

Abstract
Objectives
Normally, VWF remains inactive unless its A1A2 domains undergo a shear stress-triggered conformational change. We demonstrated the capacity of a recombinant A2 domain of VWF to bind and to affect fibrin formation, altering the fibrin clot structure. The data indicated that VWF contains an additional binding site for fibrin in the A2 domain that plays a role in the incorporation of VWF to the polymerizing fibrin. This study is to examine the hypothesis that active plasma VWF directly influence fibrin polymerization and the structure of fibrin clots.
Methods
The study used healthy and type 3 VWD plasma, purified plasma VWF, fibrin polymerization assays, confocal microscopy and scanning electron microscopy.
Results
The exposed A2 domain in active VWF harbors additional binding sites for fibrinogen, and significantly potentiates fibrin formation (p<0.02). Antibody against the A2 domain of VWF significantly decreased the initial rate of change of fibrin formation (p<0.002). Clot analyses revealed a significant difference in porosity between normal and type 3 VWD plasma (p<0.008), further supported by scanning electron microscopy, which demonstrated thicker fibrin fibers in the presence of plasma VWF (p<0.0003). Confocal immunofluorescence microscopy showed punctate VWF staining along fibrin fibrils, providing visual evidence of the integration of plasma VWF into the fibrin matrix.
Conclusion
The study with type 3 VWD plasma supports the hypothesis that plasma VWF directly influences fibrin polymerization and clot structure. Additionally, a conformational change in the A1A2 domains exposes a hidden fibrin(ogen) binding site, indicating that plasma VWF determines the fibrin clot structure.
Keywords: von Willebrand factor, von Willebrand disease, Fibrinogen, Fibrin Clot, scanning electron microscopy
Introduction:
von Willebrand factor (VWF) is a plasma glycoprotein that mediates platelet adhesion at sites of vascular injury and contributes to the process of hemostasis [1, 2]. The interaction between VWF and platelets under high shear stress has been shown to be critical in arterial thrombosis [3–5] and recent evidence suggests that VWF also plays a role in venous thrombosis [6, 7].
In normal conditions, VWF does not interact with circulating platelets in plasma. However, under conditions of high shear stress the A1 domain of VWF undergoes a conformational change which results in its activation [8, 9]. Note that this conformational change in the A1 domain is coupled to an additional change in its neighboring A2 domain [10]. Conformational changes in these two A domains can also be induced via immobilization of VWF, gain-of-function mutations or, biochemically by ristocetin [8, 10–12].
Fibrinogen is another essential protein for hemostasis and thrombosis. The conversion of fibrinogen to fibrin by thrombin results in an insoluble end-product of the coagulation pathway [13]. Several plasma proteins have been identified which contribute to the formation, stability, and structure of the fibrin mesh within a clot [14–16]. Clinical studies suggest that alterations in the fibrin clot profile might be directly related to several clinical pathologies associated with bleeding and thrombosis [17, 18].
It is well established that VWF interacts with fibrin(ogen). Previous studies have demonstrated VWF’s capacity for mediating platelet adhesion to immobilized fibrin under conditions of high shear stress [19, 20]. Further studies have indicated that VWF binds to immobilized fibrin at binding sites located on VWF’s C domains [20]. Patients suffering from von Willebrand Disease (VWD) present with qualitative and/or quantitative deficiencies in VWF [21]. Interestingly, clinical studies have reported that a number of these patients exhibit diminished capacity for fibrin formation [22, 23], suggesting a direct involvement of VWF in fibrin polymerization [24].
A number of naturally occurring mutations associated with VWD have been identified within the A1A2 domains of VWF [21]. We previously demonstrated that the recombinant A2 domain of VWF binds to fibrin, plays an active role in fibrin formation, and can alter the fibrin clot structure [25, 26]. These outcomes implied that VWF contains an additional binding site for fibrin in the A2 domain, which may play a role in the incorporation of VWF to the polymerizing fibrin. Since the involvement of VWF in fibrin clot formation is still uncertain, we employed plasma from individuals with type 3 VWD to examine the hypothesis that VWF has a direct impact on fibrin polymerization and the structure of fibrin clots. Additionally, we utilized the recombinant wild-type A1A2A3 (WT) domains protein and the R1450E gain-of-function A1A2A3 (GOF) variant, (which consistently exposes A1A2 domains, mimicking the active form of VWF [27]), to demonstrate the interaction between fibrinogen and VWF, and fibrin polymerization. Here we demonstrate that a conformational change occurs in the A1A2 domains of VWF that leads to the exposure of a cryptic fibrin(ogen) binding site that is determinant in fibrin clot structure.
Methods
Type 3 VWD and healthy plasma donors
The plasma samples from type 3 VWD used in this study were obtained from a human donor that has markedly reduced levels of factor VIII (FVIII) and VWF (George King Bio-Medical, Inc, cat# 1403, lot 6825, FVIII = 4%, VWF = <1%). To obtain healthy human blood, informed consent was provided based on the recommendations of the Declaration of Helsinki. Approval was attained from the Baylor College of Medicine institutional review board (IRB) the Research & Development Committee of the Michael E. DeBakey VA Medical Center for these studies.
Reagents
Human fibrinogen from Calbiochem (Gibbstown, NJ), Anti-A2 (MAB27642, detects sequence D1596-Y1605 of VWF-A2 domain, R&D) and thrombin from Sigma (St. Louis, MO) was used for fibrin polymerization. For fibrin degradation we used tPA (Tissue plasminogen activator) from Cathflo Activase. Ristocetin was obtained from Bio/Data (Horsham, PA). Plasma VWF was obtained as described [28].
Binding assays.
The analyses of the interaction of purified VWF or A1A2A3 proteins with fibrin(ogen), which exposes fibrin-specific sequences upon surface adsorption, were performed by enzyme-linked immunosorbent assay (ELISA) as described elsewhere [25, 26]. Briefly, the wells of microtiter plate were coated with fibrinogen (5 μg/ml) in 50 mM carbonate buffer, pH 9.6, and blocked with 3% (w/v) bovine serum albumin (BSA). Following incubation and washing, the bound proteins were detected by using either rabbit polyclonal anti-VWF antibody horseradish peroxidase (HRP) conjugate (Dako) or rabbit polyclonal anti-VWF antibody (Abcam) followed by a secondary antibody horseradish peroxidase (HRP) conjugate.
Fibrin porosity
Fibrin clot was visualized with confocal microscopy and porosity was quantified using Image J software as previously detailed [29], [26].
Fibrin polymerization assays
Fibrin formation and degradation were performed as we described [26]. Anti-A2 was supplemented to fibrin formation reaction as a concentration of 20 ug/ml to monitor the effect in fibrin formation.
Imaging of the fibrin-clot structure
Briefly, plasma was supplemented with 2 % (w/w) of human fibrinogen conjugated to Alexa Fluor 488 (Thermo Scientific). Clot formation was initiated with the addition of 1 U thrombin (EMD Millipore) in the presence of 2.4 mM calcium as described[26].
For VWF interaction, plasma was supplemented with 2 % (w/w) of human fibrinogen conjugated to Alexa Fluor 647 (Thermo Scientific) and 1 U thrombin (EMD Millipore) in the presence of 2.4 mM calcium as described[26]. Fibrin clots were washed one time with 1X phosphate buffered saline (PBS) and incubated immediately with anti-human VWF FITC (ab8822) conjugate for 45 min. Then, fibrin-clot was washed three times with PBS and fixed with 4 % paraformaldehyde for 10 min and washed three times for visualization by confocal microscopy.
Scanning Electron Microscopy
For imaging via scanning electron microscopy (SEM), clots were fixed in 2.5% fresh glutaraldehyde in 100 mM sodium cacodylate buffer for 2 days at 4°C. Samples were then washed in cacodylate buffer prior to dehydration through an acetone series before transferring to hexamethyldisilizane (HMDS, Sigma-Aldrich, Saint Louis, MO, USA) in a fume hood. Samples were then dried overnight prior to gold-coating using a Denton Desk-II Vacuum Sputtering Device equipped with a standard gold foil target. Samples were imaged using a Mira 3 Field Emission Scanning Electron Microscope (Tescan, Pittsburg, PA). Imaging was conducted under a high vacuum (0.047 Pa) using a Schottky emitter and an accelerating voltage of 15 keV resulting in an imaging resolution (spot size) of 3.9 nm. The fibrin clot diameter analysis was performed using ImageJ.
Statistical analysis
GraphPad Prism 8 software (San Diego, CA) was used to perform statistical analyses. Comparisons between groups were conducted by t-test or ANOVA. P values were 2-sided, and statistical significance was determined by a P value <0.05.
Results
Exposure of the A2 domain increments the binding of VWF to immobilized fibrin(ogen) and enhances fibrin formation potential.
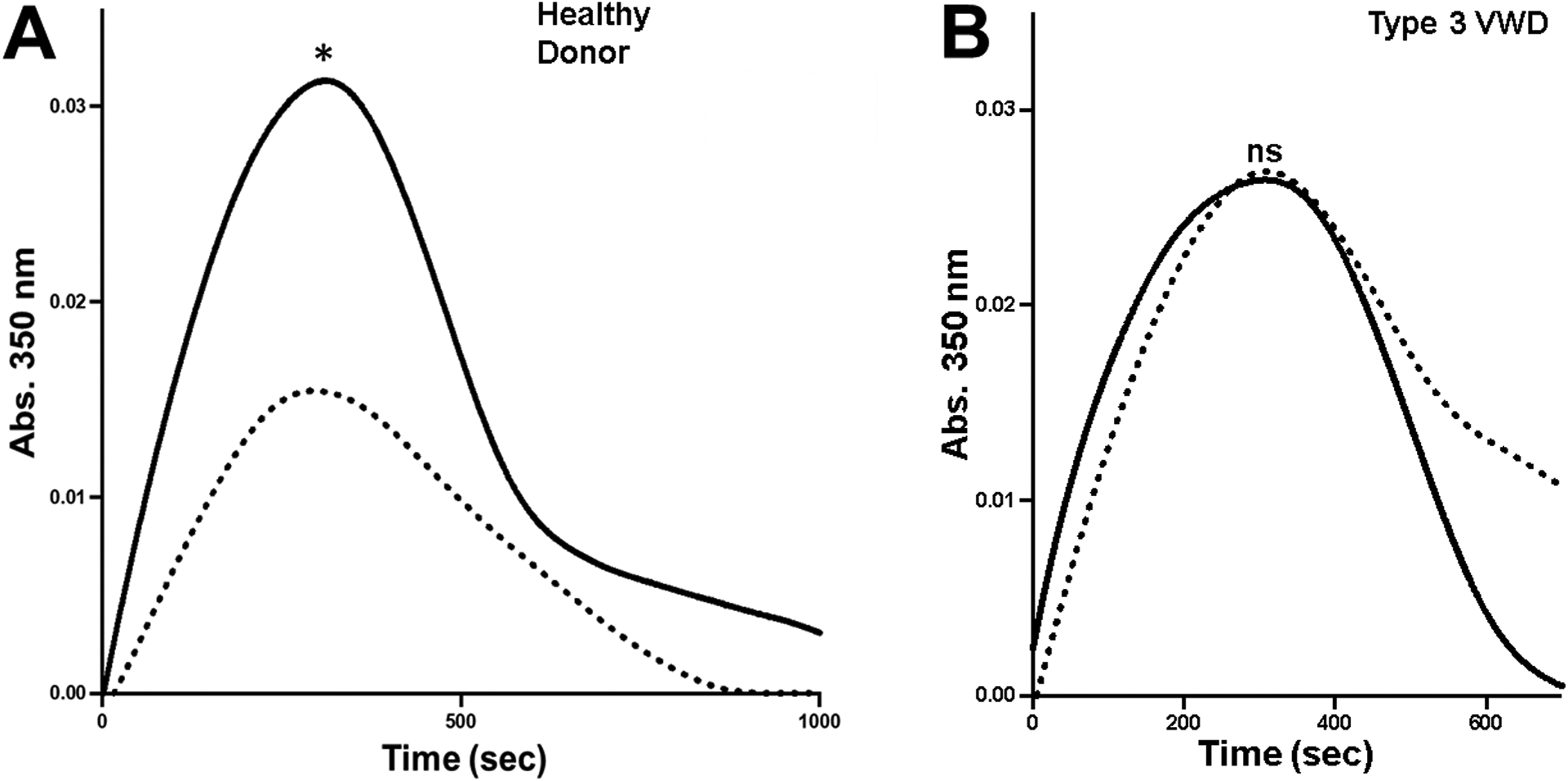
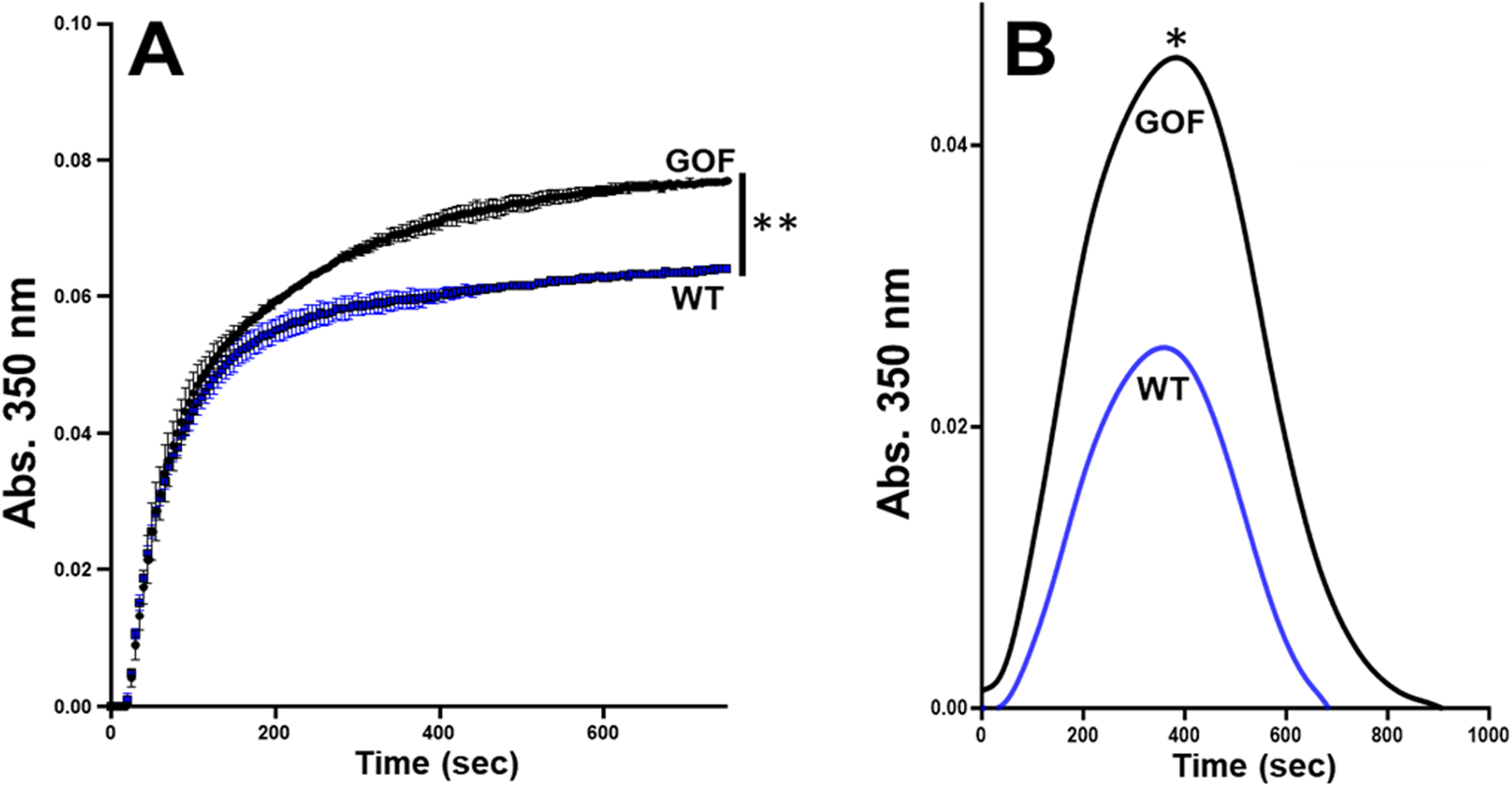
First, we examined the effect of ristocetin on the interaction between plasma VWF and fibrinogen-coated surface. As shown in figure 1A, ristocetin increased the reactivity of VWF in plasma for fibrinogen. Moreover, figure 1B shows that purified plasma VWF in the presence of ristocetin bound to fibrinogen in a concentration-dependent and saturable manner and attained a half-maximal binding at ~1.0 μg/ml (or 4 nM, based on the MW of a VWF subunit), comparable to purified plasma VWF in the absence of ristocetin (~0.8 μg/ml). Next, we tested the effect of ristocetin on VWF in fibrin formation and degradation with healthy plasma. As shown in figure 2A, ristocetin significantly potentiated fibrin formation (increased maximal absorbance) by ~100% over control. In contrast, ristocetin did not affect fibrin formation in the VWF deficient plasma (Fig. 2B), indicating that the effect of ristocetin in healthy plasma (Fig. 2A) was not potentiated via a direct interaction with fibrinogen. To further exclude the artificial effect of ristocetin, which flocculates proteins [30], and the VWF C domains that express a binding site for fibrin, we employed our recombinant gain-of-function (GOF) A1(R1450E)A2A3 mutant. This mutant is expressed and purified as a soluble, non-aggregated monomer; constitutively exposes the A1A2 domains; and binds to platelet GPIb without the need of ristocetin [27]. The mutation R1450E reduced inter-domain interaction among the three A domains, exposing the A2 domain. Figures S1A and S1B in Supplemental Materials show that both wild type (WT) and GOF proteins bound to fibrinogen with comparable half-maximal binding constant (87 ± 97 nM SD vs. 63 ± 32 nM SD, respectively, n=3, p=0.71). However, the A1A2A3 GOF protein significantly increased fibrin formation potential by ~ 60% (maximal absorbance) over WT protein in a purified system (Fig. S1C, in supplemental materials). We further tested the effect of the A1A2A3 GOF protein in fibrin formation and degradation ex vivo in type 3 VWD plasma without ristocetin. In comparison to WT protein, figures 3A and 3B show that the mutant protein significantly increased fibrin polymerization (max absorbance) in a ristocetin-independent manner. Although it was less pronounced, the effect of the mutant protein in the VWF-deficient plasma was like that of ristocetin in healthy plasma (Fig. 2A). Next, we tested the hypothesis of functional interaction between A2 domain of VWF and fibrinogen by measuring fibrin formation in the presence of ristocetin and anti-A2 domain antibody. In comparison with IgG, figure 4 shows that the anti-A2 antibody significantly decreased the initial rate of change (slope), 2.5 × 10−4 vs. 1.9 × 10−4, p<0.002. The initial rate of change was determined from the slope of the line at the midpoint between initial baseline and maximum absorbance as described [26]. In contrast, although the polyclonal anti-VWF antibody did not change the initial rate of change, it slightly reduced (not significant) the maximal absorbance in healthy plasma (Fig. S2A, in supplemental materials). As expected, the same antibody had no effect in fibrin formation in type 3 VWD plasma (Fig. S2B, in supplemental materials). Together, these results demonstrated that the exposed A2 domain in VWF contains another contact site(s) for fibrinogen that appears to positively affect fibrin formation.
Figure 1. Binding of VWF to fibrinogen.
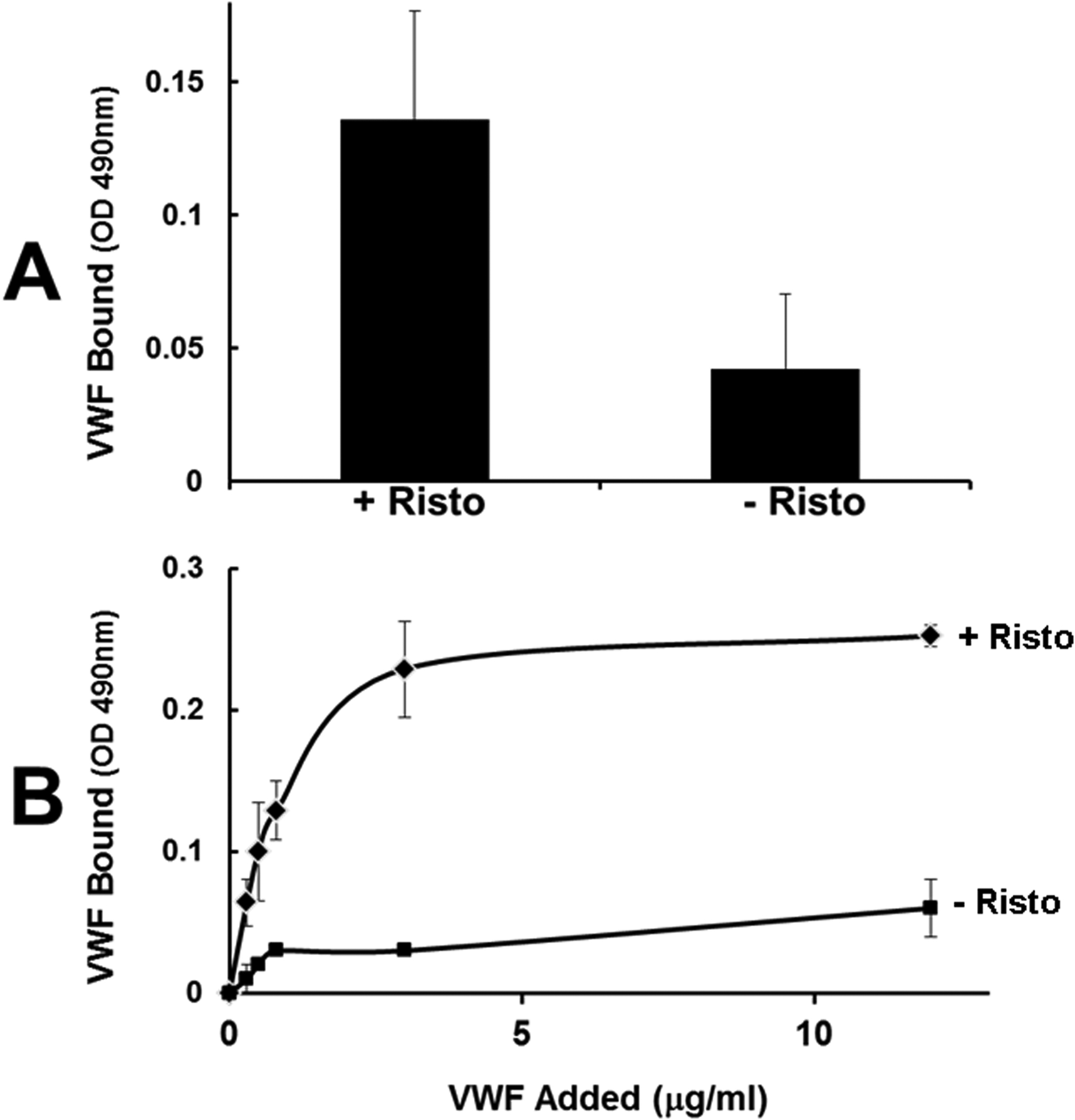
(A) Wells of microtiter plates were coated with fibrinogen (5 μg/ml) and incubated with diluted plasma (1:10) in the presence or absence of ristocetin (0.5 mg/ml) for 1 hour. After washing, the bound VWF was detected by an ELISA using peroxidase-labeled anti VWF (p< 0.05). (B) Purified plasma VWF at indicated concentrations with (top curve, solid diamonds) or without (lower curve, solid squares) ristocetin were added to fibrinogen-coated plates, and bound VWF was quantified by ELISA. The binding isotherm represents mean and standard deviations of three separate determinations.
Figure 2. Active VWF enhances fibrin formation potential.
(A) Fibrin formation and fibrinolysis using healthy donor was initiated with thrombin, calcium, and in the presence of recombinant tissue-type plasminogen activator, with (0.5 mg/ml) (solid lines) or without ristocetin (dash lines). Turbidity was measured at 350 nm. Curves are representative for two separate experiments in duplicate reactions and each curve is the mean of duplicate reactions. Measurements of both fibrin formation and fibrinolysis were presented as the area under the curve. The addition of ristocetin significantly increased fibrin formation in the healthy plasma (*p<0.02). (B) Fibrin formation and fibrinolysis using type 3 VWD plasma with (0.5 mg/ml) (solid lines) or without ristocetin (dash lines) as in figure 2A. The graph represents curves of two separate experiments and each curve is the mean of duplicate reactions, p=0.5.
Figure 3. A1A2A3 GOF protein enhances fibrin formation potential in type 3 VWD plasma.
(A) The effect of the A1A2A3 proteins (40 ug/ml) on fibrin formation in the absence of ristocetin was examined ex vivo in VWF-deficient plasma. Turbidity was measured at 350 nm. Each point in represents the mean S.E.M. (error bars) of values obtained from each curve. **p=0.0027. (B) As described in figure 2, the A1A2A3 proteins were examined for their effect in fibrin formation and fibrinolysis in VWF-deficient plasma in the absence of ristocetin. The GOF protein increased maximal absorbance, *p=0.051. Graphs A and B are representative curves of three separate experiments of triplicate reactions.
Figure 4. Anti-A2 domain antibody decreases fibrin formation.

Healthy donor plasma was used to examine the effect of anti-A2 antibody (20 μg/ml) on fibrin formation in the presence of ristocetin (0.5 mg/ml). Turbidity was measured at 350 nm. Representative curves for fibrin formation and each curve is the mean of triplicate reactions. The antibody significantly decreased the initial rate of change (slope), 2.5 × 10−4 vs. 1.9 × 10−4, p<0.002.
Deficiency of plasma VWF alters the fibrin clot structure.
We then investigated the effect of VWF on the resultant fibrin network structure. We utilized plasma deficient in VWF (type 3 VWD) to assess the biological function of VWF in fibrin structure. Figure 5A shows representative clot structures generated from plasma of healthy subjects or type 3 VWD without ristocetin. An evident and significant difference in the porosity area (area unoccupied by fibrin formed from plasma) was observed between clots formed in healthy or type 3 VWD plasma (Fig. 5B). Additionally, for a clearer understanding of these fibrin structures, we performed scanning electron microscopy (SEM) analysis. Figure 5C illustrates that plasma VWF provoked the formation of thicker fibrin fiber diameter compared to that of VWF-deficient plasma (Fig. 5D). The changes observed in the clot architecture suggested that VWF could bind fibrin(ogen) and be incorporated into the resultant network. To test this postulate, we visualized fibrin associated VWF from healthy plasma by confocal immunofluorescence microscopy. Punctate staining for VWF was found distributed along the length and branching points of the fibrin fibrils (Fig. 6, center panel), illustrating the incorporation of plasma VWF into fibrin.
Figure 5. Deficiency of plasma VWF alters porosity and fibrin fibers diameter of the fibrin network.
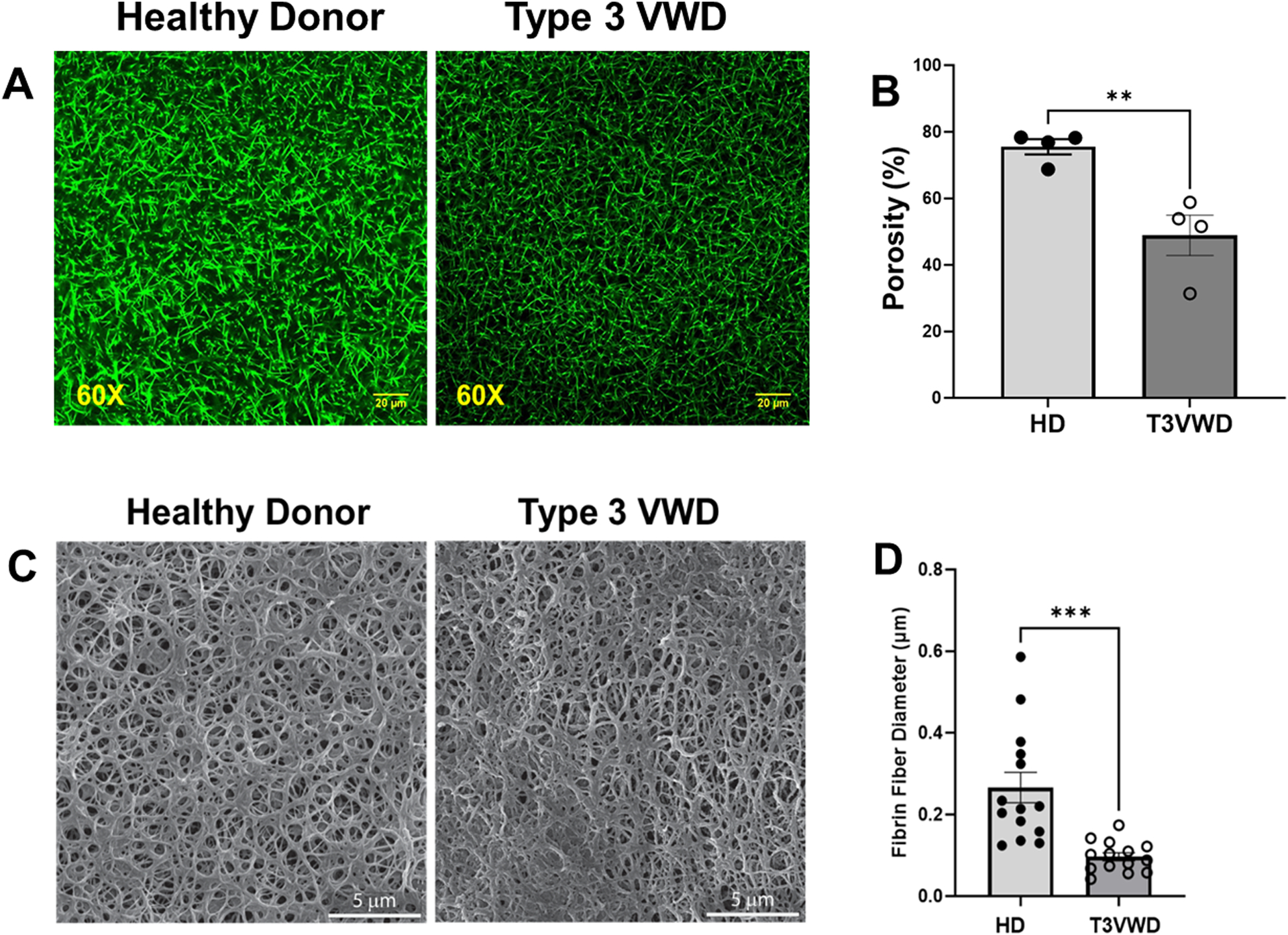
(A) Representative confocal microscopy images of fibrin clots at magnification of 60× formed in plasma from healthy donor (HD, n=4) or type 3 VWD (T3VWD, n=4) patient without ristocetin. In the absence of VWF, the fibrin pore size was decreased in the resultant fibrin meshwork (right panel). Scale bar, 20 μm. (B) Quantification of porous area as percentage of image field using Image J (n = 3 per condition). **p<0.008. (C). Scanning electron microscopy of fibrin clots from Healthy donors (HD) and type 3 VWD plasma in absence of ristocetin. Representative images from three different analysis, VWF affects the clot structure for both type 3 VWD and healthy (Scale bar = 5 μm, SEM MAG= 27.7 kx). (D) Quantification of fibrin fiber diameters was performed using Image J. T-test was performed by using GraphPad Prism, ***p< 0.0003.
Figure 6. Localization of endogenous VWF in plasma fibrin clot.

The confocal microscopy images show fibrin clots at magnification of 234X formed in plasma from healthy donor. Plasma was supplemented with human fibrinogen conjugated to Alexa Fluor 647. The left panel shows the fibrin mesh, and the middle panel shows the optical section through the plasma clot stained for VWF with anti-VWF FITC-conjugated antibody. The right panel shows a merge of fibrinogen and VWF stained in the clot. Note the staining pattern for VWF along the fibrin fibrils (white arrows). Representative of three experiments. Scale bar, 5 μm.
Discussion:
It is established that VWF interacts with polymerizing fibrin [24, 31] with the VWF C domains reported to contain the determinant residues for VWF-fibrin interaction in supporting flow-dependent platelet adhesion to fibrin [20]. However, we previously described the effectiveness of a recombinant A2 domain of VWF to block both ristocetin-induced VWF binding to fibrinogen and flow-dependent platelet adhesion to immobilized fibrinogen at high shear stress [25]. This study examined the hypothesis that the interaction of full length VWF with fibrinogen also occurs via the exposed A2 domain.
Ristocetin induces simultaneous conformational changes in A1A2 domains of VWF that leads to the exposure of the cryptic sites for platelet GPIb (A1 domain) and ADAMTS-13 (A2 domain)[10]. We have shown that VWF binds to immobilized fibrinogen in a ristocetin dependent and independent manner with a half maximal binding of ~ 4.0 nM. This value was comparable to that reported by Keuren et. al. [20], who reported an apparent KD of 8.8 nM without ristocetin. The fibrin contact site located in the C domains may explain why VWF retained binding activity for immobilized fibrinogen in the absence of ristocetin since surface-adsorbed fibrinogen possibly adopts a conformation that functionally differs from soluble fibrinogen [32]. Moreover, it may also explain why recombinant A2 blocked only ~65% the binding of VWF to fibrinogen when treated with ristocetin [25]. We reason that the contribution of multiple VWF domains (A2 for immobilized fibrinogen and C for fibrin) increase the overall avidity of VWF for fibrin(ogen), which allows it to withstand the hydrodynamic forces while still allowing the recruitment of additional circulating platelets and the growth of a fibrin-rich hemostatic plug.
The notion that a cryptic site for immobilized fibrinogen or a soluble fibrinogen-to-fibrin intermediate is in the A2 domain of VWF is also supported by the fact that ristocetin and the A1A2A3 GOF protein, which constitutively exposes the A1A2 domains [27, 33], significantly augments fibrin formation (maximal absorbance) ex vivo in healthy and type 3 VWD plasma, respectively. These outcomes indicate that a conformational change within the A1A2 domains in VWF increases the avidity for fibrin(ogen). We reason that the ultra large (UL)VWF multimers, which constitutively expose A1A2 domains [28], have a differential binding activity for a cryptic site(s) in fibrinogen as compared to that of plasma VWF multimers. This speculation is interesting because endothelial cells secrete ULVWF multimers upon cell stimulation [34, 35], and therefore, further studies are required to investigate the role of these ULVWF multimers in fibrin polymerization during platelet aggregation (thrombus growth) [36, 37] or fibrin deposition on inflamed endothelium [38, 39]. A recent study reported the lack of VWF-fibrinogen interaction at a single molecule level and as indicated by the authors, the contradictory outcome may be due to the different methodology that they employed [40]. However, the same study also demonstrated a positive effect of endothelial-derived ULVWF in promoting fibrin formation with thrombin-treated fibrinogen on engineered microvessels. Thus, the results of our study may have clinical implications in conditions associated with systemic inflammation where elevated antigen levels of VWF reflects the active release of ULVWF, being a potential contributor to the pathogenesis of thromboinflammation via fibrin polymerization.
A previous study used purified fibrinogen and VWF to study the effect of VWF in fibrin polymerization and clot structure [41]. To our knowledge, our study is the first to employ VWF deficient plasma (type 3 VWD) to determine the biological function of VWF in fibrin formation and fibrin clot structure. The results of this study are in agreement with the observations reported by the group that used the purified system [41]. First, in comparison to healthy plasma, the failure of ristocetin or anti-VWF antibody to alter fibrin formation ex vivo in type 3 VWD plasma demonstrated the direct association that occurs between VWF and fibrin(ogen) in polymerizing fibrin. Additionally, we have shown that deficiency of plasma VWF significantly reduced both pore size of the fibrin network and fibrin fibers diameter as compared to the clot in healthy plasma. It is accepted that changes in the fibrin clot structure, including density, pore size, and fiber diameter are associated with a high risk for bleeding and thrombosis in certain diseases [42]. Therefore, structural modifications, absence, or abundance of plasma VWF may cause alterations in the fibrin clot structure, negatively influencing hemostasis.
We have reported the characterization of the recombinant VWF-A1A2A3 proteins and their similarities with full length VWF, including their capacity of mediating flow-dependent platelet adhesion and binding activity for A1 domain of VWF and vimentin [27, 33, 43]. Particularly, the A1A2A3 GOF protein, like ULVWF, bound to platelet GPIb without the need of ristocetin and it was readily cleaved by ADAMTS-13, demonstrating that it mimics the active form of VWF [27, 33]. We utilized the A1A2A3 GOF protein to demonstrate the presence of an additional cryptic contact site for fibrin(ogen) in the A2 domain of the active form of VWF that appears to be a determinant for fibrin formation. On the other hand, although the observations with GOF are consistent with polymerizing fibrinogen interacting with active VWF [40], multimeric VWF has a more robust, apparent response in fibrin formation possibly due to additional binding sites. This differential effect most likely is because the presence of the C domains in the full length VWF [20], the binding to fibrin is multimers size dependent [44] and full length VWF is cross-linked by factor (F)XIII to fibrin via amino acid residues located outside the A domains of VWF [45]. Altogether, the results obtained from using type 3 VWD plasma provides another potential explanation of why deficient or defective VWF increases the risk of a weak fibrin clot formation as described in patients with VWD [46].
Low FVIII levels accompany severe VWF deficiency as confirmed by the supplier of the type 3 VWD plasma (see Methods). A previous study demonstrated that FVIII binds to thrombin-treated, immobilized fibrinogen and that fibrin promotes tenase activity [47]. One can argue that FVIII may affect fibrin formation by multiple mechanisms. Although we bypassed tenase-mediated activation of prothrombin with direct addition of thrombin (Figures 2–6), we cannot exclude the possibility that insufficient FVIII within polymerizing fibrinogen contributes to a weak fibrin structure independently of its cofactor activity in type 3 VWD. However, there are no reports describing a direct role of FVIII in fibrin polymerization. This intriguing possibility deserves further investigation.
The binding capacity of the A2 domain of VWF for fibrin(ogen) has been established by several means. First, the purified recombinant A2 domain blocked ristocetin-induced VWF binding to fibrin(ogen), modulated fibrin polymerization, and altered fibrin network formation [25, 26]. Furthermore, like seen in healthy plasma (Figs. 5 and 6), the isolated A2 domain caused the formation of larger pores in the fibrin clot structure, and it incorporated directly into the fibrin clot network or fibrils [26]. Moreover, the effect of the anti-A2 antibody in decreasing the initial rate of change in fibrin formation and in moderately reducing the maximal absorbance ex vivo in healthy plasma demonstrated the role of the A2 domain in VWF in binding to fibrin(ogen) (Fig. 4). Lastly, we reported that an A2 (E1567A) mutant protein impaired the binding activity of recombinant A2 domain for fibrin. The result suggested that the alpha2-helix of the A2 domain may contain a fibrin(ogen)-binding site [26]. More studies are necessary to describe the putative binding site for fibrin in the A2 domain in the context of full-length VWF protein.
In summary, the results of this study validate the function of plasma VWF in fibrin polymerization and fibrin clot structure. Furthermore, the outcomes suggest the presence of a cryptic contact site for fibrin(ogen) within the A2 domain of VWF. Additionally, the data indicate that deficiency of plasma VWF causes structural changes in the fibrin clot network. One can propose that infusion of VWF to VWD patients not only stabilizes the circulating coagulation factor VIII and restores platelet adhesion to vascular injuries but also normalizes the polymerizing fibrin process independently of FVIII.
Supplementary Material
Acknowledgements
The Alkek Foundation, the Fondren Foundation (M.A.C.), American Society of Hematology Scholar Award (A.Y.), and NIH-NIGMS R01 GM112806 (M.A.C.). NIH-NHLBI R01 HL154688 (M.A.C. and A.Y.) and T32 HL139425 (M.M-V. and J.C.) and a Merit Review Award I01 BX002551 from the Department of Veterans Affairs Biomedical Laboratory Research & Development (R.E.R. and M.A.C.). The content is solely the responsibility of the authors and does not represent the official views of National Institutes of Health, Department of Veterans Affairs or the United States government.
Footnotes
Conflicts of interest
M.A. Cruz is the founder and CSO of A2 Therapeutics, Inc.
References
- 1.Wu YP, Vink T, Schiphorst M, van Zanten GH, Ijsseldijk MJ, de Groot PG, et al. : Platelet thrombus formation on collagen at high shear rates is mediated by von Willebrand factor-glycoprotein Ib interaction and inhibited by von Willebrand factor-glycoprotein IIb/IIIa interaction. Arterioscler Thromb Vasc Biol 2000, 20:1661–1667. [DOI] [PubMed] [Google Scholar]
- 2.Fressinaud E, Federici AB, Castaman G, Rothschild C, Rodeghiero F, Baumgartner HR, et al. : The role of platelet von Willebrand factor in platelet adhesion and thrombus formation: a study of 34 patients with various subtypes of type I von Willebrand disease. Br J Haematol 1994, 86:327–332. [DOI] [PubMed] [Google Scholar]
- 3.Shahidi M: Thrombosis and von Willebrand Factor. Adv Exp Med Biol 2017, 906:285–306. [DOI] [PubMed] [Google Scholar]
- 4.Dhanesha N, Prakash P, Doddapattar P, Khanna I, Pollpeter MJ, Nayak MK, et al. : Endothelial Cell-Derived von Willebrand Factor Is the Major Determinant That Mediates von Willebrand Factor-Dependent Acute Ischemic Stroke by Promoting Postischemic Thrombo-Inflammation. Arterioscler Thromb Vasc Biol 2016, 36:1829–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nieswandt B, Pleines I, Bender M: Platelet adhesion and activation mechanisms in arterial thrombosis and ischaemic stroke. J Thromb Haemost 2011, 9 Suppl 1:92–104. [DOI] [PubMed] [Google Scholar]
- 6.Michels A, Lillicrap D, Yacob M: Role of von Willebrand factor in venous thromboembolic disease. JVS Vasc Sci 2022, 3:17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edvardsen MS, Hansen ES, Ueland T, Aukrust P, Brækkan SK, Morelli VM, et al. : Impact of the von Willebrand factor-ADAMTS-13 axis on the risk of future venous thromboembolism. J Thromb Haemost 2023, 21:1227–1237. [DOI] [PubMed] [Google Scholar]
- 8.Miyata S, Goto S, Federici AB, Ware J, Ruggeri ZM: Conformational changes in the A1 domain of von Willebrand factor modulating the interaction with platelet glycoprotein Ibalpha. J Biol Chem 1996, 271:9046–9053. [DOI] [PubMed] [Google Scholar]
- 9.Hillery CA, Mancuso DJ, Evan SJ, Ponder JW, Jozwiak MA, Christopherson PA, et al. : Type 2M von Willebrand disease: F606I and I662F mutations in the glycoprotein Ib binding domain selectively impair ristoc. Blood 1998, 91:1572–1581. [PubMed] [Google Scholar]
- 10.Chen J, Ling M, Fu X, Lopez JA, Chung DW: Simultaneous Exposure of Sites in von Willebrand Factor for Glycoprotein Ib Binding and ADAMTS13 Cleavage: Studies With Ristocetin. Arterioscler Thromb Vasc Biol 2012, 32:2625–2630. [DOI] [PubMed] [Google Scholar]
- 11.Fu X, Chen J, Gallagher R, Zheng Y, Chung DW, Lopez JA: Shear stress-induced unfolding of VWF accelerates oxidation of key methionine residues in the A1A2A3 region. Blood 2011, 118:5283–5291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Savage B, Saldivar E, Ruggeri ZM: Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell 1996, 84:289–297. [DOI] [PubMed] [Google Scholar]
- 13.Kattula S, Byrnes JR, Wolberg AS: Fibrinogen and Fibrin in Hemostasis and Thrombosis. Arterioscler Thromb Vasc Biol 2017, 37:e13–e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.King RJ, Schuett K, Tiede C, Jankowski V, John V, Trehan A, et al. : Fibrinogen interaction with complement C3: a potential therapeutic target to reduce thrombosis risk. Haematologica 2021, 106:1616–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Podor TJ, Campbell S, Chindemi P, Foulon DM, Farrell DH, Walton PD, et al. : Incorporation of vitronectin into fibrin clots. Evidence for a binding interaction between vitronectin and gamma A/gamma’ fibrinogen. J Biol Chem 2002, 277:7520–7528. [DOI] [PubMed] [Google Scholar]
- 16.Gaffney PJ: Fibrin degradation products. A review of structures found in vitro and in vivo. Ann N Y Acad Sci 2001, 936:594–610. [PubMed] [Google Scholar]
- 17.Zabczyk M, Ariens RAS, Undas A: Fibrin clot properties in cardiovascular disease: from basic mechanisms to clinical practice. Cardiovasc Res 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lisman T, Ariens RA: Alterations in Fibrin Structure in Patients with Liver Diseases. Semin Thromb Hemost 2016, 42:389–396. [DOI] [PubMed] [Google Scholar]
- 19.Endenburg SC, Hantgan RR, Lindeboom-Blokzijl L, Lankhof H, Jerome WG, Lewis JC, et al. : On the role of von Willebrand factor in promoting platelet adhesion to fibrin in flowing blood. Blood 1995, 86:4158–4165. [PubMed] [Google Scholar]
- 20.Keuren JF, Baruch D, Legendre P, Denis CV, Lenting PJ, Girma JP, et al. : von Willebrand factor C1C2 domain is involved in platelet adhesion to polymerized fibrin at high shear rate. Blood 2004, 103:1741–1746. [DOI] [PubMed] [Google Scholar]
- 21.Keeney S, Cumming AM: The molecular biology of von Willebrand disease. Clin Lab Haematol 2001, 23:209–230. [DOI] [PubMed] [Google Scholar]
- 22.Favaloro EJ: Von Willebrand disease: local diagnosis and management of a globally distributed bleeding disorder. Semin Thromb Hemost 2011, 37:440–455. [DOI] [PubMed] [Google Scholar]
- 23.Turitto VT, Weiss HJ, Baumgartner HR: Platelet interaction with rabbit subendothelium in von Willebrand’s disease: altered thrombus formation distinct from defective platelet adhesion. J Clin Invest 1984, 74:1730–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beguin S, Keularts I, al Dieri R, Bellucci S, Caen J, Hemker HC: Fibrin polymerization is crucial for thrombin generation in platelet-rich plasma in a VWF-GPIb-dependent process, defective in Bernard-Soulier syndrome. J Thromb Haemost 2004, 2:170–176. [DOI] [PubMed] [Google Scholar]
- 25.Nguyen TC, Gushiken F, Correa JI, Dong JF, Dasgupta SK, Thiagarajan P, et al. : A recombinant fragment of von Willebrand factor reduces fibrin-rich microthrombi formation in mice with endotoxemia. Thromb Res 2015, 135:1025–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Valladolid C, Martinez-Vargas M, Sekhar N, Lam F, Brown C, Palzkill T, et al. : Modulating the rate of fibrin formation and clot structure attenuates microvascular thrombosis in systemic inflammation. Blood Adv 2020, 4:1340–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Auton M, Sowa KE, Smith SM, Sedlak E, Vijayan KV, Cruz MA: Destabilization of the A1 domain in von Willebrand factor dissociates the A1A2A3 tri-domain and provokes spontaneous binding to glycoprotein Ibalpha and platelet activation under shear stress. J Biol Chem 2010, 285:22831–22839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arya M, Anvari B, Romo GM, Cruz MA, Dong JF, McIntire LV, et al. : Ultralarge multimers of von Willebrand factor form spontaneous high- strength bonds with the platelet glycoprotein Ib-IX complex: studies using optical tweezers. Blood 2002, 99:3971–3977. [DOI] [PubMed] [Google Scholar]
- 29.Brubaker LS, Saini A, Nguyen TC, Martinez-Vargas M, Lam FW, Yao Q, et al. : Aberrant Fibrin Clot Structure Visualized Ex Vivo in Critically Ill Patients With Severe Acute Respiratory Syndrome Coronavirus 2 Infection. Crit Care Med 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scott JP, Montgomery RR, Retzinger GS: Dimeric ristocetin flocculates proteins, binds to platelets, and mediates von Willebrand factor-dependent agglutination of platelets. J Biol Chem 1991, 266:8149–8155. [PubMed] [Google Scholar]
- 31.Loscalzo J, Inbal A, Handin RI: von Willebrand protein facilitates platelet incorporation in polymerizing fibrin. J Clin Invest 1986, 78:1112–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mosesson MW: Fibrinogen and fibrin structure and functions. J Thromb Haemost 2005, 3:1894–1904. [DOI] [PubMed] [Google Scholar]
- 33.Yago T, Lou J, Wu T, Yang J, Miner JJ, Coburn L, et al. : Platelet glycoprotein Ibalpha forms catch bonds with human WT vWF but not with type 2B von Willebrand disease vWF. J Clin Invest 2008, 118:3195–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McGrath RT, van den Biggelaar M, Byrne B, O’Sullivan JM, Rawley O, O’Kennedy R, et al. : Altered glycosylation of platelet-derived von Willebrand factor confers resistance to ADAMTS13 proteolysis. Blood 2013, 122:4107–4110. [DOI] [PubMed] [Google Scholar]
- 35.Dong JF, Moake JL, Nolasco L, Bernardo A, Arceneaux W, Shrimpton CN, et al. : ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood 2002, 100:4033–4039. [DOI] [PubMed] [Google Scholar]
- 36.Parker RI, Gralnick HR: Fibrin monomer induces binding of endogenous platelet von Willebrand factor to the glycocalicin portion of platelet glycoprotein IB. Blood 1987, 70:1589–1594. [PubMed] [Google Scholar]
- 37.Kumar RA, Moake JL, Nolasco L, Bergeron AL, Sun C, Dong JF, et al. : Enhanced platelet adhesion and aggregation by endothelial cell-derived unusually large multimers of von Willebrand factor. Biorheology 2006, 43:681–691. [PubMed] [Google Scholar]
- 38.Palabrica T, Lobb R, Furie BC, Aronovitz M, Benjamin C, Hsu YM, et al. : Leukocyte accumulation promoting fibrin deposition is mediated in vivo by P-selectin on adherent platelets. Nature 1992, 359:848–851. [DOI] [PubMed] [Google Scholar]
- 39.Massberg S, Enders G, Matos FC, Tomic LI, Leiderer R, Eisenmenger S, et al. : Fibrinogen deposition at the postischemic vessel wall promotes platelet adhesion during ischemia-reperfusion in vivo. Blood 1999, 94:3829–3838. [PubMed] [Google Scholar]
- 40.Rayner SG, Scholl Z, Mandrycky CJ, Chen J, LaValley KN, Leary PJ, et al. : Endothelial-derived von Willebrand factor accelerates fibrin clotting within engineered microvessels. J Thromb Haemost 2022, 20:1627–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marchi R, Rojas H: Effect of von Willebrand factor on clot structure and lysis. Blood Coagul Fibrinolysis 2015, 26:533–536. [DOI] [PubMed] [Google Scholar]
- 42.Mihalko E, Brown AC: Clot Structure and Implications for Bleeding and Thrombosis. Semin Thromb Hemost 2020, 46:96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Da Q, Behymer M, Correa JI, Vijayan KV, Cruz MA: Platelet adhesion involves a novel interaction between vimentin and von Willebrand factor under high shear stress. Blood 2014, 123:2715–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ribes JA, Francis CW: Multimer size dependence of von Willebrand factor binding to crosslinked or noncrosslinked fibrin. Blood 1990, 75:1460–1465. [PubMed] [Google Scholar]
- 45.Takagi J, Aoyama T, Ueki S, Ohba H, Saito Y, Lorand L: Identification of factor-XIIIa-reactive glutaminyl residues in the propolypeptide of bovine von Willebrand factor. Eur J Biochem 1995, 232:773–777. [PubMed] [Google Scholar]
- 46.De Wee EM, Klaij K, Eikenboom HC, Van Der Bom JG, Fijnvandraat K, Laros-Van Gorkom BA, et al. : Effect of fibrinolysis on bleeding phenotype in moderate and severe von Willebrand disease. Haemophilia 2012, 18:444–451. [DOI] [PubMed] [Google Scholar]
- 47.Gilbert GE, Novakovic VA, Shi J, Rasmussen J, Pipe SW: Platelet binding sites for factor VIII in relation to fibrin and phosphatidylserine. Blood 2015, 126:1237–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.