

Summary
Mycena s.s. is a ubiquitous mushroom genus whose members degrade multiple dead plant substrates and opportunistically invade living plant roots. Having sequenced the nuclear genomes of 24 Mycena species, we find them to defy the expected patterns for fungi based on both their traditionally perceived saprotrophic ecology and substrate specializations. Mycena displayed massive genome expansions overall affecting all gene families, driven by novel gene family emergence, gene duplications, enlarged secretomes encoding polysaccharide degradation enzymes, transposable element (TE) proliferation, and horizontal gene transfers. Mainly due to TE proliferation, Arctic Mycena species display genomes of up to 502 Mbp (2–8× the temperate Mycena), the largest among mushroom-forming Agaricomycetes, indicating a possible evolutionary convergence to genomic expansions sometimes seen in Arctic plants.
Overall, Mycena show highly unusual, varied mosaic-like genomic structures adaptable to multiple lifestyles, providing genomic illustration for the growing realization that fungal niche adaptations can be far more fluid than traditionally believed.
Keywords: fungal genomics, fungal guild, genome size diversity, saprotrophs, Arctic biology, plant-fungus interactions, TE proliferation, biotrophy–saprotrophy evolution, root-associations, carbon degradation
Graphical abstract
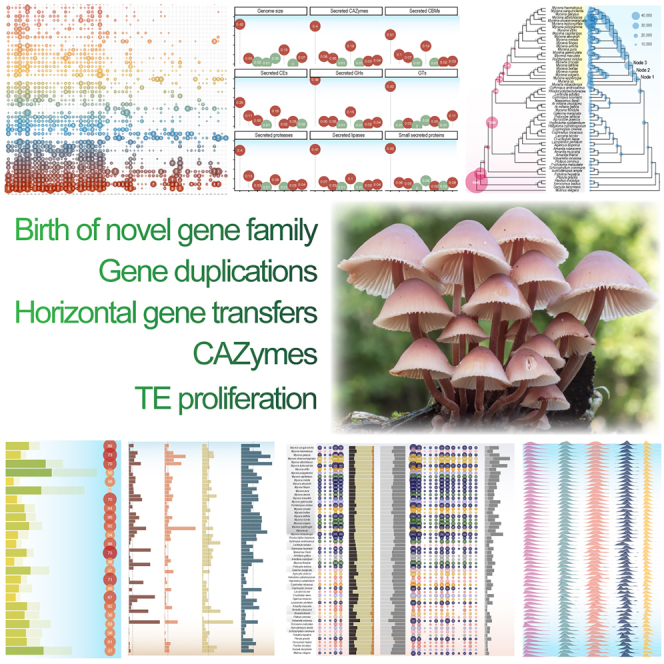
Highlights
-
•
Mycena have the largest mushroom genomes hitherto found
-
•
Caused by TE proliferation, novel gene families, duplications, and enlarged secretomes
-
•
Their genome structures are fit for multiple lifestyles
-
•
Arctic Mycena strain genomes are particularly large, mimicking Arctic plant genomes
Mycena genome sizes and structure do not follow the expectations from their supposed saprotrophic ecological specializations. Instead, they are overall highly expanded in size in all aspects with multiple genes enabling them for different lifestyles. Certain Arctic Mycena strains have the largest mushroom genomes hitherto found.
Introduction
Among Agaricomycetes, a wide range of genomic architectures has been identified for saprotrophic and biotrophic species.1 Ancestral clades of this class most likely had a genome resembling that of extant wood decayers2,3 harboring a broad battery of plant cell-wall-degrading enzymes (PCWDEs) for decomposing plant biomass. White rot fungi later evolved via the emergence of lignin-degrading capabilities, mostly attributable to the expansion of class-II peroxidases (PODs).3,4,5 Brown rot decayers evolved mostly from within white rot lineages and have lost genes for metabolizing lignin, while most biotrophs (ectomycorrhizal, lichenized, or parasitic), which are mainly reliant on their hosts for obtaining carbon, have a greatly reduced repertoire of PCWDEs but have in return evolved other strategies that facilitate symbiosis with plant hosts (e.g., control of plant immunity by secreted effector proteins).6,7,8,9 The genomes of biotrophic species, such as rust fungi and ectomycorrhizal symbionts, often contain abundant transposable elements (TEs), leading to an increased genome size.10 Litter-decaying saprotrophs share enzymatic mechanisms with white rot wood decayers11,12 However, a recent detailed comparison of litter and soil decayers with white and brown rot decayers13 showed that they have different gene repertoires related to hemicellulose and lignin degradation. Overall, they presented high diversity of PCWDEs, suggesting a high degree of functional versatility. The observed genomic idiosyncrasies within Agaricomycetes are consistent with other biological features showing that several fungal species and genera transgress their known ecological boundaries14,15,16,17 and suggesting that fungal ecological niches may be far more fluid than the traditional categories suggested.18 Although the genomic blueprints of several fungal lifestyles have been characterized, there are ecologically important clades whose genomic landscapes remain uncharted.
The genus Mycena sensu stricto19 includes species with diverse lifestyles. It is one of the largest genera in the Agaricales with >500 known species and is traditionally believed to contain major litter decomposers or late-phase degraders of already softened wood, with various degrees of specialization on grass, conifer, or broadleaf litter/wood substrates.20,21,22,23 They are common across all climate zones, including polar regions,24,25,26,27 and biotrophic interactions have been reported in several species of the genus. Some species are orchid mycorrhizal symbionts,28,29 endophytes,30,31,32 mycorrhiza-like partners, and parasites.16,33 Several Mycena species have also been documented in the roots of multiple plant host species in temperate and Arctic regions.34,35,36,37 Nonetheless, stable isotopic data37 suggest that the predominant nutritional mode among most Mycena species is saprotrophy.
Despite their importance in terrestrial habitats, the genomic resources for Mycena species are scarce. A previous study described the genomes of five Mycena species, with a focus on genome methylation and bioluminescence-related pathways,38 and reported large (+100 Mbp) genomic assemblies for the sequenced Mycena species. However, overall genomic trends and possible correlations between gene content and lifestyle, particularly their secretome,39 remained to be explored.
In the present study, we sequenced the genomes of 24 Mycena s.s. species and Atheniella floridula, formerly named Mycena s.l.19 (i.e., 25 new genomes). Ecologically, they represent six broad decayer categories: wood generalists, broadleaf wood decayers, grass litter generalists, broadleaf litter decayers, coniferous litter decayers, and overall litter generalists. Three of our genomes came from Arctic collections. In our comparative analysis, we also included the published Mycena galopus genome10 and 33 additional Agaricomycetes genomes from wood/litter decayers, ectomycorrhizal symbionts, and parasites2,8,9,10,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54 for a total of 58 fungal genomes included in the analyses. Thus, whenever we analyze Mycena vs. non-Mycena in our comparative analyses, the 25 Mycena genomes refer to those belonging to Mycena s.s. (i.e., our 24 new Mycena s.s. genomes plus the already published M. galopus). Conversely, our newly sequenced Atheniella (Mycena) floridula genome is analyzed with the 33 non-Mycena genomes.
We aimed to understand (1) do Mycena species differ in their PCWDE repertoire according to their wide ecological niche and substrate preferences? (2) What factors—genome duplications, TE proliferations, novel genes, secretome genes, or others—are key in driving genomic enlargement and evolution in this group, and in specific species? (3) Could particular genome-increasing factors such as TE proliferation be linked to their biotrophic ability and/or adaptation to oligotrophic environments, as described for other organisms55,56
Results
Mycena genomes, especially of the Arctic strains, represent the largest mushroom genomes known
We sequenced 24 new genomes using long-read technology supplemented with RNA sequencing (RNA-seq) for supporting gene prediction. For a summary of genomic features, see Table S1. The Benchmarking Universal Single-Copy Orthologs (BUSCO) values of all genomes were above 90%, suggesting that the genome assemblies captured most of the conserved, single-copy genes commonly present in fungi (Figure 1). The quality of the Mycena genome assemblies varied, as the number of scaffolds ranged from 118 for Mycena capillaripes to 4,647 for Mycena leptocephala (Figure 1), with an average of 1,187 scaffolds. We found that Mycena species had significantly larger genomes than all other non-Mycena taxa represented in our study (two-sample permutation test with 10,000 permutations, p < 0.0001); the average genome size and the number of genes of Mycena are approximately 153.9 Mb and 38,739, respectively, whereas those of the non-Mycena genomes analyzed here were 59.8 Mb and 17,791, respectively (Figure 1). The Mycena species had the highest number of predicted genes across all analyzed fungi, with up to 75,904 and 97,030 predicted protein-coding genes in M. leptocephala and Mycena olivaceomarginata, respectively (Figure 1). These high gene numbers were obtained after accounting for potential allelic differences in the heterokaryons. We included Atheniella (Mycena) floridula27 as a representative of this genus of former members of Mycena s.l. As it is now regarded as a member of Porotheleaceae57 or Cyphellaceae,58 which are considered to be related to "marasmioid" fungi, its phylogenetic placement here as more closely related to Armillaria (Physalacriaceae) than to Marasmius irrespective of phylogenetic method (Figures 1 and S1) and could merit further investigation by systematic mycologists.
Figure 1.

Genomic features and statistics for 58 Agaricomycete species
Fungal lifestyle in color. The median values are indicated by dotted lines. Genome, genome size; TE content, coverage of TE in the genome; genes, number of genes; secreted, number of predicted SSPs (see STAR Methods). Scaffolds, number of scaffolds; L50, N50 length; BUSCO, genome completeness. Further details are provided in Table S6.
We found no significant differences in genome size due to the degree of specialization in their mode of nutrition among narrow specialists, broader specialists, and generalists in Mycena (Kruskal-Wallis test, p = 0.126) (Figure S2; Table S2). However, the differences in genome size between the three species sampled in the Arctic and the 22 non-Arctic Mycena genomes were significant when doing two-sample permutation test with 10,000 permutations to correct for the skewedness in sample sizes (p = 0.0028). This was true also for the differences in TE content (p = 0.0005), total CAZyme gene copy number (p = 0.0095), and small secreted protein (SSP) gene copy number (p = 0.0087), which are all tightly correlated to overall genome size. The Arctic-temperate Mycena differences were also robust when comparing the three Arctic species with only the 14 temperate Mycena generalist litter decayers for both genome sizes and TE contents, excluding specialists with smaller genomes (p = 0.0015) and TE content (0.006). To explain this unprecedented genome expansion, we next evaluated the potential role of whole-genome duplications (WGDs), loss of genome surveillance systems, correlation with lifestyle, origins of novel and duplicated genes, TE bursts, and proximity to and overlapping with coding genes of TEs.
TE proliferation is particularly high in Mycena genomes
A global view of TE coverage in the 58 fungi showed that the genome coverage of TE was higher in Mycenaceae, Omphalotaceae, and Physalacriaceae than in other fungal families, except for ectomycorrhizal fungi (Figure 2), indicating that TE expansion occurred in specific lineages. The nuclear genome size showed similar trends to the number of TE copies (Figure 1). Mycena species had significantly more repetitive elements than other Agaricomycetes (Welch two-sample t test, p = 0.001), with on average 30%–50% of their genomes made up of TEs, whereas, in other Agaricomycetes, this was 10%–25% (except certain mycorrhizal species). In particular, the coverage of DNA transposons significantly contributed to genome size (15% variance explained; permutational multivariate analysis of variance [PERMANOVA] p < 0.05; see gene size in Figure 2). More specifically, long interspersed nuclear elements (LINEs) were abundant in Mycena species and in a few non-Mycena fungi (Figure 2). Long terminal repeat (LTR) transposons tended to be evenly distributed among the 58 fungi. DNA transposons and unclassified repeats were also more frequent in the Mycena species. The coverage of LTR retrotransposons and DNA transposons influenced fungal separation (Figure S3A). Notably, Arctic Mycena species possessed a higher content of DNA transposons (Figure S3B).
Figure 2.

Distribution of TEs in the 58 genomes
LTR, long terminal repeat retrotransposons; non-LTR, non-long terminal repeat retrotransposons; DNA, DNA transposons; LINE, long interspersed nuclear element; unknown, unclassified repeated sequences. The bubble size is proportional to the coverage of each TE (shown inside the bubbles). The right bars show the total coverage per genome. Fungal ecology is indicated by color codes. See Tables S1 and S7.
We examined the age of TE insertions in the genomes of 58 fungi (Figure S4). Overall, the 25 Mycena species showed a sharply left-skewed distribution compared to the rest of the fungi, suggesting that Mycena species contain younger TEs (more recently inserted) than the other species. Notably, we observed no strong patterns in TE proportions between the different Mycena ecological groups (Figure 2).
TE proliferation in proximity to transcription factors
We then determined TE families in proximity to these genes (Figures S5 and S6). Mycena species contained a large number of DNA transposons near the end and within the gene space (Figures S7 and S8). The TE-gene distances of the Mycena species were significantly shorter than those of the non-Mycena group (Figures 3A and S9). We further investigated TEs inserted into the gene-coding space (TE-overlapping genes). We identified that the selected TE-overlapping genes were transcribed in Mycena (Figure S10).
Figure 3.

DNA transposons near the ends and within the gene spaces
(A) Short gene-repeat distances of Mycena. Mycena species were compared to non-Mycena fungi based on estimated mean distances. Cold-adapted Mycena species (n = 3, in blue), other Mycena species (n = 22, in red), and the remaining fungi (n = 33, in green). Different letters represent significantly different groups (Dunn test; FDR-adjusted p < 0.05).
(B) Selected genes containing repeats among Mycena fungi. A high number of repeat elements overlapping genes present in Mycena fungi were selected. The following TE families were included in the analysis: DNA transposons (DNA), retrotransposons with long terminal repeats (LTR), retrotransposons with long interspersed nuclear elements (LINEs), retrotransposons with short interspersed nuclear elements (SINEs), rolling-circle/helitron transposons (RC), and unclassified repeats (unknown). See Table S3.
Mycena species might have developed species-specific transcriptional regulation networks. The fungi showed transcribed genes harboring repeated sequences (Figures S10, S11, and S12). Most of them are not classified as TEs (Figures S7B and S8B). Such unclassified repeats in the genes possibly function as transcription factor binding sites for signal transduction and gene regulations (Figure S12C). Also, we found no obvious evolutionary pattern in the unclassified repeats (Figure S13).
Carboxylesterase-related protein (GGGX) is a secreted lipase with repeat sequences. Some genes coding for zinc-dependent DNA-binding sites (Zn(2)-C6 fungal-type DNA-binding domain), tetratricopeptide repeats, which mediate protein-protein interactions in cytoskeleton, and repeats involved in carbohydrate transport and metabolism (major facilitator superfamily; Figures 3B and S10; Table S3). The cold-adapted species M. olivaceomarginata, M. leptocephala, and Mycena sp. 59 distinguished themselves by containing a particularly high number of basic-leucine zipper domains acting as a transcription regulator (Figures 3B and S10; Table S3).
Rampant emergence of de novo gene families in Mycena species
To assess the contribution of novel genes arising from gene duplications, de novo gene birth, and/or segmental or WGD in Mycena species, we characterized the predicted proteomes in the 58 genomes. First, we found that Mycena species differ from other Agaricomycetes not only in gene content but also in their much lower proportion of protein-coding genes with a predicted function (Figure 4A) (i.e., Mycena species have a disproportionately lower percentage of genes with known Pfam/InterPro domains). In species with large genomes (e.g., M. leptocephala and M. olivaceomarginata), >70% of the genes had no known conserved domains. This suggests that the expansion of the protein-coding gene repertoire in Mycena is mostly related to the recently evolved genes with unknown functions.
Figure 4.

Patterns of gene repertoire expansion in Mycena
(A) Relationship between genome size and number of genes containing known conserved protein motifs (InterPro terms).
(B) Origins of novel gene families (left) across 58 species.
(C) Differences in singleton genes between Mycena and non-Mycena species (Welch two-sample t test).
(D) Patterns of gene duplication (right) across the 58 species.
Similarity-based analyses reveal a burst of gene family origin in Mycena
Genes without known Pfam/InterPro domains can result from the birth of novel gene families44 and/or spurious gene predictions. To distinguish between these two scenarios, the predicted proteins were grouped into similarity-based clusters using Markov clustering. If the unannotated genes have prediction errors, we expect no similarity among their sequences; therefore, they should not form large clusters or gene families. In contrast, most genes with no known function formed clusters that not only harbored multiple genes per species but were also conserved across multiple or all Mycena species (Table S4). These results suggested that the propensity of genes with unknown functions in Mycena can be explained by the evolution of novel, functional, and conserved gene families in this genus. We next analyzed gene family origins in our Agaricomycetes dataset. Figure 4B shows the locations of the gene family origins along the phylogenetic tree of Agaricomycetes. Most gene families originated early in the evolution, consistent with their conservation across Agaricomycetes. However, we observed an unusually high number of novel gene families in three basal nodes of Mycena species having 1,719, 849, and 1,845 novel gene families, 10–200 times more than in other genera included in the dataset (Figure 4C). This clearly indicates that most genes with unknown functions form gene families that are conserved across the genus Mycena, excluding the possibility of annotation errors and suggesting that these are de novo gene families that originated primarily in three of the basal nodes of the Mycena genus.
We also noted that, in addition to conserved genes with unknown functions, Mycena genomes harbored a significantly higher copy number of orphan genes (defined as having no homologs with detectable similarity) than non-Mycena Agaricomycetes (p = 0.00019, Welch two-sample t test; Figure 4C). This further supports the propensity for de novo gene births in Mycena. The number of orphan genes ranges from 2,534 to 16,570. About 45% to 75% of such genes showed the presence of unique transcripts (see Figure S14). We found the number of orphan genes were significantly correlated with the genome statistics (p < 0.05; Figure S15). The genome size was strongly correlated with such genes (R = 0.92), suggesting the birth of species-specific genes is associated with the genome enlargement.
N50 and BUSCO scores were also correlated (R = 0.79 and −0.51), indicating there were tendencies of (1) a larger number of orphan genes leading to higher N50 (i.e., many fragmented scaffolds) and (2) a smaller number of orphan genes in genomes giving higher BUSCO scores.
However, the BUSCO score of all Mycena genomes was high (>94%). The difference in BUSCO is only 5% between the highest score with fewest orphan genes (99%; Mycena sanguinolenta) and lowest scores with a largest number of orphan genes (94%; Mycena haematopus).
To assess the function of novel gene families, we identified MCL (Markov Cluster Algorithm) clusters in which Mycena species were overrepresented (Fisher’s exact test) and functionally characterized them based on InterPro conserved domains. The results of the enrichment analysis support a rather unspecific expansion with no clear ecological function among the Mycena-expanded families. Clusters of genes with unknown functions were common among the Mycena-enriched clusters (green rows in Table S4). Nevertheless, some functionally relevant genes were identified among Mycena-enriched clusters. For example, these overrepresented clusters included a class-II-peroxidase-like family (cluster 134), which could be related to wood decay, multiple kinesin clusters (related to intracellular transport), and a cluster containing germins (cluster 375), which are plant glycoproteins related to the extracellular matrix, mostly functioning in development and defense. We also detected a cluster of AA10 lytic polysaccharide monooxygenases (LPMOs) (cluster 8,299), enzymes that primarily act on chitin (or cellulose). Because germins and AA10 LPMO are not widespread in fungi, it is possible that they were obtained from Mycena species through horizontal gene transfer (HGT), similar to that reported in Armillaria.59 Indeed, Alien Index60 calculations revealed that Mycena species possess 129–836 genes that showed considerably higher similarity to Ascomycota proteins across their lengths (80% bidirectional coverage) (Table S5), indicative of potential HGT events. To more rigorously test the possibility of HGT, we created clusters from the top 50 Ascomycota, Mycena, and non-Mycena hits of each potentially HGT-derived protein and inferred gene trees for them. Next we filtered for Mycena proteins for which the phylogeny supports an origin via HGT (see STAR Methods). This resulted in an overall 2,562 phylogenetically verified HGT-derived Mycena genes (31–263 genes/species; Table S5). Most proteins encoded by HGT-derived genes show the highest similarity to proteins from the Dothideomycetes (811), Sordariomycetes (752), Eurotiomycetes (583), and Leotiomycetes (494) (Table S5), suggesting that the donor taxon or taxa probably belonged to these classes. InterPro domain enrichment analysis (Fisher exact test, Benjamini-Hochberg-adjusted p ≤ 1e−3) for HGT-derived genes/proteins showed that Mycena species acquired several transporters, CAZymes, and proteases from Ascomycota (Table S5), some of them being present in >50% of Mycena species. A notable protein encoded by an HGT-derived gene is present in 11 Mycena species and harbors a necrosis-inducing protein domain (IPR008701) that is reported to be responsible for necrosis induction on host plants by biotrophic pathogenic fungi.61 It is noteworthy that eight of these 11 species that harbor this necrosis-inducing HGT-derived protein are also found in the category of Mycena cultures that induced poor growth in the birch hosts they invaded.16
Gene duplications are rampant in the Mycena
We then reconstructed gene duplication/loss histories using reconciled gene trees as described previously62 to obtain a fully resolved view of gene family evolution in Mycena and related Agaricomycetes. The protein-coding gene repertoire and its evolutionary dynamics in the 58 species showed a clear peak in the Mycena clade (Figures 4D and S16). This finding indicates that gene duplication also contributes to the inflation of the proteome of Mycena species. The expansion event started with 23,591 reconstructed ancestral genes in the last common ancestor of the marasmioid clade (sensu Matheny 200663 marked with Armillaria, Gymnopus, and Mycena, as shown in Figure 4D) and expanded to 29,195 genes in the last common ancestor of the Mycena clade (8,107 gains and 2,503 losses). In the following two nodes (#2 and # 3 in Figure 4D), the genome was further expanded to 33,542 (4,521 gains and 174 losses) and 42,713 genes (9,701 gains and 380 losses), respectively (Figure S16), which was three to four times greater than the number of other Agaricomycetes genomes in the dataset. It is noteworthy that ancestral genomes expanded by 81% across these three nodes, which raises the possibility that we see signals of an ancestral WGD spread across these nodes (see below). Ancestral gene counts remained approximately at this level in the Mycena clade, with gene numbers fluctuating between 42,000 and 45,000 genes in the Mycena clade. We also inferred a large number of more recent gene duplication events that contributed to the further expansion of certain exceptionally large Mycena genomes.
No clear-cut evidence for WGDs
It is perplexing that the expansion of protein-coding gene content localizes to only three nodes in the genus Mycena. Therefore, we examined the possibility that the genome expansions were related to an ancient WGD event and subsequent perturbed genome dynamics. Analyses of dS (synonymous substitution) values between gene duplicates revealed a second peak of gene propensity around dS ∼ 2 both in highly duplicated (M. olivaceomarginata, M. leptocephala, Mycena galericulata, Mycena polygramma, and Mycena sp.) and not highly duplicated (Mycena pura and Mycena belliae) (Figure S17); however, this signal was weaker than in two other cases of documented fungal WGD events64,65 and was comparable to the pattern in Schizophyllum commune, but not Laccaria bicolor (Figure S17). The second peak may correspond to the signal of WGD,66 with its lower height compared to Saccharomyces cerevisiae and Phycomyces blakesleeanus likely being abundant small-scale duplications following the potential WGD event. However, in light of all comparisons, we interpret these patterns as inconclusive for the presence of an ancient WGD in Mycena, requiring further research with other methods in the future.
The loss of genomic control mechanisms cannot explain the genome expansion in Mycena
We hypothesized that Mycena genomes may have expanded following the potential loss of genes related to the control mechanisms that could maintain TE and/or gene proliferation. To this end, we examined the presence of genes involved in the DNA methylation (TE silencing) machinery and RNA-mediated silencing (quelling and meiotic silencing). Based on an orthology analysis using gene sequences from the well-characterized model Neurospora crassa as queries, we found that each of the genes appeared to be conserved throughout the phylogeny of the 58 Mycena and non-Mycena species (Table S6). The sole exception was sad-1, an RNA-dependent RNA polymerase, the key component of the meiotic silencing by unpaired DNA (MSUD) pathway (NCU02178 in Neurospora crassa), which was present in 24 out of 25 Mycena species and was missing in the other examined Agaricomycetes (except for two Armillaria species). Thus, Mycena species appear to retain nearly all genes related to the maintenance of genomic integrity, suggesting that its genome expansion cannot be explained by the loss of known components of the machinery that suppress TE activity and/or proliferation.
Large variation in CAZyme production and secretome irrespective of ecological specialization
Although comparative genomic analyses suggested nonspecific genome expansion in Mycena, we assessed whether the known ecological versatility of the genus correlates with genes playing a role in organic matter decomposition and/or plant interactions. Hereafter, we will focus on the secretome, including CAZymes, in particular, PCWDE, lipases, proteases, and SSPs (Figures 5, S18, S19, and S20). The predicted secretome of the Mycena species was considerably larger than that of other fungi, regardless of their ecology (Figure 5). These differences correlated with phylogeny, and PERMANOVA analyses suggested that approximately 50%–55% of the observed variance was explained by the phylogenetic relationships for secreted CAZymes, proteases, lipases, and SSP (Figure S21).
Figure 5.

Secretomic profiles of 58 fungi
Bubble plots (left) show the number of secreted genes for CAZymes, lipases, proteases, and other proteins that were not present in the first three groups. SSPs are a subcategory showing the number of small secreted proteins (<300 amino acids). The size of the bubbles corresponds to the number of genes. Fungal lifestyle is indicated by colors. The first bar plots (in the middle) represent the ratio of CAZymes, lipases, and proteases to all secreted proteins (left), and the ratio of SSPs in the entire secretome (right). The second bubble plot (on the right) shows CAZymes grouped according to their functions, including PCWDEs and fungal cell-wall-degrading enzymes (FCWDEs); peptidoglycan (i.e., bacterial membrane)-degrading enzymes (BMDEs); trehalose-, starch-, glycogen-degrading enzymes (storage); lytic polysaccharide monooxygenase (LPMO); substrate-specific enzymes for cellulose, hemicellulose, lignin, and pectin (plant cell walls); and chitin, glucan, and mannan (fungal cell walls). The second bar plots (far right) show the total number of genes, including PCWDE, microbial cell wall degrading enzymes (MCWDEs) and BMDE (left), and the proportions of PCWDE, MCWDE, and BMDE (right)
Across all 58 species, ecology (i.e., the mode of nutrition) significantly contributed to the distribution of secreted CAZymes (ecology effect was 19%; PERMANOVA p < 0.05; Figure S21). For the various functional groups (auxiliary activities [AAs], carbohydrate-binding domains [CBMs], carbohydrate esterases [CEs], glycoside hydrolases [GHs], and polysaccharide lyases ]PLs]) within CAZymes, the effects of ecology on the distribution of each family ranged from 6% to 21%, but we note that most of this signal is provided by ectomycorrhizal (ECM) species vs. others, not separation within Mycena. Thus, our results confirmed that ectomycorrhizal fungi differed significantly from other fungal guilds, with a low content of CAZymes, SSPs, lipases, and proteases (false discovery rate [FDR]-adjusted p < 0.05; Kruskal-Wallis and Dunn tests; Figure S22). Among the six wood/litter decayer categories, only the conifer litter decayers and broadleaf wood decayers were significantly different from each other (FDR-adjusted p < 0.05; Kruskal-Wallis and Dunn tests), indicating that ecology has a low predictive value for extracellular CAZyme composition within Mycena species.
Mycena species displayed a high number of genes encoding enzymes involved in lignocellulose degradation (Figure 5), including those involved in decomposing all components of wood.67,68,69 We also identified CAZyme families that are mostly or exclusively found in Mycena spp. within Agaricomycetes (GH54, GH67, GH106, GH81, GH5_31, GH32, CBM42, GH5_51, and GH43_14; Figure S18). The conservation of these families in Mycena species, but lack thereof in other Agaricomycetes, also suggests a possibility that these species might have acquired these genes by HGT.
Ecology-wise analyses of Mycena CAZymes highlighted that genome size in general is strongly correlated with CAZyme copy number (Pearson coefficient = 0.835; p < 0.05), with PC1 heavily influenced by genome size and explaining 18%–49% of the variance (PERMANOVA p < 0.05; Table S7). For example, this was reflected in low CAZyme counts in the small-genome species M. belliae, Mycena crocata, or M. haematopus, and a high count in M. leptocephala, M. olivaceomarginata, M. galopus, and Mycena rebaudengoi, which have larger genomes. In the phylogenetic principal-component analysis (PCA) of secreted CAZyme counts (Figure 6), even as this method applies a correction for phylogenetic relatedness, the first PC still largely separated Mycena from non-Mycena, and explained 70.6% of the observed secreted CAZyme variation among the 58 fungi included in this study. The second PCA explained only 5.5% and mainly separated ECM fungi from saprotrophs, with no clear effect of ecological decay guilds. Thus, our findings indicate that substrate ecology has low predictive power for CAZyme gene content. The same pattern was also broadly observed for proteases, lipases, and SSPs (Figures S23, S24, and S25). Similar to the CAZyme gene content, ecology also had a low explanatory power for enzyme activity (Figures S26 and S27). These observations are consistent with those above, reinforcing the view that secretome expansion in the Mycena genus has not been shaped by the species’ lifestyle/mode of nutrition. Among the Mycena species analyzed, the most frequent invaders of living plant roots were generalist litter degraders (Figure S28; Welch two-sample t test, p = 0.02). However, in the secretome analysis, we found no strong correlation between the number of plant species a Mycena species is known to regularly invade37 and the gene counts of effector-like SSPs or CAZymes, or with other genes and genomic features (Figure S29). All correlations were insignificant when excluding the two outlying genomes M. olivaceomarginata and M. leptocephala.
Figure 6.

Phylogenetic PCA of secreted CAZyme profiles
All 58 species were placed according to the first and second principal components calculated by the count of secreted CAZymes, with phylogenetic distances of the species taken into account. CAZymes with high loadings are shown in red with arrows. Fungal ecology is in color, and Mycena and non-Mycena fungi are in different shapes. The blue circle shows a cluster of Mycena species.
Discussion
The new data presented here complement genomic resources of plant biomass-degrading Agaricomycetes, an ecologically important group of saprobes that recently proved considerably more heterogeneous than traditional views assumed.3,43,47,50 The all-encompassing genome-wide increased size of Mycena spp. both in terms of assembly size and number of genes encoded, and particularly the large number of apparently Mycena-specific gene families, and the multiple instances of HGTs make Mycena genomes quite deviant in their structure from the other Agaricomycetes irrespective of their ecologies.
Our analysis of the secretome of Mycena spp., with its special emphasis on PCWDEs and its connection to species lifestyles, showed that Mycena species have a large repertoire of CAZyme genes that encode enzymes that act on all lignocellulosic components of plant cell walls, with similarities to other litter degraders, as recently outlined.13 Their genomes also encode a large set of genes encoding lipases and proteases. These copy numbers are consistent with the genome expansion of the genus and indicate that these fungi have robust machinery to decay organic matter. Nevertheless, the gene copy numbers of CAZymes were not related to the ecology of the investigated Mycena species. These observations could be explained by a number of factors that are not mutually exclusive: (1) the confounding effects of genome expansion may blur true ecology-related signals, (2) lifestyle may act through the transcriptional regulation of these CAZyme genes, or (3) bias and subjectivity in the traditional textbook assignment of species to discrete lifestyles.
Similarly, compared to other Basidiomycota70 species, Mycena also showed high interspecific variability in the production of degrading enzymes, yet again without obvious links to known substrate preferences. The differences in exocellulase and beta-glucosidase activity activities are surprising, as all Mycena would be expected to utilize cellulose as the main plant polymer, regardless of substrate preference. The secreted CAZyme gene content does not correspond to higher enzyme activity either, as would be expected.71 Biochemically, different CAZyme family members encode enzymes with the same activity.72 Conversely, individual enzymes that are products of the same gene may catalyze several different biochemical activities.73 Overall, Mycena has a wide genomic potential to decompose lignocellulose and other biopolymers, and the variation in enzyme activity in Mycena cultures may indicate the ability to efficiently regulate expression and adapt it to environmental conditions.74 Nonetheless, the answer to our first research question (1) must be negative, i.e., that even though species do differ in their PCWDE repertoire, their supposed ecological niches and substrate preferences cannot be identified as key determining factors.
Turning to our research question (2) (what are the main causes of genome expansions?), other parts of the protein-coding repertoire of Mycena species have also expanded notably. The >4,000 novel conserved Mycena-specific gene families (and the large number of gene duplications) with unknown functions is very striking. Such families are known from other fungi, but (with the possible exception of rust fungi75,76) usually orders of magnitude smaller, as in the 400 such families described in the genus Armillaria.42 Assessing the specific function of Mycena-specific proteins, in the context of ecological adaptation or other, will hopefully cast some light on the role of unannotated genes in fungi. So far, their conservation across the Mycena clade suggests that their role is important.
Furthermore, our data are consistent with an ancestral WGD event in Mycena in the observed genome expansion patterns, the very high number of genes, a large number of gene duplications that affected all gene groups equally, and the rapid expansion of the ancestral genome after the origin of the genus. Unfortunately, however, the confounding effects of likely abundant subsequent small-scale duplications as also suggested by Figure S17 makes it very hard to discern such a potential WGD event conclusively.
The major quantitative cause of the Mycena genome expansion was TE proliferation. This is known to promote both gene duplications and the emergence of new gene families, in addition to increasing transcription rates.77The proliferation of TEs and gene duplications is a common driving force of genome expansion reported in other fungal groups.9,10,38,42 TEs are a key drivers of rapid shifts in eukaryotic genome size.78 In Mycena, up to 60% of the genomes were composed of TEs, similar to the prevalence reported in some highly repetitive genomes of ectomycorrhizal species.9,10 Thus, the answer to research question (2) is that the main reasons for genome expansions is (1) increases in coding regions containing the secretomes and PCWDEs, (2) a huge set of novel gene families, and (3) multiple duplications of existing genes, and particularly an intense TE proliferation—perhaps all on a background of an ancestral WGD event.
In plant-parasitic/associated fungi, genes overlapping or associated with TEs that are enriched are often direct virulence factors and effector genes (e.g., SSPs).10,79 In Mycena, the enriched genes with overlapping TEs (C66 fungal-type DNA-binding domains, tetratricopeptide repeats, the major facilitator superfamily, or the basic-leucine zipper domain) instead mainly have functions as transcription regulation mechanisms,80,81,82 perhaps suggesting a more general adaptive difference in gene regulation/transcriptomics in Mycena in addition to their genomic peculiarities. TE sequences in genes could bind with non-coding RNAs, which create complex regulatory networks of epigenetic control.83 It could be imagined that TE insertions accumulated in non-coding regions became novel DNA sequences with repeated sequences acting as molecule-binding sites, which were subsequently promoted to new exons over time. In this way, Mycena fungi would incorporate TEs in the genomes, building up efficient regulatory networks that add to their ecological plasticity.
The basic-leucine zipper domain transcription factors (bZIPs) are particularly pronounced in the TEs of the three cold-adapted strains collected in the Arctic (Svalbard). In filamentous fungi, the bZIP factors are known to be key oxidative, temperature-related, and cell wall integrity stress response factors.84
Thus, this might suggest that adaptations to stressful, nutrient-poor environments are a potential driving force for TE-mediated genome enlargement, which helps answer our research question (3) on adaptive roles of TE proliferations. Arctic fungi must survive a short growing season, with extraordinary environmental stress from frequent exposure to freezing temperatures and associated hyphal damage. TE proliferation coupled with genes for rebuilding cell walls and damaged hyphae is one possible evolutionarily advantageous explanation for the particularly large genome increases observed in Arctic Mycena. It should be noted that, while major facilitator superfamily expansion is known to be a general feature on the kingdom level separating non-flagellated fungi from the ophistokonts,85 TE proliferation has happened multiple times independently throughout the fungal kingdom and appears to be generally non-adaptive, with specific instances of adaptive functions and TE proximity mainly appearing to be taxon specific.86,87 Thus, it should be investigated if a putative adaptive link between TEs and bZIP transcription factors in Arctic Mycena is more of a specific response by one particularly versatile genus with flexible genomes than a general Arctic effect on Basidiomycetes and mushrooms more widely.
However, irrespective of the possible adaptive explanations, TE proliferation is the main reason that two of the Arctic strains, M. olivaceomarginata and M. leptocephala, stand out with genomes of 501 and 377 Mb, respectively. This is a further TE proliferation on top of the already large and TE-rich genomes of all other Mycena, which makes those two strains the largest mushroom genomes hitherto found, two to three times larger than the largest of the non-Arctic Mycena genomes. This effects bears resemblance to that in several Arctic plants that have also been shown to evolutionarily advantageously increase in genome size both by associated TE proliferation and polyploidy in response to environmental stress.88,89 This is then often associated with speciation and reduction in effective populations.90,91 However, the Arctic Mycena show no signs of independent WGDs/polyploidy (Figure S17), and M. olivaceomarginata and M. leptocephala morphospecies are widely found in temperate regions, suggesting that their effective population sizes are large. It remains to be seen if temperate strains of M. olivaceomarginata and M. leptocephala would also have the extreme genome size of their Arctic counterparts. Recent research has found that fungi are much more prone to mutations and TE proliferations in the monokaryotic rather than the dikaryotic stage.92 A non-adaptive reason for the extreme Arctic genome sizes (and TE proliferations) might thus be that Arctic mushrooms simply experience a much longer fraction of their lifetime as monokaryons waiting for a compatible partner to land, compared to temperate mushrooms with larger population sizes. Both adaptive and non-adaptive Arctic effects on Mycena specifically and mushrooms more generally should be tested more directly in future research on Arctic mushroom genomes compared to close non-Arctic relatives.
While previous studies have suggested that narrower ecological specialization conversely often leads to reduced genome size and gene content,93,94 we found no such significant association between the level of specialization in Mycena and genome size, however. The exclusively reed-stalk specialist M. belliae (56 Mb) and the predominantly Fagus-wood decayer M. haematopus (63 Mb) indeed had the smallest genomes; however, the conifer litter specialists Mycena rosella (129 Mb) and Mycena vulgaris (193 Mb) both had larger genomes than the two most widespread broad generalists M. pura (97 Mb) and Mycena epipterygia (119 Mb). Similarly, we found no robust correlations between the number of plant species invaded37 and the gene counts of SSPs or CAZymes, or other genes and genomic features.
Finally, Ke et al.38 previously noted that certain gene groups at the origin of the mycenoid lineage had closer homologs in the Ascomycota instead of the expected Basidiomycota. We further corroborate this finding by our HGT prediction, which indicated the unexpected similarity of hundreds of Mycena proteins to those from Ascomycota. In a recent study in Armillaria,59 multiple horizontally transferred genes were shown to represent a wide variety of cellular functions, and, in Mycena, we similarly find transporters, signaling and membrane proteins, and metabolic CAZymes and proteases (Table S5). Collectively, these observations provide evidence for potential HGTs with raw material from Ascomycota sharing the same ecological habitats, similar to the recent observations in Armillaria.59 The occurrence of HGT and the presence of bioluminescence in both Mycena and Armillaria is a striking correlation, sparking speculations about complex gene flow patterns between these two genera and certain Ascomycota that should be explored in future additional phylogenetic analyses.
Limitations of the study
While our study does find the Arctic Mycena strains to display the biggest mushroom genomes found and a significant size difference compared to the temperate Mycena, we cannot directly prove that they are larger because of a specific Arctic effect. Nor can we hypothesize much as to such an effect in mushrooms more broadly. To do this, more Arctic fungal genomes need to be sequenced, and preferably in pairs of conspecific strains from species that occur both in the Arctic and in other climate zones.
Conclusions
We find that the genomes of the Mycena genus do not conform to other studied fungal lineages in terms of the effects of their traditionally perceived saprobic lifestyles, narrowness of specializations, or propensity to invade plant roots as obvious driving factors of their genomic content.
Instead, Mycena is chiefly characterized by an uncharacteristic overarching genome expansion on all parts of its genome, due to TEs, whole novel gene families, and gene duplications, with no loss of the genes maintaining genomic stability. Furthermore, the genus contains evidence of several HGTs from Ascomycetes, including proteins known to induce necrosis in plant hosts upon invasion by fungi. Several enriched genes with overlapping TEs are associated with general transcription regulation mechanisms, suggesting a corresponding adaptive difference in the gene regulation/transcriptomics of Mycena, which could be hypothesized to explain the surprising ecological plasticity of the genus.
More specifically, and in a convergent evolution with Arctic plants with huge genomes, Arctic Mycena strains contain the largest mushroom genome hitherto found with a genome size of up to 501 Mbp, two to three times the size of the already large non-Arctic Mycena strains. Unlike the frequent polyploid evolution in Arctic plants, the expansion in the Arctic Mycena is mainly driven by further TE proliferation added on top of the already TE-rich Mycena genomes, and several of those Arctic TE sequences are inserted into genes related to oxidative, temperature-related, and cell wall integrity stress response factors.
Our resources and data presented here provide a solid foundation for expanding our knowledge on genome dynamics, novel and duplicating gene families, secretome composition, PCWDE evolution in Agaricomycetes, and of adaptation to harsh Arctic/polar environments. With genomes seemingly fit for different lifestyles, Mycena species provide genomic illustrations for the growing realization that fungal niche adaptations may be much more complex and broader than traditionally believed.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Species names of whole fungal genomes used in this paper Mycena albidolilacea |
This paper | All identifiers represent GenBank accession numbers unless otherwise noted. JARIHO000000000 |
| Mycena alexandri | This paper | JARJCM000000000 |
| Mycena amicta | This paper | JARIHP000000000 |
| Mycena belliae | This paper | JARJCN000000000 |
| Mycena capillaripes | This paper | JARHKZ000000000 |
| Mycena crocata | This paper | JARJCP000000000 |
| Mycena epipterygia | This paper | JARJCQ000000000 |
| Mycena filopes | This paper | JARIHR000000000 |
| Mycena galopus | Miyuachi, S. et al. (2020)1 | WIQG00000000 |
| Mycena galericulata | This paper | JARKIA000000000 |
| Mycena haematopus | This paper | JARJCV000000000 |
| Mycena latifolia | This paper | JARJLE000000000 |
| Mycena leptocephala | This paper | JARJLF000000000 |
| Mycena maculata | This paper | JARJLG000000000 |
| Mycena metata | This paper | JARKIB000000000 |
| Mycena olivaceomarginata | This paper | JARKIC000000000 |
| Mycena polygramma | This paper | JARKID000000000 |
| Mycena pura | This paper | JARJCW000000000 |
| Mycena rebaudengoi | This paper | JARJCX000000000 |
| Mycena rosella | This paper | JARKIE000000000 |
| Mycena sanguinolenta | This paper | JARGYH000000000 |
| Mycena sp. CBHHK59 | This paper | JARGXW000000000 |
| Mycena vitilis | This paper | JARGYF000000000 |
| Mycena vulgaris | This paper | JARGYA000000000 |
| Roridomyces roridus | This paper | JARKIF000000000 |
| Rhodocollybia butyracea | Ruiz-Dueñas, F.J. et al.2 | JADNRY000000000 |
| Gymnopus androsaceus | Barbi, F. et_al.3 | VKGB00000000 |
| Lentinula edodes | Sakamoto, Y. et al.4 | JANVFS010000000 |
| Gymnopus luxurians | Kohler, A. et al.5 | JJNP00000000 |
| Marasmius fiardii | Miyuachi, S. et al.1 | WIOQ00000000 |
| Armillaria gallica | Sipos, G. et al.6 | NKEW00000000 |
| Armillaria cepistipes | Sipos_G. et al.6 | FTRY00000000 |
| Atheniella (Mycena) floridula (Fr.) | This_study | JARIHS000000000 |
| Psilocybe serbica | Fricke, J. et al.7 | Genome can be downloaded from the JGI portal: (https://genome.jgi.doe.gov/portal/Psiser1/download/Psiser1_AssemblyScaffolds_Repeatmasked.fasta.gz) |
| Galerina marginata | Riley_R. et al.8 | AYUM00000000 |
| Agrocybe praecox | Unpublished (1000 fungal genomes project) | Information about the project and accessing the data can be found at the JGI page (https://mycocosm.jgi.doe.gov/Agrpra2/Agrpra2.home.html) |
| Hebeloma cylindrosporum | Kohler, A. et al.5 | JMDQ00000000 |
| Hypholoma sublateritium | Kohler, A. et al.5 | JMSJ01000000 |
| Coprinellus micaceus | Varga, T. et al.9 | QPFP00000000 |
| Coprinopsis cinerea | Stajich, J.E. et al.10 | AACS00000000 |
| Laccaria bicolor | Martin, F. et al.11 | ABFE00000000 |
| Crucibulum laeve | Varga, T. et al.9 | QPFR00000000 |
| Agaricus bisporus | Morin, E et al.12 | AEOL00000000 |
| Lycoperdon perlatum | Unpublished (1000 fungal genomes project) | Information about the project and accessing the data can be found at the JGI page (https://mycocosm.jgi.doe.gov/Lycper1/Lycper1.home.html) |
| Amanita muscaria | Kohler_A et al.5 | JMDV00000000 |
| Amanita rubescens | Miyuachi_et_al.1 | WION00000000 |
| Amanita thiersii | Hess, J. et al.13 | JDWF00000000 |
| Pluteus cervinus | Varga, T. et al.9 | QPFM00000000 |
| Volvariella volvacea | Bao, D. et al.14 | AMXZ00000000 |
| Tricholoma matsutake | Miyuachi, S. et al.1 | WIUY00000000 |
| Auriculariopsis ampla | Almasi, E. et al.15 | VDMD00000000 |
| Schizophyllum commune | Ohm, R.A. et al.16 | ADMJ00000000 |
| Fistulina hepatica | Floudas, D. et al.17 | JYFI00000000 |
| Pterula gracilis | Varga, T. et al.9 | QPFO00000000 |
| Xerocomus badius | Miyuachi, S. et al.1 | WIUX00000000 |
| Paxillus involutus | Kohler, A. et al.5 | JOMD00000000 |
| Serpula lacrymans | Eastwood, D.C. et al.18 | AEQC00000000 |
| Mutinus elegans | Unpublished (1000 fungal genomes project) | Information about the project and accessing the data can be found at the JGI page (https://mycocosm.jgi.doe.gov/Mutel1/Mutel1.home.html) |
| Deposited data on sequencing platform, bases, reads, and NCBI SRA accession numbers | https://doi.org/10.6084/m9.figshare.25423936.v1 | |
| Critical commercial assays | ||
| Mycena sanguinolenta (Mycelial culture) | This paper | All new cultures for this paper are deposited in the Belgian culture collection of microorganisms BCCM/MUCL in Leuven (https://bccm.belspo.be/about-us/bccm-mucl) with the MUCL initial and the numeric identifier. MUCL (Mycothèque de l'Université Catholique de Louvain) is the specific fungal section of the microbial culture collections at Louvain). MUCL_056397 |
| Mycena haematopus (Mycelial culture) | This paper | MUCL_056386 |
| Atheniella (Mycena) floridula | This paper | MUCL_056381 |
| Mycena olivaceomarginata (Mycelial culture) | This paper | MUCL_056391 |
| Mycena albidolilacea (Mycelial culture) | This paper | MUCL_056374 |
| Mycena leptocephala (Mycelial culture) | This paper | MUCL_056387 |
| Mycena vitilis (Mycelial culture) | This paper | MUCL_056401 |
| Mycena polygramma (Mycelial culture) | This paper | MUCL_056373 |
| Mycena capillaripes (Mycelial culture) | This paper | MUCL_056404 |
| Mycena metata (Mycelial culture) | This paper | MUCL_056390 |
| Mycena alexandri (Mycelial culture) | This paper | MUCL_056408 |
| Mycena filopes (Mycelial culture) | This paper | MUCL_056380 |
| Mycena pura (Mycelial culture) | This paper | MUCL_056407 |
| Mycena amicta (Mycelial culture) | This paper | MUCL_056405 |
| Mycena maculata (Mycelial culture) | This paper | MUCL_056388 |
| Mycena galericulata (Mycelial culture) | This paper | MUCL_056383 |
| Roridomyces_roridus (Mycelial culture) | This paper | MUCL_056393 |
| Mycena crocata (Mycelial culture) | This paper | MUCL_056378 |
| Mycena belliae (Mycelial culture) | This paper | MUCL_056377 |
| Mycena latifolia (Mycelial culture) | This paper | MUCL_056406 |
| Mycena rosella (Mycelial culture) | This paper | MUCL_056394 |
| Mycena vulgaris (Mycelial culture) | This paper | MUCL_056402 |
| Mycena epipterygia (Mycelial culture) | This paper | MUCL_056372 |
| Mycena sp. CBHHK59 (Mycelial culture) | This paper | MUCL_056398 |
| Mycena rebaudengoi (Mycelial culture) | This paper | MUCL_056392 |
| Deposited data | ||
| PRINGO | Miyuachi et al.1 | https://github.com/ShingoMiyauchi/PRINGO |
| COMPARE | Nagy et al.19 | https://github.com/laszlognagy/COMPARE |
| MAFFT (version 7.4.07) | Katoh and Standley20 | https://mafft.cbrc.jp/alignment/software/ |
| Notung 2.9 | Darby et al.21 | https://bio.tools/notung |
| RAxML | Stamatakis22 | https://apolo-docs.readthedocs.io/en/latest/software/applications/raxml/raxml-8.2.12/index.html |
| IQ Tree | Nguyen et al.23 | http://www.iqtree.org/ |
| OrthoFinder | Emms and Kelly24 | https://github.com/davidemms/OrthoFinder |
| BUSCO | Simão et al.26 | https://busco.ezlab.org/ |
| JGI Mycocosm | Grigoriev et al.27 | https://mycocosm.jgi.doe.gov/mycocosm/home |
| RepeatScout | Smit et al.28 | https://github.com/mmcco/RepeatScout |
| RepeatModeler | Smit and Hubley29 | https://www.repeatmasker.org/RepeatModeler/ |
| TINGO | Morin et al.30 | https://github.com/ShingoMiyauchi/TINGO |
| ASTRAL | Emms and Kelly31 | https://github.com/smirarab/ASTRAL |
| Gblocks | Castresana32 | https://github.com/TGAC/earlham-galaxytools/tree/master/tools/gblocks |
| WGDdetector | Yang et al.33 | https://github.com/yongzhiyang2012/WGDdetector |
| Trinity | Grabherr et al.34 |
https://github.com/trinityrnaseq/trinity rnaseq/releases/tag/v2.8.6 |
| FALCON | Chin et al.35 | https://github.com/PacificBiosciences/pb-assembly |
| FastOrtho | Wattam et al.36 | http://enews.patricbrc.org/fastortho/ |
| PCAtools | Blighe and Al37 | https://www.bioconductor.org/packages/release/bioc/html/PCAtools.html |
| Vegan 2.5–4 | Oksanen et al.38 | https://github.com/vegandevs/vegan |
| DescTools | Signorell et al.39 | https://cran.r-project.org/web/packages/DescTools/ |
| ape | Paradis et al.40 | https://cran.r-project.org/web/packages/ape |
| phylobase | Hackathon41 | https://cran.r-project.org/web/packages/phylobase/ |
| adephylo | Jombart and Dray42 | https://cran.r-project.org/web/packages/adephylo/ |
| ggplot2 | Wickham et al.43 | https://cran.r-project.org/web/packages/ggplot2/ |
| regioneR | Gel44 | https://bioconductor.org/packages/release/bioc/html/regioneR |
| Synteny-Governed Overview | Looney et al.45 | https://github.com/ShingoMiyauchi/SynGO |
| CLC Genomics Workbench | QIAGEN | Cat. No: 832021 |
| Fasttree | Price et al.46 | www.microbesonline.org/fasttree/ |
| Software and algorithms | ||
| RNeasy Mini Kit | QIAGEN | Cat. No. 74104 |
| Qubit dsDNA BR assay | ThermoFisher Scientific | Cat. No. Q32850 |
| Qubit RNA BR assay | ThermoFisher Scientific | Cat. No. Q10210 |
| RNAse A | ThermoFisher Scientific | Cat. No. 12091021 |
| Genomic tip | QIAGEN | Cat. No. 10223 |
| SMRTbell Template Prep | Pacific Biosciences | Cat. No. 100-938-900 |
| TruSeq Stranded mRNA HT sample prep | Illumina | Cat. No. 20020594 |
| HiSeq TruSeq SBS sequencing kits | Illumina | Cat. No. TG-410-1002 |
| BluePippin System | Sage Science | Cat. No. BLU0001 |
Resource availability
Lead contact
Further information on the fungal strains, the laboratory work, and the bioinformatics/statistics can be directed to the lead contact, Christoffer Bugge Harder (cbharder@bio.ku.dk).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
Genomes have been deposited at GenBank and are publicly available from there. Accession numbers are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
We developed cultures by spore dispersion on malt extract agar (MEA) plates overnight, after which the strains were maintained and grown for harvesting on solid MEA agar medium with ampicillin (100 mg/L) and benomyl (50 mg/L). For DNA extraction, fungi were incubated in the dark at 20°C for 2–6 weeks, depending on their growth rate prior to harvesting. For RNA extraction, fungi were incubated in the dark at 20°C for 2–4 weeks.
All new cultures were deposited in the Belgian Coordinated Collection of Microorganisms/MUCL at the Université de Louvain, and the strain numbers are listed in the Key resources table. Further information about each species can be found at the JGI homepage (https://genome.jgi.doe.gov/portal/).
Method details
DNA and RNA extraction
Harvested mycelia were harvested, ground in liquid nitrogen, and subjected to DNA and RNA extraction. For DNA, we used a phenol/chloroform protocol (including RNAse A treatment (Thermo Fisher Scientific)), as in Skrede et al. 2011.95 After extraction, all samples were cleaned using Qiagen Genomic tip 100/G columns according to the manufacturer’s protocols (Qiagen, Valencia, CA, USA). RNA was extracted using the standard protocol of the RNeasy Mini Kit (Qiagen) and β-mercaptoethanol was added to the lysis buffer. The quantity and quality of the samples were determined by gel electrophoresis, Qubit BR assay kits for RNA/DNA (Thermo Fisher Scientific)), and a NanoDrop 1000 spectrophotometer.
Enzyme analysis
We grew all Mycena s.s. species in triplicate stationary cultures on liquid malt agar at 20°C for 21 days and analyzed their extracellular enzyme activity as described by.96 All strains used were the same as those sequenced for this study, except for the already sequenced M. galopus (ATCC)), where we used another strain (M. galopus Telemark).16 We measured activities for the broad CAZyme types endocellulase (with Azo-Carboxymethylcellulose), endoxylanase (with Azo-Xylan from birch), laccase (with ABTS), Mn-peroxidase (with DMAB and MBTH), as well as the following hydrolases using methylumbelliferol-based substrates: α-glucosidase, α-galactosidase, exocellulase, β-glucosidase, β-mannosidase, N-acetylglucosaminidase, phosphomonoesterase, galacturonidase and β-xylosidase, as well as for the overall lipase and protease activity.
Genome sequencing and assembly
The 25 new genomes reported in this study were sequenced using the Pacific Biosciences platform (PacBio), assembled with FALCON,97 and annotated using the JGI annotation pipeline1,98 aided by the 25 matching transcriptomes. For genome sequencing, five μg gDNA was used to generate each library, except for M. olivaceomarginata, for which 20 μg gDNA was used. The DNA was sheared to >10 kb fragments using Covaris g-TUBE, except for the DNA of M. olivaceiomarginata, which was sheared to >20 kb fragments using 26G blunt needles (VWR). The sheared DNA was treated with exonuclease to remove single-stranded ends and DNA damage repair mix, followed by end-repair and ligation of blunt adapters using SMRTbell Template Prep or Express Kit 1.0 (Pacific Biosciences). The final library was then purified using AMPure PB beads. The M. olivaceomarginata library was additionally size selected with BluePippin (Sage Science) at >15 kb cutoff size. For all but five exceptions, the PacBio Sequencing primer was then annealed to the SMRTbell template library, and the sequencing polymerase was bound to them using the Sequel Binding kit 2.0. The prepared SMRTbell template libraries were then sequenced on a Pacific Biosystems Sequel sequencer using v3 sequencing primers, 1M v2 SMRT cells, and Version 2.0, sequencing chemistry with 1 × 360 and 1 × 600 sequencing movie run times. For the 5 exceptions (M. capillaripes, M. haematopus, M. leptocephala, M. rosella, and Mycena sp. CBHHK59/15), the PacBio Sequencing primer was annealed to the SMRTbell template library, and Version P6 sequencing polymerase was bound to them. The prepared SMRTbell template libraries were then sequenced on a Pacific Biosciences RSII sequencer using Version C4 chemistry and 1 × 240 sequencing movie run times.
Transcriptome sequencing and assembly
Sequenced transcriptomes were used to assess the completeness of genome assemblies and to seed and assess genome annotations. All 25 Mycena transcriptomes were sequenced using Illumina RNA-Seq with polyA selection; the reads were assembled into RNA contigs using Trinity99
For all but five exceptions, plate-based RNA sample prep was performed on the PerkinElmer Sciclone NGS robotic liquid handling system using Illumina’s TruSeq Stranded mRNA HT sample prep kit utilizing poly-A selection of mRNA following the protocol outlined by Illumina in their user guide: https://www.illumina.com/products/by-type/sequencing-kits/library-prep-kits/truseq-stranded-mrna.html, and with the following conditions: total RNA starting material was 1 μg per sample and 8 cycles of PCR was used for library amplification. For the 5 exceptions (M. galericulata, M. metata, M. olivaceomarginata, M. rosella, and Mycena sp. CBHHK59/15 and stranded cDNA libraries were generated using the Illumina Truseq Stranded mRNA Library Prep Kit. mRNA was purified from 1 μg of total RNA using magnetic beads containing poly T oligos. mRNA was fragmented using divalent cations at high temperatures. The fragmented RNA was reverse-transcribed using random hexamers and SSII (Invitrogen) followed by second-strand synthesis. The fragmented cDNA was treated with an end pair, A-tailing, adapter ligation, and eight cycles of PCR. For all 25 transcriptomes, the prepared libraries were quantified using the KAPA Biosystem’s next-generation sequencing library qPCR kit and run on a Roche LightCycler 480 real-time PCR instrument. The quantified libraries were then prepared for sequencing on the Illumina HiSeq sequencing platform using a TruSeq paired-end cluster kit v4. Sequencing of the flow cell was performed on an Illumina HiSeq2500 sequencer using HiSeq TruSeq SBS sequencing kits v4 following a 2 × 150 indexed run recipe.
Genome annotation
Each genome was annotated using the JGI Annotation Pipeline, which detects and masks repeats and transposable elements and predicts genes based on transcriptome or nr protein evidence, characterizes each conceptually translated protein with sub-elements such as domains and signal peptides, chooses the best gene model at each locus to provide a filtered working set, clusters the filtered sets into draft gene families, discovers higher-order structure structures such as segmental duplications, ascribes functional descriptions such as GO terms and EC numbers, and creates a JGI genome portal with tools for public access and community-driven curation of the annotation.1,98
Detecting whole-genome duplication
Whole-genome duplication (WGD) was detected using WGDdetector,100 a pipeline that discovers WGD in a genome by finding pairs of paralogs and estimating the rates of synonymous substitutions per synonymous site (dS) between paralogs. A WGD is expressed as two peaks in the distribution of the dS values compared to a single peak without a WGD. We further explored this by plotting the distribution of dS values between duplicate pairs of segmental duplications and showed that these are consistent with small-scale duplication events.66 We ran seven selected Mycena genomes through this computationally intensive pipeline: the three Arctic M. sp, M. leptocephala and M. olivaceomarginata, the two highly duplicated M. galericulata and M. polygramma; as well as M. belliae and M. pura with lowest duplication levels.
Tree reconstruction
Orthologous genes among the selected fungi were identified using FastOrtho, with the parameters set to 50% identity, 50% coverage, and inflation of 3.0.101 The protein sequences used were genome-wide protein annotations from the JGI fungal portal, MycoCosm.1 1,516 orthologous gene clusters containing 87,928 of single copy genes among 58 fungi were identified and aligned with MAFFT 7.221,102 ambiguous regions (containing gaps and poorly aligned) were eliminated using Gblocks 0.91b,103 and single-gene alignments were concatenated. A phylogenetic tree was inferred with RAxML 7.7.2,104 using the standard algorithm under the PROTGAMMAWAG model of sequence evolution and 1,000 bootstrap replicates. We made additional two different species trees based on gene trees inferred from orthologous groups using STAG and ParGenes with ASTRAL.105,106,107,108,109 Orthologous genes groups were identified with OrthoFinder.110 We confirmed the evolutionary distance of M. floridula from the Mycena group by comparing three phylogenomic trees using single-copy gene concatenation (RAxML), coalesence (ASTRAL), and multi-gene (STAG) approaches.
Analysis of Mycena species-specific genes
For correlations between the count of Mycena species-specific genes and genome size and other metrics, orthologous genes were identified using OrthoFinder.110 We determined species-specific genes from 202,351 orthogroups. Mycena species-specific genes were counted. Pearson correlation was calculated with variables including the number of species-specific genes, scaffold N50, genome size, and BUSCO. Custom R scripts were used for the process.
For verifying the presence of transcription in the 24 Mycena species specific genes, they were examined with JGI RNA-seq data (The 25th species, Mycena pura, had insufficient RNA data for this purpose). Genes showing more than one uniquely mapped read were counted as transcribed genes. The process was performed with custom R scripts.
Analyses of genome contractions/expansions, cluster enrichment analysis and genome integrity tests
We used the COMPARE method to analyze evolutionary changes in gene family sizes.62 Protein sequences of gene families were aligned with L-INS-I or auto algorithm of MAFFT (version 7.4.07)102 and trimmed with TrimAL_1.2rev59 (gt −0.4).111 Gene trees were inferred with RAxmlHPC-PTHREADS-AVX2 8.2.12 under the PROTGAMMAWAG model, and the estimated branch robustness was estimated using SH-like support.104 The gene trees were rooted and reconciled with the species tree using Notung 2.9 with 95% SH-like support as the edge-weight threshold for topological rearrangements.112 Duplication/loss events were mapped onto the species tree by Dollo parsimony using the COMPARE pipeline.62
Identification of horizontally transferred genes
To identify horizontally transferred genes in Mycena species we calculated the alien index60 for each Mycena protein. In order to do that, we merged the 58 Basidiomycota proteomes with 12 Ascomycota whole proteomes and performed mmseqs analysis (with a bidirectional query coverage cutoff of 0.8). Hits were assigned to the taxonomic groups of Mycena, non-Mycena Basidiomycota and Ascomycota. Alien index was calculated as the difference of the bitscore of the best Ascomycota hit and the bitscore of the best non-Mycena Basidiomycota hit both divided by the bitscore of the best Mycena hit. Proteins with an alien index >0 were considered as potentially horizontally transfred proteins.113
In order to phylogenetically verify horizontal gene transfer events, we used the following strategy: we extracted the top 50 Mycena, non-Mycena and Ascomycota hits for each potentially HGT-derived Mycena proteins from the mmseqs results. Proteins showing ≥60% similarity were clustered together. Proteins in each cluster were aligned using mafft in L-INS-i mode. Gene trees were inferred using IQ-tree 114 under the LG + G model. Next we identified nodes of the gene trees that met the following criteria: they show at least 70% bootstrap support, they only contain Mycena proteins, have a sister clade that contains Ascomycota proteins exclusively and the preceding clade contains at least 70% Ascomycota proteins. Mycena proteins that met these criteria were accepted as horizontally transferred genes. To identify the donor taxa we took the phylogenetically verified HGT-derived Mycena proteins as queries and performed a similarity search against a target database that contained the original 58 proteomes supplemented with 151 Ascomycota species using mmseqs as above and identified the best hits for each query.
TE-gene distance analysis and transcription levels
We measured the mean TE-gene distances with statistical support by comparing the locations of the observed genes and TEs and 10,000 null hypothesis genome models created by randomly reshuffling the locations of genes. The probability (p value) of the mean TE-gene distances was calculated using the R package regioneR.115 Across the entire genomic space, the proximity of the coding genes to repeat elements was calculated. The distance of genes to the nearest repeat elements was defined as the length from the mid-point of genes to a) the start of nearest repeats downstream, and b) the end of nearest repeats upstream. We used such tactics to estimate repeat elements located outside genes as well as overlapping genes. The distance of genes to the nearest repeat elements within 2.5 kb (from the mid-point of the genes) was defined as “close proximity,” and coding genes tend to be smaller than 4 kb. The process was conducted using the visual pipeline Synteny-Governed Overview.116
To confirm the presence of transcription in such TE-overlapping genes, we examined the transcription level of TE overlapping genes for three cold-adapted Mycena species and eight other Mycena species from temperate areas for comparison. Transcriptomic data were obtained from the JGI Mycocosm. Transcripts were mapped to the genomes and counted per gene using CLC Genomics Workbench (QIAGEN). Genes mapped with over ten transcript reads were selected.
For an analysis of the unknown TE repeats, we used unknown repeats overlapping genes coding for major facilitator superfamily domain involved in carbohydrate transport and metabolic pathway. We made a phylogenetic tree with the unknown repeat sequences identified from RepeatModeller and RepeatMasker.117,118 Sequences were aligned with Mafft102 and a tree was built with Fasttree.119
Root invasion and species ecologies
We assessed the root invasion ability simply on a scale from 0 to 10 based on the number of ten plant species in a meta-analysis of Mycena inside plant roots,37 where a particular Mycena species constituted at least 5% of the sample. Species ecologies/lifestyles were inferred based on multiple sources.26,27,94,120,121 Mycena species were further grouped into the three specialization ranks of narrow specialists (exclusively/predominantly on one or two species of substrate), broader specialists (exclusively/predominantly associated with either coniferous, broadleaf, or grass substrates)), and generalists that are regularly found on all three broad substrate types.
Quantification and statistical analysis
Comparative genomic feature analyses
Statistics of JGI genome assemblies (i.e., N50, number of genes and scaffolds, and genome size) were obtained from the JGI Mycocosm.1 Genome completeness with single-copy orthologs was assessed using BUSCO v3.0.2 and basidiomycota_odb9 as a reference, with default parameters.122 The TE coverage of the TE in the genomes was identified using RepeatScout117 and RepeatModeler.118 The above information was combined and visualized using the visual-omic pipeline Transposon Identification Nominative Genome Overview (TINGO).123 Secretomes were predicted as described previously.124 We calculated, visualized, and compared the counts and ratios of total (present in the genome) and secreted CAZymes, lipases, proteases, and SSPs (<300 amino acids) as subcategories. We calculated the total count of the followings using both all and secreted PCWDE and MCWDEs. Global trends in ecological groups were evaluated using non-metric multidimensional scaling (NMDS) with counts of total and secreted CAZymes. The dissimilarities among the ecological groups were calculated, and the relationship was converted into distances in two-dimensional space with the metaMDS function in the R package Vegan 2.5–4.125 We examined statistically significant variables in genomic features using permutational multivariate analysis of variance (PERMANOVA). The percentage of variance (R2) contributing to genomic data was estimated for variables including ecological groups, phylogenetic distances, and the genome coverage. Detailed. The detailed procedures have been previously described.10 The output files generated above were combined and visualized using the visual-omic pipeline Proteomic Information Navigated Genomic Outlook (PRINGO).10
We evaluated the associations of genomic features using the R package PCAtools.126 We statistically tested differences among various groups using the Kruskal-Wallis test with post hoc Dunn test with the R package DescTools.127 For the phylogenetic PCA (Figure S19), our phylogenomic tree of 58 fungi was imported using the R package ape.128 The tree and counts of secreted CAZymes from PRINGO analyses were combined using the R package phylobase.129 Principal components considering phylogenetic distances were calculated using the R package adephylo.130 All R graphical outputs were visualized using ggplot2131 software.
Acknowledgments
This project was part of the JGI CSP “1KFG: Deep sequencing of ecologically-relevant Dikarya” (# 10.46936/10.25585/60001060) (PI: F.M.), conducted by the US DOE Joint Genome Institute, a DOE Office of Science User Facility, supported by the Office of Science of the US DOE under contract no. DE-AC02-05CH11231. The European Commission (MSCA grant no. 658849), the Carlsberg Research Grant Foundation (of Denmark, grant no. CF18-0809), and the Crafoord Foundation (of Southern Sweden, grant no. 20201043) are acknowledged for their grants to C.B.H., and he was further funded by a grant from the Danish Independent Research Fund DFF/FNU 2032-00064B (grant name SapMyc) at the time of writing. This work was also supported by a grant from the French National Research Agency (ANR) as part of the “Investissements d’Avenir” program (ANR-11-LABX-0002-01, Lab of Excellence ARBRE), and by the Momentum Program of the Hungarian Academy of Sciences (LP2019-13/2019 to L.G.N.).
Besides the funding, we are highly grateful to Thomas Kehlet, Arne Aronsen, and Thomas Læssøe for very valuable help in locating several of the Mycena specimens for culturing. We furthermore wish to thank Cecilie Mathiesen, Jacqueline Hess, and Anna Sterngren for their laboratory assistance and very useful practical suggestions.
Author contributions
C.B.H., H.K., I.S., A.T., F.M., and L.G.N. conceived and designed the study. F.M. is coordinating the JGI CSP “1KFG: Deep sequencing of ecologically-relevant Dikarya.” C.B.H. and H.K. conducted fieldwork and collected new Mycena cultures. C.B.H. isolated and grew Mycena cultures, and A.T. and H.W. provided additional cultures. C.B.H., E.T., and D.L. performed DNA/RNA extraction. P.B. and M.S. analyzed the enzyme cultures. A.L., K.B., N.B., K.L., A. Kuo, and I.V.G. handled nucleic acids and sequenced, assembled, and annotated the genomes. S.M. performed the comparative secretomic, TE, and genomic analyses. A. Kuo, V.M., Z.M., and L.G.N. analyzed the genome evolution and genomic stability genes. C.B.H., V.M., Z.M., and L.G.N. performed standard/multivariate statistical tests. E.M. and A. Kuo helped analyze the TE annotation. C.B.H., L.G.N., F.M., and S.M. wrote the first version of the manuscript and crafted the revision. All authors helped finalize the manuscript and read and approved it before submission.
Declaration of interests
The authors declare no competing interests.
Published: June 27, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2024.100586.
Contributor Information
Christoffer Bugge Harder, Email: cbharder@bio.ku.dk.
Francis Martin, Email: francis.martin@inrae.fr.
Supplemental information
References
- 1.Grigoriev I.V., Nikitin R., Haridas S., Kuo A., Ohm R., Otillar R., Riley R., Salamov A., Zhao X., Korzeniewski F., et al. MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res. 2014;42:D699–D704. doi: 10.1093/nar/gkt1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eastwood D.C., Floudas D., Binder M., Majcherczyk A., Schneider P., Aerts A., Asiegbu F.O., Baker S.E., Barry K., Bendiksby M., et al. The Plant Cell Wall-Decomposing Machinery Underlies the Functional Diversity of Forest Fungi. Science. 2011;333:762–765. doi: 10.1126/science.1205411. [DOI] [PubMed] [Google Scholar]
- 3.Floudas D., Binder M., Riley R., Barry K., Blanchette R.A., Henrissat B., Martínez A.T., Otillar R., Spatafora J.W., Yadav J.S., et al. The Paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science. 2012;336:1715–1719. doi: 10.1126/science.1221748. [DOI] [PubMed] [Google Scholar]
- 4.Nagy L.G., Riley R., Bergmann P.J., Krizsán K., Martin F.M., Grigoriev I.V., Cullen D., Hibbett D.S. Genetic Bases of Fungal White Rot Wood Decay Predicted by Phylogenomic Analysis of Correlated Gene-Phenotype Evolution. Mol. Biol. Evol. 2017;34:35–44. doi: 10.1093/molbev/msw238. [DOI] [PubMed] [Google Scholar]
- 5.Lebreton A., Zeng Q., Miyauchi S., Kohler A., Dai Y.C., Martin F.M. Evolution of the mode of nutrition in symbiotic and saprotrophic fungi in forest ecosystems. Annu. Rev. Ecol. Evol. Syst. 2021;52:385–404. doi: 10.1146/annurev-ecolsys-012021-. [DOI] [Google Scholar]
- 6.Murat C., Payen T., Noel B., Kuo A., Morin E., Chen J., Kohler A., Krizsán K., Balestrini R., Da Silva C., et al. Pezizomycetes genomes reveal the molecular basis of ectomycorrhizal truffle lifestyle. Nat. Ecol. Evol. 2018;2:1956–1965. doi: 10.1038/s41559-018-0710-4. [DOI] [PubMed] [Google Scholar]
- 7.Hess J., Skrede I., Chaib De Mares M., Hainaut M., Henrissat B., Pringle A., Gojobori J. Rapid Divergence of Genome Architectures Following the Origin of an Ectomycorrhizal Symbiosis in the Genus Amanita. Mol. Biol. Evol. 2018;35:2786–2804. doi: 10.1093/molbev/msy179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kohler A., Kuo A., Nagy L.G., Morin E., Barry K.W., Buscot F., Canbäck B., Choi C., Cichocki N., Clum A., et al. Convergent losses of decay mechanisms and rapid turnover of symbiosis genes in mycorrhizal mutualists. Nat. Genet. 2015;47:410–415. doi: 10.1038/ng.3223. [DOI] [PubMed] [Google Scholar]
- 9.Martin F., Aerts A., Ahrén D., Brun A., Danchin E.G.J., Duchaussoy F., Gibon J., Kohler A., Lindquist E., Pereda V., et al. The genome of Laccaria bicolor provides insights into mycorrhizal symbiosis. Nature. 2008;452:88–92. doi: 10.1038/nature06556. [DOI] [PubMed] [Google Scholar]
- 10.Miyauchi S., Kiss E., Kuo A., Drula E., Kohler A., Sánchez-García M., Morin E., Andreopoulos B., Barry K.W., Bonito G., et al. Large-scale genome sequencing of mycorrhizal fungi provides insights into the early evolution of symbiotic traits. Nat. Commun. 2020;11:5125. doi: 10.1038/s41467-020-18795-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Osono T. Ecology of ligninolytic fungi associated with leaf litter decomposition. Ecol. Res. 2007;22:955–974. doi: 10.1007/s11284-007-0390-z. [DOI] [Google Scholar]
- 12.Cooke R.C., Rayner A.D. Longman; 1984. Ecology of Saprotrophic Fungi. [Google Scholar]
- 13.Floudas D., Bentzer J., Ahrén D., Johansson T., Persson P., Tunlid A. Uncovering the hidden diversity of litter-decomposition mechanisms in mushroom-forming fungi. ISME J. 2020;14:2046–2059. doi: 10.1038/s41396-020-0667-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bödeker I.T.M., Clemmensen K.E., de Boer W., Martin F., Olson Å., Lindahl B.D. Ectomycorrhizal Cortinarius species participate in enzymatic oxidation of humus in northern forest ecosystems. New Phytol. 2014;203:245–256. doi: 10.1111/nph.12791. [DOI] [PubMed] [Google Scholar]
- 15.Clemmensen K.E., Lindahl B.D., Bahr A., Ovaskainen O., Dahlberg A., Ekblad A., Wallander H., Stenlid J., Finlay R.D., Wardle D.A. Roots and associated fungi drive long-term carbon sequestration in boreal forest. Science. 2013;339:1615–1618. doi: 10.1126/science.1231923. [DOI] [PubMed] [Google Scholar]
- 16.Thoen E., Harder C.B., Kauserud H., Botnen S.S., Vik U., Taylor A.F.S., Menkis A., Skrede I. In vitro evidence of root colonization suggests ecological versatility in the genus Mycena. New Phytol. 2020;227:601–612. doi: 10.1111/nph.16545. [DOI] [PubMed] [Google Scholar]
- 17.Rineau F., Shah F., Smits M.M., Persson P., Johansson T., Carleer R., Troein C., Tunlid A. Carbon availability triggers the decomposition of plant litter and assimilation of nitrogen by an ectomycorrhizal fungus. ISME J. 2013;7:2010–2022. doi: 10.1038/ismej.2013.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Selosse M.A., Schneider-Maunoury L., Martos F. Time to re-think fungal ecology? Fungal ecological niches are often prejudged. New Phytol. 2018;217:968–972. doi: 10.1111/nph.14983. [DOI] [PubMed] [Google Scholar]
- 19.Moncalvo J.-M., Vilgalys R., Redhead S.A., Johnson J.E., James T.Y., Catherine Aime M., Hofstetter V., Verduin S.J.W., Larsson E., Baroni T.J., et al. One hundred and seventeen clades of euagarics. Mol. Phylogenet. Evol. 2002;23:357–400. doi: 10.1016/S1055-7903(02)00027-1. [DOI] [PubMed] [Google Scholar]
- 20.Boberg J., Finlay R., Stenlid J., Nasholm T., Lindahl B. Glucose and ammonium additions affect needle decomposition and carbon allocation by the litter degrading fungus Mycena epipterygia. Soil Biol. Biochem. 2008;40:995–999. doi: 10.1016/j.soilbio.2007.11.005. [DOI] [Google Scholar]
- 21.Baldrian P., Kolařík M., Stursová M., Kopecký J., Valášková V., Větrovský T., Zifčáková L., Snajdr J., Rídl J., Vlček C., Voříšková J. Active and total microbial communities in forest soil are largely different and highly stratified during decomposition. ISME J. 2012;6:248–258. doi: 10.1038/ismej.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kyaschenko J., Clemmensen K.E., Hagenbo A., Karltun E., Lindahl B.D. Shift in fungal communities and associated enzyme activities along an age gradient of managed Pinus sylvestris stands. ISME J. 2017;11:863–874. doi: 10.1038/ismej.2016.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harder C.B., Læssøe T., Kjøller R., Frøslev T.G. A comparison between ITS phylogenetic relationships and morphological species recognition within Mycena sect. Calodontes in Northern Europe. Mycol. Prog. 2010;9:395–405. doi: 10.1007/s11557-009-0648-7. [DOI] [Google Scholar]
- 24.Kühner R. Le genre Mycena: étude cytologique et systèmatique des espèces d’Europe et d’Amérique du nord. Encycl. Mycol. 1938;10:1–710. [Google Scholar]
- 25.Rexer, K.H. (1994). Die Gattung Mycena s. l. - Studien zu ihrer Anatomie, Morphologie und Systematik. Dissertation (Universität Tübingen).
- 26.Maas Geesteranus R.A. North-Holland; 1992. Mycenas of the Northern Hemisphere. 2 Vvols. [Google Scholar]
- 27.Robich, G. (2003). Mycena D’Europa. A.M.B., Fondazione Centro Studi Micologici. Trento, Vicenza.
- 28.Ogura-Tsujita Y., Gebauer G., Hashimoto T., Umata H., Yukawa T. Evidence for novel and specialized mycorrhizal parasitism: the orchid Gastrodia confusa gains carbon from saprotrophic Mycena. Proc. Biol. Sci. 2009;276:761–767. doi: 10.1098/rspb.2008.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang L., Chen J., Lv Y., Gao C., Guo S. Mycena sp., a mycorrhizal fungus of the orchid Dendrobium officinale. Mycol. Prog. 2012;11:395–401. doi: 10.1007/s11557-011-0754-1. [DOI] [Google Scholar]
- 30.Davey M.L., Heimdal R., Ohlson M., Kauserud H. Host- and tissue-specificity of moss-associated Galerina and Mycena determined from amplicon pyrosequencing data. Fungal Ecology. 2013;6:179–186. doi: 10.1016/j.funeco.2013.02.003. [DOI] [Google Scholar]
- 31.Glynou K., Nam B., Thines M., Maciá-Vicente J.G. Facultative root-colonizing fungi dominate endophytic assemblages in roots of nonmycorrhizal Microthlaspi species. New Phytol. 2018;217:1190–1202. doi: 10.1111/nph.14873. [DOI] [PubMed] [Google Scholar]
- 32.Roy B.A., Thomas D.C., Soukup H.C., Peterson I.A.B. Mycena citrinomarginata is associated with roots of the perennial grass Festuca roemeri in Pacific Northwest prairies. Mycologia. 2021;113:693–702. doi: 10.1080/00275514.2021.1884814. [DOI] [PubMed] [Google Scholar]
- 33.Grelet G.A., Ba R., Goeke D.F., Houliston G.J., Taylor A.F.S., Durall D.M. A plant growth-promoting symbiosis between Mycena galopus and Vaccinium corymbosum seedlings. Mycorrhiza. 2017;27:831–839. doi: 10.1007/s00572-017-0797-5. [DOI] [PubMed] [Google Scholar]
- 34.Botnen S., Vik U., Carlsen T., Eidesen P.B., Davey M.L., Kauserud H. Low host specificity of root-associated fungi at an Arctic site. Mol. Ecol. 2014;23:975–985. doi: 10.1111/mec.12646. [DOI] [PubMed] [Google Scholar]
- 35.Lorberau K.E., Botnen S.S., Mundra S., Aas A.B., Rozema J., Eidesen P.B., Kauserud H. Does warming by open-top chambers induce change in the root-associated fungal community of the arctic dwarf shrub Cassiope tetragona (Ericaceae)? Mycorrhiza. 2017;27:513–524. doi: 10.1007/s00572-017-0767-y. [DOI] [PubMed] [Google Scholar]
- 36.Kohout P., Charvátová M., Štursová M., Mašínová T., Tomšovský M., Baldrian P. Clearcutting alters decomposition processes and initiates complex restructuring of fungal communities in soil and tree roots. ISME J. 2018;12:692–703. doi: 10.1038/s41396-017-0027-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harder C.B., Hesling E., Botnen S.S., Lorberau K.E., Dima B., von Bonsdorff-Salminen T., Niskanen T., Jarvis S.G., Ouimette A., Hester A., et al. Mycena species can be opportunist-generalist plant root invaders. Environ. Microbiol. 2023;25:1875–1893. doi: 10.1111/1462-2920.16398. [DOI] [PubMed] [Google Scholar]
- 38.Ke H.-M., Lee H.-H., Lin C.-Y.I., Liu Y.-C., Lu M.R., Hsieh J.-W.A., Chang C.-C., Wu P.-H., Lu M.J., Li J.-Y., et al. Mycena genomes resolve the evolution of fungal bioluminescence. Proc. Natl. Acad. Sci. USA. 2020;117:31267–31277. doi: 10.1073/pnas.2010761117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang Y., Wang Y., Mondo S.J., Ahrendt S., Andreopoulos W., Barry K., et al. Fungi Are What They Secrete: Evolution of Zygomycete Secretomes and the Origins of Terrestrial Fungal Ecologies. 2022. https://ssrn.com/abstract=4047252or10.2139/ssrn.4047252 SSRN: [DOI] [PMC free article] [PubMed]
- 40.Morin E., Kohler A., Baker A.R., Foulongne-Oriol M., Lombard V., Nagy L.G., Ohm R.A., Patyshakuliyeva A., Brun A., Aerts A.L., et al. Genome sequence of the button mushroom Agaricus bisporus reveals mechanisms governing adaptation to a humic-rich ecological niche. Proc. Natl. Acad. Sci. USA. 2012;109:17501–17506. doi: 10.1073/pnas.1206847109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hess J., Skrede I., Wolfe B.E., LaButti K., Ohm R.A., Grigoriev I.V., Pringle A. Transposable Element Dynamics among Asymbiotic and Ectomycorrhizal Amanita Fungi. Genome Biol. Evol. 2014;6:1564–1578. doi: 10.1093/gbe/evu121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sipos G., Prasanna A.N., Walter M.C., O’Connor E., Bálint B., Krizsán K., Kiss B., Hess J., Varga T., Slot J., et al. Genome expansion and lineage-specific genetic innovations in the forest pathogenic fungi Armillaria. Nat. Ecol. Evol. 2017;1:1931–1941. doi: 10.1038/s41559-017-0347-8. [DOI] [PubMed] [Google Scholar]
- 43.Almási É., Sahu N., Krizsán K., Bálint B., Kovács G.M., Kiss B., Cseklye J., Drula E., Henrissat B., Nagy I., et al. Comparative genomics reveals unique wood-decay strategies and fruiting body development in the Schizophyllaceae. New Phytol. 2019;224:902–915. doi: 10.1111/nph.16032. [DOI] [PubMed] [Google Scholar]
- 44.Varga T., Krizsán K., Földi C., Dima B., Sánchez-García M., Sánchez-Ramírez S., Szöllősi G.J., Szarkándi J.G., Papp V., Albert L., et al. Megaphylogeny resolves global patterns of mushroom evolution. Nat. Ecol. Evol. 2019;3:668–678. doi: 10.1038/s41559-019-0834-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stajich J.E., Wilke S.K., Ahrén D., Au C.H., Birren B.W., Borodovsky M., Burns C., Canbäck B., Casselton L.A., Cheng C.K., et al. Insights into evolution of multicellular fungi from the assembled chromosomes of the mushroom Coprinopsis cinerea (Coprinus cinereus) Proc. Natl. Acad. Sci. USA. 2010;107:11889–11894. doi: 10.1073/pnas.1003391107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Floudas D., Held B.W., Riley R., Nagy L.G., Koehler G., Ransdell A.S., Younus H., Chow J., Chiniquy J., Lipzen A., et al. Evolution of novel wood decay mechanisms in Agaricales revealed by the genome sequences of Fistulina hepatica and Cylindrobasidium torrendii. Fungal Genet. Biol. 2015;76:78–92. doi: 10.1016/j.fgb.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Riley R., Salamov A.A., Brown D.W., Nagy L.G., Floudas D., Held B.W., Levasseur A., Lombard V., Morin E., Otillar R., et al. Extensive sampling of basidiomycete genomes demonstrates inadequacy of the white-rot/brown-rot paradigm for wood decay fungi. Proc. Natl. Acad. Sci. USA. 2014;111:9923–9928. doi: 10.1073/pnas.1400592111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barbi F., Kohler A., Barry K., Baskaran P., Daum C., Fauchery L., Ihrmark K., Kuo A., LaButti K., Lipzen A., et al. Fungal ecological strategies reflected in gene transcription - a case study of two litter decomposers. Environ. Microbiol. 2020;22:1089–1103. doi: 10.1111/1462-2920.14873. [DOI] [PubMed] [Google Scholar]
- 49.Fricke J., Blei F., Hoffmeister D. Enzymatic Synthesis of Psilocybin. Angew. Chem. Int. Ed. Engl. 2017;56:12352–12355. doi: 10.1002/anie.201705489. [DOI] [PubMed] [Google Scholar]
- 50.Ruiz-Dueñas F.J., Barrasa J.M., Sánchez-García M., Camarero S., Miyauchi S., Serrano A., Linde D., Babiker R., Drula E., Ayuso-Fernández I., et al. Genomic Analysis Enlightens Agaricales Lifestyle Evolution and Increasing Peroxidase Diversity. Mol. Biol. Evol. 2021;38:1428–1446. doi: 10.1093/molbev/msaa301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ohm R.A., de Jong J.F., Lugones L.G., Aerts A., Kothe E., Stajich J.E., de Vries R.P., Record E., Levasseur A., Baker S.E., et al. Genome sequence of the model mushroom Schizophyllum commune. Nat. Biotechnol. 2010;28:957–963. doi: 10.1038/nbt.1643. [DOI] [PubMed] [Google Scholar]
- 52.Bao D., Gong M., Zheng H., Chen M., Zhang L., Wang H., Jiang J., Wu L., Zhu Y., Zhu G., et al. Sequencing and Comparative Analysis of the Straw Mushroom (Volvariella volvacea) Genome. PLoS One. 2013;8 doi: 10.1371/journal.pone.0058294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sakamoto Y., Nakade K., Sato S., Yoshida K., Miyazaki K., Natsume S., Konno N., Cullen D. Lentinula edodes Genome Survey and Postharvest Transcriptome Analysis. Appl. Environ. Microbiol. 2017;83 doi: 10.1128/aem.02990-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marian I.M., Vonk P.J., Valdes I.D., Barry K., Bostock B., Carver A., Daum C., Lerner H., Lipzen A., Park H., et al. The transcription factor Roc1 is a key regulator of cellulose degradation in the wood-decaying mushroom Schizophyllum commune. mBio. 2022;13 doi: 10.1128/mbio.00628-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Casacuberta E., González J. The impact of transposable elements in environmental adaptation. Mol. Ecol. 2013;22:1503–1517. doi: 10.1111/mec.12170. [DOI] [PubMed] [Google Scholar]
- 56.Zhang H.Y., Bissett A., Aguilar-Trigueros C.A., Liu H.W., Powell J.R. Fungal genome size and composition reflect ecological strategies along soil fertility gradients. Ecol. Lett. 2023;26:1108–1118. doi: 10.1111/ele.14224. [DOI] [PubMed] [Google Scholar]
- 57.Matheny P.B., Hughes K.W., Kalichman J., Lebeuf R. Pulverulina, a New Genus of Agaricales for Clitocybe ulmicola. SE. Nat. 2020;19:447. doi: 10.1656/058.019.0301. [DOI] [Google Scholar]
- 58.Vizzini A., Consiglio G., Marchetti M., Borovička J., Campo E., Cooper J., Lebeuf R., Ševčíková H. New data in Porotheleaceae and Cyphellaceae: epitypification of Prunulus scabripes Murrill, the status of Mycopan Redhead, Moncalvo & Vilgalys and a new combination in Pleurella Horak emend. Mycol. Prog. 2022;21 doi: 10.1007/s11557-022-01795-z. [DOI] [Google Scholar]
- 59.Sahu N., Indic B., Wong-Bajracharya J., Merényi Z., Ke H.-M., Ahrendt S., Monk T.-L., Kocsubé S., Drula E., Lipzen A., et al. Vertical and horizontal gene transfer shaped plant colonization and biomass degradation in the fungal genus Armillaria. Nat. Microbiol. 2023;8:1668–1681. doi: 10.1038/s41564-023-01448-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gladyshev E.A., Meselson M., Arkhipova I.R. Massive Horizontal Gene Transfer in Bdelloid Rotifers. Science. 2008;320:1210–1213. doi: 10.1126/science.1156407. [DOI] [PubMed] [Google Scholar]
- 61.Oome S., Raaymakers T.M., Cabral A., Samwel S., Böhm H., Albert I., Nürnberger T., Van den Ackerveken G. Nep1-like proteins from three kingdoms of life act as a microbe-associated molecular pattern in Arabidopsis. Proc. Natl. Acad. Sci. USA. 2014;111:16955–16960. doi: 10.1073/pnas.1410031111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nagy L.G., Ohm R.A., Kovács G.M., Floudas D., Riley R., Gácser A., Sipiczki M., Davis J.M., Doty S.L., de Hoog G.S., et al. Latent homology and convergent regulatory evolution underlies the repeated emergence of yeasts. Nat. Commun. 2014;5:4471. doi: 10.1038/ncomms5471. [DOI] [PubMed] [Google Scholar]
- 63.Matheny P.B., Curtis J.M., Hofstetter V., Aime M.C., Moncalvo J.M., Ge Z.W., Slot J.C., Ammirati J.F., Baroni T.J., Bougher N.L., et al. Major clades of Agaricales: a multilocus phylogenetic overview. Mycologia. 2006;98:982–995. doi: 10.3852/mycologia.98.6.982. [DOI] [PubMed] [Google Scholar]
- 64.Kellis M., Birren B.W., Lander E.S. Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature. 2004;428:617–624. doi: 10.1038/nature02424. [DOI] [PubMed] [Google Scholar]
- 65.Corrochano L.M., Kuo A., Marcet-Houben M., Polaino S., Salamov A., Villalobos-Escobedo J.M., Grimwood J., Álvarez M.I., Avalos J., Bauer D., et al. Expansion of Signal Transduction Pathways in Fungi by Extensive Genome Duplication. Curr. Biol. 2016;26:1577–1584. doi: 10.1016/j.cub.2016.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Roelofs D., Zwaenepoel A., Sistermans T., Nap J., Kampfraath A.A., Van de Peer Y., Ellers J., Kraaijeveld K. Multi-faceted analysis provides little evidence for recurrent whole-genome duplications during hexapod evolution. BMC Biol. 2020;18 doi: 10.1186/s12915-020-00789-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kameshwar A.K.S., Qin W. Systematic review of publicly available non-Dikarya fungal proteomes for understanding their plant biomass-degrading and bioremediation potentials. Bioresources and Bioprocessing. 2019;6:1–19. doi: 10.1186/s40643-019-0264-6. [DOI] [Google Scholar]
- 68.Sützl L., Laurent C.V.F.P., Abrera A.T., Schütz G., Ludwig R., Haltrich D. Multiplicity of enzymatic functions in the CAZy AA3 family. Appl. Microbiol. Biotechnol. 2018;102:2477–2492. doi: 10.1007/s00253-018-8784-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Blackman L.M., Cullerne D.P., Hardham A.R. Bioinformatic characterisation of genes encoding cell wall degrading enzymes in the Phytophthora parasitica genome. BMC Genom. 2014;15:785. doi: 10.1186/1471-2164-15-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Eichlerová I., Homolka L., Žifčáková L., Lisá L., Dobiášová P., Baldrian P. Enzymatic systems involved in decomposition reflects the ecology and taxonomy of saprotrophic fungi. Fungal Ecology. 2015;13:10–22. doi: 10.1016/j.funeco.2014.08.002. [DOI] [Google Scholar]
- 71.Park S.-G., Yoo S.i., Ryu D.S., Lee H., Ahn Y.J., Ryu H., Ko J., Hong C.P. Long-read transcriptome data for improved gene prediction in Lentinula edodes. Data Brief. 2017;15:454–458. doi: 10.1016/j.dib.2017.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Drula E., Garron M.-L., Dogan S., Lombard V., Henrissat B., Terrapon N. The carbohydrate-active enzyme database: functions and literature. Nucleic Acids Res. 2022;50:D571–D577. doi: 10.1093/nar/gkab1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Baldrian P., Valásková V. Degradation of cellulose by basidiomycetous fungi. FEMS Microbiol. Rev. 2008;32:501–521. doi: 10.1111/j.1574-6976.2008.00106.x. [DOI] [PubMed] [Google Scholar]
- 74.Huberman L.B., Liu J., Qin L., Glass N.L. Regulation of the lignocellulolytic response in filamentous fungi. Fungal Biology Reviews. 2016;30:101–111. doi: 10.1016/j.fbr.2016.06.001. [DOI] [Google Scholar]
- 75.Duplessis S., Cuomo C.A., Lin Y.C., Aerts A., Tisserant E., Veneault-Fourrey C., Joly D.L., Hacquard S., Amselem J., Cantarel B.L., et al. Obligate biotrophy features unraveled by the genomic analysis of rust fungi. Proc. Natl. Acad. Sci. USA. 2011;108:9166–9171. doi: 10.1073/pnas.1019315108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gupta Y.K., Marcelino-Guimarães F.C., Lorrain C., Farmer A., Haridas S., Ferreira E.G.C., Lopes-Caitar V.S., Oliveira L.S., Morin E., Widdison S., et al. The soybean rust pathogen Phakopsora pachyrhizi displays transposable element proliferation that correlates with broad host-range adaptation on legumes. bioRxiv. 2022 doi: 10.1101/2022.06.13.495685. https://www.biorxiv.org/content/10.1101/2022.06.13.495685v1.full [DOI] [Google Scholar]
- 77.Chuong E.B., Elde N.C., Feschotte C. Regulatory activities of transposable elements: from conflicts to benefits. Nat. Rev. Genet. 2017;18:71–86. doi: 10.1038/nrg.2016.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hawkins J.S., Kim H., Nason J.D., Wing R.A., Wendel J.F. Differential lineage-specific amplification of transposable elements is responsible for genome size variation in Gossypium. Genome Res. 2006;16:1252–1261. doi: 10.1101/gr.5282906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rouxel T., Balesdent M.H. Life, death and rebirth of avirulence effectors in a fungal pathogen of Brassica crops, Leptosphaeria maculans. New Phytol. 2017;214:526–532. doi: 10.1111/nph.14411. [DOI] [PubMed] [Google Scholar]
- 80.Pao S.S., Paulsen I.T., Saier M.H., Jr. Major facilitator superfamily. Microbiol. Mol. Biol. Rev. 1998;62:1–34. doi: 10.1128/mmbr.62.1.1-34.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Blatch G.L., Lässle M. The tetratricopeptide repeat: a structural motif mediating protein-protein interactions. Bioessays. 1999;21:932–939. doi: 10.1002/(SICI)1521-1878(199911)21:11<932::AID-BIES5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 82.MacPherson S., Larochelle M., Turcotte B. A Fungal Family of Transcriptional Regulators: the Zinc Cluster Proteins. Microbiol. Mol. Biol. Rev. 2006;70:583–604. doi: 10.1128/mmbr.00015-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mustafin R.N. The Relationship between Transposons and Transcription Factors in the Evolution of Eukaryotes. J. Evol. Biochem. Physiol. 2019;55:14–23. doi: 10.1134/s0022093019010022. [DOI] [Google Scholar]
- 84.Kocsis B., Lee M.-K., Yu J.-H., Nagy T., Daróczi L., Batta G., Pócsi I., Leiter É. Functional analysis of the bZIP-type transcription factors AtfA and AtfB in Aspergillus nidulans. Front. Microbiol. 2022;13 doi: 10.3389/fmicb.2022.1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Merényi Z., Krizsán K., Sahu N., Liu X.-B., Bálint B., Stajich J.E., Spatafora J.W., Nagy L.G. Genomes of fungi and relatives reveal delayed loss of ancestral gene families and evolution of key fungal traits. Nat. Ecol. Evol. 2023;7:1221–1231. doi: 10.1038/s41559-023-02095-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Arkhipova I.R. Neutral Theory, Transposable Elements, and Eukaryotic Genome Evolution. Mol. Biol. Evol. 2018;35:1332–1337. doi: 10.1093/molbev/msy083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Muszewska A., Steczkiewicz K., Stepniewska-Dziubinska M., Ginalski K. Transposable elements contribute to fungal genes and impact fungal lifestyle. Sci. Rep. 2019;9 doi: 10.1038/s41598-019-40965-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Baduel P., Quadrana L., Hunter B., Bomblies K., Colot V. Relaxed purifying selection in autopolyploids drives transposable element over-accumulation which provides variants for local adaptation. Nat. Commun. 2019;10 doi: 10.1038/s41467-019-13730-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Niu S., Li J., Bo W., Yang W., Zuccolo A., Giacomello S., Chen X., Han F., Yang J., Song Y., et al. The Chinese pine genome and methylome unveil key features of conifer evolution. Cell. 2022;185:204–217.e14. doi: 10.1016/j.cell.2021.12.006. [DOI] [PubMed] [Google Scholar]
- 90.Lynch M., Conery J.S. The Origins of Genome Complexity. Science. 2003;302:1401–1404. doi: 10.1126/science.1089370. [DOI] [PubMed] [Google Scholar]
- 91.Stajich J.E. Fungal Genomes and Insights into the Evolution of the Kingdom. Microbiol. Spectr. 2017;5:619–633. doi: 10.1128/microbiolspec.FUNK-0055-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hiltunen M., Ament-Velásquez S.L., Ryberg M., Johannesson H. Stage-specific transposon activity in the life cycle of the fairy-ring mushroom Marasmius oreades. Proc. Natl. Acad. Sci. USA. 2022;119 doi: 10.1073/pnas. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hess J., Balasundaram S.V., Bakkemo R.I., Drula E., Henrissat B., Högberg N., Eastwood D., Skrede I. Niche differentiation and evolution of the wood decay machinery in the invasive fungus Serpula lacrymans. ISME J. 2021;15:592–604. doi: 10.1038/s41396-020-00799-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Krah F.-S., Bässler C., Heibl C., Soghigian J., Schaefer H., Hibbett D.S. Evolutionary dynamics of host specialization in wood-decay fungi. BMC Evol. Biol. 2018;18:119. doi: 10.1186/s12862-018-1229-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Skrede I., Murat C., Hess J., Maurice S., Sønstebø J.H., Kohler A., Barry-Etienne D., Eastwood D., Högberg N., Martin F., Kauserud H. Contrasting demographic histories revealed in two invasive populations of the dry rot fungus Serpula lacrymans. Mol. Ecol. 2021;30:2772–2789. doi: 10.1111/mec.15934. [DOI] [PubMed] [Google Scholar]
- 96.Baldrian P. Microbial enzyme-catalyzed processes in soils and their analysis. Plant Soil Environ. 2009;55:370–378. [Google Scholar]
- 97.Chin C.-S., Peluso P., Sedlazeck F.J., Nattestad M., Concepcion G.T., Clum A., Dunn C., O'Malley R., Figueroa-Balderas R., Morales-Cruz A., et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods. 2016;13:1050–1054. doi: 10.1038/nmeth.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kuo A., Bushnell B., Grigoriev I.V. In: Advances In Botanical Research. Martin F., editor. Elsevier Academic Press; 2014. Ecological Genomics of Fungi; pp. 1–52. [Google Scholar]
- 99.Grabherr M.G., Haas B.J., Yassour M., Levin J.Z., Thompson D.A., Amit I., Adiconis X., Fan L., Raychowdhury R., Zeng Q., et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yang Y., Li Y., Chen Q., Sun Y., Lu Z. WGDdetector: a pipeline for detecting whole genome duplication events using the genome or transcriptome annotations. BMC Bioinf. 2019;20:75. doi: 10.1186/s12859-019-2670-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wattam A.R., Abraham D., Dalay O., Disz T.L., Driscoll T., Gabbard J.L., Gillespie J.J., Gough R., Hix D., Kenyon R., et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 2014;42:D581–D591. doi: 10.1093/nar/gkt1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Katoh K., Standley D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 104.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Emms D.M., Kelly S. STRIDE: Species Tree Root Inference from Gene Duplication Events. Mol. Biol. Evol. 2017;34:3267–3278. doi: 10.1093/molbev/msx259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Emms D.M., Kelly S. STAG: Species Tree Inference from All Genes. bioRxiv. 2018 doi: 10.1101/267914. [DOI] [Google Scholar]
- 107.Kozlov A.M., Darriba D., Flouri T., Morel B., Stamatakis A., Wren J. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics. 2019;35:4453–4455. doi: 10.1093/bioinformatics/btz305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Morel B., Kozlov A.M., Stamatakis A., Schwartz R. ParGenes: a tool for massively parallel model selection and phylogenetic tree inference on thousands of genes. Bioinformatics. 2019;35:1771–1773. doi: 10.1093/bioinformatics/bty839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang C., Rabiee M., Sayyari E., Mirarab S. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinf. 2018;19:153. doi: 10.1186/s12859-018-2129-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Emms D.M., Kelly S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 2019;20:1–14. doi: 10.1186/s13059-019-1832-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Capella-Gutierrez S., Silla-Martinez J.M., Gabaldon T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Darby C.A., Stolzer M., Ropp P.J., Barker D., Durand D. Xenolog classification. Bioinformatics. 2017;33:640–649. doi: 10.1093/bioinformatics/btw686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shen X.-X., Opulente D.A., Kominek J., Zhou X., Steenwyk J.L., Buh K.V., Haase M.A.B., Wisecaver J.H., Wang M., Doering D.T., et al. Tempo and Mode of Genome Evolution in the Budding Yeast Subphylum. Cell. 2018;175:1533–1545.e20. doi: 10.1016/j.cell.2018.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nguyen L.-T., Schmidt H.A., von Haeseler A., Minh B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015;32:268–274. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gel B., Díez-Villanueva A., Serra E., Buschbeck M., Peinado M.A., Malinverni R. regioneR: an R/Bioconductor package for the association analysis of genomic regions based on permutation tests. Bioinformatics. 2016;32:289–291. doi: 10.1093/bioinformatics/btv562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Looney B., Miyauchi S., Morin E., Drula E., Courty P.E., Kohler A., Martin F.M. Evolutionary priming and transition to the ectomycorrhizal habit in an iconic lineage of mushroom-forming fungi: is preadaptation a requirement? bioRxiv. 2021 doi: 10.1101/2021.02.23.432530. [DOI] [Google Scholar]
- 117.Smit A.F.A., Hubley R., Green P. RepeatMasker Open-4.0. 2013–2015. 2015. http://www.repeatmasker.org
- 118.Smit A., Hubley R. RepeatModeler Open-1.0. 2008-2015. 2015. http://www.repeatmasker.org
- 119.Price M.N., Dehal P.S., Arkin A.P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5 doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nguyen N.H., Song Z., Bates S.T., Branco S., Tedersoo L., Menke J., Schilling J.S., Kennedy P.G. FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecology. 2016;20:241–248. doi: 10.1016/j.funeco.2015.06.006. [DOI] [Google Scholar]
- 121.Aronsen A., Læssøe T. Fungi of Northern Europe. Danish Mycological Society. 2016;5 [Google Scholar]
- 122.Simão F.A., Waterhouse R.M., Ioannidis P., Kriventseva E.V., Zdobnov E.M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 123.Morin E., Miyauchi S., San Clemente H., Chen E.C.H., Pelin A., de la Providencia I., Ndikumana S., Beaudet D., Hainaut M., Drula E., et al. Comparative genomics of Rhizophagus irregularis, R. cerebriforme, R. diaphanus and Gigaspora rosea highlights specific genetic features in Glomeromycotina. New Phytol. 2019;222:1584–1598. doi: 10.1111/nph.15687. [DOI] [PubMed] [Google Scholar]
- 124.Pellegrin C., Morin E., Martin F.M., Veneault-Fourrey C. Comparative Analysis of Secretomes from Ectomycorrhizal Fungi with an Emphasis on Small-Secreted Proteins. Front. Microbiol. 2015;6 doi: 10.3389/fmicb.2015.01278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Oksanen J., Kindt R., Legendre P. Vegan: community ecology package. R package version. 2019;2 http://cran.r-project.org/ 2.5-4. [Google Scholar]
- 126.Blighe K., Al P. PCAtools: Everything Principal Components Analysis. R package version 2.0. 0 2020. https://github.com/kevinblighe/PCAtools
- 127.Signorell A., Aho K., Alfons A., Anderegg N., Aragon T., Arachchige C., et al. Bolker B. R Foundation for Statistical Computing; 2016. DescTools: Tools for Descriptive Statistics. R Package Version 0.99. 18. [Google Scholar]
- 128.Paradis E., Schliep K., Schwartz R. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics. 2019;35:526–528. doi: 10.1093/bioinformatics/bty633. [DOI] [PubMed] [Google Scholar]
- 129.Hackathon R. 2020. R Package phylobase.https://github.com/fmichonneau/phylobase [Google Scholar]
- 130.Jombart T., Dray S. Adephylo: exploratory analyses for the phylogenetic comparative method. Bioinformatics. 2010;26:1–21. doi: 10.1093/bioinformatics/btq292. [DOI] [PubMed] [Google Scholar]
- 131.Wickham H., Chang W., Wickham M.H. R package ‘ggplot2’. Create elegant data visualisations using the grammar of graphics. Version. 2016;2:1–189. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Genomes have been deposited at GenBank and are publicly available from there. Accession numbers are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.