vessel by syringe. The reaction mixture was stirred for an additional 4 h, after which time the dry ice bath was removed, and brine (200 mL) was added dropwise to quench the reaction. The reaction contents were allowed to gradually warm up to room temperature with vigorous stirring, transferred to a separatory funnel, and then extracted twice with an aqueous solution of 1 M HCl (100 mL). The organic layer was separated from the aqueous layer and subsequently washed with brine (100 mL), dried (MgSO4), and filtered. The solvent was then removed by rotary evaporation to yield the crude reaction product as a tan solid, which was then recrystallized using hexanes and ethyl acetate to afford the pure product as a white, crystalline solid (11.73 g, 25.45 mmol, 87% yield). Mp: 184–185 °C. [α]D = −105.8 (c = 1.10, CHCl3). 1H NMR (400 MHz, CDCl3): δ 7.38–7.17 (m, 11H), 6.86–6.80 (m, 4H), 5.70 (dd, J = 9.2, 5.9 Hz, 1H), 4.85 (apparent triplet, J = 9.2 Hz, 1H), 4.55–4.51 (m, 1H), 4.48 (dd, J = 9.2, 5.9 Hz, 1H), 3.76 (s, 3H), 3.06–2.94 (m, 2H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 185.4, 170.4, 154.7, 151.1, 137.3, 137.0, 129.7, 129.2, 129.1, 128.5, 126.7, 126.5, 116.3, 114.9, 77.5, 74.7, 73.2, 62.6, 55.7, 40.4 ppm. IR (CHCl3): 3424, 1717, 1216, 757 cm–1. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C26H25NNaO5S 486.1346; Found, 486.1349. Suitable crystals for single-crystal X-ray diffraction analyses were grown by vapor diffusion of pentane into a dichloromethane solution of (R,S,R)-10.

(2S,3R)-3-Hydroxy-N-isobutyl-2-(p-methoxyphenoxy)-4-phenylbutanamide (15)28
To a flame-dried, nitrogen-purged 500 mL round-bottom flask equipped with a stir bar were added the aldol adduct (R,S,R-10) (10.0 g, 21.6 mmol), dichloromethane (70 mL), and imidazole (4.40 g, 64.7 mmol). Once the reaction had stirred for 60 min, isobutylamine (4.30 mL, 43.0 mmol) was added by syringe, and the reaction was stirred overnight. The reaction was diluted with dichloromethane (80 mL), transferred to a separatory funnel, and treated with an aqueous solution of 2 M NaOH (2 × 40 mL). The layers were separated, and the organic layer was treated with an aqueous solution of 1 M HCl (2 × 40 mL). The organic layer was then washed with brine (40 mL), dried (MgSO4), and gravity-filtered. The solvent was then removed by rotary evaporation to yield the crude reaction product, which was then recrystallized using hexanes and ethyl acetate to afford the pure product as a white solid (6.33 g, 17.7 mmol, 82% yield). Multiple reactions were conducted to build up material for the following reaction. Mp: 102–103 °C. [α]D = −6.51 (c = 1.04, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.27–7.16 (m, 5H), 6.88 (d, J = 9.3 Hz, 2H), 6.84 (d, J = 9.3 Hz, 2H), 6.60 (broadened triplet, 1H), 4.49 (d, J = 3.0 Hz, 1H), 4.30 (broad singlet, 1H), 3.78 (s, 3H), 3.17–3.09 (m, 2H), 2.97 (dd, J = 13.8, 5.2 Hz, 1H), 2.90 (dd, J = 13.8, 8.4 Hz, 1H), 2.77 (d, J = 7.5 Hz, 1H), 1.80–1.70 (m, 1H), 0.86 (d, J = 6.7 Hz, 3H), 0.85 (d, J = 6.7 Hz, 3H) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ 170.3, 155.1, 151.3, 137.7, 129.4, 128.5, 126.6, 116.6, 115.0, 80.7, 73.0, 55.7, 46.4, 39.6, 28.5, 20.0, 19.9 ppm. IR (nujol): 3310, 1653, 1215 cm–1. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C21H27NO4Na 380.1832; Found, 380.1840.

(2S,3R)-N-(2-Ethylbutyl)-3-hydroxy-2-(4-methoxyphenoxy)-4-phenylbutanamide (18)29
To a stirred solution of 10 (7.00 g, 15.1 mmol) in dichloromethane (151 mL) was added imidazole (3.08 g, 45.2 mmol), and after 1 h, 2-ethylbutylamine (2.20 mL, 16.1 mmol) was added to the mixture. The reaction mixture was stirred overnight, diluted with dichloromethane, washed with an aqueous solution of 2 M NaOH (30 mL) and brine (30 mL), and dried over MgSO4, and the solvent was removed by rotary evaporation. The crude residue was purified by flash column chromatography (3% methanol in dichloromethane) to afford 18 (5.07 g, 13.1 mmol, 87% yield) as a clear viscous oil that crystallizes with time. [α]D = −9.3 (c = 0.562, CHCl3,). 1H NMR (500 MHz, CDCl3): δ 7.27–7.17 (m, 5H), 6.88 (d, J = 9.3 Hz, 2H), 6.83 (d, J = 9.3 Hz, 2H), 6.50 (m, 1H), 4.49 (d, J = 3.0 Hz, 1H), 4.31–4.27 (m, 1H), 3.77 (s, 3H), 3.25 (t, J = 6.3 Hz, 2H), 2.97 (dd, J = 13.8, 5.0 Hz, 1H), 2.90 (dd, J = 13.8, 8.7 Hz, 1H), 1.39–1.32 (m, 1H), 1.29–1.16 (m, 4H), 0.84 (t, J = 7.4 Hz, 6H), ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 170.3, 155.1, 151.3, 137.7, 129.4, 128.5, 126.6, 116.6, 115.0, 80.6, 73.0, 55.7, 41.5, 40.9, 39.5, 23.6, 10.9, 10.8 ppm. IR (CDCl3): 3421, 3355, 1655, 1506, 1223, 1037, 826 cm–1. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C23H38N3O7SNa 386.2326; Found, 386.2328.

(2R,3R)-2-Hydroxy-4-(isobutylamino)-3-(p-methoxyphenoxy)-1-phenylbutane (14)28
To a flame-dried, nitrogen-purged, 1000 mL round-bottom flask fitted with a Claisen adapter equipped with a condenser and pressure-equalizing addition funnel were added β-hydroxyamide (15) (13.3 g, 37.1 mmol) and THF (180 mL). Sodium borohydride (3.37 g, 89.0 mmol) was added incrementally to the flask and stirring was initiated. A solution of iodine (10.4 g, 40.8 mmol) in THF (70 mL) was prepared and carefully added to the addition funnel, whereafter dropwise addition of this solution into the reaction flask was completed over the course of 15 min. The reaction was heated to reflux using a heating mantle controlled by a variable transformer and was subsequently stirred for 16 h. At the end of the reaction time, the reaction was cooled to 0 °C and quenched by the dropwise addition of methanol (50 mL) via the addition funnel. The mixture was concentrated under reduced pressure and then extracted with EtOAc (150 mL) and an aqueous solution of 1 M NaOH (3 × 50 mL). The organic layer was washed with brine (50 mL), dried over magnesium sulfate, and gravity-filtered. The solvent was then removed by rotary evaporation to afford the corresponding amino alcohol as a colorless oil that was used in the next step without further purification (11.4 g, 33.1 mmol, 89% yield). [α]D = −52.5 (c = 1.17, CDCl3). 1H NMR (500 MHz, CDCl3): δ 7.26–7.14 (m, 5H), 6.88–6.82 (m, 4H), 4.19 (td, J = 7.0, 1.9 Hz, 1H), 4.08–4.06 (m, 1H), 3.78 (s, 3H), 3.27 (dd, J = 12.7, 4.3 Hz, 1H), 3.03–2.93 (m, 2H), 2.73 (dd, J = 12.7 Hz, 2.4 Hz, 1H), 2.42 (dd, J = 11.5, 6.5 Hz, 1H), 2.32 (dd, J = 11.5, 7.0 Hz, 1H), 1.73–1.65 (m, 1H), 0.89 (d, J = 6.7 Hz, 3H), 0.88 (d, J = 6.7 Hz, 3H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 154.4, 151.7, 138.7, 129.5, 128.4, 126.2, 117.3, 114.9, 76.6, 75.9, 58.0, 55.7, 50.6, 39.8, 28.2, 20.6, 20.5 ppm. IR (CHCl3): 3168, 1218 cm–1. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C21H30NO3 344.2220; Found, 344.2220.

(2R,3R)-4-((2-Ethylbutyl)amino)-3-(p-methoxyphenoxy)-1-phenyl-2-butanol (19)29
To a flame-dried, nitrogen-purged 2 L round-bottom flask were added β-hydroxyamide 18 (4.23 g, 11.0 mmol) and THF (275 mL). To this mixture was added a borane-dimethylsulfide complex (3.10 mL). The reaction was heated to reflux 18 h using a heating mantle controlled by a variable transformer. The reaction mixture was cooled to ambient temperature and was then cooled with an ice bath. The reaction was quenched with 20 mL of methanol and allowed to stir for 30 min in an ice bath. The quenched reaction was diluted with diethyl ether (100 mL) and then washed with an aqueous solution of 1 M NaOH (2 × 30 mL). The organic layer was washed one more time with brine, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (7:3, diethyl ether: hexanes) to afford 19 (2.52 g, 6.82 mmol, 62% yield) as a colorless viscous oil. 1H NMR (500 MHz, CDCl3): δ 7.26–7.15 (m, 5H), 6.87 (d, J = 9.3 Hz, 2H), 6.83 (d, J = 9.3 Hz, 2H), 4.19 (td, J = 7.1, 2.0 Hz, 1H), 4.06 (p, 1H), 3.78 (s, 3H), 3.25 (dd, J = 12.7, 4.3 Hz, 1H), 2.97 (m, 2H), 2.71 (dd, J = 12.7, 2.3 Hz, 1H), 2.48 (dd, J = 11.6, 4.8 Hz, 1H), 2.42 (dd, J = 11.6, 5.4 Hz, 1H), 1.30 (m, 5H), 0.83 (m, 6H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 154.4, 151.6, 138.6, 129.5, 128.4, 126.2, 117.3, 114.8, 76.9, 75.6, 55.7, 52.8, 51.0, 40.8, 39.7, 24.0, 23.9, 10.9, 10.8 ppm. IR CDCl3: 3321, 3036, 1226, 1107, 1040, 827 cm–1. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C23H34NO3SNa 372.2533; Found, 372.2536.

(2R,3R)-4-(N-(2-Isobutyl)-p-nitrophenylsulfonamido)-3-(p-methoxyphenoxy)-1-phenyl-2-p-nitrobenzenesulfonylbutane (13)28
To a flame-dried, nitrogen-purged 250 mL round-bottom flask equipped with a stir bar were added β-amino alcohol 14 (6.74 g, 19.6 mmol) and dichloromethane (64 mL). Once the substrate had fully dissolved, p-nitrobenzenesulfonyl chloride (10.9 g, 40.3 mmol), DMAP (2.64 g, 21.6 mmol), and triethylamine (11 mL, 79 mmol) were added. The reaction was vigorously stirred overnight, and the reaction contents were diluted with dichloromethane (80 mL), transferred to a separatory funnel, and washed with an aqueous solution of 1 M HCl (2 × 50 mL). The organic layer was then washed with brine (50 mL), dried (MgSO4), gravity-filtered, and concentrated under reduced pressure (7.30 g, 10.2 mmol, 52% yield). Mp: 161–162 °C. [α]D = +113.1 (c = 1.02, CHCl3). 1H NMR (500 MHz, CDCl3): δ 8.19 (d, J = 8.9 Hz, 2H), 7.98 (d, J = 8.9 Hz, 2H), 7.96 (d, J = 8.9 Hz, 2H), 7.50 (d, J = 8.9 Hz, 2H), 7.06 (t, J = 7.4 Hz, 1H), 6.96 (t, J = 7.4 Hz, 2H), 6.77 (d, J = 9.2 Hz, 2H), 6.73 (d, J = 7.4 Hz, 2H), 6.69 (d, J = 9.2 Hz, 2H), 4.80–4.77 (m, 1H), 4.68–4.65 (m, 1H), 3.87 (d, J = 15.5 Hz, 1H), 3.78 (s, 3H), 3.62 (dd, J = 15.5, 9.4 Hz, 1H), 3.28 (dd, J = 13.6, 8.6 Hz, 1H), 3.11 (dd, J = 13.6, 6.6 Hz, 1H), 3.05 (dd, J = 14.6, 2.2 Hz, 1H), 2.64 (dd, J = 14.6, 10.8 Hz, 1H), 2.15–2.06 (m, 1H), 0.96 (d, J = 6.7 Hz, 3H), 0.95 (d, J = 6.7 Hz, 3H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 155.0, 150.4, 149.8, 149.7, 146.0, 140.5, 135.6, 129.0, 128.7, 128.6, 128.3, 126.9, 124.3, 124.0, 116.3, 114.9, 82.5, 75.6, 56.2, 55.7, 46.7, 34.1, 26.3, 19.9, 19.8 ppm. IR (CHCl3): 1531, 1350 cm–1. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C33H35N3NaO11S2 736.1605; Found, 736.1606.

(2R,3R)-4-(N-(2-Ethylbutyl)-p-nitrophenylsulfonamido)-3-(p-methoxyphenoxy)-1-phenyl-2-p-nitrobenzenesulfonylbutane (20)29
To a stirred solution of β-amino alcohol 19 (2.16 g, 5.80 mmol) dissolved in dichloromethane (19 mL) were added p-nitrobenzenesulfonyl chloride (3.22 g, 14.5 mmol) and DMAP (0.782 g, 6.40 mmol). Lastly, triethylamine (3.20 mL, 23.1 mmol) was then added dropwise to the reaction mixture via an addition funnel. The reaction mixture was stirred overnight at room temperature. After completion, the reaction mixture was diluted with dichloromethane and washed with aqueous solution of 1 M HCl (2 × 30 mL). The organic layer was treated once with brine, dried over MgSO4, and concentrated under reduced pressure to afford a crude yellow viscous oil. The crude viscous oil was crystallized from diethyl ether and hexanes to yield 20 (3.72 g, 4.99 mmol, 86% yield) as yellow crystals. Mp: 152.3–153.6 °C. 1H NMR (500 MHz, CDCl3): δ 8.22 (d, J = 9.0 Hz, 2H), 7.99 (d, J = 9.0 Hz, 2H), 7.95 (d, J = 9.0 Hz, 2H), 7.50 (d, J = 9.0 Hz, 2H), 7.05 (t, J = 7.3 Hz, 1H), 6.96 (t, J = 7.8 Hz, 2H), 6.77 (d, J = 9.2 Hz, 2H), 6.72 (d, J = 8.5 Hz, 2H), 6.69 (d, J = 9.2 Hz, 2H), 4.82 (dq, J = 9.6, 4.1, 1.5 Hz, 1H),.82 (dq, J = 10.8, 4.1, 2.3 Hz, 1H), 3.83 (dd, J = 15.5, 1.2 Hz, 1H), 3.79 (s, 3H), 3.61 (dd, J = 15.4, 9.5 Hz, 1H), 3.40 (dd, J = 13.6, 8.5 Hz, 1H), 3.13 (dd, J = 13.6, 6.2 Hz, 1H), 3.05 (dd, J = 14.6, 2.2 Hz, 1H), 2.64 (dd, J = 14.6, 10.8 Hz, 1H), 1.77–1.70 (m, 2H), 1.48–1.27 (m, 4H), 0.91 (t, J = 7.4 Hz, 3H), 0.85 (t, J = 7.4 Hz, 3H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 155.0, 150.3, 149.72, 149.70, 145.9, 140.5, 135.6, 129.0, 128.7, 128.6, 128.3, 126.9, 124.3, 124.0, 116.3, 114.9, 82.5, 75.4, 55.7, 52.4, 46.5, 37.9, 34.2, 22.9, 22.6, 10.6, 10.2 ppm. IR (CDCl3): 3108, 1531, 1506, 1350, 855 cm–1. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C35H40N3NaO11S2 742.2099; Found, 742.2082.

N-((2R,3S)-3-[Azido-2-(p-methoxyphenoxy)-4-phenylbutyl]-N-(2-isobutyl)-p-nitrobenzene sulfonamide (21)28
To a flame-dried, nitrogen-purged 100 mL round-bottom flask equipped with a stir bar were added bis-sulfonylated substrate (13) (2.00 g, 2.80 mmol) and DMSO (12 mL). Once the substrate had fully dissolved, sodium azide (0.551 g, 8.41 mmol) was added, and the system was allowed to stir overnight. The system was transferred to a separatory funnel, diethyl ether (150 mL) was added, and the material was extracted with 1 M HCl (2 × 40 mL). The organic layer was washed with brine (30 mL), dried over magnesium sulfate, and gravity-filtered. The solvent was then removed under reduced pressure to afford the target azide product as a yellow oil that was used in the next step without further purification (1.55 g, 2.79 mmol, near quantitative yield). [α]D = +3.51 (c = 1.05, CHCl3). 1H NMR (500 MHz, CDCl3): δ 8.11 (d, J = 8.8 Hz, 2H), 7.84 (d, J = 8.8 Hz, 2H), 7.40–7.25 (m, 5H), 6.64 (d, J = 9.1 Hz, 2H), 6.37 (d, J = 9.1 Hz, 2H), 4.34 (dt, J = 9.3, 2.5 Hz, 1H), 3.94 (td, J = 7.6 Hz, 2.5 Hz, 1H), 3.75 (dd, J = 15.5, 2.3 Hz, 1H), 3.72 (s, 3H), 3.49 (dd, J = 15.5, 9.3 Hz, 1H), 3.23 (dd, J = 13.6, 8.1 Hz, 1H), 3.02 (dd, J = 13.6, 7.0 Hz, 1H), 2.86 (dd, J = 14.0, 7.9 Hz, 1H), 2.79 (dd, J = 14.0, 7.4 Hz, 1H), 2.08–2.00 (m, 1H), 0.91 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 6.7 Hz, 3H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 154.6, 149.8, 149.6, 146.0, 136.6, 129.3, 128.9, 128.1, 127.3, 124.2, 116.5, 114.7, 77.9, 63.7, 57.4, 55.6, 47.6, 37.2, 26.7, 20.0, 19.9 ppm. IR (CHCl3): 2119, 1532, 1350 cm–1. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C27H32N5NaO6S 554.2068; Found, 554.2064.

N-((2R,3S)-3-[Azido-2-(p-methoxyphenoxy)-4-phenylbutyl]-N-(2-ethylbutyl)-p-nitrobenzene sulfonamide (22)29
To a stirring solution of sulfonamide 20 (3.35 g, 4.52 mmol) in DMSO (30 mL) was added sodium azide (1.17 g, 18.0 mmol). The reaction mixture was stirred overnight at ambient temperature. After completion, the reaction mixture was diluted with diethyl ether (100 mL) and treated with an aqueous solution of 1 M HCl (2 × 30 mL). The organic layer was collected and treated with brine (30 mL), dried over MgSO4, and concentrated under reduced pressure. The residue was purified by flash column chromatography eluting with dichloromethane to afford 22 (2.52 g, 4.34 mmol, 96% yield) as a yellow viscous oil. 1H NMR (500 MHz, CDCl3): δ 8.13 (d, J = 8.9 Hz, 2H), 7.84 (d, J = 8.9 Hz, 2H), 7.40–7.24 (m, 5H), 6.64 (d, J = 9.1 Hz, 2H), 6.38 (d, J = 9.1 Hz, 2H), 4.36 (dt, J = 9.4, 2.6 Hz, 1H), 3.95 (td, J = 7.6, 2.6 Hz, 1H), 3.72 (s, 3H), 3.73 (dd, J = 15.6, 2.5 Hz, 1H), 3.49 (dd, J = 15.6, 9.4 Hz, 1H), 3.35 (dd, J = 13.6, 8.1 Hz, 1H), 3.05 (dd, J = 13.5, 6.7 Hz, 1H), 2.85 (dd, J = 14.1, 8.1 Hz, 1H), 2.78 (dd, J = 14.0, 7.3 Hz, 1H), 1.69–1.61 (m, 1H), 1.43–1.34 (m, 1H), 1.32–1.25 (m, 3H), 0.83 (q, J = 7.4 Hz, 6H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 154.7, 149.8, 149.7, 145.8, 136.5, 129.2, 128.9, 128.1, 127.3, 124.2, 116.5, 114.7, 77.9, 63.6, 55.6, 53.5, 47.4, 38.5, 37.2, 23.0, 22.9, 10.6, 10.3 ppm. IR (CDCl3): 3105, 2119, 1607, 1530, 1506, 1350, 1224, 826 cm–1. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C29H35N5NaO6S 604.2200; Found, 604.2205.

N-((2R,3S)-3-Azido-2-hydroxy-4-phenylbutyl)-N-(2-isobutyl)-4-nitrobenzenesulfonamide (12)28
To a flame-dried, nitrogen-purged 250 mL round-bottom flask equipped with a stir bar was added azide 21 (1.89 g, 3.41 mmol). Acetonitrile (56 mL) and deionized water (14 mL) were added in a 4:1 ratio to achieve an overall concentration of 0.049 M. Ceric ammonium nitrate (7.48 g, 13.7 mmol) was added, and the reaction was allowed to stir for 90 min. The reaction was then diluted with brine (20 mL), and the solvent was then removed by rotary evaporation. The concentrate was diluted with diethyl ether (150 mL), transferred to a separatory funnel, and extracted with brine (2 × 20 mL). The organic layer was dried over MgSO4, gravity-filtered, and concentrated under reduced pressure. The resulting crude residue was purified over silica using a mobile phase gradient (95:5, 80:20, hexanes:EtOAc) to afford the target azido alcohol as a dark, viscous oil (0.683 g, 1.53 mmol, 45% yield). [α]D = −3.2 (c = 1.19, CHCl3). 1H NMR (500 MHz, CDCl3): δ 8.38 (d, J = 8.8 Hz, 2H), 7.99 (d, J = 8.8 Hz, 2H), 7.35–7.32 (m, 2H), 7.29–7.25 (m, 3H), 3.79–3.75 (m, 1H), 3.66–3.62 (m, 1H), 3.31 (dd, J = 15.2, 9.3 Hz, 1H), 3.20 (dd, J = 15.2, 2.4 Hz, 1H), 3.09–3.04 (m, 3H), 2.93 (dd, J = 13.5, 6.9 Hz, 1H), 2.83 (dd, J = 14.1, 8.9 Hz, 1H), 1.89–1.80 (m, 1H), 0.92 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 150.1, 144.7, 137.1, 129.3, 128.8, 128.6, 127.0, 124.5, 71.6, 66.6, 58.0, 52.0, 36.8, 26.9, 20.0, 19.8 ppm. IR (CHCl3): 3515, 2111, 1531, 1350 cm–1. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C20H25N5NaS 470.1469; Found, 470.1480.

N-((2R,3S)-3-Azido-2-hydroxy-4-phenylbutyl)-N-(2-ethylbutyl)-4-nitrobenzenesulfonamide (23)29
To a stirred solution of 22 (2.01 g, 3.46 mmol) in acetonitrile (52.5 mL) was added ceric ammonium nitrate (7.58 g, 13.8 mmol), followed by deionized water (17.5 mL). The reaction mixture was stirred at room temperature for 1 h. The reaction mixture was then diluted with diethyl ether (100 mL), washed with brine (2 × 30 mL), dried over MgSO4, and concentrated under reduced pressure to afford a dark-red viscous oil. The residue was purified by flash column chromatography (2:8 diethyl ether: hexane) to afford 23 (0.661 g, 5.52 mmol, 40% yield) as a dark-red viscous oil. 1H NMR (500 MHz, CDCl3): δ 8.36 (d, J = 8.9 Hz, 2H), 7.99 (d, J = 8.9 Hz, 2H), 7.34–7.24 (m, 5H), 3.78–3.73 (broad multiplet, 1H), 3.66–3.32 (m, 1H), 3.30 (dd, J = 15.2, 9.2 Hz, 1H), 3.19 (dd, J = 15.3, 2.4 Hz, 1H), 3.16–3.11 (m, 2H), 3.05 (dd, J = 14.2, 4.7 Hz, 1H), 2.99 (dd, J = 13.4, 6.7 Hz, 1H), 2.83 (dd, J = 14.2, 8.8 Hz, 1H), 1.51–1.43 (m, 1H), 1.43–1.34 (m, 1H), 1.32–1.24 (m, 3H), 0.82 (q, J = 7.5 Hz, 6H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 150.2, 144.3, 136.9, 129.3, 128.7, 128.6, 127.1, 124.5, 71.5, 66.5, 54.3, 52.2, 38.7, 36.8, 23.0, 22.7, 10.5, 10.3 ppm. IR (CHCl3): 3516, 3106, 2110, 1606, 1531, 1456, 1350, 1159, 1089, 856 cm–1. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C22H29N5NaO5S 498.1782; Found, 498.1782.

Darunavir (1)28
To a nitrogen-purged 250 mL round-bottom flask equipped with a stir bar were added azido alcohol (12) (0.680 g, 1.52 mmol), methanol (15 mL), and activated palladium on carbon (70 mg, 10% w/w). The flask was equipped with a hydrogen balloon, and the system was stirred for 17 h. The mixture was then filtered through Celite with ethyl acetate, concentrated under reduced pressure, and reconstituted in THF (15 mL). Commercially available carbonate 19 (0.412 g, 1.53 mmol) and triethylamine (0.32 mL, 2.3 mmol) were added, and the system was stirred overnight. The material was concentrated under reduced pressure and subsequently purified over silica using 50% EtOAc in dichloromethane as the eluent to yield darunavir (1) as an amorphous solid (0.43 g, 0.78 mmol, 52% yield). Mp: 74–75 °C. [α]D = −2.40 (c = 1.04, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.55 (d, J = 8.0 Hz, 2H), 7.29–7.19 (m, 5H), 6.68 (d, J = 8.0 Hz, 2H), 5.64 (d, J = 5.1 Hz, 1H), 5.03–4.95 (m, 2H), 4.34–4.08 (broad singlet, 2H), 3.96–3.92 (m, 1H), 3.90–3.82 (m, 3H), 3.73–3.65 (m, 3H), 3.19–2.75 (m, 7H), 1.87–1.77 (m, 1H), 1.67–1.58 (m, 1H), 1.54–1.44 (m, 1H), 0.93 (d, J = 6.6 Hz, 3H), 0.88 (d, J = 6.6 Hz, 3H) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ 155.5, 151.0, 137.8, 129.5, 129.4, 128.5, 126.5, 125.9, 114.1, 109.3, 73.4, 72.9, 70.9, 69.6, 58.8, 55.2, 53.7, 45.4, 35.7, 27.3, 25.8, 20.2, 20.0 ppm. IR (CHCl3): 3492, 3415, 1717 cm–1. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C27H38N3NaO7S, 548.2425; Found, 548.2418.

(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl((2S,3R)-4-(4-amino-N-(2-ethylbutyl)phenyl sulfonamido)-3-hydroxy-1-phenyl-2-butyl)carbamate (27)29
A round-bottom flask containing 23 (0.356 g, 0.749 mmol), 5% palladium on carbon (0.004 g), 2,5-dioxopyrrolidin-1-yl((3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl)carbonate (0.162 g, 0.598 mmol), and THF (25 mL) was flushed with N2 gas for 10 min. A balloon filled with hydrogen gas was then connected to the reaction flask, and the mixture was allowed to stir overnight at ambient temperature. After completion, the reaction mixture was diluted with chloroform, filtered through a 1:1 mixture of Celite and MgSO4, and concentrated under reduced pressure. The crude residue was purified by flash column chromatography (diethyl ether) to afford the protease inhibitor 27 (0.217 g, 0.350 mmol, 50% yield) as a pale-yellow viscous oil. [α]D = +2.6 (c = 1.00, CDCl3). 1H NMR (500 MHz, CDCl3): δ 7.54 (d, J = 8.7 Hz, 2H), 7.29–7.18 (m, 5H) 6.68 (d, J = 8.7 Hz, 2H), 5.63 (d, J = 5.1 Hz, 1H), 5.00 (dd, J = 13.8, 6.5 Hz, 1H), 4.91 (d, J = 8.8 Hz, 2H), 4.17 (broad singlet, 2H), 3.94 (dd, J = 9.4, 6.4 Hz, 1H), 3.90–3.79 (m, 3H), 3.76 (broad singlet, 1H), 3.71–3.66 (m, 2H), 3.15–3.06 (m, 2H), 3.02 (dd, J = 13.4, 8.0 Hz, 1H), 2.96 (dd, J = 15.3, 2.3 Hz, 1H), 2.92–2.87 (m, 1H), 2.84–2.77 (m, 2H), 2.67–2.58 (m, 2H), 1.51–1.39 (m, 3H), 0.82 (td, J = 7.4, 2.3 Hz, 6H) ppm.13C{1H} NMR (100 MHz, CDCl3): δ 155.5, 151.3, 129.5, 129.4, 128.4, 126.5, 125.2, 114.0, 109.3, 73.3, 73.1, 70.8, 69.6, 55.3, 54.7, 53.6, 45.5, 39.0, 35.8, 25.8, 23.0, 22.8, 10.5, 10.3 ppm. IR (CHCl3): 3469, 3368, 3251, 1706, 832 cm–1. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C29H42N3NaO7S 576.2738; Found, 576.2739.

(4R)-3-[(2S’,3R’)-3-Hydroxy-2-(p-methoxyphenoxy)-4-methyl-pentanoyl]-1,3-oxazolidine-2-thione (28)29
A flame-dried 3 L round-bottom flask fitted with a Claisen adapter and an addition funnel was fitted onto the Claisen adapter was charged with (R)-11 (10.0 g, 29.1 mmol) and dichloromethane (1 L). The reaction mixture was cooled to −78 °C and titanium tetrachloride (1 M in dichloromethane, 32.0 mL, 32.0 mmol) was added via the addition funnel. The reaction mixture was stirred for 30 min, and then, triethylamine (8.9 mL, 64 mmol) was added dropwise by syringe. After an additional 30 min of stirring, the second batch of titanium tetrachloride (32 mL) was added as before. After another 30 min of stirring, isobutyraldehyde (5.8 mL, 64 mmol) was added dropwise to the reaction mixture by syringe. Finally, the reaction mixture was warmed to −40 °C for 3 h and then quenched with brine (80 mL). The reaction mixture was diluted with dichloromethane and then washed with an aqueous solution of 1 M HCl (2 × 80 mL). The extracted organic layer was washed with brine (80 mL), dried over MgSO4, and concentrated under reduced pressure. The resulting crude viscous oil was crystallized from ethyl acetate and hexanes to afford compound 28 as a white crystalline solid (8.89 g, 21.5 mmol, 74% yield). Mp: 149.5–151.5 °C. [α]D = −102.9 (c = 0.46, CDCl3). 1H NMR (500 MHz, CDCl3): δ 7.39–7.29 (m, 5H), 7.02 (d, J = 1.8 Hz, 1H), 6.85–6.79 (m, 4H), 5.73 (dd, J = 9.3, 6.5 Hz, 1H), 4.88 (t, J = 9.3 Hz, 1H), 4.50 (dd, J = 9.3, 6.5 Hz, 1H), 3.93 (ddd, J = 10.2, 8.4, 1.8 Hz, 1H), 3.74 (s, 3H), 2.05–1.95 (m, 1H), 1.60 (d, J = 10.2 Hz, 1H), 1.06 (d, J = 6.7 Hz, 3H), 1.01 (d, J = 6.7 Hz, 3H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 185.5, 171.1, 154.6, 151.2, 136.7, 129.2, 129.1, 126.6, 116.0, 114.8, 77.5, 76.7, 74.7, 62.8, 55.7, 32.4, 19.2, 19.1 ppm. IR (CDCl3): 3454, 1727, 1228, 825 cm–1. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C22H26NNaO5S 416.1526; Found, 416.1523.

(2S,3R)-3-Hydroxy-N-isobutyl-2-(p-methoxyphenoxy)-4-methylpentanamide (30)29
To a stirring solution of aldol adduct 28 (5.36 g, 12.9 mmol) in dichloromethane (129 mL) was added imidazole (2.63 g, 38.6 mmol). After 1 h, isobutylamine (1.30 mL, 13.1 mmol) was added to the reaction mixture, and the reaction was stirred overnight. The reaction mixture was diluted with dichloromethane (30 mL) and washed twice with an aqueous solution of NaOH (2 × 40 mL). The organic layer was then washed with brine (40 mL), dried over MgSO4, and concentrated under reduced pressure to afford compound 33a (3.80 g, 12.2 mmol, 95% yield) as pure white crystals. The crystals obtained were pure and did not require further purification. Mp: 84–86 °C. 1H NMR (500 MHz, CDCl3): δ 6.88 (d, J = 9.3 Hz, 2H), 6.83 (d, J = 9.3 Hz, 2H), 6.49 (broad triplet, 1H), 4.56 (d, J = 2.6 Hz, 1H), 4.30 (broad singlet, 1H), 3.77 (s, 3H), 3.73 (dd, J = 7.8, 2.6 Hz, 1H), 3.12 (t, J = 6.6 Hz, 2H), 2.01–1.92 (m, 1H), 1.78–1.70 (m, 1H), 1.06 (d, J = 6.7 Hz, 3H), 0.89 (d, J = 6.7 Hz, 3H), 0.84 (d, J = 3.7 Hz, 3H), 0.83 (d, J = 3.7 Hz, 6H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 170.7, 154.9, 151.6, 116.2, 114.9, 80.6, 55.7, 46.4, 31.0, 28.5, 19.94, 19.90, 19.3, 18.7 ppm. IR (CDCl3): 3470, 3338, 1634, 1508, 1465, 1224, 816.5 cm–1. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C17H28NO4, 310.2013; Found, 310.2018.

(2R,3R)-1-(Isobutylamino)-2-(p-methoxyphenoxy)-4-methyl-3-pentanol (31)29
To a round-bottom flask connected to nitrogen lines and mounted on a jacked stir plate were added compound 30 (3.27 g, 10.6 mmol) and THF (265 mL). A borane dimethylsulfide complex (3.00 mL) was then added to the reaction mixture while stirring at room temperature. The reaction was then heated to reflux overnight using a heating mantle controlled by a variable transformer. The reaction was then cooled with an ice bath and quenched by the addition of methanol (15 mL). After stirring for 30 min, the solvent was removed via rotary evaporation, and the reaction mixture was diluted with diethyl ether (120 mL) and then washed with an aqueous solution of 1 M NaOH (2 × 30 mL) and brine (30 mL). The organic layer was dried over MgSO4 and concentrated under reduced pressure. The residue was purified by flash column chromatography (7:3, diethyl ether:hexanes) to afford compound 31 (2.56 g, 8.6 mmol, 82% yield) as a colorless viscous oil. 1H NMR (500 MHz, CDCl3): δ 6.92 (d, J = 9.2 Hz, 2H), 6.83 (d, J = 9.2 Hz, 2H), 4.34–4.33 (m, 1H), 3.77 (s, 3H), 3.51 (dd, J = 8.4, 2.0 Hz, 1H), 3.33 (dd, J = 12.6, 4.2 Hz, 1H), 2.79 (dd, J = 12.6, 2.4 Hz, 1H), 2.44 (dd, J = 11.5, 6.4 Hz, 1H), 2.35 (dd, J = 11.5, 7.1 Hz, 1H), 2.03–1.96 (m, 1H), 1.75–1.67 (m, 1H), 1.05 (d, J = 6.7 Hz, 3H), 0.89 (d, J = 4.3 Hz, 3H), 0.88 (d, J = 4.3 Hz, 3H), 0.87 (d, J = 6.7 Hz, 3H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 154.2, 151.8, 117.1, 114.8, 81.0, 75.7, 58.1, 55.7, 51.0, 30.7, 28.2, 20.5, 19.1, 19.0 ppm. IR (CDCl3): 3325, 3224, 1226, 1039, 827 cm–1. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C17H30NO3 296.2220; Found, 296.2220.

(2R,3R)-1-(N-Isobutyl-P-nitrophenylsulfonamido)-2-(p-methoxyphenoxy)-p-methyl-3-pentyl-p-nitrobenzenesulfonate (32)29
To a vigorously stirring solution of 31 (1.92 g, 6.50 mmol) in dichloromethane (23 mL) in a round-bottom flask was added p-nitrobenzenesulfonyl chloride (3.60 g, 16.2 mmol), followed by DMAP (0.874 g, 7.15 mmol). Triethylamine (3.60 mL, 25.8 mmol) was then added dropwise to the reaction mixture via an addition funnel. The reaction mixture was then stirred at ambient temperature overnight. The reaction mixture was diluted with dichloromethane (50 mL) and washed with an aqueous solution of 1 M HCl (25 mL) and brine (25 mL). The organic layer was then isolated and dried over MgSO4 and concentrated under reduced pressure to afford a crude yellow viscous oil that was crystallized from diethyl ether and hexanes to afford 32 (2.45 g, 57% yield) as yellow crystals. Mp: 132.6–133.9 °C. 1H NMR (500 MHz, CDCl3): δ 8.19 (dd, J = 14.1, 8.9 Hz, 4H), 7.98 (d, J = 8.9 Hz, 2H), 7.87 (d, J = 8.9 Hz, 2H), 6.62 (d, J = 9.1 Hz, 2H), 6.51 (d, J = 9.1 Hz, 2H), 4.66–4.63 (m, 1H), 4.55 (t, J = 5.9 Hz, 1H), 3.73 (s, 3H), 3.67 (dd, J = 15.3, 1.8 Hz, 1H), 3.4 (dd, J = 15.3, 9.3 Hz, 1H), 3.11 (dd, J = 13.8, 8.3 Hz, 1H), 2.99 (dd, J = 13.8, 6.9 Hz, 1H), 2.06–1.93 (m, 2H), 1.02 (d, J = 6.7 Hz, 3H), 0.89 (d, J = 6.7 Hz, 3H) 0.83 (d, J = 6.7 Hz, 6H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 154.7, 150.6, 150.5, 149.8, 145.6, 142.0, 129.0, 128.1, 124.3, 124.1, 116.2, 114.5, 86.8, 57.0, 55.6, 48.5, 29.0, 26.5, 19.8, 19.7, 18.2 ppm. IR (CDCl3): 3108, 1607, 1350, 855 cm–1. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C29H35N3NaO11S2 688.1605; Found, 688.1605.

N-((2R,3S)-3-Azido-2-(p-methoxyphenoxy)-4-methylpentyl)-N-isobutyl-p-nitrobenzene sulfonamide (33)29
To a solution of 32 (2.00 g, 3.00 mmol) in DMSO (20 mL) was added sodium azide (0.781 g). The reaction mixture was stirred overnight at ambient temperature, diluted with diethyl ether (100 mL), and washed twice with an aqueous solution of 1 M HCl (2 × 30 mL). The organic layer was then washed with brine (30 mL), dried over MgSO4, and concentrated under reduced pressure. The residue was purified by flash column chromatography eluting using dichloromethane to afford azide 33 (1.40 g, 2.75 mmol, 92% yield) as a yellow viscous oil. 1H NMR (500 MHz, CDCl3): δ 8.23 (d, J = 8.9 Hz, 2H), 7.94 (d, J = 8.9 Hz, 2H), 6.77 (d, J = 9.1 Hz, 2H), 6.69 (d, J = 9.1 Hz, 2H), 4.68 (dt, J = 9.7, 2.4 Hz, 1H), 3.75 (s, 3H), 3.57 (dd, J = 15.6, 2.1 Hz, 1H), 3.42 (dd, J = 15.6, 9.6 Hz, 1H), 3.33 (dd, J = 9.6, 2.7 Hz, 1H), 3.27 (dd, J = 13.5, 8.7 Hz, 1H), 3.30 (dd, J = 13.5, 6.4 Hz, 1H), 2.11–2.03 (m, 1H), 1.67–1.60 (m, 1H), 1.05 (d, J = 6.7 Hz, 3H), 1.01 (d, J = 6.7 Hz, 3H), 0.89 (d, J = 6.7 Hz, 3H), 0.84 (d, J = 6.7 Hz, 3H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 154.7, 150.0, 149.8, 145.8, 128.2, 124.2, 116.9, 114.9, 78.2, 69.5, 57.8, 55.6, 48.0, 30.2, 26.6, 20.6, 20.0, 19.9, 19.2, 18.6 ppm. IR (CDCl3): 3105, 1606, 1350, 856 cm–1. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C23H31N5NaO6S 528.1887; Found, 528.1895.

N-((2R,3S)-3-Azido-2-hydroxy-4-methylpentyl)-N-isobutyl-p-nitrobenzenesulfonamide (34)29
To a stirred solution of compound 33 (1.11 g, 2.20 mmol) in acetonitrile (33 mL) were added cerium (IV) ammonium nitrate (4.83 g, 8.81 mmol) and deionized water (11 mL). The reaction mixture was stirred at ambient temperature for 1 h. After completion, the reaction mixture was diluted with diethyl ether, washed twice with brine, dried over MgSO4, and concentrated under reduced pressure to afford a dark-red viscous oil. The residue was purified by flash column chromatography (2:8, diethyl ether:hexane) to afford β-azido alcohol 34 (0.399 g, 0.99 mmol, 45% yield) as a dark-red viscous oil. 1H NMR (500 MHz, CDCl3): δ 8.39 (d, J = 8.9 Hz, 2H), 8.02 (d, J = 8.9 Hz, 2H), 3.88 (broad triplet, J = 7.9 Hz, 1H), 3.31 (dd, J = 15.2, 9.6 Hz, 1H), 3.19 (t, J = 6.2, Hz, 1H), 3.15 (dd, J = 15.2, 2.2 Hz, 1H), 3.09 (dd, J = 13.6, 8.2 Hz, 1H), 2.98 (dd, J = 13.6, 7.1 Hz, 2H), 2.0–1.87 (m, 2H), 1.04 (d, J = 6.7 Hz, 3H), 1.00 (d, J = 6.7 Hz, 3H), 0.95 (d, J = 6.7 Hz, 3H), 0.90 (d, J = 6.7 Hz, 3H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 150.2, 144.6, 128.6, 124.4, 72.2, 70.1, 58.3, 52.4, 29.9, 27.1, 20.1, 20.0, 19.8, 18.0 ppm. IR (CDCl3): 3519, 3107, 2104, 1607, 1531, 1350, 856 cm–1. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C16H25N5NaO5S 422.1469; Found, 422.1469.

(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl((2R,3S)-1-(4-amino-N-isobutylphenyl sulfonamido)-2-hydroxy-4-methylpentan-3-yl)carbamate (35)29
A round-bottom flask containing compound 34 (0.100 g, 0.250 mmol), 5% palladium on carbon (0.001 g), 2,5-dioxopyrrolidin-1-yl((3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl)carbonate (0.0611 g), and THF (8.30 mL) was flushed with N2 gas for 10 min. A balloon filled with H2 gas was then connected to the reaction flask, and the mixture was allowed to stir overnight at room temperature. After completion, the reaction mixture was diluted with chloroform, filtered through a 1:1 mixture of Celite and MgSO4, and concentrated under reduced pressure. The crude residue was purified by flash column chromatography (diethyl ether) to afford the protease inhibitor 35 (0.0908 g, 73% yield) as a rusty viscous oil. [α]D = +10.1 (c = 0.490, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.55 (d, J = 8.6 Hz, 2H), 6.68 (d, J = 8.6 Hz, 2H), 5.71 (d, J = 5.1 Hz, 1H), 5.12 (dd, J = 14.6, 6.7 Hz, 1H), 4.84 (d, J = 10.0 Hz, 2H), 4.20 (br.s, 2H), 4.02 (dd, J = 9.5, 6.5 Hz, 1H) 3.98 (td, J = 8.4, 2.6 Hz, 1H) 3.92–3.87 (m, 1H), 3.78–3.71 (m, 2H), 3.48–3.43 (m, 1H), 3.36 (d, J = 2.5 Hz, 1H), 3.07 (dd, J = 15.2, 8.5 Hz, 2H), 3.01 (dd, J = 15.4, 2.7 Hz, 1H), 2.91 (dd, J = 13.1, 7.6 Hz, 1H), 2.82 (dd, J = 13.3, 7.1 Hz, 1H), 2.29–2.22 (m, 1H), 2.07–2.01 (m, 1H), 1.95–1.78 (m, 2H), 0.93 (dd, J = 6.7, 4.3 Hz, 6H), 0.89 (d, J = 6.7 Hz, 6H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 156.1, 151.1, 129.5, 125.5, 114.0, 109.3, 73.4, 71.6, 70.8, 69.5, 58.8, 58.5, 54.3, 53.9, 45.3, 27.3, 27.1, 25.8, 20.2, 20.1, 20.0, 15.9 ppm. IR (CDCl3): 3466, 3368, 3252, 1708, 1632,1149, 1091, 830 cm–1. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C23H38N3O7S 500.2425; Found, 500.2426.

(4S)-[(2S’,3R’)-3-Hydroxy-2-(p-methoxyphenoxy)-5,5-dimethylhexanoyl]-4-phenyl-1,3-oxazolidine-2-thione (39)29
A flame-dried, nitrogen-purged 5 L round-bottom flask fitted with a Claisen adapter for an additional funnel was charged with acylated oxazolidine-2-thione (S)-11 (10.0 g, 29.1 mmol) and dichloromethane (1 L). The reaction mixture was cooled to −78 °C, and titanium tetrachloride (1 M in dichloromethane, 32 mL, 32 mmol) was added via the addition funnel. The reaction mixture was stirred for 30 min, then treated with triethylamine (8.9 mL, 64 mmol), and added dropwise by syringe. After another 30 min of stirring, 3,3-dimethylbutyraldehyde (6.9 mL, 64 mmol) was added dropwise to the reaction mixture by syringe. Finally, the reaction mixture was warmed to −40 °C for 3 h and then quenched with brine (80 mL). The reaction mixture was diluted with dichloromethane and treated with an aqueous solution of 1 M HCl (2 × 100 mL). The recovered organic layer was treated with brine (100 mL), dried over MgSO4, and concentrated under reduced pressure. The crude residue was purified by flash column chromatography on silica gel (15:85, diethyl ether:hexanes) to afford aldol adduct 39 (10.62 g, 25.0 mmol, 82% yield) as a pale-yellow viscous oil. [α]D = +70.4 (c = 1.00, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.34–7.29 (m, 3H), 7.23–7.21 (m, 2H), 6.67 (d, J = 2.0 Hz, 1H), 6.66–6.61 (m, 4H), 5.68 (dd, J = 8.3, 2.5 Hz, 1H), 4.84 (t, J = 8.7 Hz, 1H), 4.53–4.47 (m, 2H), 3.70 (s, 3H), 1.71 (dd, J = 14.7, 9.5 Hz, 1H), 1.61 (dd, J = 14.6, 2.0 Hz, 1H), 1.02 (s, 9H) ppm. 13C{1H} NMR (100 MHz, CDCl3): 185.4, 169.6, 154.5, 151.3, 138.3, 129.2, 128.9, 125.8, 116.2, 114.6, 80.2, 74.7, 70.2, 62.8, 55.7, 47.8, 30.5, 30.2 ppm. IR (CDCl3): 3422, 1716, 1507, 1228, 1196, 824 cm–1. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C24H30NO5S, 444.1839; Found, 444.1838.

(2S,3R)-3-Hydroxy-N-isobutyl-2-(p-methoxyphenoxy)-5,5-dimethylhexanamide (41)29
To a stirred solution of oxazolidine-2-thione 39 (10.1 g, 21.8 mmol) in dichloromethane (218 mL) was added imidazole (4.45 g, 65.4 mmol). After 1 h, isobutylamine (2.40 mL, 24.0 mmol) was added, and the reaction mixture was allowed to stir overnight at room temperature. After completion, the reaction mixture was diluted with dichloromethane and then washed with an aqueous solution of 2 M NaOH (2 × 50 mL). The organic layer was treated with brine (50 mL), dried over MgSO4, and concentrated under reduced pressure. The crude residue was purified by flash column chromatography on silica gel (dichloromethane) to afford 41 (7.08 g, 20.9 mmol, 96% yield) as a colorless viscous oil. 1H NMR (500 MHz, CDCl3): δ 6.89 (d, J = 9.3 Hz, 2H), 6.83 (d, J = 9.3 Hz, 2H), 6.62 (broad triplet, 1H), 4.46 (d, J = 3.8 Hz, 1H), 4.11 (dq, J = 9.1, 2.2 1H), 3.77 (s, 3H), 3.19–3.08 (m, 1H), 1.81–1.73 (m, 1H), 1.51 (dd, J = 14.4, 2.1 Hz, 1H), 1.45 (dd, J = 14.4, 9.2 Hz, 1H), 0.95 (s, 9H), 0.89 (d, J = 2.5 Hz, 3H), 0.87 (d, J = 2.5 Hz, 3H) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ 170.7, 155.0, 151.2, 116.8, 114.9, 82.2, 69.4, 55.7, 46.3, 46.0, 30.1, 30.0, 28.4, 20.0, 19.9 ppm. IR (CDCl3): 3430, 3354, 3072, 1224, 1058, 827 cm–1. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C19H32NO4 338.2326; Found, 338.2317.

(2R,3R)-1-(Isobutylamino)-2-(p-methoxyphenoxy)-5,5-dimethyl-3-hexanol (42)29
To a flame-dried, nitrogen-purged 2 L round-bottom flask equipped with a magnetic stir bar were added amide 41 (6.64 g, 19.7 mmol) and THF (493 mL). A borane dimethylsulfide complex (5.60 mL) was added to the reaction mixture, and the reaction was then heated to reflux overnight using a heating mantle controlled by a variable transformer. The reaction was then cooled using an ice bath and quenched by the dropwise addition of methanol (30 mL). The reaction mixture was stirred for an additional 30 min, and the reaction solvent was removed via rotary evaporation. The concentrated reaction mixture was then diluted with diethyl ether (150 mL) and treated with an aqueous solution of 1 M NaOH (2 × 50 mL). The organic layer was washed one more time with brine (50 mL), dried over MgSO4, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (7:3, diethyl ether:hexanes) to afford 42 (5.95 g, 18.3 mmol, 93% yield) as a colorless viscous oil. 1H NMR (500 MHz, CDCl3): δ 6.91 (d, J = 9.2 Hz, 2H), 6.82 (d, J = 9.2 Hz, 2H), 4.11–4.06 (m, 2H), 3.76 (s, 3H), 3.19 (dd, J = 12.5, 5.2 Hz, 1H), 2.85 (dd, J = 12.5, 3.2 Hz, 1H), 2.43–2.36 (m, 2H), 1.75–1.64 (m, 1H), 1.59 (dd, J = 14.4, 8.5 Hz, 1H), 1.45 (dd, J = 14.5, 2.2 Hz, 1H), 0.97 (s, 9H), 0.89 (d, J = 2.8 Hz, 1H), 0.88 (d, J = 2.8 Hz, 1H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 154.3, 152.1, 117.4, 114.7, 79.9, 71.8, 58.2, 55.7, 50.7, 47.0, 30.2, 30.1, 28.2, 20.5, 20.5 ppm. IR (CDCl3): 3322, 1227, 1042, 827 cm–1. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C19H34NO3 324.2533; Found, 324.2538.

N-((2R,3R)-3-Hydroxy-2-(p-methoxyphenoxy)-5,5-dimethylhexyl)-N-isobutyl-4-nitrobenzene sulfonamide (43)29
To a stirred solution of compound 42 (5.20 g, 16.1 mmol) and dichloromethane (54 mL) in a round-bottom flask was added p-nitrobenzenesulfonyl chloride (8.91 g, 40.2 mmol), followed by DMAP (2.16 g, 17.7 mmol). Once all the reaction components had dissolved, triethylamine (8.80 mL, 63.1 mmol) was then added dropwise via an addition funnel. The reaction mixture was then allowed to stir overnight at room temperature. After completion, the reaction mixture was diluted with dichloromethane and treated with an aqueous solution of 1 M HCl (2 × 30 mL). The organic layer was treated with brine (30 mL), dried over MgSO4, and concentrated under reduced pressure to afford a crude yellow viscous oil. The residue was purified by flash column chromatography on silica gel (3:7, diethyl ether:hexanes) to afford 43 (4.132 g, 50% yield) as a yellow viscous oil. 1H NMR (500 MHz, CDCl3): δ 8.25 (d, J = 9.0 Hz, 2H), 7.93 (d, J = 9.0 Hz, 2H), 6.81–6.77 (m, 4H), 4.35 (td, J = 6.3, 2.7 Hz, 1H), 3.95–3.90 (m, 1H), 3.77 (s, 3H), 3.55 (dd, J = 15.1, 5.8 Hz, 1H), 3.25 (dd, J = 15.1, 6.8 Hz, 1H), 3.05 (dd, J = 13.6, 7.6 Hz, 1H), 2.96 (dd, J = 13.6, 7.5 Hz, 1H), 2.06–1.95 (m, 1H), 1.84 (d, J = 7.6 Hz, 1H), 1.47–1.45 (m, 2H), 0.95 (s, 9H), 0.89 (d, J = 6.7 Hz, 3H), 0.86 (d, J = 6.7 Hz, 3H)ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 154.5, 151.6, 149.9, 144.9, 128.5, 124.2, 117.0, 114.7, 80.7, 67.9, 57.9, 55.7, 48.5, 46.7, 30.2, 30.0, 26.6, 20.0, 19.9 ppm. IR (CDCl3): 3542, 3106, 1606, 1531, 1350, 1226, 1160, 855 cm–1. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C25H37N2O7S 509.2316; Found, 509.2309.

(2R,3R)-1-(N-Isobutyl-N-p-nitrophenylsulfonamido)-2-(p-methoxyphenoxy)-5,5-dimethyl-3-hexyl-p-nitrobenzenesulfonate (44)29
To a stirred solution of sulfonamide 43 (3.76 g, 7.40 mmol) and dichloromethane (25 mL) in a flame-dried, nitrogen-purged round-bottom flask was added 4-nitrobenzenesulfonyl chloride (1.97 g, 8.89 mmol), followed by DMAP (0.904 g, 7.40 mmol). Triethylamine (4.10 mL, 29.4 mmol) was then added dropwise via an addition funnel. The reaction mixture was then allowed to stir overnight at room temperature. After completion, the reaction mixture was diluted with dichloromethane and treated with an aqueous solution of 1 M HCl (2 × 30 mL). The organic layer was treated with brine (30 mL), dried over MgSO4, and concentrated under reduced pressure to afford a crude yellow viscous oil. The residue was purified by flash column chromatography on silica gel (7:3, diethyl ether: hexanes) to afford 44 (3.95 g, 5.70 mmol, 77% yield) as a yellow viscous oil. 1H NMR (500 MHz, CDCl3): δ 8.38 (d, J = 9.0 Hz, 2H), 8.12 (dd, J = 9.0, 5.0 Hz, 4H), 7.92 (d, J = 9.0 Hz, 2H), 7.50 (d, J = 9.0 Hz, 2H), 6.71 (d, J = 9.1 Hz, 2H), 6.59 (d, J = 9.1 Hz, 2H), 4.95 (ddd, J = 9.0, 4.1, 1.1 Hz, 1H), 4.77 (ddd, J = 9.7, 4.1, 1.5 Hz, 2H), 3.75 (s, 3H), 3.72 (d, J = 15.1, 1.4 Hz, 1H), 3.51 (dd, J = 15.5, 9.7 Hz, 1H), 3.28 (dd, J = 13.7, 8.3 Hz, 1H), 3.12 (dd, J = 13.7, 6.8 Hz, 1H), 2.10–2.02 (m, 1H), 1.71 (dd, J = 15.3, 1.1 Hz, 1H), 1.43 (dd, J = 15.3, 9.2 Hz, 1H), 0.93 (dd, J = 6.7, 1.8 Hz, 1H), 0.74 (s, 9H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 154.8, 150.9, 149.6, 149.5, 146.3, 142.0, 129.3, 128.0, 124.5, 124.1, 116.2, 114.7, 77.7, 75.4, 56.2, 55.6, 46.2, 40.6, 29.9, 29.3, 26.4, 19.8, 19.7 ppm. IR (CDCl3): 3108, 1607, 1532, 1350, 829 cm–1. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C31H42N3O9S2 694.2099; Found, 694.2090.

N-((2R,3S)-3-Azido-2-(p-methoxyphenoxy)-5,5-dimethylhexyl)-N-isobutyl-4-nitrobenzene sulfonamide (45)29
To a stirring solution of compound 44 (3.73 g, 5.37 mmol) in DMSO (36 mL) was added sodium azide (1.40 g). The reaction mixture was allowed to stir overnight at room temperature. The reaction mixture was diluted with diethyl ether (100 mL) and washed with an aqueous solution of 1 M HCl (2 × 30 mL). The organic layer was washed one more time with brine, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by flash column chromatography eluting with dichloromethane to afford azide 45 (2.615 g, 91% yield) as a yellow viscous oil. 1H NMR (500 MHz, CDCl3): δ 8.24 (d, J = 8.9 Hz, 2H), 7.95 (d, J = 8.9 Hz, 6.77 (d, J = 9.2 Hz, 2H), 6.69 (d, J = 9.2 Hz, 2H), 4.41 (dt, J = 9.3, 2.4 Hz, 2H), 3.76 (s, 3H), 3.71 (dt, J = 8.1, 2.6 Hz, 1H), 3.56 (dd, J = 15.6, 2.0 Hz, 1H), 3.41 (dd, J = 15.6, 9.1 Hz, 1H), 3.25 (dd, J = 13.5, 8.6 Hz, 1H), 2.97 (dd, J = 13.5, 6.5 Hz, 1H), 2.09–2.01 (m, 1H), 1.33 (dd, J = 14.5, 2.7 Hz, 1H), 1.29 (dd, J = 14.5, 8.1 Hz, 1H), 0.93 (s, 9H), 0.89 (d, J = 6.6 Hz, 3H), 0.85 (d, J = 6.6 Hz, 3H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 154.8, 149.9, 149.8, 145.7, 128.2, 124.2, 116.9, 114.9, 81.7, 59.4, 57.5, 55.6, 48.1, 43.8, 30.4, 29.4, 26.6, 20.0, 19.9 ppm. IR (CDCl3): 3106, 2115, 1606, 1531, 1350, 827 cm–1. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C25H35N5NaO6S 556.2200; Found, 556.2205.

N-((2R,3S)-3-Azido-2-hydroxy-5,5-dimethylhexyl)-N-isobutyl-p-nitrobenzenesulfonamide (46)29
To a stirred solution of 45 (2.12 g, 3.96 mmol) in CH3CN (46.5 mL) was added cerium (IV) ammonium nitrate (4.35 g, 7.93 mmol), followed by deionized water (15.5 mL). The reaction mixture was allowed to stir at room temperature for 1 h. The reaction mixture was then diluted with diethyl ether (100 mL), washed with brine (2 × 30 mL), dried over MgSO4, and concentrated under reduced pressure to afford a dark-red viscous oil. The residue was purified by flash column chromatography on silica gel (2:8, diethyl ether: hexane) to afford azide 46 (0.6406 g, 38% yield) as a dark-red viscous oil. 1H NMR (500 MHz, CDCl3): δ 8.39 (d, J = 8.9 Hz, 2H), 8.02 (d, J = 8.9 Hz, 2H), 3.81 (broad singlet, 1H), 3.39 (ddd, J = 9.1, 4.8, 2.0 Hz, 1H), 3.33 (t, J = 15.3, 9.5 Hz, 1H), 3.13–3.04 (m, 3H), 2.97 (dd, J = 13.6, 7.0 Hz, 1H), 1.95–1.86 (m, 1H), 1.44 (dd, J = 14.6, 2.0 Hz, 1H), 1.31 (dd, J = 14.6, 9.2 Hz, 1H), 0.98 (s, 9H), 0.95 (d, J = 6.7 Hz, 3H), 0.90 (d, J = 6.7 Hz, 3H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 150.2, 144.6, 128.6, 124.5, 73.7, 62.7, 58.2, 51.7, 43.3, 30.1, 29.6, 27.1, 20.1, 19.8 ppm. IR (CDCl3): 3524, 2115, 1531,1350, 1159, 1089, 856 cm–1. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C18H29N5NaO5S 450.1782; Found, 450.1786.

(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl((2R,3S)-1-(-N-p-aminophenyl-N-isobutyl sulfonamido)-2-hydroxy-5,5-dimethyl-3-hexyl)carbamate (38)29
A flame-dried 250 mL round-bottom flask containing compound 46 (0.424 g, 0.992 mmol), 5% palladium on carbon (0.005 g), 2,5-dioxopyrrolidin-1-yl((3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl)carbonate (0.242 g, 0.890 mmol), and THF (33 mL) was flushed with nitrogen gas for 10 min. A balloon filled with hydrogen gas was then connected to the reaction flask, and the mixture was allowed to stir overnight at room temperature. The reaction mixture was diluted with chloroform, filtered through a 1:1 mixture of Celite and MgSO4, and concentrated under reduced pressure. The crude residue was purified by flash column chromatography on silica gel with diethyl ether as the mobile phase to afford the desired 38 (0.4139 g, 0.784 mmol, 79% yield) as a pale-yellow viscous oil. [α]D = −19.3 (c = 1.00, CDCl3). 1H NMR (500 MHz, CDCl3): δ 7.56 (d, J = 8.8 Hz, 2H), 6.69 (d, J = 8.8 Hz, 2H), 5.71 (d, J = 5.2 Hz, 1H), 5.12–5.16 (m, 1H), 5.01 (d, J = 9.3 Hz, 1H), 4.20 (br.s, 2H), 4.28 (dd, J = 9.5, 6.4 Hz, 1H), 3.94 (d, J = 8.3, 2.75 Hz, 1H), 3.85–3.90 (m, 1H), 3.74 (d, J = 9.5, 6.7 Hz, 2H), 3.59–3.65 (m, 1H), 3.37 (d, J = 3.0 Hz, 1H), 3.13–3.02 (m, 2H), 2.96–2.90 (m, 2H), 2.81 (d, J = 13.4, 6.9 Hz, 1H), 2.02–1.97 (m, IH), 1.89–1.80 (m, 2H), 1.57 (d, J = 14.6 Hz, 1H), 1.32–1.25 (m, 1H), 9.33 (s, 9H), 0.92 (d, J = 6.5 Hz, 3H), 0.89 (d, J = 6.5 Hz, 3H) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ 155.3, 151.5, 129.3, 125.3, 113.9, 109.2, 74.0, 73.2, 70.7, 69.4, 58.2, 52.9, 51.8, 45.2, 432.0, 30.1, 29.7, 27.0, 25.8, 20.1, 20.0 ppm. IR (CDCl3): 3459, 3364, 3254, 1706, 1147, 1090, 831 cm–1. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C25H42N3O7S 528.2738; Found, 528.2743.
Acknowledgments
The authors thank Dr. B. A. Freeman and D. M. Casper for useful discussions. The authors also thank Drs. M. H. Nantz, K. F. Albizati, V. Martin, and D. Murray for their support. The authors thank the NSF for funding the X-ray diffractometer (NSF-CHE grant #1039689). The authors also thank the Department of Chemistry at Illinois State University for material support of this research.
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.4c01057.
1H and 13C NMR spectroscopic data for all reported compounds. X-ray crystallographic data for (R,S,R)-10 are provided (PDF)
Author Contributions
# J.M.W. and E.A. contributed equally to the work. C.J.S. and J.B.S. made important contributions to original project development. All authors contributed to the development of this work. G.M.F. and I.G.R. contributed to the SCXRD analyses for structure (R,S,R)-10.
The authors declare no competing financial interest.
Supplementary Material
References
- a MacArthur R. D. Darunavir: promising initial results. Lancet 2007, 369, 1143–1144. 10.1016/S0140-6736(07)60499-1. [DOI] [PubMed] [Google Scholar]; b Ghosh A. K.; Kincaid J. F.; Choi W.; Walters D. E.; Krishnan K.; Hussain K. A.; Koo Y.; Cho H.; Rudall C.; Holland L.; et al. Potent HIV protease inhibitors incorporating high-affinity P2-ligands and (R)-(hydroxyethylamino)sulfonamide isostere. Bio. Med. Chem. Lett. 1998, 8, 687–690. 10.1016/S0960-894X(98)00098-5. [DOI] [PubMed] [Google Scholar]
- Koh Y.; Nakata H.; Maeda K.; Ogata H.; Bilcer G.; Devasamudram T.; Kincaid J. F.; Boross P.; Wang Y.-F.; Tie Y.; Volarath P.; Gaddis L.; Harrison R. W.; Weber I. T.; Ghosh A. K.; Mitsuya H. Novel bis -Tetrahydrofuranylurethane-Containing Nonpeptidic Protease Inhibitor (PI) UIC-94017 (TMC114) with Potent Activity against Multi-PI-Resistant Human Immunodeficiency Virus In Vitro. Antimicrob. Agents Chemother. 2003, 47, 3123–3129. 10.1128/AAC.47.10.3123-3129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montaner J. S.; Lima V. D.; Barrios R.; Yip B.; Wood E.; Kerr T.; Shannon K.; Harrigan P. R.; Hogg R. S.; Daly P.; et al. Association of Highly Active Antiretroviral Therapy Coverage, Population Viral Load, and Yearly New HIV Diagnoses in British Columbia, Canada: A Population-Based Study. Lancet 2010, 376 (9740), 532–539. 10.1016/S0140-6736(10)60936-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a ″Drug Approval Package: Prezista (Darumavir) NDA #021976″. U.S. Food and Drug Administration (FDA). 6 September 2006.; b ″Prezista EPAR″. European Medicines Agency (EMA). 17 September 2018.; c ″FDA Approves New HIV Treatment for Patients Who Do Not Respond to Existing Drugs″. US FDA (Press release). Archived from the original on 13 November 2016.
- Ghosh A. K.; Markad S. B.; Robinson W. L. The Chiron Approach to (3R,3aS,6aR)-Hexahydro[2,3-b]furan-3-ol, a Key Subunit of HIV-1 Protease Inhibitor Drug, Darunavir. J. Org. Chem. 2021, 86, 1216–1222. 10.1021/acs.joc.0c02396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girigani S.; Singh H.; Kola S. R.; Yelmeli V. D.; Regula V. G.; Shah S.; Jain N.; Kumar P. An improved and robust scale-up process aided with identification and control of critical process impurities in darunavir ethanolate. Res. Chem. Int. 2020, 46, 267–281. 10.1007/s11164-019-03948-4. [DOI] [Google Scholar]
- Rapolu R. K.; Areveli S.; Raju V. V. N. K. V. P.; Navuluri S.; Chavali M.; Mulakayala N. An Efficient Synthesis of Darunavir Substantially Free from Impurities: Synthesis and Characterization of Novel Impurities. ChemistrySelect 2019, 4, 4422–4427. 10.1002/slct.201803825. [DOI] [Google Scholar]
- Moore G. L.; Stringham R. W.; Teager D. S.; Yue T.-Y. Practical Synthesis of the Bicyclic Side Chain: (3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-ol from Monopotassium Isocitrate. Org. Proc. Res. Dev. 2017, 21, 98–106. 10.1021/acs.oprd.6b00377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiummiento L.; Funicello M.; Lupattelli P.; Tramutola F. Ligand-Free Suzuki Coupling of Arylboronic Acids with Methyl (E)-4-Bromobut-2-enoate: Synthesis of Unconventional Cores of HIV-1 Protease Inhibitors. Org. Lett. 2012, 14, 3928–3931. 10.1021/ol3016786. [DOI] [PubMed] [Google Scholar]
- Graham B. J.; Windsor I. W.; Raines R. T. Inhibition of HIV-1 Protease by a Boronic Acid with High Oxidative Stability. ACS Med. Chem. Lett. 2023, 14, 171–175. 10.1021/acsmedchemlett.2c00464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockbaum G. J.; Rusere L. N.; Henes M.; Kosovrasti K.; Rao D. N.; Spielvogel E.; Lee S. K.; Nalivaika E. A.; Swanstrom R.; Yilmaz N. K.; et al. HIV-1 protease inhibitors with a P1 phosphonate modification maintain potency against drug-resistant variants by increased interactions with flap residues. Eur. J. Med. Chem. 2023, 257, 115501. 10.1016/j.ejmech.2023.115501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krolo T.; Bhattacharyya A.; Reiser O. Accessing HIV-1 Protease Inhibitors through Visible-Light-Mediated Sequential Photoctalytic Decarboxylative Radical Conjugate Addition-Elimination-Oxa-Michael Reactions. Org. Lett. 2021, 23, 6283–6287. 10.1021/acs.orglett.1c01964. [DOI] [PubMed] [Google Scholar]
- Ghosh A. K.; Kovela S.; Osswald H.; Amano M.; Aoki M.; Agniswamy J.; Wang Y.-F.; Weber I. T.; Mitsuya H. Structure-Based Design of highly Potent HIV-1 Protease Inhibitors Containing New Tricyclic Ring P2-Ligands: Design, Synthesis, Biological, and X-ray Structural Studies. J. Med. Chem. 2020, 63, 4867–4879. 10.1021/acs.jmedchem.0c00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A. K.; Nyalapatla P. R.; Kovela S.; Rao K. V.; Brindisi M.; Osswald H. L.; Amano M.; Aoki M.; Agniswamy J.; Wang Y.-F.; Weber I. T.; Mitsuya H. Design and Synthesis of Highly Potent HIV-1 Protease Inhibitors Containing Tricyclic Fused Ring Systems as Novel P2 Ligands: Structure-Activity Studies, Biological and X-ray, Structural Analysis. J. Med. Chem. 2018, 61, 4561–4577. 10.1021/acs.jmedchem.8b00298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ghosh A. K.; Osswald H. L.; Glauninger K.; Agniswamy J.; Wang Y. F.; Hayashi H.; Aoki M.; Weber I. T.; Mitsuya H. Probing Lipophilic Adamantyl Group as the P1-Ligand for HIV-1 Protease Inhibitors: Design, Synthesis, Protein X-ray Structural Studies, and Biological Evaluation. J. Med. Chem. 2016, 59, 6826–6837. 10.1021/acs.jmedchem.6b00639. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ghosh A. K.; Yu X.; Osswald H. L.; Agniswamy J.; Wang Y. F.; Amano M.; Weber I. T.; Mitsuya H. Structure-Based Design of Potent HIV-1 Protease Inhibitors with Modified P1-Biphenyl Ligands: Synthesis, Biological Evaluation, and Enzyme-Inhibitor X-ray Structural Studies. J. Med. Chem. 2015, 58, 5334–5343. 10.1021/acs.jmedchem.5b00676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.; Yang Y.-Q.; Hu Q. A Facile Access to Chiral 4-Isopropyl-, 4-Benzyl, and 4-Phenyloxazolidine-2-thione. J. Org. Chem. 2004, 69, 3990–3992. 10.1021/jo049799d. [DOI] [PubMed] [Google Scholar]
- The use of phenylacetic acid and related acetic acid derivatives with α-substituted undero a Claisen-type condensation that yields symmetrical ketones in good yield. See:Bhandari S.; Ray S. A. Novel Synthesis of Bisbenzyl Ketones by DCC Induced Condensation of Phenylacetic Acid. Synth. Commun. 1998, 28, 765–771. 10.1080/00032719808006472. [DOI] [Google Scholar]
- a Witte J. M.; Service J.; Addo M. A.; Semakieh B.; Collins E. S.; Sams C. J.; Dorsey T. R.; Garrelts E.; Blumenshine C. A.; Cooper T. A.; et al. Diastereoselective and enantioselective synthesis of α-p-methoxyphenoxy-β-lactones: Dependence on the stereoelectronic properties of the β-hydroxy-α-p-methoxyphenoxycarboxylic acid precursors. J. Org. Chem. 2022, 87, 9619–9634. 10.1021/acs.joc.2c00608. [DOI] [PubMed] [Google Scholar]; b Crimmins M. T.; King B. W.; Tabet E. A.; Chaudhary K. Asymmetric Aldol Additions: Use of Titanium Tetrachloride and (−)-Sparteine for the Soft Enolization of N- Acyl Oxazolidinones, Oxazolidinethiones, and Thiazolidinethiones. J. Org. Chem. 2001, 66, 894–902. 10.1021/jo001387r. [DOI] [PubMed] [Google Scholar]
- Ritter S. K. Where has all the sparteine gone?. Chem. Eng. News 2017, 95, 18–20. 10.1021/cen-09523-scitech2. [DOI] [Google Scholar]
- Zhang W.; Carter R. G.; Yokochi A. F. T. Unified Synthesis of C19-C26 Subunits of Amphidinolides B1, B2, and B3 by Exploiting Unexpected Stereochemical Differences in Crimmins’ and Evans’ Aldol Reactions. J. Org. Chem. 2004, 69, 2569–2572. 10.1021/jo035829l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flack H. D. On Enantiomorph-Polarity Estimation. Acta Crystallogr. 1983, A39, 876–881. 10.1107/S0108767383001762. [DOI] [Google Scholar]
- McKennon M. J.; Meyers A. I.; Drauz K.; Schwarm M. A convenient reduction of amino acids and their derivatives. J. Org. Chem. 1993, 58, 3568–3571. 10.1021/jo00065a020. [DOI] [Google Scholar]
- Bhaskar Kanth J. V.; Perisasamy M. Selective reduction of carboxylic acids into alcohols using sodium borohydride and iodine. J. Org. Chem. 1991, 56, 5964–5965. 10.1021/jo00020a052. [DOI] [Google Scholar]
- Liu Z.; Byun H.-S.; Bittmann R. Asymmetric Synthesis of D-ribo-Phytosphingosine from 1-Tetradecyne and (4-Methoxyphenoxy)acetaldehyde. J. Org. Chem. 2010, 75, 4356–4364. 10.1021/jo100707d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquette L. A.; Tae J.; Gallucci J. C. Synthesis and Crystal Structure of a Unique Linear Homoditopic Ligand Bifacially Complexed to Lithium Picrate. Org. Lett. 2000, 2, 143–146. 10.1021/ol9903436. [DOI] [PubMed] [Google Scholar]
- Ramachandran P. V.; Chandra J. B.; Reddy M. V. R. Stereoselective Syntheses of (+)-Goniodiol, (-)-8-Epigoniodiol, and (+)-9-Deoxygonipypyrone via Alkoxyallylboration and Ring-Closing Metathesis. J. Org. Chem. 2002, 67, 7547–7550. 10.1021/jo0259358. [DOI] [PubMed] [Google Scholar]
- Déziel R.; Favreau D. Synthesis of 1-b-Methylcarbapenem Key Intermediates Involving the Labile Acyl Auxiliary 4,4-Dimethyl-1,3-oxazolidine-2-thione. Tetrahedron Lett. 1989, 30, 1345–1348. 10.1016/S0040-4039(00)99461-9. [DOI] [Google Scholar]
- Jordan M. W.An (R)-4-Phenyl-1,3-oxazolidine-2-Thione Mediated Approach to 2, 3-Disubstituted Oxetanes and the Key Hydroxyethyl Isostere of the HIV Protease Inhibitor Darunavir. Preparation of Non-Evans Syn-Glycolate Aldol Addition Products. A New Direction for the Curtius Rearrangement in the Dehomologation of Alpha-alkoxycarboxylic Acids M.S. Thesis; Illinois State University: Normal, IL, USA, 2023. [Google Scholar]
- Ayim E.The Development of an Effective Synthetic Pathway to the HIV Protease Inhibitor Darunavir and Its Structural Derivatives for Sar Studies via an Asymmetric Glycolate Aldol Addition Reaction Approach M.S. Thesis; Illinois State University: Normal, IL, USA, 2023. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.