

Abstract
BACKGROUND
Diagnosing genetic disorders requires extensive manual curation and interpretation of candidate variants, a labor-intensive task even for trained geneticists. Although artificial intelligence (AI) shows promise in aiding these diagnoses, existing AI tools have only achieved moderate success for primary diagnosis.
METHODS
AI-MARRVEL (AIM) uses a random-forest machine-learning classifier trained on over 3.5 million variants from thousands of diagnosed cases. AIM additionally incorporates expert-engineered features into training to recapitulate the intricate decision-making processes in molecular diagnosis. The online version of AIM is available at https://ai.marrvel.org. To evaluate AIM, we benchmarked it with diagnosed patients from three independent cohorts.
RESULTS
AIM improved the rate of accurate genetic diagnosis, doubling the number of solved cases as compared with benchmarked methods, across three distinct real-world cohorts. To better identify diagnosable cases from the unsolved pools accumulated over time, we designed a confidence metric on which AIM achieved a precision rate of 98% and identified 57% of diagnosable cases out of a collection of 871 cases. Furthermore, AIM’s performance improved after being fine-tuned for targeted settings including recessive disorders and trio analysis. Finally, AIM demonstrated potential for novel disease gene discovery by correctly predicting two newly reported disease genes from the Undiagnosed Diseases Network.
CONCLUSIONS
AIM achieved superior accuracy compared with existing methods for genetic diagnosis. We anticipate that this tool may aid in primary diagnosis, reanalysis of unsolved cases, and the discovery of novel disease genes. (Funded by the NIH Common Fund and others.)
Introduction
Millions of children worldwide are born each year with severe genetic disorders, predominantly Mendelian diseases caused by one or a few genetic variants in a single gene1–3 (Fig. S1A in the Supplementary Appendix). Each individual’s exome typically carries tens of thousands of variants compared with the reference genome. Even after applying sophisticated bioinformatic tools to remove common and low-quality variants, hundreds of variants remain.4 Identifying the causative variant(s) (referred to as diagnostic variants in this paper) from this list is therefore time-consuming and requires broad domain knowledge.4,5 Hence, there is a need for efficient, systematic, and comprehensive approaches to enhance the accuracy and speed of diagnosis.6,7
The current diagnostic rate for patients with genetic disorders is estimated between 30 and 40%.4,8,9 Every year, hundreds of novel disease genes are reported, aiding in the diagnosis of previously unsolved cases.10,11 Therefore, periodic reanalysis of the remaining undiagnosed cases could result in new molecular diagnoses over time.12,13 However, the high cost of implementing routine reanalysis also poses a significant barrier for most large clinical laboratories.11,12 Bioinformatics-based reanalysis presents a cost-effective approach. To this end, several bioinformatic tools have been developed to prioritize genes and variants, including VAAST,14 Phevor,15 Phen-Gen,16 PhenIX,17 Exomiser,15 Phenolyzer,18 Genomiser,19 Xrare,20 LIRICAL,21 AMELIE,22 GEM,23 MOON,24 Emedgene,25 and others (Table S1). However, these tools often have limited accuracy, difficulty in prioritizing non-coding variants, and use simulated data.26–30
To address these limitations, we developed a new artificial intelligence (AI) system called AI-MARRVEL (AIM, MARRVEL: Model organism Aggregated Resources for Rare Variant ExpLoration)31 to prioritize causative genes/variants for Mendelian disorders (Fig. 1A) based on patients’ clinical features and sequencing profiles. AIM was trained using high-quality samples that were clinically diagnosed and curated by American Board of Medical Genetics and Genomics–certified experts along with additional expert-engineered features that encode prior knowledge, such as genetic principles and the knowledge of clinical genetics experts. We evaluated AIM on three independent patient datasets across several application scenarios including dominant, recessive, trio diagnosis, large scale reanalysis, and novel disease gene discovery.
Figure 1.
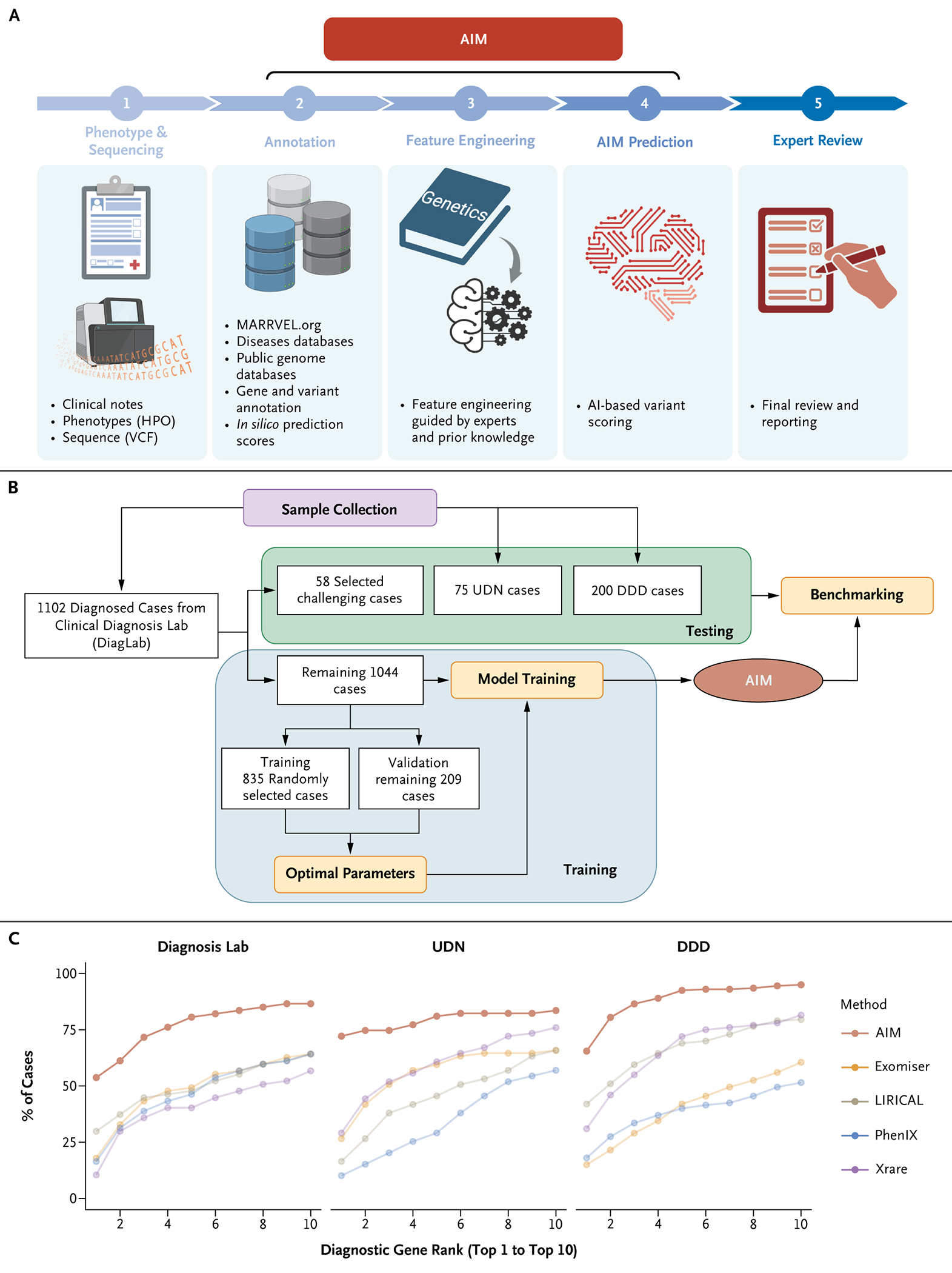
AIM Outperforms State-of-the-Art Methods. (Panel A) The workflow of AI-MARRVEL (AIM). (Panel B) Summary of the sample collection; see the Supplementary Appendix for details. (Panel C) AIM outperforms four state-of-the-art methods, including Exomiser, LIRICAL, PhenIX, and Xrare in three independent datasets: Clinical Diagnosis Lab (DiagLab), Undiagnosed Diseases Network (UDN), and Deciphering Developmental Disorders project (DDD). The graph shows how the diagnostic genes rank within top-1 to top-10 positions among the four methods. AI denotes artificial intelligence; HPO, Human Phenotype Ontology; and VCF, variant call format.
Methods
DATA COLLECTION
We compiled exome sequencing data and Human Phenotype Ontology (HPO) terms from three distinct patient groups: 1102 patients from the Clinical Diagnostic Lab (DiagLab), 75 from the Undiagnosed Disease Network (UDN),32 and 200 from the Deciphering Developmental Disorders project (DDD).33,34 The DiagLab group was divided into a training set of 1044 patients and a testing set of 58. Additionally, both the UDN and DDD groups were used as separate testing sets (Fig. 1B). Each dataset includes Variant Call Format files and phenotypes annotated with HPO terms and a diagnostic variant curated by clinical experts.
TRAINING AND TESTING FOR THE DEFAULT AIM MODEL
Knowledge-Based Feature Engineering
To gather raw features, patient variants were annotated using VEP35 and additional databases summarized by marrvel.org31 (including DGV: the Database of Genomic Variants,36 DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources,37 ClinVar,38 and OMIM: Online Mendelian Inheritance in Man39). To guide AIM with genetic principles and clinical expertise, we performed knowledge-based feature engineering. Fifty-six raw features were selected, encompassing disease database, minor allele frequency, variant impact, evolutionary conservation, inheritance pattern, phenotype matching, gene constraint, variant pathogenicity prediction scores, splicing prediction, and sequencing quality. To incorporate prior knowledge into the AIM model, we developed six modules that cover different aspects of genetic diagnosis decision-making. Module 1 evaluates whether the candidate variant or corresponding gene is curated in disease databases such as OMIM,39 ClinVar,38 or others. Module 2 focuses on the evolutionary conservation and frequency of the candidate gene/variant. Module 3 categorizes the variant based on mutation type, and Module 4 assesses the functional impact of the variant based on prediction algorithms. Module 5 determines the functional distance of the candidate variant and gene to known disease genes in a biological network. Module 6 evaluates the inheritance pattern of the candidate variant.
Each module offers an independent analysis of the pathogenicity of a variant or gene, producing new features that emulate the decision-making of human experts. We engineered these modules to incorporate diverse aspects of expert logic, generating 47 additional features on top of the 56 raw features.
Default AIM Model Training
The random forest algorithm provided by scikit-learn40 was used as the backbone machine-learning algorithm. To determine the optimal parameters, 20% of the cases (n=209) were randomly selected as a validation set. These validation samples were used for tuning parameters, such as the number of trees and the maximum tree depths. The remaining 80% of the cases (n=835) were employed for training multiple random-forest models, each with different parameter combinations. To avoid overfitting due to a 7791:1 negative-to-diagnostic variant ratio of training sample, we weighted the minority class more heavily when calculating impurity scores during tree construction.
We performed parameter selection based on a formula that assigns higher weight to the top-1 accuracy. The formula, denoted as , consisted of weighted contributions that is defined as , where defines the top-k accuracy given the parameter set Λ. Using this objective function, the parameters that yielded the highest performance in ranking diagnostic variants with the top-1, top-5, and top-10 were selected (see the Supplementary Appendix for details).
Results
AIM OUTPERFORMS ESTABLISHED ALGORITHMS ON THREE INDEPENDENT DATASETS
To compare AIM with other algorithms, we considered recent benchmarking papers28,29,41 and selected top performers that provide free and local programmable access as the benchmarking algorithms (Table S1). This includes Exomiser,15 LIRICAL,21 PhenIX,17 and Xrare.20 AIM outperformed all four algorithms in ranking the diagnostic genes across three independent datasets (Fig. 1C). Direct assessment of AMELIE was not feasible due to its lack of local programmatic access.22 However, according to Birgmeier et al.,22 AIM and AMELIE have a similar top-1 accuracy on the DDD datasets, whereas AIM outperforms AMELIE by 7.6% in top 5 accuracy (Fig. S2A).
Approximately 10.7% of the diagnosed variants in the training dataset are noncoding (Fig. S1C). To enhance AIM’s ability to prioritize splicing variants, we integrated splicing-related features such as SpliceAI into the initial design.42 Given that Exomiser is primarily tailored for coding variants, we compared Genomiser and AIM for cases diagnosed with noncoding variants. AIM outperforms Genomiser, with a noticeable difference in the DiagLab group and only a slight edge in the UDN cohort (Fig. S2B).
ACCURATE AIM DIAGNOSIS REQUIRES HIGH-QUALITY LABELING AND FEATURE ENGINEERING
On average, each individual carries 15.5 genetic variants categorized as pathogenic in ClinVar (Fig. 2A). Approximately 5.6 of them are exclusively associated with recessive diseases, according to OMIM (Fig. S3A). Nonetheless, in all three cohorts, 94% of the cases were diagnosed with one or two variant(s) (Fig. S1A). Specifically, among all diagnosed cases, there are 1586 listed as pathogenic by at least one submitter in ClinVar. However, only 8% (n=128) were identified by clinical experts as diagnostic variants (Fig. 2B). Moreover, we observed that one third of the diagnostic variants were not annotated as pathogenic in ClinVar (Fig. 2B). These findings collectively suggest that relying solely on ClinVar information is not sufficient for an accurate diagnosis. Complementing this, the AIM algorithm has successfully differentiated between diagnostic and nondiagnostic pathogenic variants listed in ClinVar (Fig. 2C; P<0.0001).
Figure 2.
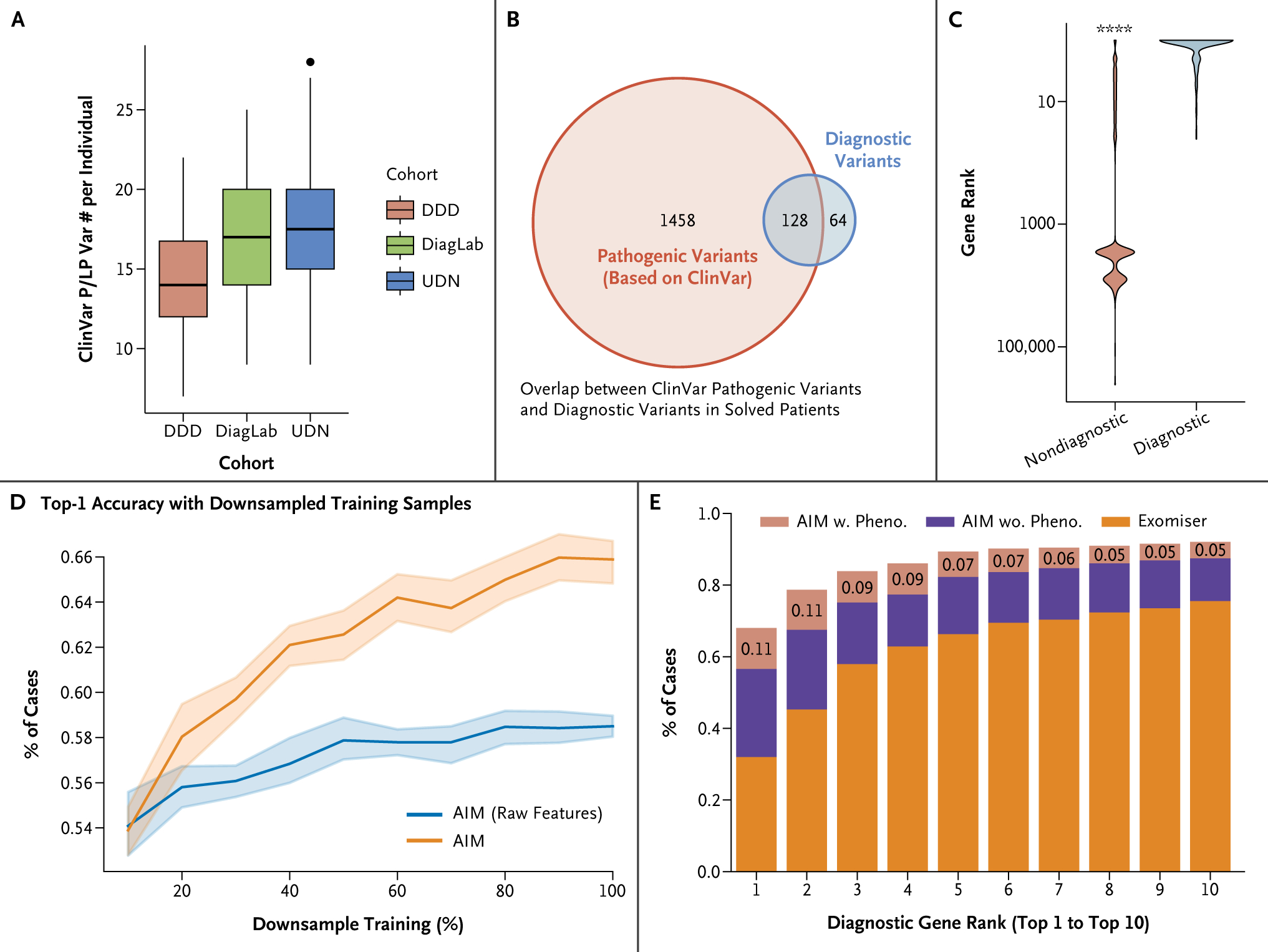
Accurate AIM Diagnosis Requires High-Quality Labeling and Feature Engineering. (Panel A) Box plot showing the number of pathogenic or likely pathogenetic (P/LP) variants per individual based on ClinVar. There are around 15 P/LP variants per individual in the three testing datasets. (Panel B) Venn diagram showing the overlap between ClinVar pathogenic variants and diagnostic variants identified in solved patients from DiagLab and UDN. The red circle represents ClinVar (likely) pathogenic variants, and the blue circle represents diagnostic variants identified in patients. The diagram shows that only 8% of ClinVar (likely) pathogenic variants are disease-causing in patients, whereas one third of diagnostic variants are not annotated as pathogenic in ClinVar. (Panel C) Violin plot of the rankings of diagnostic and nondiagnostic ClinVar P/LP variants. AI-MARRVEL (AIM) separates these two groups. (Panel D) Line plot showing the percentage of cases in which AIM ranks the diagnostic variant as top-1 with down-sampling. The orange line represents AIM trained with both raw and engineered features, and the blue line represents AIM trained with raw features only. (Panel E) Comparing AIM’s performances between data using or ignoring phenotype information. DDD denotes Deciphering Developmental Disorders project; DiagLab, Clinical Diagnosis Lab; and UDN, Undiagnosed Diseases Network.
The use of variant curation status from ClinVar in AIM raises concerns about overreliance on this information. To explore this, we created a model (AIM-withoutVarDB) removing all features related to variant curation status. It exhibited only a slight performance decrease compared with AIM but still outperformed other benchmarked methods (Fig. S3B). This shows that AIM learned a diagnostic logic beyond the ClinVar features.
EXPERT FEATURE ENGINEERING ENHANCES AIM MODEL ACCURACY AND DELAYS TRAINING SATURATION
When only 20% of the training samples were used, AIM’s performance was consistent at 54% top-1 accuracy, regardless of whether it was trained with or without the engineered features (Fig. 2D). However, as the volume of training samples increased, AIM trained with engineered features improved its performance to 66%, whereas AIM without the engineered features plateaued at 58% (Fig. 2D). This suggests that expert-engineered features are more effective in capturing the underlying patterns within the data and help to delay the onset of training saturation.
MOLECULAR EVIDENCE, PHENOTYPIC DATA, AND DIAGNOSTIC PERFORMANCE
A molecular diagnosis is usually achieved by joint consideration of two factors: molecular evidence and phenotype matching. To assess the impact of inaccurate phenotype information on AIM’s prediction, we set phenotype-related features to minimum values, mimicking an extreme scenario where the phenotype information is completely irrelevant to the diagnostic gene. We observed an 11% decrease in the top-1 accuracy (Fig. 2E), highlighting that accurate phenotype annotation provides an important although albeit relatively modest contribution. The difference in the top-k accuracy was smaller when k was greater than 5, suggesting that considering a few more top-ranked genes per patient can mitigate the issue of inaccurate phenotype annotation (Fig. 2E). Additionally, even with completely irrelevant phenotype information, AIM still achieves a top-5 accuracy of 78%, outperforming all the other benchmark approaches, and showing that molecular evidence is paramount.
INTERPRETABILITY OF THE AIM MODEL IN CLINICAL GENETIC DIAGNOSIS
To understand how AIM arrives at its predictions, we developed a “feature climbing” method to evaluate the contribution of each feature by perturbing the feature value and re-running the predictions (Fig. 3A). We quantify the effect size of each feature as the maximum difference between the prediction score and the minimum value achievable through perturbation. All features are grouped into different classes based on their biological meaning (Fig. 3B). The biggest effect size was seen when perturbing the minor allele frequency (MAF), followed by the variant curation status in disease databases and phenotypic matching (Fig. 3B). However, all these factors present similar effect size. These findings collectively indicate that no single factor is decisive in establishing the final diagnosis.
Figure 3.
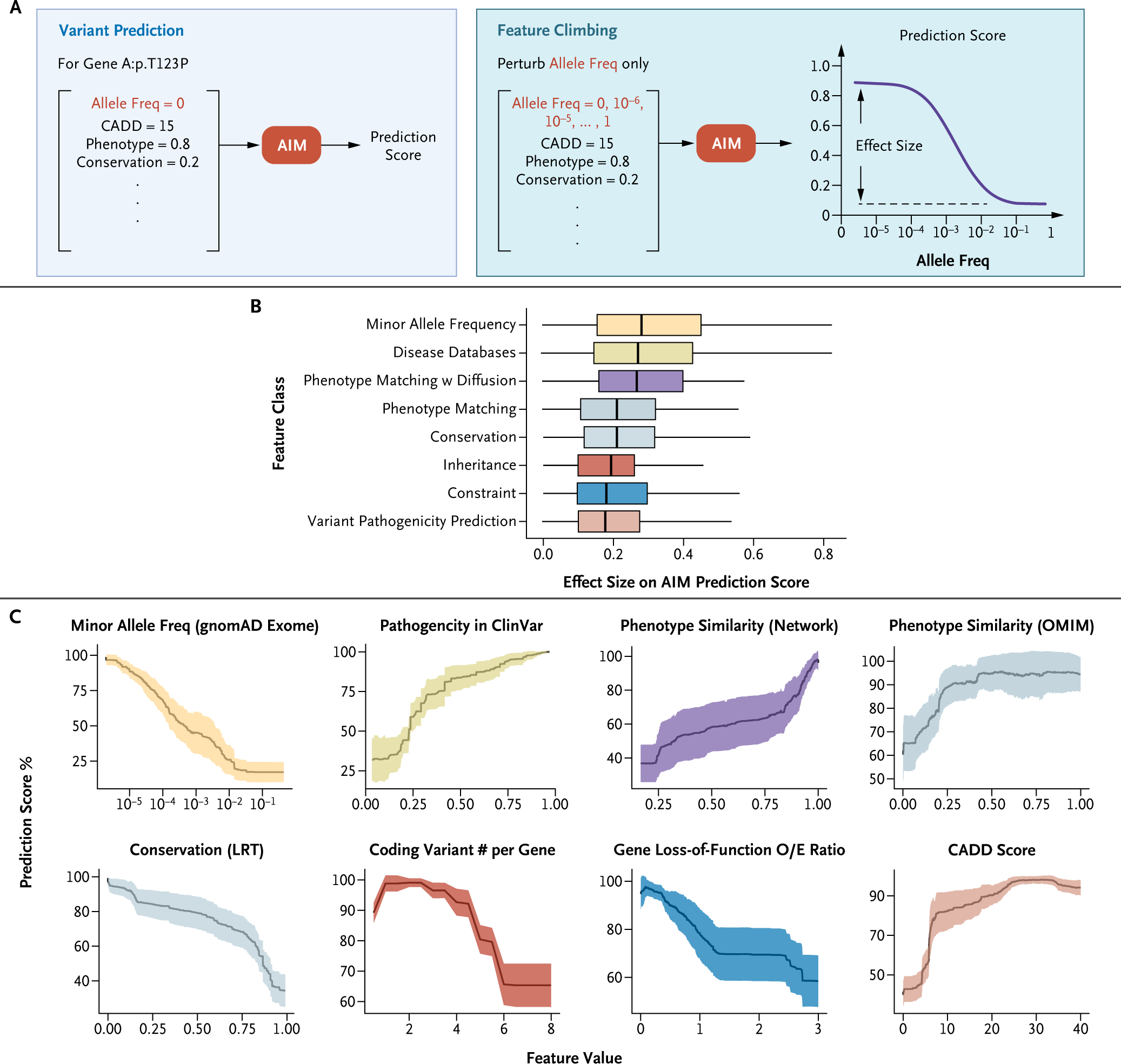
Enhancing Interpretability of AI Models in Clinical Genetic Diagnosis: Analyzing Feature Contributions through Feature Climbing to Demystify AIM’s Random-Forest Model. (Panel A) Schematic representation of the process of feature climbing. (Panel B) Box plot of features’ importance grouped by their types. All the features are grouped into different classes based on biological meaning (color-coded). Conservation: evolutionary conservation; Constraint: population genetic constraint metric; IMPACT: variant impact in gene function, Inheritance: mode of inheritance. (Panel C) Perturbation curves as line plots showing the percentage of prediction score after perturbing each feature. The x axis represents the feature values after perturbing and the y axis shows the percentage of prediction scores with the perturbed features. Line plots display the mean (black, solid lines) and standard deviation (colored, shaded lines) of the performances for diagnostic variants in two testing datasets: Diagnosis Lab and Undiagnosed Diseases Network. Representative features of each class are shown and colored accordingly. AI denotes artificial intelligence; AIM, AI-MARRVEL; CADD, Combined Annotation Dependent Depletion; Freq, frequency; LRT, likelihood ratio test; O/E, observed-to-expected; OMIM, Online Mendelian Inheritance in Man; and w, with.
The phenotype similarity score, based on OMIM, ranges from 0 (no similarity) to 1 (high similarity). We observed that when the score increased from none (0) to low (0.25) similarity, the prediction scores sharply increased from 60% to 90% (Fig. 3C). However, further increases in phenotypic similarity above 0.25 only yielded a moderate increase (Fig. 3C), suggesting that an exact match with OMIM phenotypes is not critical. Patients with identical diagnostic genes/variants might display different disease manifestations due to environmental influences, incomplete penetrance, or variations in genetic background.
Furthermore, the engineered feature “Coding Variant # per Gene,” which counts the occurrences of coding variants identified within a candidate gene in a patient, also revealed an intriguing pattern (Fig. 3C). The prediction score peaked when the feature’s value was between 1 and 2, aligning precisely with the number of variants necessary for a gene to follow a dominant or recessive inheritance pattern. Conversely, the prediction score for a variant sharply decreased when this feature value is above 3 (Fig. 3C). This outcome suggests that AIM has learned that genes with a high number of coding variants per individual are less likely to be associated with rare genetic disorders.
CROSS-SAMPLE CONFIDENCE SCORE FOR HIGHTHROUGHPUT REANALYSIS
The 30 to 40% diagnostic rate in clinical genetic diagnosis leads to an extensive backlog of unresolved cases, making manual regular review of all unresolved cases costly and labor-intensive.4,8,9 Updates to disease databases present opportunities to diagnose previously unsolved cases. This necessitates an automated method to re-evaluate unsolved cases periodically, to pinpoint those that are now diagnosable. Consequently, we designed a cross-sample score to represent the likelihood that a diagnostic variant can be correctly identified in a patient using AIM. Patients are then stratified into two categories: those in the high-confidence group are forwarded for manual review, and those in the lower confidence bracket are deferred to the subsequent reanalysis cycle (Fig. 4A).
Figure 4.
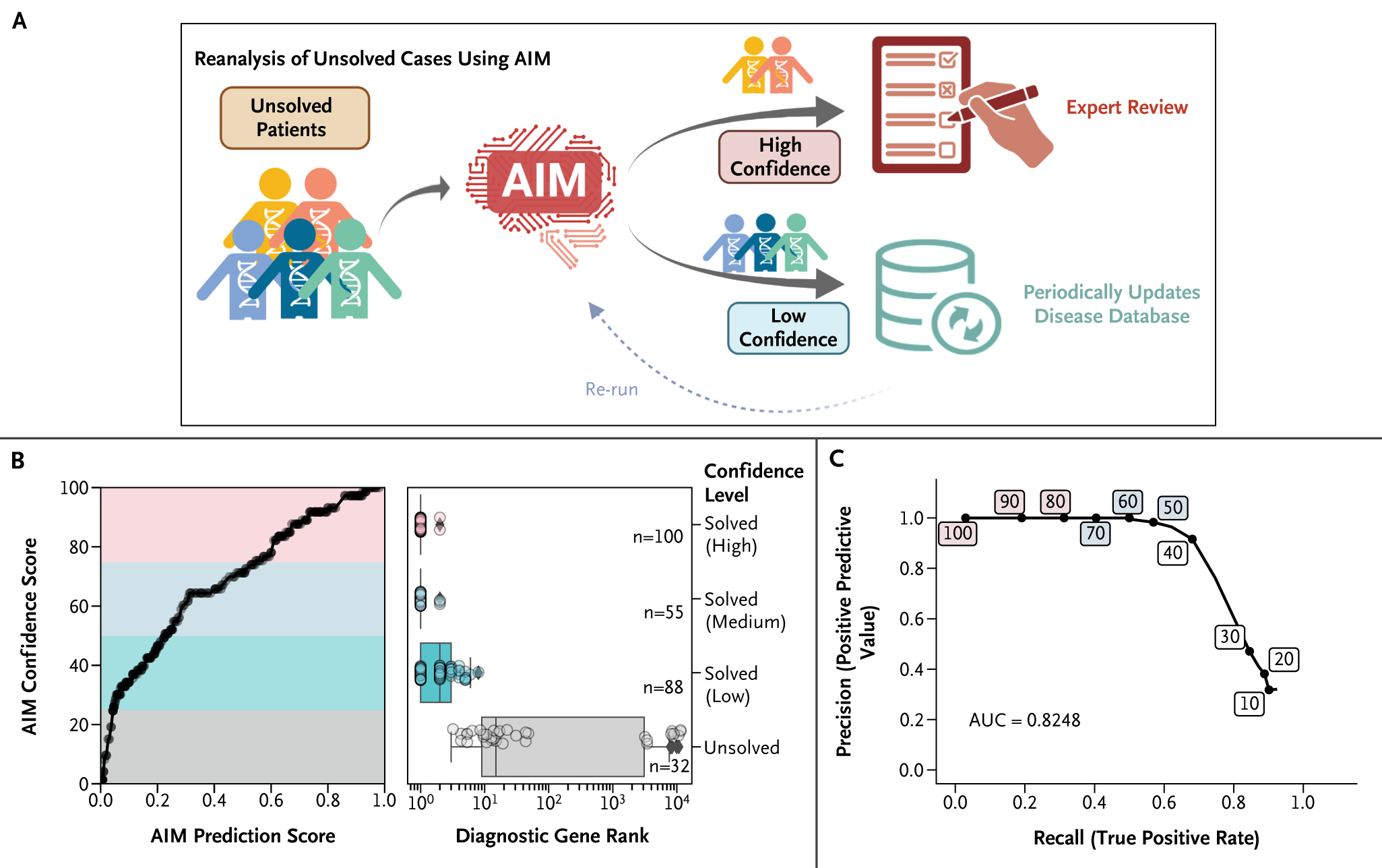
Cross-Sample Confident Scoring for High-Throughput Reanalysis. (Panel A) Schematic diagram illustrating the reanalysis process. For undiagnosed patients, we employed AI-MARRVEL (AIM) to determine the likelihood of a diagnosis. Cases with a high level of confidence are referred for manual review by a trained clinical geneticist, and those with a low level of confidence are reanalyzed periodically after updates to the disease database. (Panel B, left) Line plot showing the relationship between confidence score vs. the AIM prediction score for diagnostic variants of UDN and DDD samples. (Panel B, right) Four levels of confidence are created based on the confidence score: high (75~100, n=108), medium (50~75, n=55), low (25~50, n=88), and unsolved (0~25, n=32). (Panel C) The precision and recall curve for reanalysis at different confident score thresholds (area under the curve [AUC] = 0.82).
We established four confidence levels — high, medium, low, and unsolved — according to the quartiles of diagnostic variants from DiagLab samples and applied them to UDN and DDD samples.43 Notably, 56% of the diagnostic variants were classified as high or medium confidence (Fig. 4B). Moreover, variants with high or medium confidence were predominantly ranked as top candidates, demonstrating the metric’s rigor (Fig. 4B). To assess the confidence score’s ability to identify diagnosable cases, we gathered diagnosed patients from DDD and UDN as positives (n=275) and unaffected relatives of patients with de novo diagnostic variants as negatives (n=596). The precision-recall curve yielded an area under the curve of 0.82, affirming effectiveness in identifying diagnosable cases (Fig. 4C).
AIM-RECESSIVE: ADDRESSING LOW PERFORMANCE FOR RECESSIVE GENETIC DISORDERS
Similar to other tools, AIM performed better for dominant cases than for recessive cases in both DiagLab and UDN datasets (Fig. 5A), likely due to the differences in genetic mechanisms between dominant and recessive diseases.44 Therefore, a single classifier will not be effective for all types of genetic disorders. Indeed, recessive-specific classifiers from Exomiser provide better accuracy for recessive cases (Fig. S5B). To this end, we designed an AIM-Recessive model (see the Supplementary Appendix and Fig. S5A). This model requires the presence of at least two variants (or a homozygous variant) in a patient in the same gene. To create the new feature matrix, we enumerated all possible pairs of variants within each candidate gene and concatenated their features. Out of 56 diagnostic variant pairs, 15 are ranked top-1 in both default and recessive models. AIM-Recessive successfully prioritized 63.4% (n=26) of the remaining 41 cases as top-1 (Fig. 5B).
Figure 5.
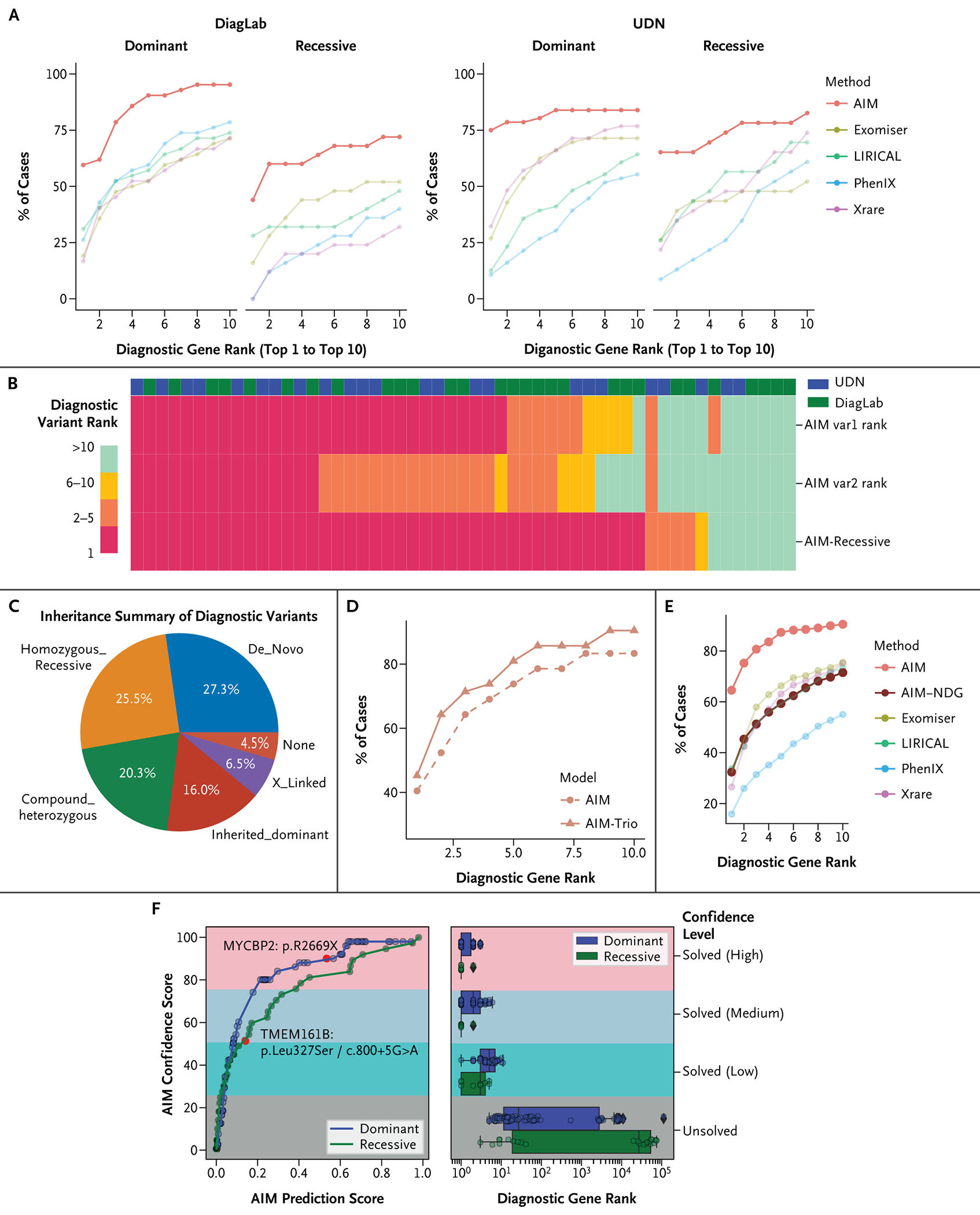
AIM Model Extensions Tailored for Diverse Diagnostic Scenarios. (Panel A) Accuracy of AI-MARRVEL (AIM) and other methods based on different inheritance modes on two independent datasets. (Panel B) Comparing recessive-specific model (AIM-Recessive) and the default model (AIM) on all recessive cases from two datasets: Clinical Diagnosis Lab (DiagLab) and Undiagnosed Diseases Network (UDN). The Heatmap shows the ranking of the diagnostic variants using AIM (AIM var1 and var2) vs. AIM-Recessive model. (Panel C) Inheritance of all diagnostic variants in all trio samples from DiagLab. (Panel D) AIM-Trio model outperformed the singleton models. We trained and compared our trio model under the same settings as the previously mentioned default. With 31 test samples (42 variants), top-k accuracies (variant level) are shown on the plots. (Panel E) Benchmarking of AIM-NDG; the y axis presents the fraction of cases that different tools rank the diagnostic genes within top 1 to top 10. (Panel F) Similar to Figure 4B, the line plot shows the confidence score vs. AIM prediction score for the AIM-NDG model (blue line for cases with diagnostic gene of dominant inheritance and green line for recessive inheritance). We highlight two recently published novel disease genes in the plot (red dots): one dominant gene, MYCBP2 (red dot on blue line), and one recessive gene, TMEM161B (red dot on green line).
AIM-TRIO: ENHANCING PERFORMANCE WITH INHERITANCE INFORMATION
Heterozygous variants that are inherited from one parent (named as “inherited dominant” here) are less likely to be diagnostic. However, approximately 16% of the diagnostic variants are “inherited dominant” in our trio training samples (243 diagnosed cases from DiagLab; Fig. 5C). This may due to factors such as incomplete penetrance, unrecorded milder phenotypes in parents, or incomplete parental phenotype annotation. Therefore, a filter to rule out all inherited dominant variants is not appropriate. Consequently, we trained a trio-specific model by incorporating inheritance-related features (see Fig. S6A). The trio classifier (AIM-Trio) performs much better than Exomiser and Genomiser Trio models (Fig. S6B). It also provides slightly better accuracy than the proband-only model (AIM) (Fig. 5D; top-1 accuracy increases from 40 to 45%).
AIM-NDG: A STEP TOWARD MORE EFFICIENT AND COST-EFFECTIVE NOVEL DISEASE GENE DISCOVERY
The genetic diagnosis process becomes more complex when the candidate gene or variant has not yet been linked to any disease. In response, we designed the AIM-NDG model, by eliminating all features that are directly or indirectly connected to established disease databases such as OMIM, ClinVar, and the Human Gene Mutation Database (HGMD).45 Consequently, 50% of the features were discarded, leading to a noticeable decrease in accuracy (Fig. 5E). Despite this reduction, the performance of AIM-NDG remains comparable to that of other benchmarked tools that do use features from these curated disease databases (Fig. 5E).
To test the efficacy of our AIM-NDG algorithm in a real-world scenario, we searched for recently published novel disease genes and variants in the UDN data after the last disease database update of AIM (October 2022). Two novel disease genes were identified: MYCBP2 and TMEM161B. MYCBP2 was recently discovered in a cohort of eight patients with neurodevelopmental disorder characterized by neurobehavioral phenotypes and corpus callosum defects.46 TMEM161B was recently identified to regulate cerebral cortical gyration, sonic hedgehog signaling, and ciliary structures in the developing central nervous system.47 We employed our AIM-NDG algorithm for these two individuals using a default model and the recessive-specific model, yielding confident scores of 88 for MYCBP2:p.R2669X and 51 for TMEM161B:p.Leu327-Ser/c.800+5G>A (Fig. 5F, left panel). Based on a reference of 275 solved cases, we estimate that these two variants should rank within the top 3 and top 2, respectively (Fig. 5F, right panel). Our results demonstrate the potential of AIM-NDG in identifying possible novel disease genes and variants, even in real-world settings with limited patient data.
Discussion
AIM is a machine-learning model trained with over 3.5 million variant data points derived from thousands of diagnosed cases. We further created a Web interface (https://ai.marrvel.org) that enables users to submit cases and interactively review the results. Our interface provides the convenience of automatic extraction of HPO terms from clinical notes using ClinPhen.48 Users have the flexibility to refine the extracted HPO terms through ontology trees, allowing for more accurate and personalized results.
AIM uses a relatively loose filter for MAF (<0.01) and avoids stringent filters such as coding variants. In addition, AIM rescues common variants (MAF >0.01) that are annotated as pathogenic by databases such as ClinVar and HGMD. Therefore, AIM is capable of prioritizing challenging cases such as intronic variants that potentially affect splicing, or common variants (MAF >0.01) that linked to diseases with much milder phenotypes.
AIM has several limitations. Although AIM can process single-nucleotide variants and small insertions or deletions, it is not equipped to analyze structural variations or copy-number variations. Furthermore, AIM has been predominantly trained on cases with coding variants, which constrains its capacity to effectively prioritize noncoding variants. The training samples used exome sequencing as opposed to whole-genome sequencing, which detects a wider array of variants that include deep intronic, copy number variations, and structural variations. At present, the AIM interface employs ClinPhen for mapping clinical notes into phenotype terms. Yet, large language models such as PhenoBCBERT49 and PhenoGPT49 have shown superior performance. These may be considered for future integration into the AIM platform.
In conclusion, AIM is a machine-learning genetic diagnosis tool with the potential to discover novel disease genes. Its capacity to run analyses on thousands of samples within days makes periodic reanalysis of unsolved cases feasible and cost-effective. We envision that the approach and methodology presented here can be expanded and adopted for clinical use, providing a valuable resource for clinicians and researchers to identify and interpret genetic variations, ultimately improving patient outcomes.
Supplementary Material
Acknowledgments
Supported by the NIH Common Fund, through the Office of Strategic Coordination/Office of the National Institutes of Health (NIH) Director under award number(s) NIH P50HD103555 to Z.L.; NIH/NHGRI R01 HG011795 to H.J.B., M.F.W., and Z.L.; NIH/NINDS U54 NS093793 and U54 NS093793–07S2 to H.J.B., S.Y., and M.F.W.; NIH/NHGRI R35HG011311 to P.L.; U01HG007709, U01HG007942, U01HG007943, U01HG007530, U01HG007672, U01HG010218, U01HG007708, U01HG007703, U01HG007674, and U54NS093793 to UDN. This research is also supported by the Chan Zuckerberg Initiative (grant no. 2023–332162 to Z.L., S.Y., and H.J.B.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Z.L., D.M., C.L., and L.W. are partially supported by the Chao Endowment and the Huffington Foundation. Z.L., D.M., C.L., L.W., and H.J.B. are also supported by Jan and Dan Duncan Neurological Research Institute at Texas Children’s Hospital.
Footnotes
Disclosures
Author disclosures and other supplementary materials are available at ai.nejm.org.
References
- 1.Ng SB, Buckingham KJ, Lee C, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet 2010;42:30–35. DOI: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Church G Compelling reasons for repairing human germlines. N Engl J Med 2017;377:1909–1911. DOI: 10.1056/NEJMp1710370. [DOI] [PubMed] [Google Scholar]
- 3.Posey JE, O’Donnell-Luria AH, Chong JX, et al. Insights into genetics, human biology and disease gleaned from family based genomic studies. Genet Med 2019;21:798–812. DOI: 10.1038/s41436-018-0408-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 2013; 369:1502–1511. DOI: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Posey JE, Rosenfeld JA, James RA, et al. Molecular diagnostic experience of whole-exome sequencing in adult patients. Genet Med 2016;18:678–685. DOI: 10.1038/gim.2015.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Posey JE, Harel T, Liu P, et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med 2017;376:21–31. DOI: 10.1056/NEJMoa1516767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoskinson DC, Dubuc AM, Mason-Suares H. The current state of clinical interpretation of sequence variants. Curr Opin Genet Dev 2017;42:33–39. DOI: 10.1016/j.gde.2017.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boycott KM, Vanstone MR, Bulman DE, MacKenzie AE. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat Rev Genet 2013;14:681–691. DOI: 10.1038/nrg3555. [DOI] [PubMed] [Google Scholar]
- 9.Trujillano D, Bertoli-Avella AM, Kumar Kandaswamy K, et al. Clinical exome sequencing: results from 2819 samples reflecting 1000 families. Eur J Hum Genet 2017;25:176–182. DOI: 10.1038/ejhg.2016.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bamshad MJ, Nickerson DA, Chong JX. Mendelian gene discovery: fast and furious with no end in sight. Am J Hum Genet 2019;105:448–455. DOI: 10.1016/j.ajhg.2019.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wright CF, FitzPatrick DR, Firth HV. Paediatric genomics: diagnosing rare disease in children. Nat Rev Genet 2018;19:253–268. DOI: 10.1038/nrg.2017.116. [DOI] [PubMed] [Google Scholar]
- 12.Liu P, Meng L, Normand EA, et al. Reanalysis of clinical exome sequencing data. N Engl J Med 2019;380:2478–2480. DOI: 10.1056/NEJMc1812033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wenger AM, Guturu H, Bernstein JA, Bejerano G. Systematic reanalysis of clinical exome data yields additional diagnoses: implications for providers. Genet Med 2017;19:209–214. DOI: 10.1038/gim.2016.88. [DOI] [PubMed] [Google Scholar]
- 14.Flygare S, Hernandez EJ, Phan L, et al. The VAAST Variant Prioritizer (VVP): ultrafast, easy to use whole genome variant prioritization tool. BMC Bioinformatics 2018;19:57. DOI: 10.1186/s12859-018-2056-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smedley D, Jacobsen JOB, Jäger M, et al. Next-generation diagnostics and disease-gene discovery with the Exomiser. Nat Protoc 2015;10:2004–2015. DOI: 10.1038/nprot.2015.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Javed A, Agrawal S, Ng PC. Phen-Gen: combining phenotype and genotype to analyze rare disorders. Nat Methods 2014;11:935–937. DOI: 10.1038/nmeth.3046. [DOI] [PubMed] [Google Scholar]
- 17.Zemojtel T, Köhler S, Mackenroth L, et al. Effective diagnosis of genetic disease by computational phenotype analysis of the disease-associated genome. Sci Transl Med 2014;6:252ra123. DOI: 10.1126/scitranslmed.3009262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang H, Robinson PN, Wang K. Phenolyzer: phenotype-based prioritization of candidate genes for human diseases. Nat Methods 2015;12:841–843. DOI: 10.1038/nmeth.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smedley D, Schubach M, Jacobsen JOB, et al. A whole-genome analysis framework for effective identification of pathogenic regulatory variants in Mendelian disease. Am J Hum Genet 2016;99:595–606. DOI: 10.1016/j.ajhg.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Q, Zhao K, Bustamante CD, Ma X, Wong WH. Xrare: a machine learning method jointly modeling phenotypes and genetic evidence for rare disease diagnosis. Genet Med 2019;21:2126–2134. DOI: 10.1038/s41436-019-0439-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robinson PN, Ravanmehr V, Jacobsen JOB, et al. Interpretable clinical genomics with a likelihood ratio paradigm. Am J Hum Genet 2020;107:403–417. DOI: 10.1016/j.ajhg.2020.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Birgmeier J, Haeussler M, Deisseroth CA, et al. AMELIE speeds Mendelian diagnosis by matching patient phenotype and genotype to primary literature. Sci Transl Med 2020;12:eaau9113. DOI: 10.1126/scitranslmed.aau9113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De La Vega FM, Chowdhury S, Moore B, et al. Artificial intelligence enables comprehensive genome interpretation and nomination of candidate diagnoses for rare genetic diseases. Genome Med 2021;13:153. DOI: 10.1186/s13073-021-00965-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Brien TD, Campbell NE, Potter AB, Letaw JH, Kulkarni A, Richards CS. Artificial intelligence (AI)-assisted exome reanalysis greatly aids in the identification of new positive cases and reduces analysis time in a clinical diagnostic laboratory. Genet Med 2022;24:192–200. DOI: 10.1016/j.gim.2021.09.007. [DOI] [PubMed] [Google Scholar]
- 25.Meng L, Attali R, Talmy T, et al. Evaluation of an automated genome interpretation model for rare disease routinely used in a clinical genetic laboratory. Genet Med 2023;25:100830. DOI: 10.1016/j.gim.2023.100830. [DOI] [PubMed] [Google Scholar]
- 26.Smedley D, Robinson PN. Phenotype-driven strategies for exome prioritization of human Mendelian disease genes. Genome Med 2015;7:81. DOI: 10.1186/s13073-015-0199-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eilbeck K, Quinlan A, Yandell M. Settling the score: variant prioritization and Mendelian disease. Nat Rev Genet 2017;18:599–612. DOI: 10.1038/nrg.2017.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kelly C, Szabo A, Pontikos N, et al. Phenotype-aware prioritization of rare Mendelian disease variants. Trends Genet 2022;38:1271–1283. DOI: 10.1016/j.tig.2022.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuan X, Wang J, Dai B, et al. Evaluation of phenotype-driven gene prioritization methods for Mendelian diseases. Brief Bioinform 2022;23:bbac019. DOI: 10.1093/bib/bbac019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jacobsen JOB, Kelly C, Cipriani V, et al. Phenotype-driven approaches to enhance variant prioritization and diagnosis of rare disease. Hum Mutat 2022;43:1071–1081. DOI: 10.1002/humu.24380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang J, Al-Ouran R, Hu Y, et al. MARRVEL: integration of human and model organism genetic resources to facilitate functional annotation of the human genome. Am J Hum Genet 2017;100:843–853. DOI: 10.1016/j.ajhg.2017.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gahl WA, Tifft CJ. The NIH undiagnosed diseases program: lessons learned. JAMA 2011;305:1904–1905. DOI: 10.1001/jama.2011.613. [DOI] [PubMed] [Google Scholar]
- 33.Firth HV, Wright CF. The Deciphering Developmental Disorders (DDD) study. Dev Med Child Neurol 2011;53:702–703. DOI: 10.1111/j.1469-8749.2011.04032.x. [DOI] [PubMed] [Google Scholar]
- 34.Wright CF, Campbell P, Eberhardt RY, et al. Genomic diagnosis of rare pediatric disease in the United Kingdom and Ireland. N Engl J Med 2023;388:1559–1571. DOI: 10.1056/NEJMoa2209046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McLaren W, Gil L, Hunt SE, et al. The Ensembl variant effect predictor. Genome Biol 2016;17:122. DOI: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.MacDonald JR, Ziman R, Yuen RKC, Feuk L, Scherer SW. The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic Acids Res 2014;42:D1: D986–D992. DOI: 10.1093/nar/gkt958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Firth HV, Richards SM, Bevan AP, et al. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet 2009;84:524–533. DOI: 10.1016/j.ajhg.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Landrum MJ, Lee JM, Riley GR, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 2014;42:D1:D980–D985. DOI: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amberger J, Bocchini CA, Scott AF, Hamosh A. McKusick’s Online Mendelian Inheritance in Man (OMIM). Nucleic Acids Res 2009; 37(Database issue):D793–D796. DOI: 10.1093/nar/gkn665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pedregosa F, Varoquaux G, Gramfort A, et al. Scikit-learn: machine learning in Python. J Mach Learn Res 2011;12:2825–2830. [Google Scholar]
- 41.Tosco-Herrera E, Muñoz-Barrera A, Jáspez D, et al. Evaluation of a whole-exome sequencing pipeline and benchmarking of causal germline variant prioritizers. Hum Mutat 2022;43:2010–2020. DOI: 10.1002/humu.24459. [DOI] [PubMed] [Google Scholar]
- 42.Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, et al. Predicting splicing from primary sequence with deep learning. Cell 2019;176:535–548.e24. DOI: 10.1016/j.cell.2018.12.015. [DOI] [PubMed] [Google Scholar]
- 43.Virtanen P, Gommers R, Oliphant TE, et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods 2020;17:261–272. DOI: 10.1038/s41592-019-0686-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tepe B, Macke EL, Niceta M, et al. Bi-allelic variants in INTS11 are associated with a complex neurological disorder. Am J Hum Genet 2023;110:774–789. DOI: 10.1016/j.ajhg.2023.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet 2014;133:1–9. DOI: 10.1007/s00439-013-1358-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.AlAbdi L, Desbois M, Rusnac DV, et al. Loss-of-function variants in MYCBP2 cause neurobehavioural phenotypes and corpus callosum defects. Brain 2023;146:1373–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Akula SK, Marciano JH, Lim Y, et al. TMEM161B regulates cerebral cortical gyration, Sonic Hedgehog signaling, and ciliary structure in the developing central nervous system. Proc Natl Acad Sci USA 2023;120:e2209964120. DOI: 10.1073/pnas.2209964120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deisseroth CA, Birgmeier J, Bodle EE, et al. ClinPhen extracts and prioritizes patient phenotypes directly from medical records to expedite genetic disease diagnosis. Genet Med 2019;21:1585–1593. DOI: 10.1038/s41436-018-0381-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang J, Liu C, Deng W, et al. Enhancing phenotype recognition in clinical notes using large language models: PhenoBCBERT and PhenoGPT. Patterns N Y 2023;5:100887. DOI: 10.1016/j.patter.2023.100887. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.