

Abstract

In this article, we report the development of ruthenium-catalyzed hydrogenolysis of epoxides to selectively give the branched (Markovnikov) alcohol products. In contrast to previously reported catalysts, the use of Milstein’s PNN-pincer-ruthenium complex at room temperature allows the conversion of enantiomerically enriched epoxides to secondary alcohols without racemization of the product. The catalyst is effective for a range of aryl epoxides, alkyl epoxides, and glycidyl ethers and is the first homogeneous system to selectively promote hydrogenolysis of glycidol to 1,2-propanediol, without loss of enantiomeric purity. A detailed mechanistic study was conducted, including experimental observations of catalyst speciation under catalytically relevant conditions, comprehensive kinetic characterization of the catalytic reaction, and computational analysis via density functional theory. Heterolytic hydrogen cleavage is mediated by the ruthenium center and exogenous alkoxide base. Epoxide ring opening occurs through an opposite-side attack of the ruthenium hydride on the less-hindered epoxide carbon, giving the branched alcohol product selectively.
Introduction
The homogeneous transition-metal-catalyzed hydrogenolysis of epoxides, first reported by Ikariya and co-workers in 2003,1 has recently emerged as a method for the selective synthesis of a variety of substituted primary, secondary, and tertiary alcohols (Scheme 1). Several reported catalysts selectively give the linear (anti-Markovnikov) alcohol isomer. For example, Scheuermann and co-workers described a PCP-pincer-iridium/triflic acid catalyst system,2 which they later showed operates via initial acid-catalyzed hydrolysis of the epoxide, followed by hydrogenolysis to the terminal alcohol catalyzed by iridium nanoparticles generated in situ.3 Yao et al. disclosed a titanium–cobalt dual-catalyst system and provided evidence for a radical-based activation of H2 and transfer to epoxide substrates.4 Beller and co-workers reported an iron/trifluoroacetic acid system5 and a cobalt/zinc triflate system,6 both of which operate through initial Meinwald rearrangement of the epoxide to the aldehyde, followed by metal-catalyzed hydrogenation to the primary alcohol.
Scheme 1. Catalytic Epoxide Hydrogenolysis.

On the other hand, several catalysts selectively produce the branched (Markovnikov) alcohol isomer. So far, all reported catalysts in this category are capable of Noyori-type7 metal–ligand cooperation, involving either RuH/NH or FeH/OH moieties (Chart 1). Ikariya’s 2003 report1 featured a combination of Cp*RuCl(1,5-cyclooctadiene), PPh2CH2CH2NH2, and KOH. Gunanathan reported that the commercially available Ru-MACHO, in combination with KOtBu, promotes the selective hydrogenolysis of a variety of substituted epoxides to give the Markovnikov product.8 In both cases, the authors proposed that the epoxide ring opens through a Noyori-type concerted transfer of Ru–H and N–H to the epoxide C and O atoms, respectively. Tadiello et al. showed that a Knölker-type iron-cyclopentadienone catalyst selectively gives the linear product if Al(OTf)3 is added as a cocatalyst, while the branched product is favored with Zr(OiPr)4 as a cocatalyst.9 Based on density functional theory (DFT) calculations, these authors proposed a competition between a Noyori-type concerted pathway and an initial Meinwald isomerization to the aldehyde followed by aldehyde hydrogenation.
Chart 1. Previously Reported Catalysts for Branched-Selective Epoxide Hydrogenolysis.

Notably, none of the above studies describe the synthesis of enantiomerically enriched alcohols via the hydrogenolysis of enantiomerically enriched epoxides. Ikariya and co-workers noted in a 2007 review10 that racemization of the secondary alcohol products was rapid and prevented the application of epoxide hydrogenolysis to the synthesis of enantiomerically enriched secondary alcohols. Gunanathan reported that attempted hydrogenolysis of (R)-glycidol gave a complex mixture of products and did not describe other attempts with enantiomerically enriched substrates.8
In 2022, we reported11 that two Noyori-type ruthenium catalysts, the commercially available Ru-MACHO-BH and RuPNNHEt, formed from Milstein’s catalyst12 by ethane loss and hydrogen addition,13 are highly active for the branched-selective hydrogenolysis of epoxides, without the requirement of strongly basic or Lewis-acidic cocatalysts (Chart 1). High yields were obtained at catalyst loadings as low as 0.03%, compared with 11,8 or 5%9 loading in prior reports. Through monitoring of the reactant and product e.e. over the course of the reaction, we showed that product racemization is rapid under the catalytic conditions, which prevented the application of this method for the synthesis of enantiomerically enriched alcohols from epoxides.
In 2023, we completed a combined experimental/computational mechanistic study of the Ru-MACHO-BH and RuPNNHEt catalysts for epoxide hydrogenolysis.14 For both catalysts, we showed that the previously proposed1,8,9 concerted, Noyori-type mechanisms for hydrogen transfer to the epoxide have implausibly high free energy barriers more than 50 kcal/mol. Instead, epoxide ring opening proceeds through an SN2-like opposite-side attack of the ruthenium hydride on the less-substituted epoxide carbon, without involvement of the pendant N–H group (Scheme 2, right). Hydrogen activation proceeds by Noyori-type metal–ligand cooperation, assisted by an alcohol functioning as a proton shuttle (Scheme 2, left).
Scheme 2. Abbreviated Mechanism for Branched-Selective Epoxide Hydrogenolysis Catalyzed by Noyori-Type Ruthenium-Pincer Complexes.

Because product racemization presumably proceeds through reversible dehydrogenation of the secondary alcohol to the ketone via a Noyori-type bifunctional mechanism,7 we hypothesized that an analogous complex lacking the pendant N–H group could potentially catalyze epoxide hydrogenolysis while avoiding product racemization. Following an extensive process of screening and optimization, we were pleased to find that the commercially available Milstein’s catalyst, in combination with KOtBu or KOiPr in iPrOH, promotes the hydrogenolysis of a range of substituted epoxides at room temperature, with extremely high-branched:linear selectivity and minimal product racemization. This article describes the discovery and optimization of this catalyst system, an exploration of the substrate scope, and a detailed mechanistic study combining computation, kinetics, and spectroscopic analysis of resting-state speciation. We conclude that epoxide ring opening proceeds through an SN2-like attack of the ruthenium hydride on the less-hindered epoxide carbon, while heterolytic hydrogen activation is mediated by an exogenous alkoxide base. This article was previously deposited to the preprint server ChemRxiv.15
Results and Discussion
Catalyst Screening and Optimization
We began our screening process with the following goals: (1) high yields of alcohol products with low catalyst loading under mild conditions; (2) high selectivity for the branched (chiral) product over the linear product; and (3) minimal racemization of the branched product. For catalyst screening, we chose (R)-styrene oxide as the model substrate because it is available commercially with 98% e.e. and because achieving high selectivity for the branched product has been challenging with aryl epoxide substrates.1,8,9,11 We began by screening a variety of known transition-metal pincer complexes (Ru, Ir, and Mn). We used preformed catalysts instead of an in situ combination of ligand and metal precursor, to avoid potential side reactions arising from incomplete metalation. Because the solvent has been shown to strongly affect both catalyst activity and selectivity in epoxide hydrogenolysis,11 we screened catalyst systems in toluene, tAmOH, iPrOH, EtOH, and MeOH. Table 1 shows highlighted experiments from this optimization process; Table S1 in the Supporting Information shows the results of all 104 screening experiments conducted.
Table 1. Catalyst Screening and Optimization.

| entry | catalyst | mol % | additive | mol % | solvent | [epoxide] (M) | yield (%) | e.e. (%) | b:l |
|---|---|---|---|---|---|---|---|---|---|
| 1 | RuCNN-dipp-Me | 1 | NaOtBu | 10 | iPrOH | 0.125 | 74 | 92 | 7.4 |
| 2 | RuCNN-dipp-Et | 1 | NaOtBu | 10 | iPrOH | 0.125 | >99 | 79 | 7.7 |
| 3 | RuCNN-Mes-Me | 1 | NaOtBu | 10 | iPrOH | 0.125 | 79 | 91 | 5.5 |
| 4 | RuCNN-Mes-Et | 1 | NaOtBu | 10 | iPrOH | 0.125 | 62 | 92 | 5.2 |
| 5 | RuCl | 1 | NaOtBu | 10 | iPrOH | 0.125 | 31 | 98 | 11.1 |
| 6 | RuCl | 1 | NaOtBu | 10 | toluene | 0.125 | 2 | 98 | >10 |
| 7 | RuCl | 1 | NaOtBu | 10 | tAmOH | 0.125 | 62 | 93 | 19.1 |
| 8 | RuCl | 1 | NaOtBu | 10 | EtOH | 0.125 | 3 | 98 | 6.7 |
| 9 | RuCl | 1 | none | iPrOH | 0.125 | 0 | |||
| 10 | RuCl | 1 | CsF | 10 | iPrOH | 0.125 | 0 | ||
| 11 | RuCl | 1 | Cs2CO3 | 10 | iPrOH | 0.125 | 24 | 98 | 11.5 |
| 12 | RuCl | 1 | KF | 10 | iPrOH | 0.125 | 3 | 0 | |
| 13 | RuCl | 1 | BEMP | 10 | iPrOH | 0.125 | 4 | 96 | 9.5 |
| 14 | RuCl | 1 | LiAlH4 | 10 | iPrOH | 0.125 | 3 | 98 | 11.6 |
| 15 | RuCl | 1 | NaBH4 | 10 | iPrOH | 0.125 | 5 | 93 | 5.1 |
| 16 | RuCl | 1 | KOAc | 10 | iPrOH | 0.125 | 0 | ||
| 17 | RuCl | 1 | K3PO4 | 10 | iPrOH | 0.125 | 8 | 98 | 9.5 |
| 18 | RuCl | 1 | KOtBu | 10 | iPrOH | 0.125 | 57 | 98 | 11.3 |
| 19 | RuCl | 0.25 | KOtBu | 10 | iPrOH | 0.5 | 36 | 98 | 12.2 |
| 20 | RuCl | 1 | KOtBu | 2.5 | iPrOH | 0.5 | >99 | 98 | 11.9 |
| 21 | RuCl | 1 | KOtBu | 10 | iPrOH | 0.5 | >99 | 98 | 12 |
| 22 | RuCl | 1 | LiOiPr | 2.5 | iPrOH | 0.5 | 52 | 98 | 11.9 |
| 23 | RuCl | 1 | NaOiPr | 2.5 | iPrOH | 0.5 | 60 | 98 | 12.0 |
| 24 | RuCl | 1 | KOiPr | 2.5 | iPrOH | 0.5 | >99 | 98 | 11.9 |
Entries 1–5 show the most promising precatalysts identified in our screening. Notably, all five are ruthenium–pincer complexes, in which the pincer ligand lacks an N–H functional group. Of these complexes, we identified Milstein’s hydridochloride precatalyst RuCl (entry 5) as the most promising because it provided the highest branched:linear selectivity and showed no observable product racemization. Switching the solvent from isopropyl alcohol (entry 5) to toluene (entry 6) or ethanol (entry 8) dramatically decreased the product yield. Catalyst activity was improved in t-amyl alcohol (entry 7), but some product racemization was observed in this solvent. Continuing with RuCl in isopropyl alcohol, we then screened a variety of basic or hydridic additives (entries 9–18). In this series, KOtBu (entry 18) emerged as the most promising additive, providing the alcohol product with a 57% yield with high-branched selectivity and no observable product racemization. We then surveyed a range of substrate, catalyst, and base concentrations (entries 19–21) and found the following: (1) higher epoxide concentration is beneficial; (2) 1 mol % loading of RuCl is necessary for full conversion; and (3) the loading of KOtBu can be lowered to 2.5 mol % with no decrease in yield or selectivity.
The improved yield with KOtBu compared to NaOtBu (entries 5 vs 18) prompted a comparison of the effect of the alkali metal cation. To avoid complexities arising from multiple alcohols and alkoxide anions in the isopropyl alcohol solvent, we compared the commercially available salts of isopropoxide (entries 22–24). We found that KOiPr was as effective as KOtBu (entries 24 vs 20), but that NaOiPr and LiOiPr were less effective, giving similar selectivities but decreased conversion to product. In the end, entries 20 and 24 represent the optimized conditions for this reaction, giving >99% yield of phenethyl alcohol with no observable racemization and a branched:linear ratio of 11.9:1. For practical synthetic applications, KOtBu is preferred as the base because of its commercial availability as a solid. KOiPr was employed in our kinetic studies described below, which were conducted in isopropyl alcohol solvent.
Substrate Scope
With optimized conditions determined for epoxide hydrogenolysis catalyzed by RuCl and KOtBu, we began to survey the reactivity of a variety of monosubstituted epoxide substrates, to assess whether the catalyst activity, selectivity, and absence of product racemization were maintained (Table 2). First, we compared differently substituted aryl epoxides, which have posed a challenge in the past in obtaining high regioselectivity for the branched product.1,8,11 (R)-Styrene oxide, the subject of the above optimization study, is reduced with 11.9:1 selectivity for the branched product, with no observable product racemization. para-Fluoro, chloro-, and bromo-substituents are all tolerated, and even higher regioselectivity for the branched product is observed. The fluoro- and bromo-substituted epoxides required a higher catalyst loading, 4 and 2% respectively, to achieve full conversion. For the chloro- and bromo-substituted epoxides, a small amount of product racemization were observed, corresponding to 3 and 2% loss of enantiomeric excess, respectively, for these substrates. While we do not know the origin of this small amount of racemization, we note that aryl-halide-containing substrates are challenging for ruthenium-catalyzed epoxide hydrogenolysis, typically requiring higher catalyst loadings for full conversion.8,11 This may stem from an unknown transformation of the catalyst through reaction with the substrate, which may contribute to product racemization.
Table 2. Substrate Scope for Epoxide Hydrogenolysis.

2 mol % RuCl and 5 mol % KOtBu were used.
4 mol % RuCl and 10 mol % KOtBu were used.
Percent yields in parentheses represent isolated yields on a 1.0 g scale.
(S)-Glycidol was used as shown and gave the (S)-1,2-propanediol product expected if the configuration of the stereocenter is retained.
3.3 mol % RuCl and 8.3 mol % KOtBu were used.
We then turned to monosubstituted epoxides with a directly attached sp3 carbon, which typically give only the branched product in hydrogenolysis catalyzed by Noyori-type complexes.1,8,11 The aliphatic epoxides 1-octene oxide and 1-tetradecene oxide were cleanly converted to the secondary alcohols in high yield with no observable linear product and no product racemization. Similar results were obtained for phenyloxy- and benzyloxy-substituted derivatives, as well as allyl benzene oxide. The hydrogenolysis of substrates containing a primary alcohol functional group poses a particular challenge, as base-promoted oligomerization competes with hydrogenolysis at higher temperatures. Ikariya reported no substrates with alcohol functional groups,1 and Gunanathan reported that a complex mixture was obtained for the hydrogenolysis of (R)-glycidol catalyzed by Ru-MACHO and KOtBu.8 With our system operating at room temperature, (S)-glycidol was hydrogenated to give the (S)-1,2-propanediol product with no loss of e.e. in 63% yield, albeit with a higher 3.3% loading of RuCl required to achieve high conversion.
The high-branched selectivity for this catalyst system likely stems from much slower reactivity at more highly substituted epoxide positions. Indeed, the 1,2-disubstituted substrate trans-stilbene oxide is unreactive under our optimized conditions. Although this method tolerates a substrate containing primary alcohol as described above, limitations were observed for other functional groups. In the attempted hydrogenolysis of allyl glycidyl ether, a complex mixture of products was obtained due to competitive hydrogenation of the C=C double bond. In the attempted hydrogenolysis of the ester-containing glycidyl methacrylate, a mixture of products was observed due to known base-promoted transesterification.16
Mechanistic Study: Background
Milstein’s catalyst precursor RuCl—and the deprotonated, dearomatized form Ru-dearom—have a rich history of application in the hydrogenation and dehydrogenation of polar bonds,17 following Milstein’s initial reports of ester hydrogenation18 and the reverse reaction, the acceptorless dehydrogenative coupling of primary alcohols to esters.12Ru-dearom is known to reversibly activate dihydrogen at room temperature to give the dihydride RuH (Scheme 3).18 This metal–ligand-cooperative cleavage of hydrogen (or its reverse in dehydrogenation reactions) was featured in many DFT studies of the reactions catalyzed by RuH or Ru-dearom,19 but experimental studies of catalyst speciation under operating conditions (≥100 °C) were not reported.
Scheme 3. Reversible Activation of Hydrogen by Ru-dearom.

In 2019, we demonstrated that Ru-dearom is catalytically inactive for ester hydrogenation but rapidly undergoes a dehydroalkylation reaction under the conditions of catalysis, releasing ethane and ultimately producing RuPNNHEt,13 which operates through a well-precedented Noyori-type mechanism requiring the nascent N–H functional group (Scheme 4, top).20 Khaskin and Gusev later showed that the closely related RuPNNbpy also forms a Noyori-type catalyst RuPNNpip under operating conditions, this time through hydrogenation of the pyridine ring (Scheme 4, bottom).21 In 2020, Gusev showed through DFT that for Milstein’s catalyst, hydrogen activation mediated by an exogenous alkoxide ion proceeds with a lower barrier than activation through the CH2 linker.22 Taken together, these recent reports call into question the involvement of aromatization/dearomatization pathways through the CH2 linkers in catalysis for complexes, such as Ru-dearom and RuPNNbpy, and emphasize that DFT calculations and studies of stoichiometric reactivity at low temperature, while informative, are most reliable when paired with experimental characterization under catalytically relevant conditions.
Scheme 4. Conversion of Precatalysts with Dearomatized Pincer Ligands to the Active, Noyori-Type Forms.

For the present catalytic transformation, dehydroalkylative catalyst activation (Scheme 4, top) can be excluded because the reaction is conducted at 25 °C, in which dehydroalkylation is known to be extremely slow.13 Although RuPNNHEt, the product of dehydroalkylative activation of Ru-dearom, is known to catalyze epoxide hydrogenolysis, it also rapidly catalyzes product racemization,11 which is not observed in this work. Based on Gusev’s recent report, we expected that the most energetically accessible pathway for hydrogen activation would involve deprotonation of ruthenium-coordinated H2 by exogenous alkoxide.22 We anticipated that the preferred pathway for epoxide ring opening by RuH would involve an SN2-like attack of the ruthenium hydride on the less-hindered epoxide carbon, as we previously demonstrated for the closely analogous complex RuPNNHEt.14 To test these hypotheses while considering plausible alternatives, we employed a combination of spectroscopic analysis of catalyst speciation under catalytically relevant conditions, kinetic analysis, and DFT calculations.
Analysis of Catalyst Resting Speciation
We began by studying the speciation of RuCl, activated with KOtBu, by NMR spectroscopy in isopropyl alcohol solvent under varying pressures of H2. In the absence of hydrogen, the hydridoalkoxide species RuOiPr is formed cleanly, in agreement with previous reports.22,23 Under hydrogen, a rapid equilibrium is established between RuOiPr and RuH (Scheme 5).
Scheme 5. Equilibrium between RuOiPr and RuH.

Figure 1 shows the mole fraction [RuH]/[Ru]total as a function of the hydrogen concentration, as measured by 1H NMR spectroscopy in nondeuterated isopropyl alcohol at 25 °C. A least-squares fit gives K1 = 89 ± 6, corresponding to ΔG° = −2.66 ± 0.04 kcal/mol (see the SI for details). The addition of 0.25 M tetradecene oxide did not produce any new ruthenium species under these conditions or alter the observed ratios, providing evidence against the involvement of additional species with a ruthenium- or ligand-bound epoxide.24
Figure 1.
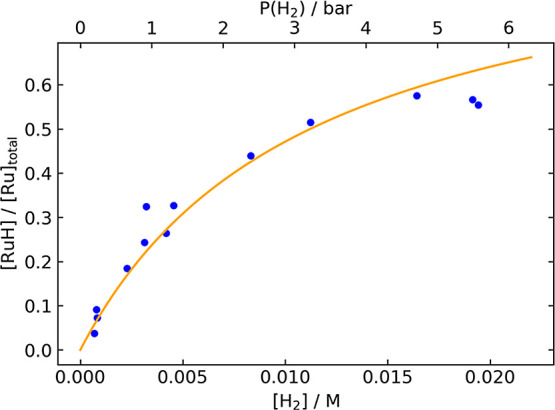
Mole fraction [RuH]/[Ru]total vs [H2], as determined by 1H NMR spectroscopy. Blue points represent values measured in independent experiments. The orange curve represents the best fit of the data to determine the equilibrium constant K1.
Using DFT, we calculated the free energies of the potential resting states RuOiPr, RuH, and Ru-dearom, in the presence and absence of explicit isopropyl alcohol solvent molecules. Scheme 6 shows the calculated relative free energies. RuH and Ru-dearom show a very small effect of explicit solvent (≤0.6 kcal/mol), but RuOiPr shows a greater effect, as RuOiPr-solv is 1.9 kcal/mol lower than RuOiPr. This is consistent with previous studies14,20 and results from the strong hydrogen-bond-accepting ability of the coordinated alkoxide oxygen in RuOiPr. The calculated standard-state free energy change of −5.7 kcal/mol for the conversion of RuOiPr to RuH agrees well with the experimentally measured value of −2.66 kcal/mol. The absence of Ru-dearom in these experiments is qualitatively consistent with its higher standard-state free energy calculated by DFT.
Scheme 6. Relative Standard-State Free Energies of Plausible Catalyst Resting States, Calculated by DFT at 298.15 K.

Minimum Energy Pathway
With experimental confirmation of the resting-state speciation calculated by DFT, we turned to elucidate the pathways for hydrogen activation and epoxide ring opening. To appropriately model the steric and electronic nature of our experimental model substrate, 1-tetradecene oxide, we chose propylene oxide as the model substrate for computations. This has the benefit of minimizing complications due to the multiple conformations of the alkyl chain in 1-tetradecene oxide. Figure 2 shows the calculated minimum energy pathway for the hydrogenolysis of propylene oxide to isopropyl alcohol, beginning with RuOiPr-solv. First, alkoxide dissociation to give a is followed by H2 coordination to give the σ-complex b. This species is deprotonated by exogenous isopropoxide in a nearly barrierless reaction through c-TS, which generates the predominant resting-state RuH-solv. This isopropoxide-mediated hydrogen activation reaction closely follows the ethoxide-mediated pathway previously reported by Gusev.22RuH-solv then forms the dispersion adduct d, after which epoxide ring opening proceeds through e-TS, representing an SN2-like attack of the ruthenium hydride on the terminal epoxide carbon. This leads directly to the C–H σ-complex f, which rearranges to regenerate RuOiPr-solv. The epoxide ring-opening pathway from RuH-solv to RuOiPr-solv is analogous to that calculated previously for the very similar complex RuPNNHEt.14
Figure 2.

Minimum energy pathway for epoxide hydrogenolysis beginning with RuOiPr-solv. Atoms in bold and blue represent the atoms primarily involved in bond-breaking and bond-forming in transition states. The energies reported are Gibbs free energies at 298.15 K, corrected to the 1.0 M standard state for all species except for the solvent isopropyl alcohol, whose standard state is 13.08 M, its neat molarity. Mass balance is ensured throughout, and energies are calculated relative to RuH-solv, the main catalyst resting state.
Because e-TS features a developing negative charge on the epoxide oxygen, we modeled this pathway including an explicit molecule of isopropyl alcohol to stabilize this negative charge through hydrogen bonding. This pathway, shown in Figure S15, had a slightly higher barrier of 26.7 kcal/mol. In contrast, the activation of hydrogen via c-TS does require the explicit isopropyl alcohol molecule shown in Figure 2; the analogous pathway without explicit solvent proceeded through a transition-state k-TS that was 6.2 kcal/mol higher in free energy (Figure S16). As hydrogen activation involving the pincer CH2 linkers has featured prominently in previous DFT studies on Milstein’s catalyst,19b,19d,19e,19g,19h,25 we considered these pathways as alternatives to the MEP shown in Figure 2. We located H2 activation transition states involving either Ru-dearom or its isomer where the NCH2 linker is deprotonated, in both cases including an isopropyl alcohol molecule as a proton shuttle. These pathways, shown in Figures S17 and S18 in the Supporting Information, proceed through barriers at least 7.4 kcal/mol higher than the alkoxide-mediated mechanism through c-TS.
Predicted Kinetics
For the MEP shown in Figure 2, the reaction kinetics can be simplified as shown in Scheme 7. The hydridoalkoxide RuOiPr and the dihydride RuH first establish a rapid pre-equilibrium, which is followed by rate-limiting epoxide ring opening through e-TS.
Scheme 7. Simplified Mechanism Determining the Kinetics for Epoxide Hydrogenolysis.

As derived in the Supporting Information, this scheme leads to the following rate law:
The rate is expected to vary in a first-order manner with the total ruthenium concentration and the epoxide concentration. The dependence of the rate on the hydrogen pressure is expected to follow saturation kinetics, exhibiting first-order dependence at low hydrogen pressure and zero-order dependence at higher pressure. In the kinetics experiments described in the following section, the hydrogen pressures employed range from 10 to 30 bar. Based on the experimentally measured K1 value of 89 (see above), this leads to predicted mole fractions [RuH]/[Ru]total ranging from 0.76 to 0.90. Because Scheme 7 pre-equilibrium has already shifted mostly toward RuH under these conditions, only a modest effect of PH2 on the catalytic rate is expected.
Kinetic Studies
We then sought to experimentally determine the effects of reactant and catalyst concentrations on the reaction rate, to compare with the predictions from computation. For kinetic studies, we monitored the hydrogenolysis of racemic 1-tetradecene oxide with varying concentrations of epoxide, ruthenium, and base, as well as varying hydrogen pressure. We chose 1-tetradecene oxide as the substrate for several reasons: (1) the reaction is very clean, as branched 2-tetradecanol is the only observed product; (2) low volatility of the reactant and product facilitate accurate quantitation; and (3) it is sterically very similar to propylene oxide, which was used in the computational studies as described above. We used KOiPr as base rather than KOtBu, to avoid potential complications resulting from mixtures of alcohols and alkoxide anions in solution.
In the standard experiment, 1-tetradecene oxide (0.25 M), RuCl (0.005 M), and KOiPr (0.0188 M) were stirred at 25 °C for 4 h under 20 bar of hydrogen, and the reaction progress was monitored by gas chromatography (Scheme 8). In all kinetic experiments, the epoxide was consumed in a pseudo-first-order manner after an induction period of approximately 15–30 min (see Table S5 for complete data). At this point, we do not have a clear explanation for these brief induction periods, but we suspect that they may arise from the heterogeneous nature of the activation of RuCl by KOiPr, which results in precipitation of KCl. In the plots below, kobs is calculated from the slope of the plot of ln[epoxide] vs time, excluding data from the first 30 min of reaction.
Scheme 8. Standard Conditions for Kinetic Experiments.

First, we examined the effect of the concentration of the precatalyst, RuCl. As Figure 3a shows, kobs increases linearly with [Ru]total, consistent with a first-order dependence of the rate on [Ru]total. This suggests a monomeric active catalyst species, consistent with the DFT calculations described earlier. Next, we varied the initial concentration of the epoxide, 1-tetradecene oxide. As Figure 3b shows, a minimal effect on kobs is observed, consistent with minimal saturation in [epoxide] or product inhibition. We then monitored the reaction under different hydrogen pressures (Figure 3c). The slight increase in kobs with increasing hydrogen pressure agrees remarkably well with the above measurements of the equilibrium between the two resting states RuOiPr and RuH. Essentially, the increase in kobs arises from a higher steady-state mole fraction of RuH at higher hydrogen pressures, which increases from 0.76 at 10 bar to 0.90 at 30 bar. Finally, we observed a slight but consistent increase in kobs with increasing [KOiPr] (Figure 3d). As described earlier, the alkoxide base is not involved in the turnover-frequency-determining sequence from RuH to e-TS and is not expected to affect the reaction rate based on the calculated minimum energy pathway. Further investigations into this effect are described in the next section.
Figure 3.

Plots of kobs vs [Ru]total (a), [epoxide]0 (b), hydrogen pressure (c), and [KOiPr] (d). Blue points represent kobs values from independent experiments. Orange lines represent the kobs value predicted from a global fit of all 18 experiments.
Using the above overall rate law and the K1 value of 89 ± 6 determined from NMR experiments, we calculated k2 as 0.0152 ± 0.0016 M–1·s–1 (see the SI for details). Applying the Eyring equation at 298.15 K gives an activation free energy ΔG‡ of 19.93 ± 0.06 kcal/mol for this step. This experimental barrier, which corresponds to the free energy difference between RuH-solv and e-TS in Figure 2, is somewhat lower than the barrier of 26.4 kcal/mol calculated by DFT, which may reflect incomplete modeling of the beneficial effect of the alkoxide base in catalysis, as described in more detail below.
Effect of the Alkali Metal Cation and Added [2.2.2]Cryptand
Because of the notable effect of the alkali metal cation on product yield during catalyst optimization (K+ > Na+ > Li+, Table 1, entries 22–24), as well as the modest increase in reaction rate with increasing KOiPr concentration (Figure 3d), we decided to examine the rate of epoxide hydrogenolysis with NaOiPr, compared to the optimal base KOiPr. To attempt to deconvolute potential activating vs inhibiting effects of the metal cation, we measured the reaction rate for both bases in the presence of varying amounts of [2.2.2]cryptand, which sequesters both cations strongly in alcohol solvents.26Figure 4 shows the dependence of kobs on the concentration of added cryptand for both bases.
Figure 4.

Dependence of kobs on the concentration of added [2.2.2]cryptand for epoxide hydrogenolysis with 18.75 mM KOiPr (blue circles) or NaOiPr (red triangles).
First, it is notable that, in the absence of added cryptand, the rate of epoxide hydrogenolysis is approximately six times larger for KOiPr vs NaOiPr. This is consistent with the results from optimization (Table 1, entries 23 vs 24). The addition of cryptand slows the reaction with KOiPr and accelerates the reaction with NaOiPr, bringing the kobs values closer to one another. Empirically, this points to a reactivity order Na+ < [M-cryptand]+ < K+.
A recent review by Dub summarizes some of the potential causes for the beneficial effect of metal alkoxides in Noyori-type hydrogenation catalysis.27 Possible effects of the metal alkoxide include catalyst activation through deprotonation of an acidic site (e.g., replacing N–H with N–K),28 substrate activation where a metal alkoxide cluster stabilizes a developing negative charge on substrate oxygen,29 and reactivation of catalysts deactivated by trace water.30 Further complicating the magnitude of the effect, metal alkoxides are known to aggregate into variably sized clusters in alcohol solvents, which changes the effective concentration of both the alkoxide anion and the metal cation.27 Pidko, Filonenko, and co-workers recently described a detailed study of the effects of KOtBu concentration on ester hydrogenation catalyzed by a Noyori-type Mn-pincer catalyst.31 In their system, the reaction rate is higher at higher base concentration, and they applied the COSMO-RS solvation model to show that the base concentration affects the free energies of on- and off-cycle Mn species, with the net effect that inhibition by the primary alcohol product is reduced at higher [KOtBu]. In our system, the data do not clearly distinguish between these potential effects. We hesitate to draw conclusions from further computation, as even the 6-fold increase in reaction rate for KOiPr vs NaOiPr amounts to only a 1.1 kcal/mol decrease in the overall free energy barrier for catalysis at 25 °C.
Absence of the NH Group Is Key To Avoid Racemization
It is noteworthy that RuCl, which lacks an NH functional group on the supporting ligand, catalyzes epoxide hydrogenolysis with little to no product racemization, while the closely related RuPNNHEt gave the racemic product in our previous study.11 Because the two catalysts only differ in the nitrogen substituents on the pincer ligand—RuCl features an NEt2 group while RuPNNHEt features an NHEt group—we suspected that the NH group was likely involved in product racemization. Because in our prior study, epoxide hydrogenolysis was conducted at 80 °C instead of room temperature and no base was added, we first compared the selectivity of the two catalysts under identical conditions. Figure 5 shows the time course of (R)-styrene oxide hydrogenolysis catalyzed by RuCl vs RuPNNHEt at 25 °C with added KOiPr. The two catalysts are similarly active: RuCl gives a yield of 65% after 4 h, while RuPNNHEt gives a yield of 79%. The branched:linear ratio for RuCl is nearly constant in the range of 11–12 throughout the reaction; the lower early values likely arise from an imprecise measurement of very low quantities of the linear product at early time points. The branched:linear ratio for RuPNNHEt is nearly constant in the range of 5.0–5.3 throughout the reaction, in line with previously reported results for this catalyst in isopropyl alcohol solvent at 80 °C with no added base.11 As expected, the branched product e.e. for the reaction catalyzed by RuCl is maintained at 99% throughout the reaction, indicating no observable racemization over this time period. In contrast, for RuPNNHEt the product e.e. quickly erodes from an initially measured value of 11% after 15 min, indicating fast product racemization by this catalyst.
Figure 5.

Comparison of (R)-styrene oxide hydrogenolysis catalyzed by RuCl vs RuPNNHEt. The top panel shows the total yield of hydrogenolysis products, the middle panel shows the e.e. of the 1-phenylethanol product, and the bottom panel shows the branched:linear ratio.
In a study on the mechanism of ester hydrogenation, we previously showed that RuPNNHEt catalyzes the hydrogenation of acetaldehyde to ethanol with a low computed barrier,20 with the ligand N–H playing a key role in stabilizing the hydride-transfer transition state. Based on this insight, we hypothesized that product racemization in the current system might occur through reversible Noyori-type dehydrogenation of the secondary alcohol products to the ketones. To examine this hypothesis, we conducted a DFT analysis of the RuPNNHEt-catalyzed dehydrogenation of 2-propanol to acetone. The MEP is shown in Figure 6. The catalyst resting state is p, a hydrogen-bonded adduct of RuPNNHEt and 2-propanol. First, the hydrogen-bonded alcohol protonates the ruthenium hydride through q-TS, giving the cationic H2 σ-complex r as an ion pair with isopropoxide. Then, the isopropoxide ion deprotonates the ligand N–H group through s-TS giving the neutral complex t. Release of H2 then gives u, which forms the C–H σ-complex v. The pincer nitrogen deprotonates the alcohol oxygen through w-TS giving the ion pair x, which abstracts hydride from the methine carbon through y-TS, giving the acetone complex z. Release of acetone completes the thermodynamically unfavorable dehydrogenation, regenerating RuPNNHEt. If a chiral secondary alcohol undergoes such a reversible dehydrogenation, racemization will result. The overall barrier for dehydrogenation is 18.3 kcal/mol, lower than both the computed (26.4 kcal/mol) and experimental (19.9 kcal/mol) barriers for epoxide hydrogenolysis catalyzed by RuCl. As stabilization by the N–H group is key in hydride abstraction from the methine carbon through y-TS,32 an analogous low-barrier mechanism for racemization is not available for RuCl.
Figure 6.

MEP for the reversible dehydrogenation of 2-propanol to acetone, catalyzed by RuPNNHEt. Free energies at 298.15 K are calculated relative to p, the calculated catalyst resting state.
Conclusions
In this work, we report the first example of a homogeneous catalyst for the selective formation of highly enantiomerically enriched secondary alcohols via hydrogenolysis of epoxides. The development of the optimized RuCl/KOtBu catalyst system, which minimizes product racemization, was substantially informed by previous mechanistic work in our group. In particular, the insight that epoxide ring opening mediated by ruthenium-hydrides does not require a Noyori-type Ru–H/N-H unit14 spurred us to focus our screening on catalysts lacking an N–H group, as shown in Tables 1 and S1. The knowledge that the PNN- and CNN-pincer-ruthenium complexes shown in Table 1 (entries 1–5), when activated by base, convert to N–H-containing catalysts at elevated temperatures13 informed the decision to screen catalysts at room temperature.
For the optimized catalyst system, our proposed mechanism is based on a detailed experimental/computational study. In the MEP calculated by DFT, heterolytic hydrogen activation is mediated by the ruthenium center and exogenous alkoxide base, as previously proposed by Gusev.22 Epoxide ring opening is facilitated by an SN2-like attack of the ruthenium hydride on the less-hindered epoxide carbon. The calculated MEP led to predictions for hydrogen-pressure-dependent catalyst speciation, validated experimentally by NMR measurements, and an overall rate law for catalysis validated experimentally by kinetics.
Experimental and Computational Methods
Catalyst Screening and Optimization
Screening and optimization experiments for catalytic epoxide hydrogenolysis were conducted in a 450 mL Parr 4760 reaction vessel with a stainless-steel insert designed to hold eight 13 mm disposable glass test tubes. First, the Parr reactor was brought into the glovebox. Each reaction mixture was prepared in the glovebox by transferring the appropriate metal catalyst, additive, solvent (2 mL total reaction volume), the appropriate amount of (R)-styrene oxide (98% ee, Sigma-Aldrich, 0.25 or 1.0 mmol, according to Table S1), and 0.20 equiv. n-tetradecane as an internal standard to an oven-dried test tube, along with a stir bar. The reaction tubes (up to eight, all with the same solvent) were transferred to the Parr reactor, which was sealed and removed from the glovebox. The reactor was then pressurized to 30 bar with hydrogen and stirred magnetically for 18 h at the appropriate temperature (25 or 80 °C). After cooling to room temperature if necessary, the reactor was carefully vented and opened. A 30 μL aliquot of each reaction mixture was removed and diluted into a GC vial with 1 mL toluene, and then analyzed by GC-FID. The reactants and products were separated using a 30 m Agilent Cyclosil-B column, first holding at 100 °C for 1 min, then ramping to 180 °C at a rate of 5 °C/min, and then holding at 180 °C for 5 min. Styrene oxide elutes at 7.00 min (R) and 7.14 min (S), 1-phenylethanol elutes at 9.72 min (R) and 9.92 min (S), 2-phenylethanol elutes at 10.42 min, and tetradecane elutes at 11.89 min. Conversion, yield, and e.e. were determined by comparing the appropriate integrations against the tetradecane standard. The results from selected experiments are shown in Table 1, and the complete list of 104 experiments is shown in Table S1.
Epoxide Hydrogenolysis End Point Analysis Experiments Shown in Table 2
In the glovebox, the epoxide (1.0 mmol), RuCl (1.0 mol %), KOtBu (2.5 mol %), and n-tetradecane (0.20 equiv) as an internal standard were added to isopropyl alcohol (2.0 mL total volume) and transferred to an oven-dried test tube with a stir bar. The test tube was placed in a stainless-steel pressure reactor, which was sealed and brought out of the glovebox. The reactor was pressurized with 30 bar H2. The reactor was then kept at room temperature (25 °C) using a water bath and was stirred magnetically for 18 h. The reactor was then carefully vented and opened to air. A 30 μL aliquot of the reaction mixture was removed, diluted in 1 mL toluene, and analyzed by GC-FID. Conversion and yields were calculated based on the calibrated response factors of the reactant and products relative to the tetradecane internal standard. Reactions were analyzed by GC-FID on either a 30 m Agilent Cyclosil-B column or a 30 m Supelco Alpha DEX 120 column. The GC temperature programs and relevant retention times are listed in Table S2 in the Supporting Information.
Gram-Scale Hydrogenolysis of (S)-Glycidyl Phenyl Ether
A 100 mL stainless-steel reactor and an oven-dried glass liner were brought into the glovebox. A stirbar, glycidyl phenyl ether (1.00 g, 6.7 mmol), KOtBu (18.7 mg, 0.17 mmol), RuCl (32.5 mg, 0.067 mmol), and isopropyl alcohol (13.3 mL) were added to the glass liner. The glass liner was placed in the reactor, which was sealed and removed from the glovebox. The reactor was pressurized with 30 bar H2. The reactor was kept at 25 °C in a water bath and was stirred magnetically for 18 h. The reactor was then carefully vented and opened to air. The reaction solution was transferred to a round-bottom flask and volatiles were removed by rotary evaporation. The crude material was then purified by silica gel chromatography using a gradient of 0–40% ethyl acetate in hexanes and transferred to a round-bottom flask, then the solvent was removed by rotary evaporation. Yield of (S)-1-phenoxypropan-2-ol: 0.896 g, 88%, >99% e.e.
Gram-Scale Hydrogenolysis of (S)-Benzyl Glycidyl Ether
A 100 mL stainless-steel reactor and an oven-dried glass liner were brought into the glovebox. A stirbar, benzyl glycidyl ether (1.00 g, 6.09 mmol), KOtBu (18.7 mg, 0.17 mmol), RuCl (32.5 mg, 0.067 mmol), and isopropyl alcohol (13.3 mL) were added to the glass liner. The glass liner was placed in the reactor, which was sealed and removed from the glovebox. The reactor was pressurized with 30 bar H2. The reactor was kept at 25 °C in a water bath and was stirred magnetically for 18 h. The reactor was then carefully vented and opened to air. The reaction solution was transferred to a round-bottom flask, and volatiles were removed by rotary evaporation. The crude material was then purified by silica gel chromatography using a gradient of 0 to 50% ethyl acetate in hexanes and transferred to a round-bottom flask, then the solvent was removed by rotary evaporation. Yield of (S)-1-benzyloxypropan-2-ol: 0.922 g, 91%, >99% e.e.
Computational Methods
DFT calculations were performed using the Gaussian 16 computational chemistry package, Revision B.01.33 The geometries and energies of all species were calculated using the dispersion-corrected hybrid functional ωB97X-D34 and the def2-SVP basis set.35 A superfine integration grid was used for all calculations, which aided convergence of structures with loosely bound fragments such as explicit isopropyl alcohol molecules. Complete structures with no truncations were used in all cases. Geometry optimization and frequency calculation were conducted in a solvent, using a polarizable continuum with radii and nonelectrostatic terms from Truhlar and co-workers’ SMD solvation model, and with dielectric constants chosen for 2-propanol.36 Geometry optimization in the solvent is important to identify ion-pair intermediates that might be missed in the gas phase.37
Frequency calculations ensured the absence of imaginary vibrational modes in intermediates and the presence of exactly one imaginary mode in transition states. Intrinsic reaction coordinate calculations were employed to verify that transition states led to the specified minima. Free energy corrections were calculated at the experimental reaction temperature of 25 °C, or 298.15 K. Standard-state corrections were applied in order to adjust from 1 atm to 1 M for solution-phase free energies, amounting to 1.89 kcal/mol added to the free energy of each isolated molecule at 298.15 K.38 For the solvent 2-propanol, the standard state is its neat molarity of 13.08 M, corresponding to a correction of 3.42 kcal/mol from 1 atm. Although the standard state for molecular hydrogen is sometimes taken as the gas at 1 atm, we have used a 1 M standard state in 2-propanol, for consistency in computing reaction kinetics from the calculated free energies. The solvation-corrected electronic energies were further refined using the same ωB97X-D functional and the larger basis set def2-TZVP.35 All energies reported in this article are standard-state free energies at 298.15 K. Tables of calculated energies are provided in the Supporting Information, and geometries in Cartesian coordinates are included in a separate, compiled.XYZ file.
Apparatus for Kinetic Experiments
Kinetic experiments were conducted in an Asynt Multicell Parallel High-Pressure Reactor, designed to allow sampling of aliquots from five hydrogenation reactions run in parallel. Our customization of this apparatus was described previously13 but is repeated here for convenience. The reactor was customized to fit in our glovebox antechamber and to include reaction sampling valves on five reactor cells. The sampling valves employ 0.8 mm ID 1/16″ stainless-steel tubing, a Swagelok Low Flow metering valve (part no. SS-SS1) to control flow, and a Swagelok ball valve (part no. SS-41GS1) to allow removal of samples. As the internal volume of the sampling valve system was measured to be 0.25 mL, removal of 0.50 mL of liquid before collecting each aliquot ensures that a fresh sample is being taken directly from the reaction mixture. A sixth reactor cell is fitted with a thermocouple to allow control of the internal reaction temperatures. For reactions at 25 °C, the reactor was partially submerged (2 cm past the internal liquid level) in a stirred water bath (a nonmagnetic stainless-steel cooking pot), whose temperature is controlled using a stirring hot plate with a temperature probe. The stirring hot plate allows stirring of the reactions and water bath.
Experimental Procedure for Kinetic Experiments
First, the water bath was preheated to 25 °C. The Asynt reactor was brought into the glovebox with oven-dried glass reactor liners and Teflon-coated stir bars. Reaction solutions, with a total volume of 10.0 mL, were prepared with the appropriate amount of ruthenium complex, racemic 1-tetradecene oxide, KOiPr solution (5% in isopropyl alcohol), optionally [2.2.2]cryptand, and tetradecane (0.20 equiv relative to epoxide) as internal standard. In selected experiments, KOiPr was replaced by NaOiPr. The reactor was closed and removed from the glovebox and allowed to incubate for 20 min in the water bath. After gently purging the H2 line for 2 min, the reactor was pressurized to the desired pressure. Aliquots were removed at predetermined times for analysis by gas chromatography. To ensure that samples represented the reaction mixture without contamination from the transfer line, 0.5 mL of the reaction mixture was discarded before one drop was collected for each aliquot. Each one-drop aliquot was diluted with 1 mL toluene in a crimp-top vial, then sealed and analyzed by GC-FID. The concentrations of reactants and products at each time point were determined by integration of their GC signals against the tetradecane standard. GC analysis employed a 30 m Agilent Cyclosil-B column. The oven temperature was initially held at 100 °C for 1 min, then increased to 220 °C at a rate of 5 °C/min, and then held at 220 °C for 5 min. With these parameters, tetradecene oxide was eluted at 18.14 min, 2-tetradecanol was eluted at 18.71 min, and the standard tetradecane was eluted at 11.77 min. Although the chiral column was used to minimize the need to switch columns repeatedly for different experiment types, the reactant and product enantiomers were not separated by this temperature program. The concentrations measured over time in these experiments are reported in Tables S5–S7.
Acknowledgments
We thank the National Science Foundation (CHE-2247229) for support of this research project.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.organomet.4c00214.
Author Contributions
O.J.B.: catalyst discovery and optimization, substrate scope. B.T.J.: DFT calculations. M.C.H.: kinetic experiments, NMR study of catalyst speciation. O.A.A.: kinetic experiments. D.E.K.: substrate scope. A.C.B.: substrate scope. J.M.K.: funding acquisition, supervision of computational work, review and editing of manuscript. A.R.C.: conceptualization, funding acquisition, supervision, writing and editing of manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Ito M.; Hirakawa M.; Osaku A.; Ikariya T. Highly Efficient Chemoselective Hydrogenolysis of Epoxides Catalyzed by a (Η5-C5(CH3)5)Ru Complex Bearing a 2-(Diphenylphosphino)Ethylamine Ligand. Organometallics 2003, 22, 4190–4192. 10.1021/om034006j. [DOI] [Google Scholar]
- Rainsberry A. N.; Sage J. G.; Scheuermann M. L. Iridium-Promoted Conversion of Terminal Epoxides to Primary Alcohols under Acidic Conditions Using Hydrogen. Catal. Sci. Technol. 2019, 9, 3020–3022. 10.1039/C9CY00791A. [DOI] [Google Scholar]
- Gitnes R. M.; Wang M.; Bao Y.; Scheuermann M. L. In Situ Generation of Catalytically Relevant Nanoparticles from a Molecular Pincer Iridium Precatalyst During Polyol Deoxygenation. ACS Catal. 2021, 11, 495–501. 10.1021/acscatal.0c03180. [DOI] [Google Scholar]
- Yao C.; Dahmen T.; Gansäuer A.; Norton J. Anti-Markovnikov Alcohols Via Epoxide Hydrogenation through Cooperative Catalysis. Science 2019, 364, 764–767. 10.1126/science.aaw3913. [DOI] [PubMed] [Google Scholar]
- Liu W.; Li W.; Spannenberg A.; Junge K.; Beller M. Iron-Catalysed Regioselective Hydrogenation of Terminal Epoxides to Alcohols under Mild Conditions. Nature Catalysis 2019, 2, 523–528. 10.1038/s41929-019-0286-7. [DOI] [Google Scholar]
- Liu W.; Leischner T.; Li W.; Junge K.; Beller M. A General Regioselective Synthesis of Alcohols by Cobalt-Catalyzed Hydrogenation of Epoxides. Angew. Chem., Int. Ed. 2020, 59, 11321–11324. 10.1002/anie.202002844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noyori R.; Ohkuma T. Asymmetric Catalysis by Architectural and Functional Molecular Engineering: Practical Chemo- and Stereoselective Hydrogenation of Ketones. Angew. Chem., Int. Ed. 2001, 40, 40–73. . [DOI] [PubMed] [Google Scholar]
- Thiyagarajan S.; Gunanathan C. Ruthenium-Catalyzed Selective Hydrogenation of Epoxides to Secondary Alcohols. Org. Lett. 2019, 21, 9774–9778. 10.1021/acs.orglett.9b03995. [DOI] [PubMed] [Google Scholar]
- Tadiello L.; Gandini T.; Stadler B. M.; Tin S.; Jiao H.; De vries J. G.; Pignataro L.; Gennari C. Regiodivergent Reductive Opening of Epoxides by Catalytic Hydrogenation Promoted by a (Cyclopentadienone)Iron Complex. ACS Catal. 2022, 12, 235–246. 10.1021/acscatal.1c03549. [DOI] [Google Scholar]
- Ito M.; Ikariya T. Catalytic Hydrogenation of Polar Organic Functionalities Based on Ru-Mediated Heterolytic Dihydrogen Cleavage. Chem. Commun. 2007, 5134–5142. 10.1039/b709704b. [DOI] [PubMed] [Google Scholar]
- Kirlin F. L.; Borden O. J.; Head M. C.; Kelly S. E.; Chianese A. R. Epoxide Hydrogenolysis Catalyzed by Ruthenium PNN and PNP Pincer Complexes. Organometallics 2022, 41, 1025–1033. 10.1021/acs.organomet.2c00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Leitus G.; Ben-david Y.; Milstein D. Facile Conversion of Alcohols into Esters and Dihydrogen Catalyzed by New Ruthenium Complexes. J. Am. Chem. Soc. 2005, 127, 10840–10841. 10.1021/ja052862b. [DOI] [PubMed] [Google Scholar]
- He T.; Buttner J. C.; Reynolds E. F.; Pham J.; Malek J. C.; Keith J. M.; Chianese A. R. Dehydroalkylative Activation of CNN- and PNN-Pincer Ruthenium Catalysts for Ester Hydrogenation. J. Am. Chem. Soc. 2019, 141, 17404–17413. 10.1021/jacs.9b09326. [DOI] [PubMed] [Google Scholar]
- Head M. C.; Joseph B. T.; Keith J. M.; Chianese A. R. The Mechanism of Markovnikov-Selective Epoxide Hydrogenolysis Catalyzed by Ruthenium PNN and PNP Pincer Complexes. Organometallics 2023, 42, 347–356. 10.1021/acs.organomet.2c00503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borden O. J.; Joseph B. T.; Head M. C.; Ammons O.; Kim D. E.; Bonino A. C.; Keith J. M.; Chianese A.. Highly Enantiomerically Enriched Secondary Alcohols Via Epoxide Hydrogenolysis. 2024, ChemRxiv. 10.26434/chemrxiv-2024-6rps5 [DOI] [Google Scholar]
- a Stanton M. G.; Gagné M. R. The Remarkable Catalytic Activity of Alkali-Metal Alkoxide Clusters in the Ester Interchange Reaction. J. Am. Chem. Soc. 1997, 119, 5075–5076. 10.1021/ja970556v. [DOI] [Google Scholar]; b Stanton M. G.; Allen C. B.; Kissling R. M.; Lincoln A. L.; Gagné M. R. ″New″ Catalysts for the Ester-Interchange Reaction: The Role of Alkali-Metal Alkoxide Clusters in Achieving Unprecedented Reaction Rates. J. Am. Chem. Soc. 1998, 120, 5981–5989. 10.1021/ja980584t. [DOI] [Google Scholar]
- a Gunanathan C.; Ben-david Y.; Milstein D. Direct Synthesis of Amides from Alcohols and Amines with Liberation of H2. Science 2007, 317, 790–792. 10.1126/science.1145295. [DOI] [PubMed] [Google Scholar]; b Gnanaprakasam B.; Ben-david Y.; Milstein D. Ruthenium Pincer-Catalyzed Acylation of Alcohols Using Esters with Liberation of Hydrogen under Neutral Conditions. Adv. Synth. Catal. 2010, 352, 3169–3173. 10.1002/adsc.201000663. [DOI] [Google Scholar]; c Balaraman E.; Gunanathan C.; Zhang J.; Shimon L. J. W.; Milstein D. Efficient Hydrogenation of Organic Carbonates, Carbamates and Formates Indicates Alternative Routes to Methanol Based on CO2 and CO. Nat. Chem. 2011, 3, 609–614. 10.1038/nchem.1089. [DOI] [PubMed] [Google Scholar]; d Gnanaprakasam B.; Balaraman E.; Ben-david Y.; Milstein D. Synthesis of Peptides and Pyrazines from Beta-Amino Alcohols through Extrusion of H2 Catalyzed by Ruthenium Pincer Complexes: Ligand-Controlled Selectivity. Angew. Chem., Int. Ed. 2011, 50, 12240–12244. 10.1002/anie.201105876. [DOI] [PubMed] [Google Scholar]; e Gnanaprakasam B.; Milstein D. Synthesis of Amides from Esters and Amines with Liberation of H2 under Neutral Conditions. J. Am. Chem. Soc. 2011, 133, 1682–1685. 10.1021/ja109944n. [DOI] [PubMed] [Google Scholar]; f Zeng H.; Guan Z. Direct Synthesis of Polyamides Via Catalytic Dehydrogenation of Diols and Diamines. J. Am. Chem. Soc. 2011, 133, 1159–1161. 10.1021/ja106958s. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Rigoli J. W.; Moyer S. A.; Pearce S. D.; Schomaker J. M. Alpha,Beta-Unsaturated Imines Via Ru-Catalyzed Coupling of Allylic Alcohols and Amines. Org. Biomol. Chem. 2012, 10, 1746–1749. 10.1039/c2ob06921k. [DOI] [PubMed] [Google Scholar]; h Huff C. A.; Sanford M. S. Catalytic CO2 Hydrogenation to Formate by a Ruthenium Pincer Complex. ACS Catal. 2013, 3, 2412–2416. 10.1021/cs400609u. [DOI] [Google Scholar]; i Chaudhari M. B.; Bisht G. S.; Kumari P.; Gnanaprakasam B. Ruthenium-Catalyzed Direct Alpha-Alkylation of Amides Using Alcohols. Org. Biomol. Chem. 2016, 14, 9215–9220. 10.1039/C6OB01786J. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Leitus G.; Ben-david Y.; Milstein D. Efficient Homogeneous Catalytic Hydrogenation of Esters to Alcohols. Angew. Chem., Int. Ed. 2006, 45, 1113–1115. 10.1002/anie.200503771. [DOI] [PubMed] [Google Scholar]
- a Yang X.; Hall M. B. Mechanism of Water Splitting and Oxygen-Oxygen Bond Formation by a Mononuclear Ruthenium Complex. J. Am. Chem. Soc. 2010, 132, 120–130. 10.1021/ja9041065. [DOI] [PubMed] [Google Scholar]; b Li H.; Wang X.; Huang F.; Lu G.; Jiang J.; Wang Z.-X. Computational Study on the Catalytic Role of Pincer Ruthenium(II)-PNN Complex in Directly Synthesizing Amide from Alcohol and Amine: The Origin of Selectivity of Amide over Ester and Imine. Organometallics 2011, 30, 5233–5247. 10.1021/om200620n. [DOI] [Google Scholar]; c Sandhya K. S.; Suresh C. H. Water Splitting Promoted by a Ruthenium(II) PNN Complex: An Alternate Pathway through a Dihydrogen Complex for Hydrogen Production. Organometallics 2011, 30, 3888–3891. 10.1021/om200046u. [DOI] [Google Scholar]; d Zeng G.; Li S. Insights into Dehydrogenative Coupling of Alcohols and Amines Catalyzed by a (PNN)-Ru(II) Hydride Complex: Unusual Metal-Ligand Cooperation. Inorg. Chem. 2011, 50, 10572–10580. 10.1021/ic200205e. [DOI] [PubMed] [Google Scholar]; e Li H.; Wen M.; Wang Z.-X. Computational Mechanistic Study of the Hydrogenation of Carbonate to Methanol Catalyzed by the (RuPNN)-P-II Complex. Inorg. Chem. 2012, 51, 5716–5727. 10.1021/ic300175b. [DOI] [PubMed] [Google Scholar]; f Ma C.; Piccinin S.; Fabris S. Reaction Mechanisms of Water Splitting and H2 Evolution by a Ru(II)-Pincer Complex Identified with Ab Initio Metadynamics Simulations. ACS Catal. 2012, 2, 1500–1506. 10.1021/cs300350b. [DOI] [Google Scholar]; g Yang X. Metal Hydride and Ligand Proton Transfer Mechanism for the Hydrogenation of Dimethyl Carbonate to Methanol Catalyzed by a Pincer Ruthenium Complex. ACS Catal. 2012, 2, 964–970. 10.1021/cs3000683. [DOI] [Google Scholar]; h Cho D.; Ko K. C.; Lee J. Y. Catalytic Mechanism for the Ruthenium-Complex-Catalyzed Synthesis of Amides from Alcohols and Amines: A DFT Study. Organometallics 2013, 32, 4571–4576. 10.1021/om4005324. [DOI] [Google Scholar]; i Hasanayn F.; Baroudi A.; Bengali A. A.; Goldman A. S. Hydrogenation of Dimethyl Carbonate to Methanol by Trans-[Ru(H)2(PNN)(CO)] Catalysts: DFT Evidence for Ion-Pair-Mediated Metathesis Paths for C–OMe Bond Cleavage. Organometallics 2013, 32, 6969–6985. 10.1021/om4005127. [DOI] [Google Scholar]; j Sandhya K. S.; Remya G. S.; Suresh C. H. Pincer Ligand Modifications to Tune the Activation Barrier for H2 Elimination in Water Splitting Milstein Catalyst. Inorg. Chem. 2015, 54, 11150–11156. 10.1021/acs.inorgchem.5b01471. [DOI] [PubMed] [Google Scholar]; k Li L.; Lei M.; Liu L.; Xie Y.; Schaefer H. F. Metal-Substrate Cooperation Mechanism for Dehydrogenative Amidation Catalyzed by a PNN-Ru Catalyst. Inorg. Chem. 2018, 57, 8778–8787. 10.1021/acs.inorgchem.8b00563. [DOI] [PubMed] [Google Scholar]
- Pham J.; Jarczyk C. E.; Reynolds E. F.; Kelly S. E.; Kim T.; He T.; Keith J. M.; Chianese A. R. The Key Role of the Latent N–H Group in Milstein’s Catalyst for Ester Hydrogenation. Chem. Sci. 2021, 12, 8477–8492. 10.1039/D1SC00703C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawe L. N.; Karimzadeh-younjali M.; Dai Z.; Khaskin E.; Gusev D. G. The Milstein Bipyridyl PNN Pincer Complex of Ruthenium Becomes a Noyori-Type Catalyst under Reducing Conditions. J. Am. Chem. Soc. 2020, 142, 19510–19522. 10.1021/jacs.0c06518. [DOI] [PubMed] [Google Scholar]
- Gusev D. G. Revised Mechanisms of the Catalytic Alcohol Dehydrogenation and Ester Reduction with the Milstein PNN Complex of Ruthenium. Organometallics 2020, 39, 258–270. 10.1021/acs.organomet.9b00542. [DOI] [Google Scholar]
- Perdriau S.; Chang M. C.; Otten E.; Heeres H. J.; De vries J. G. Alkene Isomerisation Catalysed by a Ruthenium PNN Pincer Complex. Chem.—Eur. J. 2014, 20, 15434–15442. 10.1002/chem.201403236. [DOI] [PubMed] [Google Scholar]
- Liang Y.; Luo J.; Milstein D. Facile Synthesis of Amides Via Acceptorless Dehydrogenative Coupling of Aryl Epoxides and Amines. Chem. Sci. 2022, 13, 5913–5919. 10.1039/D2SC01959K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hou C.; Zhang Z.; Zhao C.; Ke Z. DFT Study of Acceptorless Alcohol Dehydrogenation Mediated by Ruthenium Pincer Complexes: Ligand Tautomerization Governing Metal Ligand Cooperation. Inorg. Chem. 2016, 55, 6539–6551. 10.1021/acs.inorgchem.6b00723. [DOI] [PubMed] [Google Scholar]; b Wang H.; Liu C.; Zhang D. Decisive Effects of Solvent and Substituent on the Reactivity of Ru-Catalyzed Hydrogenation of Ethyl Benzoate to Benzyl Alcohol and Ethanol: A DFT Study. Mol. Catal. 2017, 440, 120–132. 10.1016/j.mcat.2017.06.026. [DOI] [Google Scholar]
- a Cox B. G.; Schneider H.; Stroka J. Kinetics of Alkali Metal Complex Formation with Cryptands in Methanol. J. Am. Chem. Soc. 1978, 100, 4746–4749. 10.1021/ja00483a019. [DOI] [Google Scholar]; b Cox B. G.; Garcia-rosas J.; Schneider H. Solvent Dependence of the Kinetics of Formation and Dissociation of Cryptate Complexes. J. Am. Chem. Soc. 1981, 103, 1054–1059. 10.1021/ja00395a010. [DOI] [Google Scholar]
- Dub P. A. Alkali Metal Alkoxides in Noyori-Type Hydrogenations. Eur. J. Inorg. Chem. 2021, 2021, 4884–4889. 10.1002/ejic.202100742. [DOI] [Google Scholar]
- a Dub P. A.; Gordon J. C. The Role of the Metal-Bound N–H functionality in Noyori-Type Molecular Catalysts. Nat. Rev. Chem. 2018, 2, 396–408. 10.1038/s41570-018-0049-z. [DOI] [Google Scholar]; b Dub P. A.; Henson N. J.; Martin R. L.; Gordon J. C. Unravelling the Mechanism of the Asymmetric Hydrogenation of Acetophenone by [RuX2(Diphosphine)(1,2-Diamine)] Catalysts. J. Am. Chem. Soc. 2014, 136, 3505–3521. 10.1021/ja411374j. [DOI] [PubMed] [Google Scholar]; c Nakane S.; Yamamura T.; Manna S. K.; Tanaka S.; Kitamura M. Mechanistic Study of the Ru-Catalyzed Asymmetric Hydrogenation of Nonchelatable and Chelatable Tert-Alkyl Ketones Using the Linear Tridentate sp3P/sp3NH/sp2N-Combined Ligand Pn(H)N: Runh- and Runk-Involved Dual Catalytic Cycle. ACS Catal. 2018, 8, 11059–11075. 10.1021/acscatal.8b02671. [DOI] [Google Scholar]
- Dub P. A.; Batrice R. J.; Gordon J. C.; Scott B. L.; Minko Y.; Schmidt J. G.; Williams R. F. Engineering Catalysts for Selective Ester Hydrogenation. Org. Process Res. Dev. 2020, 24, 415–442. 10.1021/acs.oprd.9b00559. [DOI] [Google Scholar]
- Nguyen D. H.; Trivelli X.; Capet F.; Swesi Y.; Favre-réguillon A.; Vanoye L.; Dumeignil F.; Gauvin R. M. Deeper Mechanistic Insight into Ru Pincer-Mediated Acceptorless Dehydrogenative Coupling of Alcohols: Exchanges, Intermediates, and Deactivation Species. ACS Catal. 2018, 8, 4719–4734. 10.1021/acscatal.8b00995. [DOI] [Google Scholar]
- Yang W.; Kalavalapalli T. Y.; Krieger A. M.; Khvorost T. A.; Chernyshov I. Y.; Weber M.; Uslamin E. A.; Pidko E. A.; Filonenko G. A. Basic Promotors Impact Thermodynamics and Catalyst Speciation in Homogeneous Carbonyl Hydrogenation. J. Am. Chem. Soc. 2022, 144, 8129–8137. 10.1021/jacs.2c00548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dub P. A.; Gordon J. C. Metal–Ligand Bifunctional Catalysis: The “Accepted” Mechanism, the Issue of Concertedness, and the Function of the Ligand in Catalytic Cycles Involving Hydrogen Atoms. ACS Catal. 2017, 7, 6635–6655. 10.1021/acscatal.7b01791. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian 16, Rev. B.01; Wallingford, CT, 2016. [Google Scholar]
- Chai J.-D.; Head-gordon M. Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. 10.1039/b810189b. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Marenich A. V.; Cramer C. J.; Truhlar D. G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- Dub P. A.; Gordon J. C. The Mechanism of Enantioselective Ketone Reduction with Noyori and Noyori-Ikariya Bifunctional Catalysts. Dalton Trans. 2016, 45, 6756–6781. 10.1039/C6DT00476H. [DOI] [PubMed] [Google Scholar]
- Cramer C. J.Essentials of Computational Chemistry, 2nd edition; Wiley: Chichester, UK, 2004; pp 378–379. [Google Scholar]
- Macrae C. F.; Edgington P. R.; Mccabe P.; Pidcock E.; Shields G. P.; Taylor R.; Towler M.; Van de streek J. Mercury: Visualization and Analysis of Crystal Structures. J. Appl. Crystallogr. 2006, 39, 453–457. 10.1107/S002188980600731X. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.