


Liver fibrosis — a consequence of excess alcohol consumption, viral infection, metabolic dysfunction, disorders due to mutations in the genetic code, and autoimmunity (when the body’s immune system cannot distinguish between self and foreign), among many etiologies — has sickened people from paupers to princes for millennia. The aforegoing opinion reflects the timelessness of the causes of liver fibrosis as well as the commonality of its mechanisms. Only recently, however, has the mechanism of liver fibrosis begun to be understood. While this has been a decidedly nonlinear process of discovery, we find ourselves today considering fibrosis at length scales ranging from the nano- (derived from the Greek word νάνος nanos for dwarf) and implying a factor of 10−9, for example, a nanometer or one thousand millionth of a meter, to the meso- (from the Greek μεσαίο mesaio meaning intermediate) and the macro- (from Greek μακρός makrós for large) levels. Time scales range from milliseconds to days to decades.
[Other extreme factors are 10−24 or yocto and 1024 for yotta , and as long as we are considering extraordinary dimensions of scale, it is amusing to recall that the proprietary name of the Internet search engine Google — a creative or accidental misspelling of googo l for the number 10100 that signifies the large amount of information that can be retrieved — has become a generified verb, and yet the Google trademark has been legally upheld and so far, at least, has avoided genericide .]
This review will provide a historical overview of liver fibrosis research, and then will highlight some of the key advances and most memorable and impactful publications in liver fibrosis in the modern era, particularly those most likely to impact the field through the next decades.
LIVER FIBROSIS AND CIRRHOSIS IN THE PRE-MODERN AND MODERN ERAS: RECOGNITION AND NAMING
Given the many etiologies of liver fibrosis and cirrhosis, including alcohol excess, it is certain that these pathological states have existed since antiquity. Greek physicians are credited with palpating the liver and recognizing hardening as abnormal; however, it wasn’t until the Renaissance that Leonardo da Vinci (1452–1519) and Andreas Vesalius (1514–1564) carried out anatomical dissections of human livers, describing the appearance of cirrhosis and, in the case of Vesalius, linking it to excess alcohol consumption.1,2
In the modern era, the great anatomic pathologist and physician of Padua, Giovanni Battista Morgagni (1682–1771), father of the mechanistic approach to medicine and the use of autopsies to advance understanding of the disease,3,4 is credited with identifying liver fibrosis and cirrhosis in some of the more than 600 autopsies he reported in his 1761 book De sedibus, et causis morborum per anatomen indagatis (The Seats and Causes of Diseases Investigated by Anatomy) (Figure 1).4
FIGURE 1.

Frontispiece from Giovanni Battista Morgagni’s book, De sedibus et causis morborum per anatomen indagatis libri quinque. Venetiis: ex Thypographia Remondiniana; 1761.3 Available at the Open Library (https://openlibrary.org/)
René Laennec gave cirrhosis its name after the Greek word κίρρωση kirrhosi, meaning tawny (orange-brown), in a footnote in an 1819 paper (De l’auscultation mediate) describing the physical signs of pulmonary diseases and the development of the stethoscope. More substantially, Laennec provided extensive descriptions of cirrhosis in an incomplete and only partially published manuscript (1804–1808), Treatise of Pathological Anatomy, that had been cited in an 1824 French medical dictionary5 and an 1839–1847 British Encyclopedia of Anatomy and Physiology6 — all of which is discussed in detail by Jacalyn M. Duffin,7 of the University of Ottawa, Canada. Cirrhosis was, however, first described across the English Channel by pathologists John Browne in 1685, in a treatise entitled “A liver appearing glandulous to the eye”8 and Matthew Baillie in 17939 but, strange to relate, he only published the accompanying illustrations (Figure 2) 6 years later.10 Cirrhosis and fibrosis were described histologically in the 18th and 19th centuries by, among others, Robert Carswell (1793–1857, Professor of Pathological Anatomy at the University College, London), who characterized cirrhosis as a state of atrophy with variable amounts and distribution of contractile fibrous tissue, cirrhotic nodules, and vascular compression (Figure 3).11 In the 20th century, the “Father of Hepatology,” Hans Popper (1903–1988, one of the founders of the American Association for the Study of Liver Diseases [AASLD]), further defined the pathological features of fibrosis and cirrhosis,12 notably in alcoholic and cholestatic liver disease, although his writings encompassed all of the major liver diseases and he was the first to describe capillarization of the sinusoids in chronic liver disease.13
FIGURE 2.

(A) John Browne’s illustration of a glandulous-appearing (ie, cirrhotic) liver. Key: Browne’s own upper case letters, A–G, are shown within the drawing of the cirrhotic liver itself): A, left lobe; B, concave part of the right lobe; C, cut surface of the right lobe; D, black spots, possibly representing divided vessels; E, gallbladder; F, portal vein together with the bile duct; G, liver tissue lying between the vena cava and the portal vein and bile duct; and H, vena cava.8 (B) Matthew Baillie’s illustration of a cirrhotic liver. From a series of engravings accompanied with explanations that are intended to illustrate the morbid anatomy of some of the most important parts of the human body, Fascicle 5, Plate II. A portion of the external surface (top) and cross-section (bottom) of the liver studded with tubercles. Line drawings by William Clift (John Hunter’s former assistant) and engraving by James Basire.10
FIGURE 3.

Plates from Carswell’s 1838 book Elementary Forms of Disease.11 (A) Plate shows various stages of cirrhosis in humans and (mid-right) a cow, with a human liver showing venous compression (bottom). The upper right shows the same liver, with lobules surrounded by fibrous tissue. (B) Section of the same human liver (above in A), showing atrophy and fibrous replacement.
STERNZELLEN USHER IN THE 20th CENTURY AND A NEW ERA OF FIBROSIS RESEARCH
While the great 19th and 20th century anatomists and pathologists provided landmark descriptions of fibrotic liver disease, it was in the late 19th century that Karl Wilhelm von Kupffer paved the way for the modern era of liver fibrosis research and specifically an understanding of the cell biology of the disease. In an 1876 letter to the anatomist Wilhelm von Waldeyer, von Kupffer provided the first description of hepatic stellate cells (HSCs), which he called “Sternzellen” after staining dog liver sections with a gold chloride method in an unsuccessful attempt to identify nerve fibers (Figure 4).15,16 We now appreciate that the gold precipitation Kupffer observed was the result of vitamin A within the cells. Although Kupffer himself later concluded that these new cells were “special endothelial cells of the sinusoids,” he had suggested in his original description of Sternzellen that they were connective tissue cells (part of the perivascular connective tissue network, or “perivasculäre Bindgewebzellen” described by Waldeyer),17 a prescient view given our present understanding of stellate cells as pericytes with a critical role in liver fibrogenesis, and his report stands as the first to appreciate the unique localization and anatomy of the HSC.
FIGURE 4.
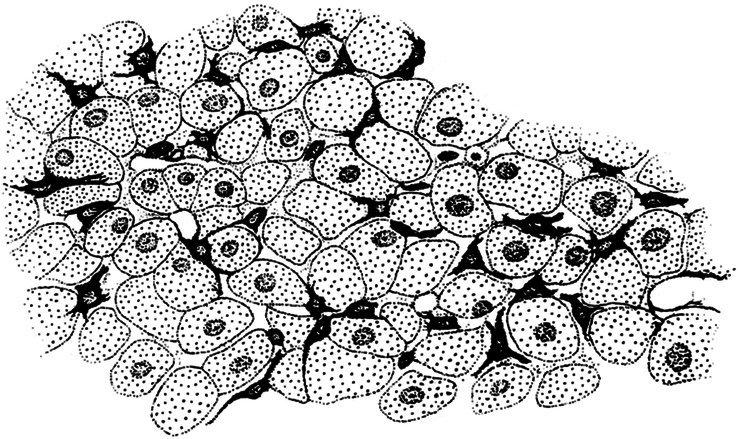
Kupffer’s drawing of Sternzellen in a dog liver, as stained by the gold chloride method.14
After Kupffer’s initial description, HSCs (also known in the literature as fat-storing cells, lipocytes, and eponymously as Ito cells15) were found to be widespread in the liver and were reidentified many times as painstakingly detailed by Kenjiro Wake (Tokyo Medical and Dental University), and the German anatomist, pathologist, and histologist Karl Wilhelm Zimmerman (1861–1935), who highlighted their pericyte nature; Toshio Ito (1904–1991)15 who noting their fat storage called them “Fettspeicherungszellen”; and by Shinsuke Suzuki (who appreciated their interstitial nature).18 The most extraordinary descriptions of stellate cells after their original identification by Kupffer were from Wake (Figure 5), who used silver impregnation to visualize their extraordinarily complex cytoplasmic processes and their anatomic relationships to the hepatic sinusoid and other components of the liver microanatomy.18 Wake also contributed significantly to the field with detailed descriptions of the history of HSC research.16,18 The recognition that the varied concepts of HSCs were unified came with the agreement by 98 investigators in 1996 on the use of the term HSC as a unified nomenclature.20 Although there was significant initial disagreement in the field, the name is now widely used.15
FIGURE 5.

Images from Kenjiro Wake, showing: (A) The complexity of HSC processes and relationship to the sinusoid. (B) Rat HSC showing complex processes around sinusoids, Golgi silver method. Reprinted from Wake.19 (https://creativecommons.org/licenses/by-nc/4.0/).
THE GOLDEN AGE OF STELLATE CELLS
What could be viewed as the “golden age” of HSCs began 40 years ago with descriptions from Knook et al21 (Figure 6) and Friedman et al,22 describing methods using density gradient centrifugation to isolate increasingly pure populations of stellate cells from rodents and then from humans.21–23 It was rapidly appreciated thereafter that HSCs undergo myofibroblastic differentiation — often referred to in the literature as “activation” or “transdifferentiation” — when cultured on standard tissue culture or glass22,23 and that the resulting cells produce collagen and a host of other matrix components (fibronectin and proteoglycans) typical of the fibrotic liver.24 The appreciation that stellate cells were fibrogenic cells occurred in the context of decades of work from Giulio Gabbiani (b. 1937) in Geneva, Switzerland, on the nature and importance of myofibroblasts, as beautifully summarized by his former trainees (now leaders in the field themselves) Alexis Desmoulière (University of Limoges) and Boriz Hinz (University of Toronto).25 The role of stellate cells, as opposed to other cells of the liver (such as hepatocytes, Kupffer cells, and sinusoidal endothelial cells) as sources of matrix was initially debated; however, a seminal paper from Jacqueline Maher, then in the laboratory of Montgomery Bissell at the University of California, San Francisco, established that stellate cells, alone among the listed liver cell types, are collagen producers,26 leading to a wide appreciation for their role in fibrosis.
FIGURE 6.
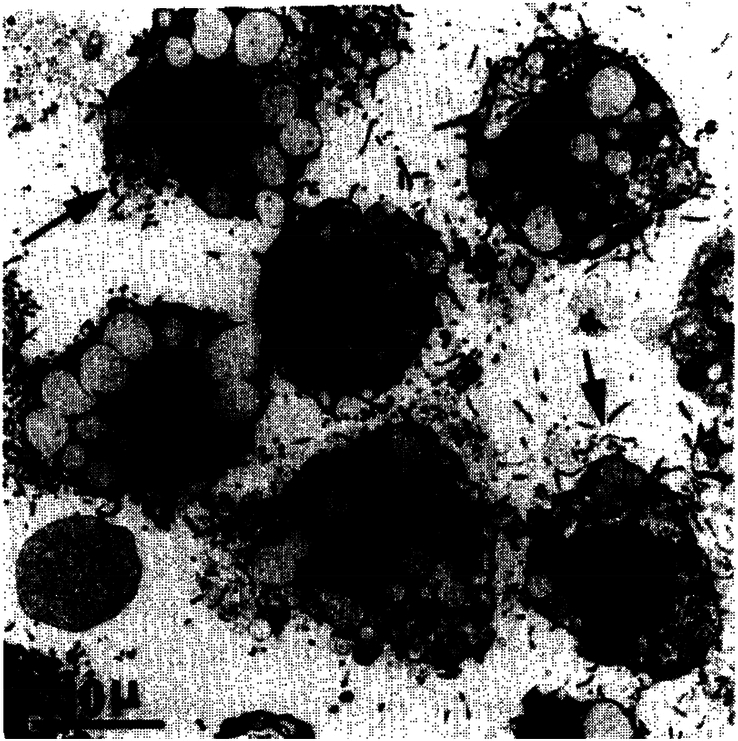
HSCs isolated from rat liver by density gradient centrifugation. Note the presence of lipid droplets that indent the nucleus and the irregular cellular outlines due to the long processes. Reprinted with permission from Knook et al.21
Thereafter followed a period of enormous productivity in fibrosis research, as evidenced by the heady days of the AASLD Fibrosis Single Topic Conferences in Airlie, Virginia (Figure 7), with significant strides made in understanding the pleotropic matrix production, the many soluble and matrix factors influencing the myofibroblast differentiation, and the variety of soluble mediators produced by stellate cells. The research from this period was meticulously summarized by Scott Friedman (now Fishberg Professor of Medicine at the Icahn School of Medicine at Mount Sinai in New York) in 200827 in a publication that aptly referred to stellate cells as “protean, multifunctional, and enigmatic cells of the liver.” One of the most important functions of stellate cells, and the best studied after reports of their isolation, is matrix production, which includes fibrillar and nonfibrillar collagens, cellular fibronectin, and other linker proteins, proteoglycans, and hyaluronic acid28,29; the list of groups and key papers (admittedly incomplete here) that contributed to this effort in the late 20th century, in addition to those noted above, includes most of the leaders in the liver fibrosis field.30–33 Similarly, the list of researchers who contributed to understanding the soluble, inflammatory, and other factors produced by and influencing stellate cell fibrogenesis, proliferation, and migration in fibrosis includes scores of liver fibrosis luminaries, as outlined in a review from 200134 by Massimo Pinzani (formerly Professor of Medicine, Director of the University College London Institute for Liver and Digestive Health, and Sheila Sherlock Chair of Medicine at the University College London; now Scientific Director at the University of Pittsburgh Medical Center- Istituto Mediterraneo per i Trapianti e Terapie ad Alta Specializzazione in Palermo Sicily [UPMC-ISMETT]) and Fabio Marra, formerly both from Florence, Italy, 2 of the investigators who pushed this aspect of fibrosis research forward. While Transforming Growth Factor-β (TGF-β) — as a profibrogenic mediator — and Platelet-Derived Growth Factor (PDGF) — as a pro-proliferative mediator — were the foci of much initial research and are still appreciated to be major determinants of stellate cell behavior, there is a complex web of interactions between different cells of the liver (including immune cells) and a variety of signals (chemokines and cytokines, lipid mediators, apoptotic cells, microRNAs and exosomes, matrix fragments, and microbial products, among many categories) now being explored in great depth through single cell and other ‘omics technologies (see below).
FIGURE 7.

Attendees at the 2006 AASLD Hepatic Fibrosis Single Topic Conference in Airlie, Virginia. Lower panels show the conference center and grounds. Photo courtesy of Hitoshi Yoshiji.
It is notable that, around the time HSC research was yielding such rich rewards, pancreatic stellate cells were identified independently by 2 groups.35,36 My first paper as a faculty member was a 1998 review of stellate cells throughout the body (with the pathologist James Crawford, currently Professor and Chair Emeritus, Department of Pathology and Laboratory Medicine, Northwell Health and the Donald and Barbara Zucker School of Medicine at Hofstra/Northwell), which was written in response to those reports about pancreatic stellate cells. It remains remarkable that, in addition to hepatic stellate cells, similar vitamin A–storing cells are found throughout the body, albeit at very low abundance.37 Pancreatic stellate cells are now appreciated as key mediators for the pancreatic stroma in health and disease, with a particularly important role in pancreatic cancer.38
LIVER FIBROSIS IN THE 21st CENTURY
The last 15–20 years, which I would term the “Modern Era of Fibrosis,” has seen the deepening and broadening of knowledge of the cellular basis of fibrosis, but also the introduction of paradigm-shifting new ideas impacting — often dramatically — both research and clinical care.
Scoring systems and one-size-fits-all
There is a tension in fibrosis research (no pun intended, see below) and clinical care between “one-size-fits-all” and disease-specific approaches. In support of the former, it is clear that in many ways fibrosis and cirrhosis represent a common pathway characterized by common active cell populations, signaling molecules, and matrix organization and that there are components of a final common pathway encompassing not just liver fibrosis of different etiologies, but tissue fibrosis in general. Indeed, there are commonalities between fibrosis and cancer and normal wound healing, as outlined by Edna Cukierman (Fox-Chase Cancer Center) and her colleagues in their work on the Wound Healing-Chronic Fibrosis-Cancer Progression triad.39
On the flip side are the many classification schemes for the progression of fibrotic diseases of the liver, including the Metavir and Ishak histological scoring systems for chronic hepatitis C40,41 (Figure 8) and the Brunt histological scoring system for nonalcoholic fatty liver disease (NAFLD), now termed metabolic dysfunction-associated steatotic liver disease (MASLD).40–43 Common to all of these systems is a focus on collagen architecture, without consideration of the deposition of other matrix components; the final stage, the architectural definition of cirrhosis, is relatively monotonic. Changes in the natural history and survival of patients with advanced liver diseases, as well as the potential for regression (see below), however, have led to a new appreciation for the diversity of clinical findings, prognoses, and histological features of cirrhosis. In 2010, a group of leading clinical investigators proposed an expansion of the standard classification schemes to incorporate a more nuanced view of cirrhosis — acknowledging that cirrhosis is highly variable in clinical presentation, histology, hemodynamics, and underlying vascular and matrix biology — with cirrhosis divided into 3 categories based on these 4 factors, better reflecting the overall state of the liver.44 In 2012, the International Liver Pathology Study Group went a step further, proposing that the term cirrhosis as applied to chronic liver disease be abandoned in favor of the term “Advanced Stage.”45 This new term can be qualified further as “End Stage,” if clinically significant portal hypertension is present (as discussed elsewhere44) and has led to the chronic liver disease becoming a systemic disorder involving other organs and systems. In cases with evidence of regression of fibrosis and architectural improvement, the qualifier “… with features of regression” can be included. Finally, this new approach allows the inclusion of a more etiology-based assessment that includes an evaluation of the risk of malignancy.45
FIGURE 8.

Schematic of Metavir scoring system for chronic hepatitis C. Reprinted with permission from Asselah et al.41
Although these are clinical staging systems, they carry mechanistic implications. Hence, it is a positive attribute that the field has evolved from a “one-size fits all” approach — at least for the end-stage of liver fibrosis — to a more nuanced view that incorporates both common mechanisms of matrix deposition and remodeling and a more individualized and etiology-specific perspective. With the emergence of transient elastography and other imaging modalities, as well as various serum panels as potential replacements for biopsies, new scoring systems are being developed, raising the possibility of even more individualized staging.
Regression of fibrosis and cirrhosis
One of the most remarkable advances in fibrosis in the modern era has been the recognition, beginning around the turn of the century, that fibrosis is a dynamic process and that even cirrhosis has the potential to regress. When I began liver fibrosis research in 1998, extant dogma held that fibrosis and cirrhosis were like the proverbial “train on a one-way track.” The dogma began to weaken in 1998 when John Iredale and colleagues in Southampton, UK, reported that fibrosis in a rat carbon tetrachloride model could regress (Figure 9) and suggested that increased HSC apoptosis and collagenase activity together contributed to recovery.46 Although highly influential in the field at that time and based on increasing numbers of reports in the literature that patients with fibrosis could improve if their primary disease was successfully treated, the study by Iredale and colleagues was with a rat model of early cirrhosis that developed over only 4 weeks, rather than the years and decades of human fibrosis and cirrhosis. It was not until 2 years later that Ian Wanless and colleagues in Toronto, Canada, proposed in the Archives of Pathology & Laboratory Medicine in the appropriately titled “Controversies in Pathology” section — complete with 4 vociferous commentaries disagreeing with Wanless et al’s conclusions — that human cirrhosis could regress.47 The image shown in Figure 10C, without which no review on regression would be complete, provided compelling histological evidence for the regression of cirrhosis in a patient with chronic hepatitis B infection over the course of 42 months of antiviral therapy.
FIGURE 9.

Regression of rat liver fibrosis, as shown in John Iredale et al’s landmark paper. Top panels show reticulin (A, left) and Sirius red (B, right) stained livers after 4 weeks of carbon tetrachloride injections. Bottom panels (reticulin [C, left], Sirius red [D, right]) show the liver after 28 days of recovery, with fibrous septae mostly gone. Reprinted with permission from Iredale et al.46
FIGURE 10.

Three biopsies over 42 months in a patient with chronic hepatitis B infection, treated after the first biopsy with lamivudine. The first biopsy (A) shows cirrhosis with sinusoidal fibrosis, while (B) the second shows enlargement of cirrhotic nodules and the absence of sinusoidal fibrosis. In (C), the third biopsy, there is no cirrhosis. Reprinted from Wanless et al.47 © 2010 College of American Pathologists.
Regression is now well accepted — fibrosis from many etiologies, including chronic Hepatitis C infection, alcohol abuse, metal overload, and biliary obstruction, decreases in response to treatment of the primary insult. Cirrhosis, too, can regress, although many vascular lesions do not; whether there is a point of no return (an actual failure to regress or a failure to regress quickly enough for the patient’s survival) remains unclear.48 The heavily cross-linked and acellular scars of some cases of advanced liver disease are almost certainly highly resistant to breakdown.
The mechanisms of fibrosis regression are still not fully understood, although there are intriguing and clinically relevant clues found in the recent literature. An underappreciated paper from Detlef Schuppan and Yury Popov (Johannes Gutenberg University Mainz and Beth Israel Deaconess Medical Center, Boston)49 showed that, in a mouse model of carbon tetrachloride–induced early cirrhosis, 36 weeks of recovery led to a decrease in collagen content per unit weight, but no significant change in total liver collagen content (Figure 11). Instead, there was a significant increase in liver weight, with loosening of the fibrous septae and apparent movement of hepatocytes into the septae, splitting them into thinner and looser strands of collagen. While these results were from rodents, the authors noted similarities between the appearance of the regressing scars in their models and the human specimens reported by Wanless et al.47 The generalizability and implications of these data remain unclear, and indeed, nearly a quarter century after the papers from Iredale et al, and Wanless et al, mechanisms of regression — in particular related to loss and reorganization of matrix — are still not fully understood.
FIGURE 11.
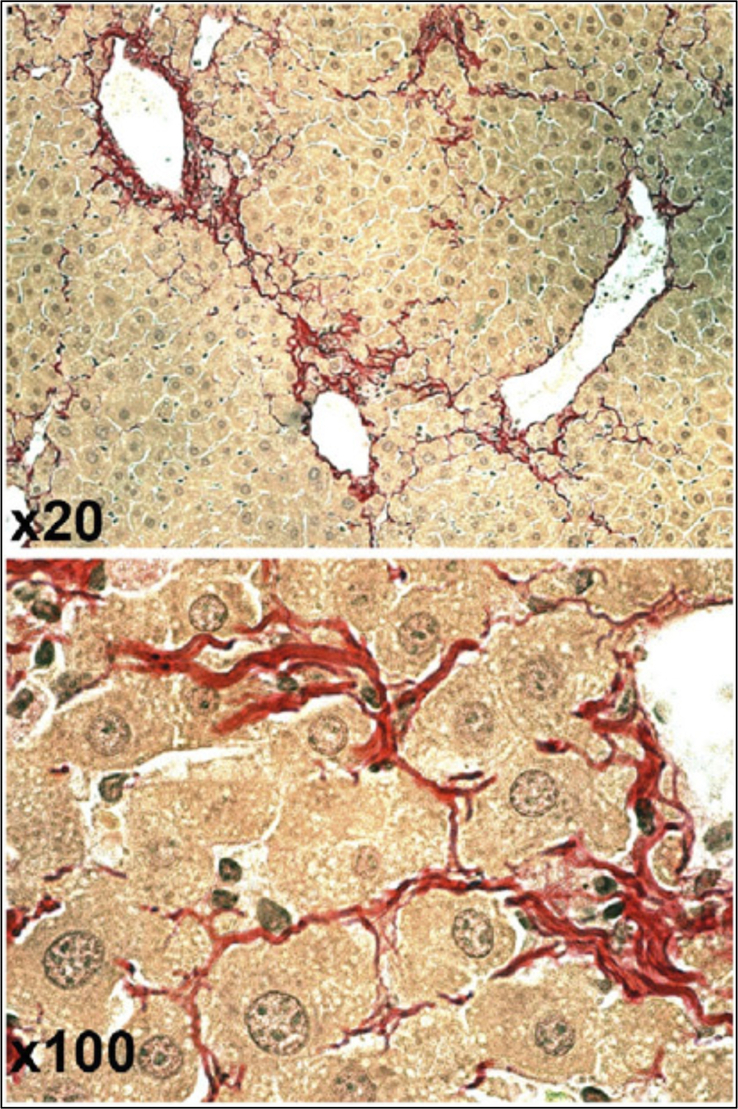
Collagen reorganization (fibril splitting) rather than proteolysis during fibrosis regression in a mouse model. The images, at 2 different magnifications, show hepatocytes “invading” fibrous septae at week 8 of recovery after 36 weeks of carbon tetrachloride treatment. Reprinted with permission from Popov et al.49
Important new clues about fibrosis regression at the cellular level, however, have come from 2 different laboratories that used different methods and approaches but reached essentially the same conclusion. The groups of Robert Schwabe (Columbia University, New York)50 and David Brenner and Tatiana Kisseleva (University of California, San Diego)51 showed that, while fibrosis regression in rodent livers resulted from the apoptosis of about 50% of activated HSCs, the remaining cells regressed to a quiescent-like but primed state capable of rapidly reactivating to fibrogenic myofibroblasts. This phenomenon, which likely resulted from persistent epigenetic changes, has important clinical applications and suggests that patients who have undergone fibrosis regression (eg, after successful treatment of hepatitis C) are at high risk for rapid, recurrent, progressive fibrosis in the setting of an additional insult — a mechanistically-interesting finding that also exemplifies the link between the bench and the bedside in fibrosis research.
The cellular basis of fibrosis
There has been significant — and often highly contentious — research activity since the turn of the century related to the cellular basis of fibrosis. Although hepatocytes, sinusoidal endothelial cells, and Kupffer cells are not collagen producers, new fibrogenic cell populations, in addition to HSCs, have been identified (and at times de-identified). Of these, portal fibroblasts, first isolated in 2002,52 are arguably the most important. These cells were initially viewed as “first responders” in biliary fibrosis, given their periportal location and responses to TGF-β.53 Their role is undoubtedly more complicated than that, and there have been competing claims and results from prominent investigators regarding the role of portal fibroblasts in fibrosis.54,55 Viewing this as a yes/no question and not considering additional roles for both cell types is likely too narrow a perspective,56 and portal fibroblasts remain viable candidates as a significant cell population in cholestatic fibrosis.57
Other cells have also had their time in the sun. Stem cells from the bone marrow contribute to a small extent to the liver myofibroblast population after injury, as shown by bone marrow transplant experiments,58–60 and additional work suggested a role in fibrosis for mesothelial cells from the region of the capsule.61 The role of the epithelial-to-mesenchymal transition in producing fibrogenic cells after liver injury has been the most contentious topic. The results in one initial report suggested that hepatocytes were major contributors to the fibrogenic myofibroblast population,62 but an extraordinary amount of work from many other laboratories failed to find any evidence in vivo that this was the case.63–66
RECENT HISTORY AND THE PROMISE OF THE 21st CENTURY
Liver fibrosis research in the 19th and 20th centuries was notable for its focus first on anatomical descriptions of the liver as a tissue and then on the identity, isolation, and function of the cells and soluble factors within the liver tissue. The 21st century, in contrast, is thus far remarkable for the new and interdisciplinary nature of much of the research being carried out and for the broadening of the length and time scales under consideration. It is likely that many new insights leading ultimately to new and effective therapies will be the result.
The increasingly open secrets of the gut microbiota in fibrosis
More than almost any other topic over the last 2 decades, the gut microbiota and its cross-talk with the liver have dominated discussions of liver disease. One of the earliest and most exciting papers on the gut-liver axis was published in 2007 by Ekihiro Seki, Robert Schwabe, and colleagues,67 showing that the products of intestinal bacteria, acting through Toll-like Receptor 4, mediate fibrosis and that, in mice, broad-spectrum antibiotic treatment resulted in a marked decline in fibrosis associated with bile duct ligation.67 Certainly, antibacterial agents are not miracle drugs in human liver fibrosis. Nonetheless, it is now clear that there are roughly 100 trillion microbes — bacteria, fungi, viruses, archaea (the latter, for the uninitiated, are a group of microorganisms that are similar to, but evolutionarily distinct from, bacteria) — that live in various parts of the luminal GI tract and participate in a bidirectional cross-talk with the liver and biliary tree (through conduits that include the portal vein, arterial circulation, and, as recently shown definitively in humans, the interstitial spaces68,69), regulating intestinal permeability, producing liver function-altering metabolites, and releasing inflammatory mediators. The effect of the microbiota in rodents is now known to go beyond fibrosis and has been clearly implicated in the progression of metabolic dysfunction–associated steatotic liver disease (MASLD), alcohol-associated liver disease (ALD), cirrhosis, hepatic encephalopathy (HE), and HCC. Moreover, there are increasingly persuasive human data and clinical trials of microbiota-altering therapies, such as fecal microbiota transplants, that show promise.70,71 We increasingly appreciate the variety of mechanisms by which the intestinal microbiota alters intestinal permeability and impacts the liver — as evidenced by a plethora of highly mechanistic presentations at the 2023 Annual Meeting of the AASLD in Boston showing that intestinal microorganisms (of all kinds) act through a variety of mechanisms to mediate fibrosis. Clearly, this is a topic that will not be going away and one that will be tapped, in increasingly sophisticated ways, for targeted antifibrotic therapy.
Matrix and mechanics
One of the sea changes [yet another turn of phrase invented by Shakespeare and spoken by Ariel the sprite to Ferdinand the Prince in The Tempest] in the clinical care of fibrosis patients in the 21st century has been the addition of stiffness measurements of the liver and spleen — through transient elastography, magnetic resonance elastography, and similar methods — to the clinical armamentarium, in some cases eliminating the need for diagnostic liver biopsies. Interestingly, the clinical use of mechanics significantly predated any understanding of the relevance of mechanics to the mechanism of fibrosis, and there continues to be little appreciation in the field of the importance of mechanical factors in driving fibrosis. In 2007, we showed in the rat that liver stiffening preceded matrix deposition and that this stiffening was secondary to lysyl oxidase (LOX)-mediated collagen cross-linking.72 Stiffening before fibrosis has since been demonstrated in other tissues, including the heart and lungs.73,74 In this context, the lack of a one-to-one relationship between collagen deposition and mechanics has been confirmed in the human liver.75 As might be predicted from these findings, many liver cell types have been shown to be stiffness-sensitive: HSCs and portal fibroblasts require a stiff environment for myofibroblast differentiation and fibrogenesis (Figure 12),76,77 while hepatocytes and sinusoidal endothelial cells de-differentiate in a stiff environment.78,79 Interestingly, HSCs are also sensitive to viscosity, as shown in a study that introduced a novel hydrogel system to enable the separate evaluation of elastic modulus (stiffness) and loss modulus (viscosity).79 More recent studies outlining the stiffness-sensing and mechanotransduction pathways of cells of the liver and the role of mechanics in specific liver diseases illustrate the exciting progress in the area of liver mechanobiology.80–84 Sebastian Mueller, at the Center for Alcohol Research, University of Heidelberg, Germany, proposed in 2016 that pressure is the cause of cirrhosis,85 a provocative hypothesis the evaluation of which will be a key area of investigation for several decades to come. Unfortunately, although the LOX family member LOXl2 was found to be specifically upregulated in pathological situations, including cancer and fibrosis,86 several clinical trials in humans failed to show evidence of improvement.87 Related work on mechanics has yielded the results of exquisite single molecule studies, demonstrating that TGF-β, a key profibrogenic mediator in most forms of fibrosis (including in the liver), can undergo activation from the latent state when in a stiff environment.88,89
FIGURE 12.

HSCs showing varying levels of myofibroblastic activation when cultured on polyacrylamide supports of varying stiffness ranging from soft (0.4 kPa) to stiff (12 kPa). The phenotypes are stable for at least 2 weeks but can change if stiffness is altered. Size bar, 50 μm. Reprinted from Olsen et al.76
Finally, no section on the recent history of matrix and mechanics in liver fibrosis would be complete without a mention of the ground-breaking work of Neil Henderson, Dean Sheppard et al (from the Lung Biology Center, Department of Medicine, University of California, San Francisco) showing a key role for the family of αv integrins in liver (and lung) fibrosis.90 In addition to the clear therapeutic implications, this work also suggests a common mechanism of fibrosis — across organs — that requires this particular integrin.
Onward march the “etics and the omics”
Perhaps nothing in the 21st century has pushed knowledge of liver fibrosis further than the “etics and omics”. Like studies of the impact of the microbiome, the addition of big data in genetics, epigenetics, and transcriptomics has led to giant leaps and paradigm shifts in understanding fibrosis. Epigenetics refers to the potentially heritable changes in gene expression that result not from changes in the underlying DNA sequence but rather from changes in chromatin conformation, including DNA methylation, histone modifications, and gene silencing mediated by noncoding RNA.91,92 These molecular mechanisms are now appreciated as major mediators in liver cells and, thereby, liver diseases. One of the most remarkable meeting talks I’ve heard was from Jelena Mann (Newcastle University, UK), detailing her work on epigenetic alterations in rats that lead to a paternally inherited decrease in susceptibility to liver fibrosis (Figure 13).93
FIGURE 13.
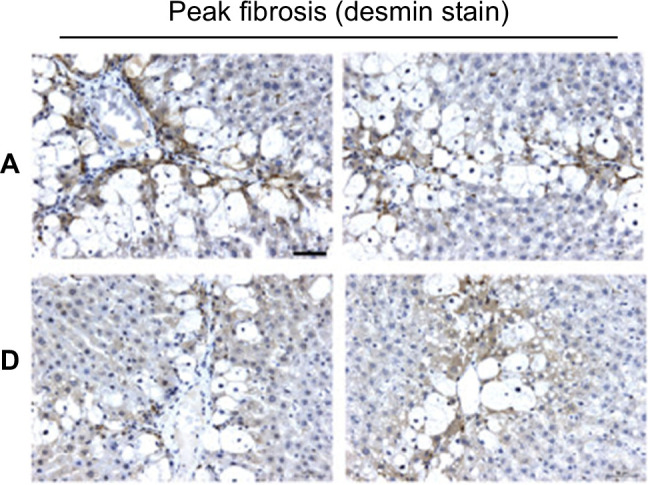
Desmin stain showing peak fibrosis in carbon-tetrachloride rats with 2 generations of male ancestors treated with vehicle (row A, 2 animals represented) or with carbon tetrachloride (row D, 2 animals), demonstrating that epigenetic adaptation over generations protects against fibrosis. Reprinted with permission from Zeybel et al.93
There were also extraordinary advances in understanding the genetics of liver fibrosis in the early part of this century. A review from the early 2000s identified a number of key genetic polymorphisms involved in liver disease, but the authors acknowledged that the field was in its infancy and called for “carefully designed, large epidemiologic studies.”94 In 2011, with the launch of the Million Veterans Program, which had the extraordinarily ambitious goal of carrying out genome sequencing of a million veterans and linking this information to disease, the recommendation of the previous authors became a reality. Important genetic links to cirrhosis and steatotic liver disease, among others, have already been identified.95,96
Transcriptomic data examining cells of the liver began with microarray technology but then moved to single-cell sequencing, which has provided a remarkable view of the different cell populations of the liver in homeostatic as well as pathological situations, including liver regeneration and fibrosis. In addition to identifying previously unimagined subsets and potential cell functions, single-cell analyses using tools, including the cleverly named CellPhoneDB, have allowed the identification of both homotypic and heterotypic ligand-receptor interactions between cells of the liver. Spatial transcriptomics, which links these cell-cell interactions to specific locations in the liver, as well as functional information from proteomics and metabolomics, is now driving forward the study of liver diseases, including fibrosis and steatotic liver disease.97,98 Neil Henderson’s group in Edinburgh, Scotland, who have made significant contributions in this area, have, among other findings, noted significant HSC zonation.99
The shifting sands of time …. and etiology
Perhaps the biggest shift in liver fibrosis understanding during my tenure in the field has been in its etiology. Although alcohol-associated injury has always been and likely will continue to be a significant contributor to liver fibrosis, most of my colleagues and I have lived through the discovery of the HCV, its rise to the fore of fibrosis etiologies and its near-complete cure — one of the most remarkable stories in modern medicine. Now, ALD, hepatitis B, and biliary fibrosis are deservedly receiving some of the attention that had gone to hepatitis C, but they’ve been joined by diseases in the NAFLD spectrum, recently renamed MASLD,100 which are the most prevalent and rapidly becoming the most morbid liver diseases worldwide.101 New etiologies of fibrosis are likely to be a major focus of the next few decades, especially environmental and toxic causes (potentially in combination with additional disease states, like ALD and MASLD).
NEW DIRECTIONS
Whither liver fibrosis from here? While others have insightfully outlined the critical clinical needs in the care of patients with liver fibrosis,102 my focus here is on the research of the future. In basic fibrosis research, single-cell proteomics and metabolomics, combined with spatial analyses, should make single-cell sequencing look like child’s play. My hope is also that single-cell mechanical measurements, incorporating the mechanics of various cell compartments (nucleus, endoplasmic reticulum, and mitochondria), extended to include measurements of single cells in tissues, will be added to this array of tools. Spatial matrisomics103 and, more fancifully, glycomics, linking the other “omics” technologies with the distribution of the hundreds of core and ancillary matrix proteins and glycoproteins, would add a new perspective to these big data. The complex heterogeneity of the regions and cells of the liver will undoubtedly become more apparent as a result of these studies. Along these lines, the role of the multiple processes extended around the sinusoid by HSCs — and the interactions of these processes with heterogeneous cells and regions — may be an expanding area of research and a potential therapeutic target.
While it is somewhat disheartening that our fibrosis cures are limited to rodents and not humans, fibrosis regression after treatment of the primary etiology — most notably in hepatitis C, but also in biliary obstruction, MASLD, and ALD — surely counts in the cure column even if not directly targeting fibrosis. Nonetheless, a better understanding of the role of the matrix in complications of cirrhosis and the development of more direct approaches to degrade matrix proteins (eg, by harnessing matrix metalloproteases and other matrix-degrading enzymes) are clearly needed for those patients whose liver disease is “beyond the point of no return” and requires more than removal of the primary insult — or for those with diseases of unclear etiology, such as many of the cholangiopathies.
It has been a privilege to be part of the liver fibrosis research community for more than a quarter century and to witness the extraordinary evolution of our collective knowledge. The new approaches of the 21st century promise an even more exciting time to come.
SERIES EDITOR’S POSTSCRIPT
Rebecca Wells’s roots are in Western Massachusetts. Following a Yale undergraduate education, schooling in Medicine at Johns Hopkins University, internship, residency, and fellowship at Harvard’s Brigham and Women’s Hospital, she undertook a postdoctoral fellowship at Harvard Medical School and the Whitehead Institute — also in Cambridge, MA. It was a few years later, while she was on the Faculty at Yale (1998–2002), that her interest in liver fibrosis was kindled and continued vigorously when she joined the Faculty of the University of Pennsylvania Perelman School of Medicine (Department of Medicine, Division of Gastroenterology and Hepatology). In Philadelphia, a chance question after Becky delivered a lecture on “Sternzellen,” led to her pursuit of “stiffness” as a factor implicated in liver fibrogenesis.
Somehow, it seems that our careers should have overlapped much earlier than they did, especially since she was studying fibrosis at Yale and I, having been on the Faculty at Yale, too, had left for South Carolina 5 years earlier, where, by chance, I began investigating other aspects of liver stellate cells. However, I had the good fortune to meet and spend time with her in 2022, when I was the 2022 Sheila Sherlock Lecturer in Hepatology.
It would be hard to do justice to Professor Wells’s mastery of every minute aspect of liver fibrosis and fibrogenesis, which this tour de force tour through the history of this topic amply justifies. It seems that no component of hepatofibromics is beyond her reach, from all the varied cells in the liver, the inert and living components of the liver matrix, substrate, enzymes, transmitters, regulators, and effectors, and their biology and pathobiology, not to overlook her keen understanding of methodology even the most exotic and obscure. I do not think there is a better-published overview of hepatic fibrogenesis.
And here, a brief digression can be justified for the sake of those readers — I wager — who might be grateful for a precise definition of exactly what the suffixes “omics and etics” mean since they started as informal cliquish jargon often used as bone fide nouns by biological scientists and other technical professionals. The branches of science known informally as “omics” are various disciplines in biology whose names end in the suffix -omics, such as genomics, proteomics, metabolomics, microbiomics, and so forth. “Omics” aims at the collective study, characterization, and quantification of pools of biological molecules that translate into the structure, function, and dynamics of an organism or organisms, with the acquisition of very large data sets along the way. The related neosuffix — ‘ome, as used in molecular biology, addresses the objects of study of such fields, such as the genome, proteome, metabolome, microbiome, and so on, respectively, which refer to a totality of some sort; they are formed by abstraction from various Greek terms (that strictly do not form an identifiable suffix in Greek). Surprisingly, the creation of such suffixes became productive in the late 19th century as back formations from mitome — the supposed fibrillar reticulum of protoplasm — of which early examples were genome and biome. And, as for “etics”, this is the study of a field or sets of molecules at a more individual level, not so much involving large data sets.
In addition to Becky exploring liver fibrosis, from soup to nuts, she and her husband own a farm (still under development) in the Upper Hudson Valley in New York State. This homestead will incorporate concepts from Restoration Agriculture and Permaculture, with an additional specific focus on maximizing native food-producing plants. It will be part of two 37-acre parcels, including ecologically important meadows, formally protected for conservation, as well as areas suitable for farming, and 25 acres of forest, a creek, and a pond. I can only suppose that the hefty weekly commute has additional eco-friendly benefits.
Acknowledgments
CONFLICTS OF INTEREST
The author has no conflicts to report.
Footnotes
Abbreviations: ALD, alcohol-associated liver disease; AASLD, American Association for the Study of Liver Diseases; HSCs, hepatic stellate cells; LOX, lysyl oxidase; MASLD, metabolic dysfunction–associated steatotic liver disease.
REFERENCES
- 1. Franken FH. History of hepatology In: Csomos G, Thaler H, eds. Clinical Hepatology. Springer-Verlag; 1983:1–15. [Google Scholar]
- 2. Robinson K, Shah VH. Alcohol-related liver disease. Clin Liver Dis (Hoboken). 2021;18(suppl 1):93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Porzionato A, Macchi V, Stecco C, Parenti A, De Caro R. The anatomical school of Padua. Anat Rec (Hoboken). 2012;295:902–916. [DOI] [PubMed] [Google Scholar]
- 4. Zampieri F, Zanatta A, Thiene G. An etymological “autopsy” of Morgagni’s title: De sedibus et causis morborum per anatomen indagatis (1761). Hum Pathol. 2014;45:12–16. [DOI] [PubMed] [Google Scholar]
- 5. Ferrus G. Foie In: Adelon, Andral, Beclard, eds. Dictionnaire de medecine. Bechet; 1824:210–212. [Google Scholar]
- 6. Erasmus WJ. Liver In: Todd RB, ed. Cyclopaedia of Anatomy and Physiology. Longman; 1839-1847:188–189. [Google Scholar]
- 7. Duffin JM. Why does cirrhosis belong to Laennec? CMAJ. 1987;137:393–396. [PMC free article] [PubMed] [Google Scholar]
- 8. Brown J. A remarkable account of a liver, appearing glandulous to the eye; communicated by Mr John Brown, Chirurgeon of St. Thomas’s Hospitall in Southwark; in a letter to one of the secretarys of the Royal Society. Philos Trans (1683-1775). 1685;15:1266–1268. [Google Scholar]
- 9. Baillie M. The Morbid Anatomy of Some of the Most Important Parts of the Human Body. J. Johnson and G. Nicol;; 1793. [Google Scholar]
- 10. Baillie M. A Series of Engravings Accompanied With Explanations Which are Intended to Illustrate the Morbid Anatomy of Some of the Most Important Parts of the Human Body. W. Bulmer and Co; 1799. [Google Scholar]
- 11. Carswell R. Elementary Forms of Disease. Longman, Orme, Brown, Green, and Longman; 1838. [Google Scholar]
- 12. Popper H. Pathologic aspects of cirrhosis. A review. Am J Pathol. 1977;87:228–264. [PMC free article] [PubMed] [Google Scholar]
- 13. Schaffner F, Popper H. Capillarization of hepatic sinusoids in man. Gastroenterology. 1963;44:239–242. [PubMed] [Google Scholar]
- 14. von Ebner. A Kolliker’s Handbuck der Gewebelehre des Mechschen, 6th ed.. Engelmann; 1902;III. [Google Scholar]
- 15. Reuben A. Ito becomes a star. Hepatology. 2002;35:503–504. [DOI] [PubMed] [Google Scholar]
- 16. Wake K. Karl Wilhelm Kupffer and his contributions to modern hepatology. Comp Hepatol. 2004;3(suppl 1):S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Geerts A. History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin Liver Dis. 2001;21:311–335. [DOI] [PubMed] [Google Scholar]
- 18. Wake K. Perisinusoidal stellate cells (fat-storing cells, interstitial cells, lipocytes), their related structure in and around the liver sinusoids, and vitamin A-storing cells in extrahepatic organs. Int Rev Cytol. 1980;66:303–353. [DOI] [PubMed] [Google Scholar]
- 19. Wake K. Hepatic stellate cells: Three-dimensional structure, localization, heterogeneity and development. Proc Jpn Acad Ser B Phys Biol Sci. 2006;82:155–§64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ahern M, Hall P, Liddle C, Olynyk J, Ramm G, Denk J, et al. Hepatic stellate cell nomenclature. Hepatology. 1996;23:193. [PubMed] [Google Scholar]
- 21. Knook D, Seffelaar A, de Leeuw A. Fat-storing cells of the rat liver. Their isolation and purification. Exp Cell Res. 1982;139:468–471. [DOI] [PubMed] [Google Scholar]
- 22. Friedman SL, Roll FJ. Isolation and culture of hepatic lipocytes, Kupffer cells, and sinusoidal endothelial cells by density gradient centrifugation with Stractan. Anal Biochem. 1987;161:207–218. [DOI] [PubMed] [Google Scholar]
- 23. Friedman SL, Rockey DC, McGuire RF, Maher JJ, Boyles JK, Yamasaki G. Isolated hepatic lipocytes and Kupffer cells from normal human liver: Morphological and functional characteristics in primary culture. Hepatology. 1992;15:234–243. [DOI] [PubMed] [Google Scholar]
- 24. Friedman SL, Roll FJ, Boyles J, Bissell DM. Hepatic lipocytes: The principal collagen-producing cells of normal rat liver. Proc Natl Acad Sci USA. 1985;82:8681–8685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Desmouliere A, Hinz B. The myofibroblast and Giulio Gabbiani: An inseparable couple celebrates their 50 years golden wedding anniversary. Wound Repair Regen. 2021;29:511514. [DOI] [PubMed] [Google Scholar]
- 26. Maher JJ, McGuire RF. Extracellular matrix gene expression increases preferentially in rat lipocytes and sinusoidal endothelial cells during hepatic fibrosis in vivo. J Clin Invest. 1990;86:1641–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Friedman SL. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schuppan D, Ruehl M, Somasundaram R, Hahn EG. Matrix as a modulator of hepatic fibrogenesis. Semin Liver Dis. 2001;21:351–372. [DOI] [PubMed] [Google Scholar]
- 29. Wells RG. Function and metabolism of collagen and other extracellular matrix proteins In: Rodes J BJ-P, Blei AT, Reichen J, Rizzetto M, eds. The Textbook of Hepatology: From Basic Science to Clinical Practice, 3rd ed.. Blackwell Publishing; 2008. [Google Scholar]
- 30. Costa AM, Tuchweber B, Rubbia-Brandt L, Peyrol S, Chevallie M, Adham M, et al. Early activation of hepatic stellate cells and perisinusoidal extracellular matrix changes during ex vivo pig liver perfusion. J Submicrosc Cytol Pathol. 2001;33:231–240. [PubMed] [Google Scholar]
- 31. Gressner AM, Haarmann R. Hyaluronic acid synthesis and secretion by rat liver fat storing cells (perisinusoidal lipocytes) in culture. Biochem Biophys Res Commun. 1988;151:222–229. [DOI] [PubMed] [Google Scholar]
- 32. Witsch P, Kresse H, Gressner AM. Biosynthesis of small proteoglycans by hepatic lipocytes in primary culture. FEBS Lett. 1989;258:233–235. [DOI] [PubMed] [Google Scholar]
- 33. Xu G, Niki T, Virtanen I, Rogiers V, De Bleser P, Geerts A. Gene expression and synthesis of fibronectin isoforms in rat hepatic stellate cells. Comparison with liver parenchymal cells and skin fibroblasts. J Pathol. 1997;183:90–98. [DOI] [PubMed] [Google Scholar]
- 34. Pinzani M, Marra F. Cytokine receptors and signaling in hepatic stellate cells. Semin Liver Dis. 2001;21:397–416. [DOI] [PubMed] [Google Scholar]
- 35. Apte MV, Haber PS, Applegate TL, Norton ID, McCaughan GW, Korsten MA, et al. Periacinar stellate shaped cells in rat pancreas: Identification, isolation, and culture. Gut. 1998;43:128–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bachem MG, Schneider E, Groß H, Weidenbach H, Schmid RM, Menke A, et al. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology. 1998;115:421–432. [DOI] [PubMed] [Google Scholar]
- 37. Wells RG, Crawford JM. Pancreatic stellate cells: The new stars of chronic pancreatitis? Gastroenterology. 1998;115:491–493. [DOI] [PubMed] [Google Scholar]
- 38. Pang TCY, Wilson JS, Apte MV. Pancreatic stellate cells: What’s new? Curr Opin Gastroenterol. 2017;33:366–373. [DOI] [PubMed] [Google Scholar]
- 39. Rybinski B, Franco-Barraza J, Cukierman E. The wound healing, chronic fibrosis, and cancer progression triad. Physiol Genomics. 2014;46:223–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bedossa P. Intraobserver and interobserver variations in liver biopsy interpretation in patients with chronic hepatitis C. The French METAVIR Cooperative Study Group. Hepatology. 1994;20:15–20. [PubMed] [Google Scholar]
- 41. Asselah T, Bieche I, Sabbagh A, Bedossa P, Moreau R, Valla D, et al. Gene expression and hepatitis C virus infection. Gut. 2009;58:846–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: A proposal for grading and staging the histological lesions. Am J Gastroenterol. 1999;94:2467–2474. [DOI] [PubMed] [Google Scholar]
- 43. Ishak K, Baptista A, Bianchi L, Callea F, De Groote J, Gudat F, et al. Histological grading and staging of chronic hepatitis. J Hepatol. 1995;22:696–699. [DOI] [PubMed] [Google Scholar]
- 44. Garcia-Tsao G, Friedman S, Iredale J, Pinzani M. Now there are many (stages) where before there was one: In search of a pathophysiological classification of cirrhosis. Hepatology. 2010;51:1445–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hytiroglou P, Snover DC, Alves V, Balabaud C, Bhathal PS, Bioulac-Sage P, et al. Beyond “cirrhosis”: A proposal from the International Liver Pathology Study Group. Am J Clin Pathol. 2012;137:5–9. [DOI] [PubMed] [Google Scholar]
- 46. Iredale JP, Benyon RC, Pickering J, McCullen M, Northrop M, Pawley S, et al. Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J Clin Invest. 1998;102:538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wanless IR, Nakashima E, Sherman M. Regression of human cirrhosis. Morphologic features and the genesis of incomplete septal cirrhosis. Arch Pathol Lab Med. 2000;124:1599–1607. [DOI] [PubMed] [Google Scholar]
- 48. Hytiroglou P, Theise ND. Regression of human cirrhosis: An update, 18 years after the pioneering article by Wanless et al. Virchows Arch. 2018;473:15–22. [DOI] [PubMed] [Google Scholar]
- 49. Popov Y, Sverdlov DY, Sharma AK, Bhaskar KR, Li S, Freitag TL, et al. Tissue transglutaminase does not affect fibrotic matrix stability or regression of liver fibrosis in mice. Gastroenterology. 2011;140:1642–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Troeger JS, Mederacke I, Gwak GY, Dapito DH, Mu X, Hsu CC, et al. Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology. 2012;143:1073–1083.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kisseleva T, Cong M, Paik Y, Scholten D, Jiang C, Benner C, et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci USA. 2012;109:9448–9453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kruglov EA, Jain D, Dranoff JA. Isolation of primary rat liver fibroblasts. J Investig Med. 2002;50:179–184. [DOI] [PubMed] [Google Scholar]
- 53. Wells RG, Kruglov E, Dranoff JA. Autocrine release of TGF-beta by portal fibroblasts regulates cell growth. FEBS Lett. 2004;559:107–110. [DOI] [PubMed] [Google Scholar]
- 54. Iwaisako K, Jiang C, Zhang M, Cong M, Moore-Morris TJ, Park TJ, et al. Origin of myofibroblasts in the fibrotic liver in mice. Proc Natl Acad Sci USA. 2014;111:E3297–E3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4:2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wells RG. The portal fibroblast: Not just a poor man’s stellate cell. Gastroenterology. 2014;147:41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kim HY, Sakane S, Eguileor A, Carvalho Gontijo Weber R, Lee W, Liu X, et al. The origin and fate of liver myofibroblasts. Cell Mol Gastroenterol Hepatol. 2023;17:93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Higashiyama R, Moro T, Nakao S, Mikami K, Fukumitsu H, Ueda Y, et al. Negligible contribution of bone marrow-derived cells to collagen production during hepatic fibrogenesis in mice. Gastroenterology. 2009;137:1459–1466.e1. [DOI] [PubMed] [Google Scholar]
- 59. Kisseleva T, Brenner DA. The phenotypic fate and functional role for bone marrow-derived stem cells in liver fibrosis. J Hepatol. 2012;56:965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kisseleva T, Uchinami H, Feirt N, Quintana-Bustamante O, Segovia JC, Schwabe RF, et al. Bone marrow-derived fibrocytes participate in pathogenesis of liver fibrosis. J Hepatol. 2006;45:429–438. [DOI] [PubMed] [Google Scholar]
- 61. Lua I, Asahina K. The role of mesothelial cells in liver development, injury, and regeneration. Gut Liver. 2016;10:166–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zeisberg M, Yang C, Martino M, Duncan MB, Rieder F, Tanjore H, et al. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem. 2007;282:23337–23347. [DOI] [PubMed] [Google Scholar]
- 63. Chu AS, Diaz R, Hui JJ, Yanger K, Zong Y, Alpini G, et al. Lineage tracing demonstrates no evidence of cholangiocyte epithelial-to-mesenchymal transition in murine models of hepatic fibrosis. Hepatology. 2011;53:1685–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kisseleva T, Brenner DA. Is it the end of the line for the EMT? Hepatology. 2011;53:1433–1435. [DOI] [PubMed] [Google Scholar]
- 65. Scholten D, Österreicher CH, Scholten A, Iwaisako K, Gu G, Brenner DA, et al. Genetic labeling does not detect epithelial-to-mesenchymal transition of cholangiocytes in liver fibrosis in mice. Gastroenterology. 2010;139:987–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Taura K, Miura K, Iwaisako K, Österreicher CH, Kodama Y, Penz-Österreicher M, et al. Hepatocytes do not undergo epithelial-mesenchymal transition in liver fibrosis in mice. Hepatology. 2010;51:1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Seki E, De Minicis S, Österreicher CH, Kluwe J, Osawa Y, Brenner DA, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13:1324–1332. [DOI] [PubMed] [Google Scholar]
- 68. de Jong IEM, Theise ND, Wells RG. The space of Mall confirmed in humans: A response to “portal venous branches as an anatomic railroad for a gut-bile duct axis”. J Hepatol. 2024;80:e126–e127. [DOI] [PubMed] [Google Scholar]
- 69. Fickert P, Lin AC, Ritschl H, Hammer N, Denk H. Portal venous branches as an anatomic railroad for a gut-bile duct-axis. J Hepatol. 2023;79:e82–e84. [DOI] [PubMed] [Google Scholar]
- 70. Bajaj JS. The role of microbiota in hepatic encephalopathy. Gut Microbes. 2014;5:397–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hsu CL, Schnabl B. The gut-liver axis and gut microbiota in health and liver disease. Nat Rev Microbiol. 2023;21:719–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Georges PC, Hui JJ, Gombos Z, McCormick ME, Wang AY, Uemura M, et al. Increased stiffness of the rat liver precedes matrix deposition: Implications for fibrosis. Am J Physiol Gastrointest Liver Physiol. 2007;293:G1147–G1154. [DOI] [PubMed] [Google Scholar]
- 73. Jones MG, Andriotis OG, Roberts JJ, Lunn K, Tear VJ, Cao L, et al. Nanoscale dysregulation of collagen structure-function disrupts mechano-homeostasis and mediates pulmonary fibrosis. Elife. 2018;7:e36354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Neff LS, Bradshaw AD. Cross your heart? Collagen cross-links in cardiac health and disease. Cell Signal. 2021;79:109889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Chin L, Theise ND, Loneker AE, Janmey PA, Wells RG. Lipid droplets disrupt mechanosensing in human hepatocytes. Am J Physiol Gastrointest Liver Physiol. 2020;319:G11–G22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Olsen AL, Bloomer SA, Chan EP, Gaça MDA, Georges PC, Sackey B, et al. Hepatic stellate cells require a stiff environment for myofibroblastic differentiation. Am J Physiol Gastrointest Liver Physiol. 2011;301:G110–G118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Li Z, Dranoff JA, Chan EP, Uemura M, Sévigny J, Wells RG. Transforming growth factor-beta and substrate stiffness regulate portal fibroblast activation in culture. Hepatology. 2007;46:1246–1256. [DOI] [PubMed] [Google Scholar]
- 78. Desai SS, Tung JC, Zhou VX, Grenert JP, Malato Y, Rezvani M, et al. Physiological ranges of matrix rigidity modulate primary mouse hepatocyte function in part through hepatocyte nuclear factor 4 alpha. Hepatology. 2016;64:261–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ford AJ, Jain G, Rajagopalan P. Designing a fibrotic microenvironment to investigate changes in human liver sinusoidal endothelial cell function. Acta Biomater. 2015;24:220–227. [DOI] [PubMed] [Google Scholar]
- 80. Charrier EE, Pogoda K, Wells RG, Janmey PA. Control of cell morphology and differentiation by substrates with independently tunable elasticity and viscous dissipation. Nat Commun. 2018;9:449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Felli E, Selicean S, Guixé-Muntet S, Wang C, Bosch J, Berzigotti A, et al. Mechanobiology of portal hypertension. JHEP Rep. 2023;5:100869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Guo T, Wantono C, Tan Y, Deng F, Duan T, Liu D. Regulators, functions, and mechanotransduction pathways of matrix stiffness in hepatic disease. Front Physiol. 2023;14:1098129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kang N. Mechanotransduction in liver diseases. Semin Liver Dis. 2020;40:84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Mitten EK, Baffy G. Mechanotransduction in the pathogenesis of non-alcoholic fatty liver disease. J Hepatol. 2022;77:1642–1656. [DOI] [PubMed] [Google Scholar]
- 85. Mueller S. Does pressure cause liver cirrhosis? The sinusoidal pressure hypothesis. World J Gastroenterol. 2016;22:10482–10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Barry-Hamilton V, Spangler R, Marshall D, McCauley S, Rodriguez HM, Oyasu M, et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat Med. 2010;16:1009–1017. [DOI] [PubMed] [Google Scholar]
- 87. Fickert P. Is this the last requiem for simtuzumab? Hepatology. 2019;69:476–479. [DOI] [PubMed] [Google Scholar]
- 88. Buscemi L, Ramonet D, Klingberg F, Formey A, Smith-Clerc J, Meister JJ, et al. The single-molecule mechanics of the latent TGF-beta1 complex. Curr Biol. 2011;21:2046–2054. [DOI] [PubMed] [Google Scholar]
- 89. Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol. 2007;179:1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Henderson NC, Arnold TD, Katamura Y, Giacomini MM, Rodriguez JD, McCarty JH, et al. Targeting of alphav integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med. 2013;19:1617–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Mann DA, Mann J. Epigenetic regulation of hepatic stellate cell activation. J Gastroenterol Hepatol. 2008;23(suppl 1):S108–S111. [DOI] [PubMed] [Google Scholar]
- 92. Moran-Salvador E, Mann J. Epigenetics and liver fibrosis. Cell Mol Gastroenterol Hepatol. 2017;4:125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zeybel M, Hardy T, Wong YK, Mathers JC, Fox CR, Gackowska A, et al. Multigenerational epigenetic adaptation of the hepatic wound-healing response. Nat Med. 2012;18:1369–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bataller R. Genetic polymorphisms and the progression of liver fibrosis: A critical appraisal. Hepatology. 2003;37:493–503. [DOI] [PubMed] [Google Scholar]
- 95. Emdin CA, Haas ME, Khera AV, Aragam K, Chaffin M, Klarin D, et al. A missense variant in Mitochondrial Amidoxime Reducing Component 1 gene and protection against liver disease. PLoS Genet. 2020;16:e1008629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Vujkovic M, Ramdas S, Lorenz KM, Guo X, Darlay R, Cordell HJ, et al. A multiancestry genome-wide association study of unexplained chronic ALT elevation as a proxy for nonalcoholic fatty liver disease with histological and radiological validation. Nat Genet. 2022;54:761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Dobie R, Henderson NC. Unravelling fibrosis using single-cell transcriptomics. Curr Opin Pharmacol. 2019;49:71–75. [DOI] [PubMed] [Google Scholar]
- 98. Wallace SJ, Tacke F, Schwabe RF, Henderson NC. Understanding the cellular interactome of non-alcoholic fatty liver disease. JHEP Rep. 2022;4:100524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Dobie R, Wilson-Kanamori JR, Henderson BEP, Smith JR, Matchett KP, Portman JR, et al. Single-cell transcriptomics uncovers zonation of function in the mesenchyme during liver fibrosis. Cell Rep. 2019;29:1832–1847 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Rinella ME, Lazarus JV, Ratziu V, Francque SM, Sanyal AJ, Kanwal F, et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. Hepatology. 2023;78:1966–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Wong VWS, Ekstedt M, Wong GLH, Hagström H. Changing epidemiology, global trends and implications for outcomes of NAFLD. J Hepatol. 2023;79:842–852. [DOI] [PubMed] [Google Scholar]
- 102. Friedman SL, Pinzani M. Hepatic fibrosis 2022: Unmet needs and a blueprint for the future. Hepatology. 2022;75:473–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Naba A, Clauser KR, Ding H, Whittaker CA, Carr SA, Hynes RO. The extracellular matrix: Tools and insights for the “omics” era. Matrix Biol. 2016;49:10–24. [DOI] [PMC free article] [PubMed] [Google Scholar]