

Abstract
In humans, FOXP gene family is involved in embryonic development and cancer progression. The FOXP4 (Forkhead box protein P4) gene belongs to this FOXP gene family. FOXP4 gene plays a crucial role in oncogenesis. Single nucleotide polymorphisms are biological markers and common determinants of human diseases. Mutations can largely affect the function of the corresponding protein. Therefore, the molecular mechanism of nsSNPs in the FOXP4 gene needs to be elucidated. Initially, the SNPs of the FOXP4 gene were extracted from the dbSNP database and a total of 23124 SNPs was found, where 555 nonsynonymous, 20525 intronic, 1114 noncoding transcript, 334 synonymous were obtained and the rest were unspecified. Then, a series of bioinformatics tools (SIFT, PolyPhen2, SNAP2, PhD SNP, PANTHER, I-Mutant2.0, MUpro, GOR IV, ConSurf, NetSurfP 2.0, HOPE, DynaMut2, GeneMANIA, STRING and Schrodinger) were used to explore the effect of nsSNPs on FOXP4 protein function and structural stability. First, 555 nsSNPs were analyzed using SIFT, of which 57 were found as deleterious. Following, PolyPhen2, SNAP2, PhD SNP and PANTHER analyses, 10 nsSNPs (rs372762294, rs141899153, rs142575732, rs376938850, rs367607523, rs112517943, rs140387832, rs373949416, rs373949416 and rs376160648) were common and observed as deleterious, damaging and diseases associated. Following that, using I-Mutant2.0 and MUpro servers, 7 nsSNPs were found to be the most unstable. GOR IV predicted that these seven nsSNPs affect protein structure by altering the protein contents of alpha helixes, extended strands, and random coils. Following DynaMut2, 5 nsSNPs showed a decrease in the ΔΔG value compared with the wild-type and were found to be responsible for destabilizing the corresponding protein. GeneMANIA and STRING network revealed interaction of FOXP4 with other genes. Finally, molecular dynamics simulation analysis revealed consistent fluctuation in RMSD and RMSF values, Rg and hydrogen bonds in the mutant proteins compared with WT, which might alter the functional and structural stability of the corresponding protein. As a result, the aforementioned integrated comprehensive bioinformatic analyses provide insight into how various nsSNPs of the FOXP4 gene change the structural and functional properties of the corresponding protein, potentially proceeding with the pathophysiology of human diseases.
Keywords: FOXP4 gene, Functional prediction, Stability prediction, Secondary and tertiary structure and interaction analysis
1. Introduction
Forkhead box protein P4 (FOXP4) protein is encoded by the FOXP4 gene [1]. This gene also known as hFKHLA plays a crucial role in oncogenesis [2]. FOXP4 gene belongs to subfamily P of the forkhead box (FOX) transcription factor family which also consists of three different proteins FOXP1 (3p14.1), FOXP2 (7q31), and FOXP3 (Xp11.23). All of subfamily P of the forkhead box are comprised of immune disorders, embryonic maturity, and cancer progression [3]. Those proteins also show multifarious and influential roles in the development and organogenesis for the regulation of metabolism and the immune system [4]. FOXP4 gene is precisely associated with FOXP1 and FOXP2 with 54 % and 60 % identity, respectively. FOXP4 gene forms a large multidomain transcriptional repressor with FOXP1 and FOXP2 [5]. It is a transcription factor of b-catenin, raises the transcription of b-catenin and leads to the malicious progress of ESCC [6]. This gene also plays a vital role in the expansion of tumors in the kidney and larynx [7]. As a transcriptional factor, this gene is also involved in the triggering of several genes that play a vital role in various biological activities for cancer progression [8].
Single nucleotide polymorphism (SNP) is the replacement (variation) of a single nucleotide in deoxyribonucleic acid [9]. It can occur in the coding regions of the gene and make a change in amino acid (nonsynonymous), or can be silent without changing in amino acid (synonymous), or can also occur in noncoding regions (5′UTR, 3′UTR, and introns) [10]. In the human genome, SNPs are the most common cause of genetic alteration. They make up nearly 90 % of human genome polymorphism [11]. Mutations can largely affect the function of corresponding proteins, hence analysis of possible modifications in protein tertiary structure can reveal new evidence for their effect on phenotype. The nsSNPs are one of the major causes of the functional diversity of proteins. The nsSNPs also known as missense variations, which change the amino acid sequence of the protein, are particularly important as they result in almost 50 % of the known genetic variations related to human inherited diseases [12].
Currently, computational methods are widely used for the analysis of possible candidates. The present study performed extensive screening for the most damaging nsSNPs in the FOXP4 gene and identified the possible pathogenic nsSNPs. This study aimed to sort out the functionally important nsSNPs in the FOXP4 gene. Additionally, MD simulation analysis was conducted to differentiate the stable and unstable nature of the native protein and nsSNPs.
Following these, the FOXP4 gene was subjected to functional and structural analysis (multi-level way) to determine their pathogenicity. Briefly, this study involves the following steps: 1) Retrieving of nsSNPs and protein sequence, 2) Functional effect prediction based on sequence homology, 3) Stability prediction of nsSNPs on protein, 4) Prediction of FOXP4 nsSNPs effect on the secondary and tertiary structure using GOR IV, 5) Evolutionary conservation analysis using ConSurf Server, 6) Surface accessibility prediction of FOXP4 gene nsSNPs, 7) Prediction of structural effects of FOXP4 mutants using HOPE Server, 8) Prediction of destabilizing effects upon nsSNPs of FOXP4 gene, 9) Prediction of alterations in protein stability and interaction upon nsSNPs, 10) Interaction of FOXP4 with other Genes and Proteins and 11) Molecular dynamic simulation analysis.
This is the first inclusive in silico analysis that conceals the total nsSNPs of FOXP4 gene. Henceforth, our findings could benefit the precision medicine discipline by acknowledging disease treatment based on these genomic changes.
2. Material and methods
2.1. Retrieving of nsSNPs and protein sequence
The National Center for Biotechnology Information, dbSNP database was used to retrieve the nsSNPs of the FOXP4 gene (Accessed March 2024). Sequence of the wild type FOXP4 protein (Q8IVH2) was obtained from UniProtKB database. The nsSNPs could change the amino acid of any protein and cause disease pathogenesis [13]. Therefore, nsSNPs have been selected for downstream analysis. Here, numerous bioinformatics tools have been used to analyze the effect of nsSNPs on the corresponding proteins.
2.2. Functional effect prediction based on sequence homology
2.2.1. SIFT
The Sorting Intolerant from Tolerant (SIFT) is a bioinformatics web server that was used to identify deleterious nsSNPs from tolerated for corresponding protein. The higher the tolerance index, the lower the functional impact of amino acid substitution is likely to have. The nsSNPs having prediction score of ≥0.05, are considered as tolerated and the same having the score of ≤0.05 are regarded as deleterious. The rsIDs of the corresponding gene were submitted as input for the analysis [14].
2.2.2. PolyPhen2
The Polymorphism Phenotyping v2 (PolyPhen2) web server was used to analyze the nsSNPs which can affect protein functionality of the corresponding protein. A position-specific independent count (PSIC) score has been calculated for each variant. PolyPhen2 sorts out the score into three categories, (1) benign (0.00–0.45), (2) possibly damaging (0.45–0.95), and 3) probably damaging (0.95–1). The nsSNPs having HumDiv and HumVar both similar scores were chosen for further analysis [15].
2.2.3. SNAP2
Screening for Deprecated Polymorphism v2 (SNAP2) is a bioinformatics web server that was used to predict the functional effect of sequence variants on protein. On the heat map, a score of +100 (dark red) or −100 (dark blue) indicates the amino acid substitution is pathogenic or neutral respectively. The prediction score ranges from 0 (very low reliability) to 9 (very high reliability) [16].
2.2.4. PhD SNP
The Predictor of Human Deleterious Single Nucleotide Polymorphisms (PhD SNP) web server utilized to predict disease-associated substitutions based on the support vector machine (SVM) method. For each mutation, the PhD SNP predicts an output score (from 0 to 1) representative probability of this nsSNP is associated with the disease. The method considers 0.5 as the threshold above which nsSNP is predicted to be associated with disease [17].
2.2.5. PANTHER
Protein Analysis Through Evolutionary Relationship (PANTHER) was utilized to assess the functional impact of amino acid substitutions on the biological function. This server is based on the Hidden Markov Model (HMM) to estimate the probability that nsSNPs variants can affect the phenotype of a protein according to its evolutionary origin [18].
2.3. Stability prediction of nsSNPs on protein
2.3.1. I-Mutant2.0
I-Mutant2.0 web server was utilized to estimate the potential effects of nsSNPs on the structural reliability of the protein and free energy change Delta Delta G (DDG). Accordingly, stability will increase when DDG is > 0 kcal/mol and decrease when DDG is < 0 kcal/mol. We have inputted the protein FASTA sequence, location AAS, and variant residue [19].
2.3.2. MUpro
The MUpro server was utilized to estimate the stability of mutated proteins due to the substitution of amino acids following both neural networks and support vector machines (SVM). The decrease in protein stability is predicted if the confidence score is < 0, and increases if the confidence score is > 0 [20].
2.4. Prediction of FOXP4 nsSNPs effect on secondary and tertiary structure using GOR IV
GOR IV is a web server, utilized to determine protein secondary structure upon nsSNPs. For each secondary structure at each amino acid location, it provides probability values. GOR IV employs all potential pair frequencies across a range of amino acid residues. GOR IV provides a sequence and predicted secondary structure in rows (including E = extended, H = helix, beta-strand, and C = coil) [21].
2.5. Evolutionary conservation analysis using ConSurf server
ConSurf server was utilized to address the evolutionary pattern of conservation at each amino acid site. By using the Bayesian calculation method, this server analyzes close sequence homologs and estimates the phylogenetic relationship. For each residue of the corresponding protein, the degree of conservation is estimated on a scale of 1–9 and categorized as 1) variable (1–4), 2) intermediate (5–6) and 3) conserved (7–9) [22].
2.6. Surface accessibility prediction of nsSNPs
The NetSurfP 2.0 server was utilized to determine the surface accessibility of the corresponding protein. This tool predicts surface accessibility, secondary structure, and structural disorder of any respective amino acid position by analyzing the amino acid sequence. Here, 25 % was set for relative surface accessibility (RSA) as the threshold. Greater than 25 % of RSA was considered as exposed [23].
2.7. Prediction of structural effects of FOXP4 mutants using HOPE server
Have (y)Our Protein Explained (HOPE) web server utilized to estimate the structural and functional effects of point mutations. It provides the 3D structural visualization of mutated proteins and gives the results by using the UniProt and DAS prediction servers. Inputted the protein sequence, wild-type, and new amino acids for the prediction [24].
2.8. Prediction of destabilizing effects upon nsSNPs of FOXP4 gene
2.8.1. mCSM
The mCSM web server is an artificial intelligence method created to anticipate the consequences of mutations in corresponding proteins. This is also taking into account the structural features of the proteins. Particularly, mCSM is employed to assess how missense variations impact the stability of FOXP4 protein. This server predicts changes in the energy required for protein folding (ΔΔG) caused by mutations. This prediction sorts into two categories, either stabilizing or destabilizing which depend on their respective ΔΔG values.
2.8.2. SDM
SDM (Site-Directed Mutagenesis) web server is a knowledge-based SVM algorithm predictor. This server is used to predict the stability of the respective protein. This prediction is based on conformationally constrained, environment-specific substitution tables derived from amino acid replacements that are tolerated within protein families.
2.8.3. DUET
DUET is also a web server that was used to predict the effect of nsSNPs on the stability of the corresponding protein. It is an integrated computational approach and its calculation is based on secondary structure. Its utilized algorithms include SVM regression with a Radial Basis Function kernel and RSA. It helps to estimate the consequence of a single mutation on protein stability and its function. This web server is utilized for applying and considering incorporated mutations and their impact on the structure of proteins by computational methodology.
2.9. Prediction of alterations in protein stability and interaction upon nsSNPs
The DynaMut web server was used for prediction of the alterations in the stability of protein and interaction upon nsSNPs. This server performed prediction by evaluating flexibility analysis and protein dynamics. We performed a single mutation prediction feature in this analysis. A mutation list and the wild-type structure in PDB format were inputted [25].
2.10. Interaction of FOXP4 with other genes and proteins
2.10.1. GeneMANIA
GeneMANIA is a flexible and user-friendly web interface for generating hypotheses about gene function, analyzing gene lists and prioritizing genes for functional analyses. GeneMANIA finds related genes to input genes, using a very large set of functional association data. Association data include protein and genetic interactions, pathways, co-expression, co-localization and protein domain similarity. It was used to find new members of a pathway, additional genes [26].
2.10.2. STRING
The predicted version of protein-protein interaction information was analyzed using the STRING server. The protein-protein interaction outcomes are important to analyze since the mutated protein continuously affects the other protein during the diseased condition. This helps to study the mechanism of the diseased condition for targeting the source protein and other corresponding proteins [27].
2.11. Molecular dynamic simulation analysis
To evaluate the structural stability of the protein, a 50 ns MD simulation was performed using the “Desmond v6.3 Program” in Schrodinger 2020-3 under the Linux framework [28]. The simulation was examined by the three-site transferrable intermolecular potential (TIP3P) water model [29]. An orthorhombic box shape with a 10 Å distance from the center was used to maintain a specific volume, and Na+ and Cl− were added to neutralize the whole system with a salt concentration of 0.15 M. An OPLS3e force field was applied [30]. The protein structure system was further minimized using a natural time and pressure (NPT) ensemble at a constant pressure of 101325 Pa's and a temperature of 300 K. RMSD (root means square deviation), RMSF (root means square fluctuation), Rg (radius of gyration), and hydrogen bonds were analyzed to evaluate the stability and dynamic characteristics of the protein.
3. Results
The complete workflow is summarized in Fig. 1.
Fig. 1.

The complete workflow of nsSNPs analysis.
3.1. Retrieving of nsSNPs and protein sequence
The nsSNPs of the FOXP4 gene were extracted from the dbSNP database. Here, about 23124 SNPs, where 555 missense (2.4 %), 20525 introns (88.76 %), 1114 non-coding transcript (4.82 %), and 334 synonymous (1.44 %) were obtained. Then the missense variants were further analyzed to explore the most deleterious.
3.2. Functional effect prediction based on sequence homology
Firstly, 555 missense nsSNPs were analyzed using SIFT, where 57 nsSNPs were found deleterious with score less than ≤0.05. Then, those nsSNPs were analyzed using PolyPhen2. During Polyphen2 analysis, HumDiv and HumVar both scores were computed. After SIFT and PolyPhen2 prediction 10 nsSNPs to be deleterious and probably/possibly damaging and benign, respectively. Finally, those nsSNPs were further analyzed using SNAP2, PhD SNP and PANTHER (Table 1). However, all 10 nsSNPs were observed as non-neutral in SNAP2 analysis. Following that, among all 10 nsSNPs, 5 and 6 SNPs were associated with disease when analyzed by PhD SNP, and PANTHER respectively. In all five analyses, the 10 nsSNPs (rs372762294, rs141899153, rs142575732, rs376938850, rs367607523, rs112517943, rs140387832, rs373949416, rs373949416 and rs376160648) were common and observed as deleterious, damaging and diseases associated (Table 1 and Supplementary Table 1).
Table 1.
Functional effect prediction of non-synonymous SNPs using numerous bioinformatics tools.
| SL | SNP ID | Amino acid change | SIFT | PolyPhen2 |
SNAP2 | PhD SNP | PANTHER | |
|---|---|---|---|---|---|---|---|---|
| HumDiv | HumVar | |||||||
| 1 | rs372762294 | G19V | D | Pro. D | Pos. D | E | Dis | NA |
| 2 | rs141899153 | Q156H | D | Pro. D | Pro. D | E | Dis | Dis |
| 3 | rs142575732 | R288W | D | Pro. D | Pro. D | E | Dis | Dis |
| 4 | rs376938850 | T330I | D | Pos. D | Benign | E | N | Dis |
| 5 | rs367607523 | E353D | D | Pro. D | Pro. D | E | Dis | Dis |
| 6 | rs112517943 | Q355H | D | Pos. D | Pos. D | E | N | Dis |
| 7 | rs140387832 | R373W | D | Pro. D | Pos. D | E | Dis | Dis |
| 8 | rs373949416 | D667H | D | Pos. D | Pos. D | E | N | NA |
| 9 | rs373949416 | D669H | D | Pos. D | Benign | E | N | NA |
| 10 | rs376160648 | L679P | D | Pos. D | Pos. D | E | N | NA |
D: Deleterious, E: Effect N: Neutral, Dis: Disease, Pro. D: Probably damaging, Pos. D: Possibly damaging, NA: Unspecified.
3.3. Stability prediction of nsSNPs on protein
I-Mutant2.0 and MUpro server estimate the free energy change DDG value and the Confidence score of the 10 candidates nsSNPs mediated mutated proteins. Among these, a number of 7 nsSNPs guided proteins were found to be the most unstable considering DDG and Confidence score (Table 2).
Table 2.
Stability prediction of nsSNPs on protein using I-Mutant2.0 and MUpro.
| Sl | SNP ID | Amino Acid Change | I-Mutant2.0 |
MUpro |
||
|---|---|---|---|---|---|---|
| Stability | DDG (Kcal/mol) | Prediction | Confidence Score | |||
| 1 | rs141899153 | Q156H | Decrease | −0.46 | Decrease | −0.71089341 |
| 2 | rs376938850 | T330I | Decrease | −0.38 | Decrease | −0.23065386 |
| 3 | rs112517943 | Q355H | Decrease | −1.35 | Decrease | −0.64577427 |
| 4 | rs140387832 | R373W | Decrease | −0.14 | Decrease | −0.78766219 |
| 5 | rs373949416 | D667H | Decrease | −0.6 | Decrease | −0.74309817 |
| 6 | rs373949416 | D669H | Decrease | −0.77 | Decrease | −1 |
| 7 | rs376160648 | L679P | Decrease | −1.13 | Decrease | −1 |
3.4. Prediction of FOXP4 nsSNPs effect on secondary and tertiary structure using GOR IV
GOR IV tool predicts the effect of nsSNPs on protein secondary and tertiary structure. Following this analysis, the contents of the normal secondary and tertiary structure of the FOXP4 protein, which include 237 alpha helixes, 58 extended strands, and 385 random coils differed from the contents of structures with the seven amino acids substitution. The results showed that the amino acid substitution of the seven nsSNPs effects protein structure by altering the protein contents of alpha helixes, extended strands, and random coils (Table 3).
Table 3.
Prediction of FOXP4 nsSNPs effect on secondary and tertiary protein structure using GOR IV.
| Parameter | Normal protein | Q156H | T330I | Q355H | R373W | D667H | D669H | L679P |
|---|---|---|---|---|---|---|---|---|
| Alpha helix | 237 (34.85 %) | 237 (34.85 %) | 241 (35.44 %) | 237 (34.85 %) | 236 (34.71 %) | 236 (34.71 %) | 236 (34.71 %) | 236 (34.71 %) |
| 310 helix | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Pi helix | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Beta bridge | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Extended strand | 58 (8.53 %) | 58 (8.53 %) | 58 (8.53 %) | 58 (8.53 %) | 58 (8.53 %) | 58 (8.53 %) | 58 (8.53 %) | 58 (8.53 %) |
| Beta turn | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Bend region | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Random coil | 385 (56.62 %) | 385 (56.62 %) | 381 (56.03 %) | 385 (56.62 %) | 386 (56.76 %) | 386 (56.76 %) | 386 (56.76 %) | 386 (56.76 %) |
| Ambiguous states | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Other states | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
3.5. Evolutionary conservation analysis using ConSurf server
A colorimetric conservation score was created for FOXP4 protein by the ConSurf server. The program identifies highly conserved structural and functional amino acid regions that are essential for biological function. From the analysis, we have found that Q156H, T330I, Q355H, R373W, D667H, D669H, L679P all are exposed residue (Fig. 2A).
Fig. 2.

(A) Prediction of evolutionary conserved amino acid residues by ConSurf server Conservation score is represented as the color coding bars. (B) Surface accessibility prediction of nsSNPs of the FOXP4 protein by NetSurfP 2.0. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.6. Surface accessibility prediction of nsSNPs
The NetSurfP 2.0 predicted the surface accessibility of individual amino acids of FOXP4 protein by a percentage score. The threshold was 25 %, which means the amino acid residues having a score of more than 25 % were predicted as exposed, and scores less than 25 % were buried. Among the 7 positions having all positions (Q156H, T330I, Q355H, R373W, D667H, D669H, L679P) scored more than 25 % and it indicate these residues are exposed. (Fig. 2B and Supplementary Table 2).
3.7. Prediction of structural effects of FOXP4 mutants using HOPE server
HOPE server guides how mutation affects the structural variation of a protein based on size, charge, hydrophobicity, and spatial structure compared to the wild type. Among the 7 predicted substitutions, 6 mutants (Q156H, T330I, Q355H, R373W, D667H, and D669H) were found to be bigger than the wild residue's consequences localization on the surface of the wild protein. However, L679P mutant residues were smaller than the wild residues, which caused a space in the core of the wild protein and loss of hydrophobic interactions. Therefore, mutation of the residue can interrupt inter/intra-molecular interactions of the protein (Table 4).
Table 4.
Structural effects prediction of FOXP4 nsSNPs using Project HOPE server.
| Sl | SNP ID | Amino acid change | Wild Type | Mutant |
|---|---|---|---|---|
| 1 | rs141899153 | Q156H |  |
|
| 2 | rs376938850 | T330I |  |
|
| 3 | rs112517943 | Q355H |  |
|
| 4 | rs140387832 | R373W |  |
|
| 5 | rs373949416 | D667H |  |
|
| 6 | rs373949416 | D669H |  |
|
| 7 | rs376160648 | L679P | ||
3.8. Prediction of destabilizing effects upon nsSNPs of FOXP4 gene
The mCSM anticipates the consequences of nsSNPs of the corresponding protein and considers the structural features of the proteins. This is employed to assess how the mutation impacts the stability of FOXP4 protein. Among 7 nsSNPs, 5 nsSNPs (Q156H, T330I, Q355H, R373W and L679P) are found destabilizing having the ΔΔG score <0. Consequently, the SDM server predicted the stability of nsSNPs of the corresponding protein and found upstream 5 nsSNPs as reduced stability. Lastly, the DUET web server also predicts the effect of nsSNPs on the stability of the corresponding protein. Here, 4 nsSNPs (Q156H, Q355H, R373W and L679P) are found to destabilize the corresponding protein having the ΔΔG score <0 (Table 5).
Table 5.
Prediction of destabilizing effects upon nsSNPs of FOXP4 gene.
| Sl | SNP ID | Amino Acid Change | mCSM |
SDM |
DUET |
|||
|---|---|---|---|---|---|---|---|---|
| Prediction | ΔΔG (kcal/mol) | Prediction | ΔΔG (kcal/mol) | Prediction | ΔΔG (kcal/mol) | |||
| 1 | rs141899153 | Q156H | Destabilizing | −0.745 | Reduced stability | −0.79 | Destabilizing | −0.705 |
| 2 | rs376938850 | T330I | Destabilizing | −0.048 | Reduced stability | −1.5 | Stabilizing | 0.434 |
| 3 | rs112517943 | Q355H | Destabilizing | −0.791 | Reduced stability | −0.77 | Destabilizing | −0.648 |
| 4 | rs140387832 | R373W | Destabilizing | −0.495 | Reduced stability | −0.15 | Destabilizing | −0.477 |
| 5 | rs373949416 | D667H | Stabilizing | 0.438 | Stabilizing | 0.56 | Stabilizing | 0.584 |
| 6 | rs373949416 | D669H | Stabilizing | 0.447 | Stabilizing | 0.8 | Stabilizing | 0.643 |
| 7 | rs376160648 | L679P | Destabilizing | −0.593 | Reduced stability | −1.44 | Destabilizing | −0.61 |
3.9. Prediction of alterations in protein stability and interaction upon nsSNPs
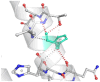
The DynaMut2 server was used to calculate general dynamic traits of the highest deleterious nsSNPs selected from the previous analysis steps. DynaMut2 represented the predictions for ΔΔG stability value among the WT and mutant protein. From 7 nsSNPs, 5 nsSNPs showed a decrease in the ΔΔG Stability value compared to the wild-type and were found to be responsible for destabilizing the protein. The free energy (ΔΔG) Stability values for Q156H, T330I, Q355H, R373W and L679P mutants were found −0.35, −0.030, −0.56, −0.4 and −1.530 kcal/mol respectively (Table 6).
Table 6.
Prediction of alterations in protein stability and interaction upon nsSNPs using DynaMut2 server.
| SL | Amino acid variants | ΔΔG ENCoM | Prediction | Wild type | Mutant type |
|---|---|---|---|---|---|
| 01 | Q156H | −0.35 kcal/mol | Destabilizing |  |
 |
| 02 | T330I | −0.030 kcal/mol | Destabilizing | 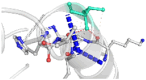 |
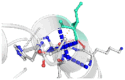 |
| 03 | Q355H | −0.56 kcal/mol | Destabilizing | 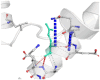 |
 |
| 04 | R373W | −0.4 kcal/mol | Destabilizing | 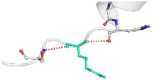 |
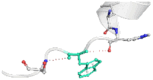 |
| 05 | L679P | −1.530 kcal/mol | Destabilizing | 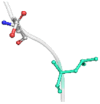 |
 |
3.10. Interaction of FOXP4 with other genes and proteins
3.10.1. GeneMANIA
Gene-gene interaction network revealed a correlation among genes for the FOXP4 gene. A composite gene-gene functional interaction network has been built by GeneMANIA. GeneMANIA results showed that the FOXP4 gene was associated with 20 other genes. It interacts more significantly with FOXP1, 2 genes, MKI67 (marker of proliferation Ki-67), GATAD2B (GATA zinc finger domain containing 2B), and others genes (Fig. 3A, Supplementary Table 3).
Fig. 3.

Interaction of FOXP4 with other genes and proteins. (A) Gene-gene interaction network of FOXP4 gene predicted by GeneMANIA. (B) Protein-protein interaction of FOXP4 gene by STRING tool.
3.10.2. STRING
Proteins work in a way that synergizes and interacts with other proteins to carry out cell signaling and other cellular functions. Therefore, amino acid variations in one protein can affect other proteins in the network. FOXP4 functional partners as predicted using the STRING tool. Forkhead box protein P4; Transcriptional inhibitors inhibit lung-specific expression. The nasal tip box protein P2 (FOXP2) plays a role in the specification and differentiation of the lung epithelium. Heart and neural crest derivatives-expressed protein 1 (HAND1) is a transcription factor that plays an essential role in both trophoblast giant cell differentiation and in cardiac morphogenesis (By similarity). Forkhead box protein P3 (FOXP3), C-terminally processed; Transcriptional regulators are important for the development and repressor function of regulatory T (Treg) cells (Fig. 3B and Supplementary Table 4) [31].
3.11. Molecular dynamic simulation analysis
A 50 ns MD simulation analysis of the predicted deleterious mutants Q156H, T330I, Q355H, R373W, and L679P and FOXP4 native protein was conducted to observe the deviation from the structural stability of native protein along with mutant protein in an artificial environment. Mutants Q156H, T330I, Q355H, R373W, and L679P showed considerable fluctuations after the simulation run to the native protein FOXP4, as shown in Fig. 4A. The RMSD values for the native protein were observed ranging from 4.12 Å to 37.79 Å while 4.31 Å to 37.41 Å for the Q156H mutant type, 4.18 Å to 27.83 Å for the T330I mutant type, 4.09 Å to 32.87 Å for the Q355H, 4.02 Å to 30.14 Å for the R373W and 4.04 Å to 29.54 Å for the L679P mutant type as shown in Fig. 4A. The average RMSD values of native FOXP4, and mutant Q156H, T330I, Q355H, R373W, and L679P are 31.48 Å, 30.73 Å, 24.80 Å, 27.92 Å, 26.42 Å, and 26.15 Å, respectively. The fluctuations of mutant proteins were more noticeable than wild-type proteins throughout the simulation run. Overall, the RMSD plot showed that the native protein was stable along the simulation run time and also demonstrated that the mutant protein has a major impact on the structural confirmation of FOXP4 protein. The RMSF of protein residue was counted to observe how these changes impact the local flexibility. In addition, RMSF analysis demonstrated considerable fluctuations between native and mutant structures throughout the simulation run. From the RMSF plot, the native protein and five mutated proteins specified by proteins Q156H, T330I, Q355H, R373W, and L679P had the highest peak fluctuation with (PRO168, ASP251, SER381, PHE404, SER627, and ALA657), (LYS232, PRO271, SER390, GLY428, ALA464, ARG506, and GLU666), (THR35, ALA98, ALA163, GLN206, GLY212, PRO266, and LEU422), (GL235, PRO282, SER392, GLY564, SER588, SER604, PRO657, GLU672, and SER680), (LYS267, SER598, LEU601, LEU603, ASP669, LEU679, and SER680), and (THR216, ASP243, THR297, PRO425, LEU600, ASP607, PRO664, andLEU670) respectively. The average RMSF values are 11.08 Å, 10.65 Å, 7.18 Å, 8.7 Å, 7.85 Å, and 8.9 Å correspondingly (Fig. 4B). In a nutshell, the mutant form of the FOXP4 protein showed higher RMSF than the native FOXP4 protein. The Rg measures how the atoms are distributed around the axis of a protein. It is an important indicator for predicting the structural activity of macromolecules and for assessing changes in the compactness of the protein structure. The native protein and five mutants (Q156H, T330I, Q355H, R373W, and L679P) revealed Rg ranges of 35.70 Å to 45.682 Å, 31.21 Å to 45.65 Å, 34.92 Å to 45.98 Å, 31.21 Å to 45.65 Å, 31.46 Å to 45.74 Å and 31.07 Å to 45.732 Å, respectively. Average fluctuation of these SNPs was 39.48 Å, 36.48 Å, 37.08 Å, 36.48 Å, 34.24 Å and 34.91 Å, respectively (Fig. 4C). The mutated protein structures were unstable in 50 ns simulations with greater fluctuation differences from lowest to highest, suggesting that the mutated proteins significantly alter the conformation of the respective protein's active site. The number of hydrogen bonds can help to characterize a protein. Therefore, hydrogen bond numbers were calculated from initial to final times during the 50 ns simulation run to observe each hydrogen bond. All the proteins formed several hydrogen bonds ranging between 300 and 500 occur simultaneously until 50 ns simulation time (Fig. 4D). These results indicated that the native protein has a higher specificity while the mutated protein has a higher fluctuating range of hydrogen bonds denoted the structural instability.
Fig. 4.

Results of 50 ns molecular dynamics simulation by Schrodinger. (A) RMSD values of native and mutant structures. (B) RMSF values of native and mutant structures. (C) Calculation of Rg over the entire simulation. (D) Calculation of H-bonds represented as a time-depend. The color scheme is as follows: native (blue), Q156H mutant (red), T330I mutant (green), Q355H mutant (violet), R373W mutant (yellow), and L679P mutant (orange). (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
4. Discussion
Forkhead box protein 4 (FOXP4) is a protein-coding gene. This gene belongs to the P subfamily of the Forkhead Box (FOX) transcription factor family [32]. It was first identified in the Drosophila forkhead gene [33] and the mammalian Hnf3 transcription factors were found to contain highly similar domains [34]. The FOXP subgroup of transcription factors includes four different proteins: FOXP1, FOXP2, FOXP3 and FOXP4. The FOXP subfamily is perhaps the most widely characterized subfamily of the fork superfamily, playing important roles in vertebrate development and homeostasis [[35], [36], [37]]. Mutation in the FOXP4 gene has previously been reported in children with growth retardation, laryngeal hypoplasia, feeding difficulties, and ventricular septal defect [38]. SNPs play an important role in gene and protein-regulated diseases in humans [39]. Several in silico studies of polymorphisms to predict nsSNPs as harmful or neutral have been performed [[40], [41], [42], [43]].
In this study, we used in-silico structural and functional analyses to identify potential nsSNPs utilizing numerous computational bioinformatics tools to predict the deleterious SNPs. Initially, the SNPs of the FOXP4 gene were extracted from the dbSNP database and a total 23124 SNPs were found, where 555 were nonsynonymous, 20525 intron, 1114 non-coding transcript, and 334 synonymous were obtained and the rest were unspecified. Then, to classify the most deleterious nsSNPs of the corresponding gene, five different prediction servers were used; SIFT, Polyphen2, SNAP2, PhD SNP and PANTHER. In all five analyses, the 10 nsSNPs (G19V, Q156H, R288W, T330I, E353D, Q355H, R373W, D667H, D669H and L679P) were common and observed as deleterious, damaging and diseases associated (Table 1). Then, the nsSNP was observed as a decrease in protein stability using the I-Mutant2.0 and MUpro. Among these, a number of 7 nsSNPs guided proteins were found to be the most unstable considering ΔΔG and Confidence score (Table 2). GOR IV tool was used to predict the effect of nsSNPs on protein secondary and tertiary structure. Following this analysis, the contents of the normal secondary and tertiary structure of the FOXP4 protein, which include 237 alpha helixes, 58 extended strands, and 385 random coils differed from the contents of structures with the seven amino acids substitutions (Table 3). Mutations affect the helix-helix interactions and disordered regions of corresponding proteins by changing their properties [44]. Hence, mutations at these positions might alter the structural and functional properties of the mutant protein. A colorimetric conservation score was created for FOXP4 protein by the ConSurf server. From the analysis, we have found that Q156H, T330I, Q355H, R373W, D667H, D669H, L679P all are exposed residues. Among them Q355H residues were found as highly conserved for playing potential biological functions (Fig. 2A). The conserved residue encounters mutation, it is more destructive compared to the less conserved one [45].
Following NetSurfP 2.0 prediction, Among the 7 positions having all positions (Q156H, T330I, Q355H, R373W, D667H, D669H, L679P) scored more than 25 % which means all are exposed (Fig. 2B and Supplementary Table 2). NetSurfP tool-based surface accessibility analysis revealed that these SNPs have potential to change surface accessibility and secondary structures following changing coil and helix structures. Significant changes in absolute surface accessibility in mutant protein might alter binding affinity of the corresponding mutant protein. HOPE provides 3D structural images of mutated proteins. Among the 7 predicted substitutions, Q156H, T330I, Q355H, R373W, D667H, and D669H mutants were found to be bigger than the wild residue's consequences localization on the surface of the wild protein. However, L679P mutant residues were smaller than the wild residues (Table 4). This space could influence the protein function, characteristics or reactivity [46]. The mCSM, SDM and DUET web server predicts the effect of nsSNPs on the stability of the corresponding protein. Following this analysis, Q156H, T330I, Q355H, R373W and L679P nsSNPs are found to destabilize the corresponding protein having the ΔΔG score <0 (Table 5). Those point mutations might play a role in disease progression. DynaMut2 tools provide Gibbs free energy change (ΔΔG) values for each protein structure; The free energy change during the unfolding of the kinetically stable protein is described by this value ΔΔG. From 7 mutants, 6 mutants showed a decrease in the ΔΔG Stability value compared to the wild-type and were found to be responsible for destabilizing the protein (Table 5). Small changes in proteins can have large phenotypic outcomes. Changes in Gibbs Free energy of folding can occur due to a myriad of factors related to incorporating these properties. Those point mutations might play a role in disease progression and pathogenesis [25]. GeneMANIA analysis revealed that FOXP4 gene significantly interacts with 20 different genes [[47], [48], [49], [50], [51], [52], [53], [54], [55], [56], [57], [58], [59], [60], [61], [62], [63], [64], [65], [66]]. FOXP4 gene showed more significant interactions with FOXP1, 2 genes, MKI67 (marker of proliferation Ki-67), GATAD2B (GATA zinc finger domain containing 2B) than others genes. Here, FOXP1 promotes cancer stem cell [47], and FOXP2 is relevant to the human ability to develop language [48]. MKI67 gene plays a vital role in the tumor microenvironment (TME) and congenital immunity [49]. ATP-dependent chromatin remodeling and histone deacetylation [50]. FOXP4 gene promotes tumor formation and progression [51]. Hence, nsSNPs in FOXP4 gene interacting with these genes might result serious health hazards. STRING tool mediated analysis resulted that FOXP4 is tightly interacting with 10 proteins [47,48,51,53,54,[67], [68], [69], [70], [71]]. Among 10 proteins, five were common interacting proteins (FOXP2, FOXP1, FOXP3, FOXP1-2 and DHDDS) in STRING and GeneMANIA analyses indicate that FOXP4 precisely interacted with the mentioned genes (Supplementary Table 3 and Table 4). An aqueous environment allows proteins to be dynamic entities. The MD simulation revealed how nsSNPs affect the stability, residual fluctuation, and compactness of the corresponding protein. We conducted a 50 ns MD simulation to examine the effect of mutation on FOXP4 protein dynamics. The protein structure is stabilized when the RMSD and RMSF values of a protein are within 1–3 Å [72]. RMSD of the Q156H, T330I, Q355H, R373W, and L679P mutants fluctuated consistently indicating that all of these mutants are likely to form an unstable structure in the physiological environment exhibited in Fig. 4A. Furthermore, the higher average RMSD value of native protein FOXP4 and mutant Q156H, T330I, Q355H, R373W, and L679P indicates a loss of stability for the mutants. According to the RMSF analysis, all mutant has the ability to modify the overall and local flexibility of corresponding protein, which affects stability and may therefore have an impact on function or interactions with other proteins (Fig. 4B). Compared to the WT, a higher Rg value represents the disassociation of the respective protein (Fig. 4C). However, the increased number of hydrogen bonds causes the structural unsteadiness [72]. Therefore, continuous fluctuations in RMSD and RMSF values, high Rg value and hydrogen bonds in mutant proteins compared to WT explore the mechanism by which these nsSNPs alter the structure and function of the native FOXP4 protein.
In the Chinese Population, rs1983891 of the FOXP4 gene associated with Prostate Cancer [73]. In Multi-Ethnic Study of Atherosclerosis Study, rs2894439 of FOXP4 gene is associated with interstitial lung disease [74]. A study in 3173 Japanese patients has found rs2495239 of FOXP4 gene were newly identified as loci associated with EGFR mutation-positive lung adenocarcinoma [75]. A study on Japan and five other Asian countries, revealed that the rs7741164 of FOXP4 gene is associated with a higher risk of lung cancer risk among never-smoking Asian females [76]. For instance, rs2494938 was associated with lung, non-cardia gastric and esophageal squamous cell carcinoma (ESCC) in Han Chinese [77], rs10484761 was associated with ESCC in a GWAS in Chinese [78] and rs1983891 in the intron of FOXP4 was associated with prostate cancer in Japanese [79]. Another study also found that rs9367106 of FOXP4 associated with both long COVID and severe COVID-19 [80].
However, the nsSNPs (rs141899153, rs376938850, rs112517943, rs140387832 and rs376160648) (Q156H, T330I, Q355H, R373W and L679P) respectively have not yet been studied, so they need to be studied with special consideration to the ethnic diversity of the different populations easily suffer from different diseases. The present study predicted high-risk nsSNPs in FOXP4 gene which might be the important candidates in the pathology and disease progression. MD simulation analysis, continuous fluctuations in RMSD and RMSF values, Rg and hydrogen bonds also confirmed that these five nsSNPs are associated with alter the stability and structural flexibility of corresponding protein. However, further in vitro and in vivo studies are needed to investigate and establish the role of these nsSNPs in various diseases.
5. Conclusion
In this prediction and assessment of deleterious and disease causing nsSNPs study, we performed a series of computational bioinformatics prediction tools analysis for the FOXP4 gene. Among the 555 nsSNPs, it is figured out that 6 nsSNPs are the most deleterious, diseases associated, decrease stability and altered stability and interaction of the corresponding protein, which is predicted using numerous prediction tools. Hence, we concluded that 6 nsSNPs (Q156H, T330I, Q355H, R373W, D667H, and L679P) might be important candidates in the pathology and disease progression. Following DynaMut2, 5 nsSNPs showed a decrease in the ΔΔG value compared with the wild-type and were found to be responsible for destabilizing the corresponding protein. Finally, molecular dynamics simulation analysis revealed continuous fluctuations in RMSD and RMSF values, Rg and hydrogen bonds in mutant proteins (Q156H, T330I, Q355H, R373W, and L679P) compared with WT, which might alter the functional and structural stability of the corresponding protein. Therefore, the key findings of the present study provide a scientific overview for the researchers to determine the significant role in these nsSNPs of these corresponding proteins. The effect of these nsSNPs in the FOXP4 gene on protein structure and function needs to be confirmed by experimental investigations. Multiple in-silico approaches are used to provide quick and inexpensive screening that may help direct additional laboratory studies.
Funding statement
This research received no external funding.
Data availability statement
Data included in article/supplementary material/referenced in article.
CRediT authorship contribution statement
Md. Mostafa Kamal: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Supervision, Visualization, Writing – original draft. Shamiha Tabassum Teeya: Formal analysis, Methodology, Writing – original draft. Md. Mahfuzur Rahman: Writing – review & editing. Md. Enamul Kabir Talukder: Formal analysis, Writing – original draft. Sonia Sarmin: Writing – review & editing. Tanveer A. Wani: Formal analysis, Resources. Md. Mahmudul Hasan: Conceptualization, Supervision, Writing – review & editing.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors extend their appreciation to supporting project number (RSP2024R357) King Saud University, Riyadh Saudi Arabia for instrumental and technical support to conduct molecular dynamics simulation in this study.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.heliyon.2024.e32791.
Contributor Information
Md. Mostafa Kamal, Email: rmkmostafa@gmail.com.
Md. Mahmudul Hasan, Email: hasanm_agb@yahoo.com.
Appendix A. Supplementary data
The following are the supplementary data to this article:
References
- 1.Xue P., Huang S., Han X., Zhang C., Yang L., Xiao W., Fu J., Li H., Zhou Y. Exosomal miR-101-3p and miR-423-5p inhibit medulloblastoma tumorigenesis through targeting FOXP4 and EZH2. Cell Death & Differentiation 2021. 2021;29(1 29):82–95. doi: 10.1038/s41418-021-00838-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu M., Shi X., Wang J., Xu Y., Wei D., Zhang Y., Yang K., Wang X., Liang S., Chen X., Yang F., Sun L., Zhu X., Zhao C., Zhu L., Tang L., Zheng C., Yang Z. Association of FOXP4 gene with prostate cancer and the cumulative effects of rs4714476 and 8q24 in Chinese men. Clin. Lab. 2015;10 doi: 10.7754/Clin.Lab.2015.150313. [DOI] [PubMed] [Google Scholar]
- 3.Kim J.H., Hwang J., Jung J.H., Lee H.J., Lee D.Y., Kim S.H. Molecular networks of FOXP family: dual biologic functions, interplay with other molecules and clinical implications in cancer progression. Molecular Cancer 2019. 2019;18(1 18):1–19. doi: 10.1186/S12943-019-1110-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jackson B.C., Carpenter C., Nebert D.W., Vasiliou V. Update of human and mouse forkhead box (Fox) gene families. Hum. Genom. 2010;4:345–352. doi: 10.1186/1479-7364-4-5-345/FIGURES/2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Santos M.E., Athanasiadis A., Leitão A.B., Dupasquier L., Sucena É. Alternative splicing and gene duplication in the evolution of the FoxP gene subfamily. Mol. Biol. Evol. 2011;28:237–247. doi: 10.1093/MOLBEV/MSQ182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Niu Y., Wang G., Li Y., Guo W., Guo Y., Dong Z. LncRNA FOXP4-AS1 promotes the progression of esophageal squamous cell carcinoma by interacting with MLL2/H3K4me3 to upregulate FOXP4. Front. Oncol. 2021;11 doi: 10.3389/FONC.2021.773864/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma T., Zhang J. Upregulation of FOXP4 in breast cancer promotes migration and invasion through facilitating EMT. Cancer Manag. Res. 2019;11:2783–2793. doi: 10.2147/CMAR.S191641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang G., Zhang G. Upregulation of foxP4 in HCC promotes migration and invasion through regulation of EMT. Oncol. Lett. 2019;17:3944–3951. doi: 10.3892/ol.2019.10049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gaaib J.N. Prediction of deleterious non-synonymous single nucleotide polymorphisms (Nssnps) of human TLR7 gene. Gaaib Iraqi Journal of Science. 2022;63:2444–2452. doi: 10.24996/ijs.2022.63.6.11. [DOI] [Google Scholar]
- 10.Ahmad T., Valentovic M.A., Rankin G.O. Effects of cytochrome P450 single nucleotide polymorphisms on methadone metabolism and pharmacodynamics. Biochem. Pharmacol. 2018;153:196–204. doi: 10.1016/J.BCP.2018.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee J.E., Choi J.H., Lee J.H., Lee M.G. Gene SNPs and mutations in clinical genetic testing: haplotype-based testing and analysis. Mutat. Res. Fund Mol. Mech. Mutagen. 2005;573:195–204. doi: 10.1016/J.MRFMMM.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 12.Krawczak M., V Ball E., Fenton I., Stenson P.D., Abeysinghe S., Thomas N., Cooper D.N. Human gene mutation database—a biomedical information and research resource. Hum. Mutat. 2000;15:45–51. doi: 10.1002/(SICI)1098-1004(200001)15:1. [DOI] [PubMed] [Google Scholar]
- 13.Thusberg J., Vihinen M. Pathogenic or not? And if so, then how? Studying the effects of missense mutations using bioinformatics methods. Hum. Mutat. 2009;30:703–714. doi: 10.1002/HUMU.20938. [DOI] [PubMed] [Google Scholar]
- 14.Sim N.L., Kumar P., Hu J., Henikoff S., Schneider G., Ng P.C. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012;40 doi: 10.1093/NAR/GKS539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adzhubei I., Jordan D.M., Sunyaev S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet Chapter. 2013;7 doi: 10.1002/0471142905.HG0720S76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hecht M., Bromberg Y., Rost B. Better prediction of functional effects for sequence variants. BMC Genom. 2015;16:1–12. doi: 10.1186/1471-2164-16-S8-S1/FIGURES/4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Capriotti E., Fariselli P. PhD-SNPg: a webserver and lightweight tool for scoring single nucleotide variants. Nucleic Acids Res. 2017;45:W247–W252. doi: 10.1093/NAR/GKX369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang H., Thomas P.D. PANTHER-PSEP: predicting disease-causing genetic variants using position-specific evolutionary preservation. Bioinformatics. 2016;32:2230–2232. doi: 10.1093/BIOINFORMATICS/BTW222. [DOI] [PubMed] [Google Scholar]
- 19.Capriotti E., Fariselli P., Casadio R. I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005;33 doi: 10.1093/NAR/GKI375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Worth C.L., Preissner R., Blundell T.L. SDM—a server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res. 2011;39:W215. doi: 10.1093/NAR/GKR363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garnier J., Gibrat J.F., Robson B. [32] GOR method for predicting protein secondary structure from amino acid sequence. Methods Enzymol. 1996;266:540–553. doi: 10.1016/S0076-6879(96)66034-0. [DOI] [PubMed] [Google Scholar]
- 22.Ashkenazy H., Erez E., Martz E., Pupko T., Ben-Tal N. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010;38 doi: 10.1093/NAR/GKQ399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klausen M.S., Jespersen M.C., Nielsen H., Jensen K.K., Jurtz V.I., Sønderby C.K., Sommer M.O.A., Winther O., Nielsen M., Petersen B., Marcatili P. NetSurfP-2.0: improved prediction of protein structural features by integrated deep learning. Proteins: Struct., Funct., Bioinf. 2019;87:520–527. doi: 10.1002/PROT.25674. [DOI] [PubMed] [Google Scholar]
- 24.Venselaar H., te Beek T.A.H., Kuipers R.K.P., Hekkelman M.L., Vriend G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinf. 2010;11:1–10. doi: 10.1186/1471-2105-11-548/FIGURES/5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodrigues C.H.M., Pires D.E.V., Ascher D.B. DynaMut2: assessing changes in stability and flexibility upon single and multiple point missense mutations. Protein Sci. 2021;30:60–69. doi: 10.1002/PRO.3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zuberi K., Franz M., Rodriguez H., Montojo J., Lopes C.T., Bader G.D., Morris Q. GeneMANIA prediction server 2013 update. Nucleic Acids Res. 2013;41:W115–W122. doi: 10.1093/NAR/GKT533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Szklarczyk D., Kirsch R., Koutrouli M., Nastou K., Mehryary F., Hachilif R., Gable A.L., Fang T., Doncheva N.T., Pyysalo S., Bork P., Jensen L.J., Von Mering C. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023;51:D638–D646. doi: 10.1093/NAR/GKAC1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Imon R.R., Kabir Talukder M.E., Akhter S., Islam M.S., Ahammad F., Anis-Ul-Haque K.M., Moniruzzaman M., Afroze M., Khan M., Hena Mostofa Jamal M.A., Wani T.A., Uddin M.J., Rahman M.M. Natural defense against multi-drug resistant Pseudomonas aeruginosa: Cassia occidentalis L. in vitro and in silico antibacterial activity. RSC Adv. 2023;13:28773–28784. doi: 10.1039/d3ra03923d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mark P., Nilsson L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A. 2001;105:9954–9960. doi: 10.1021/jp003020w. [DOI] [Google Scholar]
- 30.Roos K., Wu C., Damm W., Reboul M., Stevenson J.M., Lu C., Dahlgren M.K., Mondal S., Chen W., Wang L., Abel R., Friesner R.A., Harder E.D. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theor. Comput. 2019;15:1863–1874. doi: 10.1021/acs.jctc.8b01026. [DOI] [PubMed] [Google Scholar]
- 31.Rabby M.G., Rahman M.H., Islam M.N., Kamal M.M., Biswas M., Bonny M., Hasan M.M. In silico identification and functional prediction of differentially expressed genes in South Asian populations associated with type 2 diabetes. PLoS One. 2023;18 doi: 10.1371/JOURNAL.PONE.0294399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Teufel A., Wong E.A., Mukhopadhyay M., Malik N., Westphal H. FoxP4, a novel forkhead transcription factor. Biochim. Biophys. Acta Gene Struct. Expr. 2003;1627:147–152. doi: 10.1016/S0167-4781(03)00074-5. [DOI] [PubMed] [Google Scholar]
- 33.Weigel D., Gerd, Irgens J., Kiittner F., Seifert E., Jlckle H. The homeotic gene fork head encodes a nuclear protein and is expressed in the terminal regions of the Drosophila embryo. Cell. 1969;57:645–658. doi: 10.1016/0092-8674(89)90133-5. [DOI] [PubMed] [Google Scholar]
- 34.Lai E., Prezioso V.R., Smith E., Litvin O., Costa R.H., Darnell J.E. 1990. HNF-3A, a Hepatocyte-Enriched Transcnpuon Factor of Novel Structure is Regulated Transcriptionally. [DOI] [PubMed] [Google Scholar]
- 35.Shu W., Yang H., Zhang L., Lu M.M., Morrisey E.E. Characterization of a new subfamily of winged-helix/forkhead (fox) genes that are expressed in the lung and act as transcriptional repressors. J. Biol. Chem. 2001;276:27488–27497. doi: 10.1074/jbc.M100636200. [DOI] [PubMed] [Google Scholar]
- 36.Lu M.M., Li S., Yang H., Morrisey E.E. Foxp4: a novel member of the Foxp subfamily of winged-helix genes co-expressed with Foxp1 and Foxp2 in pulmonary and gut tissues. Mech. Dev. 2002;119:S197–S202. doi: 10.1016/S0925-4773(03)00116-3. [DOI] [PubMed] [Google Scholar]
- 37.Hori S., Nomura T., Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. J. Immunol. 2017;198:981–985. doi: 10.1126/SCIENCE.1079490/SUPPL_FILE/HORI.SOM.PDF. [DOI] [PubMed] [Google Scholar]
- 38.Charng W.L., Karaca E., Coban Akdemir Z., Gambin T., Atik M.M., Gu S., Posey J.E., Jhangiani S.N., Muzny D.M., Doddapaneni H., Hu J., Boerwinkle E., Gibbs R.A., Rosenfeld J.A., Cui H., Xia F., Manickam K., Yang Y., Faqeih E.A., Al Asmari A., Saleh M.A.M., El-Hattab A.W., Lupski J.R. Exome sequencing in mostly consanguineous Arab families with neurologic disease provides a high potential molecular diagnosis rate. BMC Med. Genom. 2016;9:1–14. doi: 10.1186/S12920-016-0208-3/FIGURES/4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hussain M.R.M., Shaik N.A., Al-Aama J.Y., Asfour H.Z., Subhani Khan F., Masoodi T.A., Khan M.A., Shaik N.S. In silico analysis of Single Nucleotide Polymorphisms (SNPs) in human BRAF gene. Gene. 2012;508:188–196. doi: 10.1016/J.GENE.2012.07.014. [DOI] [PubMed] [Google Scholar]
- 40.Hasan M.A., Hakim F.T., Islam Shovon M.T., Islam M.M., Islam M.S., Islam M.A. The investigation of nonsynonymous SNPs of human SLC6A4 gene associated with depression: an in silico approach. Heliyon. 2021;7 doi: 10.1016/J.HELIYON.2021.E07815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kamal M.M., Islam M.N., Rabby M.G., Zahid M.A., Hasan M.M. In silico functional and structural analysis of non-synonymous single nucleotide polymorphisms (nsSNPs) in human paired box 4 gene. Biochem. Genet. 2023:1–24. doi: 10.1007/S10528-023-10589-1/METRICS. [DOI] [PubMed] [Google Scholar]
- 42.Agrahari A.K., Pieroni E., Gatto G., Kumar A. The impact of missense mutation in PIGA associated to paroxysmal nocturnal hemoglobinuria and multiple congenital anomalies-hypotonia-seizures syndrome 2: a computational study. Heliyon. 2019;5 doi: 10.1016/j.heliyon.2019.e02709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rabi L.T., Valente D.Z., de Souza Teixeira E., Peres K.C., de Oliveira Almeida M., Bufalo N.E., Ward L.S. Potential new cancer biomarkers revealed by quantum chemistry associated with bioinformatics in the study of selectin polymorphisms. Heliyon. 2024;10 doi: 10.1016/J.HELIYON.2024.E28830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ahmed S.S., Rifat Z.T., Lohia R., Campbell A.J., Dunker A.K., Rahman M.S., Iqbal S. Characterization of intrinsically disordered regions in proteins informed by human genetic diversity. PLoS Comput. Biol. 2022;18 doi: 10.1371/JOURNAL.PCBI.1009911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bappy M.N.I., Roy A., Rabbi M.G.R., Jahan N., Chowdhury F.A., Hoque S.F., Sajib E.H., Khan P., Hossain F.M.A., Zinnah K.M.A. Scrutinizing deleterious nonsynonymous SNPs and their effect on human POLD1 gene. Genet. Res. 2022;2022:e61. doi: 10.1155/2022/1740768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Strnad O., Vilém Šustr V., Kozlíková B., Sochor J. 2013. Real-time Visualization of Protein Empty Space with Varying Parameters; pp. 65–70. [Google Scholar]
- 47.E. Jung Choi, E. Jin Seo, D. Kyoung Kim, S. In Lee, Y. Woo Kwon, I. Ho Jang, K.-H. Kim, D.-S. Suh, J. Ho Kim, FOXP1 functions as an oncogene in promoting cancer stem cell-like characteristics in ovarian cancer cells, n.d. www.impactjournals.com/oncotarget. [DOI] [PMC free article] [PubMed]
- 48.Enard W., Przeworski M., Fisher S.E., Lai C.S.L., Wiebe V., Kitano T., Monaco A.P., Pääbo S. Molecular evolution of FOXP2, a gene involved in speech and language. Nature. 2002;418:869–872. doi: 10.1038/nature01025. [DOI] [PubMed] [Google Scholar]
- 49.yi Wu S., Liao P., yu Yan L., yi Zhao Q., yu Xie Z., Dong J., tao Sun H. Correlation of MKI67 with prognosis, immune infiltration, and T cell exhaustion in hepatocellular carcinoma. BMC Gastroenterol. 2021;21 doi: 10.1186/s12876-021-01984-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abad C., Robayo M.C., Muñiz-Moreno M. del M., Bernardi M.T., Otero M.G., Kosanovic C., Griswold A.J., Pierson T.M., Walz K., Young J.I. Gatad2b, associated with the neurodevelopmental syndrome GAND, plays a critical role in neurodevelopment and cortical patterning. Translational Psychiatry 2024. 2024;14(1 14):1–13. doi: 10.1038/s41398-023-02678-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu A., Pang J., Xiong G., Liu Q., Chen L. Forkhead Box P4 promotes the proliferation of cells in colorectal adenocarcinoma. Oncologie. 2023;25:543–552. doi: 10.1515/oncologie-2023-0009. [DOI] [Google Scholar]
- 52.Lin P.-J., Chiu F.-Y., Wang S.-C., Li C.-Y. The oncogenic role of ARG2 in hepatocellular carcinoma. 2020. 10.1200/JCO.2020.38.15_suppl.E16713 38. [DOI]
- 53.Galosi S., Edani B.H., Martinelli S., Hansikova H., Eklund E.A., Caputi C., Masuelli L., Corsten-Janssen N., Srour M., Oegema R., Bosch D.G.M., Ellis C.A., Amlie-Wolf L., Accogli A., Atallah I., Averdunk L., Barañano K.W., Bei R., Bagnasco I., Brusco A., Demarest S., Alaix A.S., Di Bonaventura C., Distelmaier F., Elmslie F., Gan-Or Z., Good J.M., Gripp K., Kamsteeg E.J., MacNamara E., Marcelis C., Mercier N., Peeden J., Pizzi S., Pannone L., Shinawi M., Toro C., Verbeek N.E., Venkateswaran S., Wheeler P.G., Zdrazilova L., Zhang R., Zorzi G., Guerrini R., Sessa W.C., Lefeber D.J., Tartaglia M., Hamdan F.F., Grabińska K.A., Leuzzi V. De novo DHDDS variants cause a neurodevelopmental and neurodegenerative disorder with myoclonus. Brain. 2022;145:208–223. doi: 10.1093/brain/awab299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim C.H. FOXP3 and its role in the immune system. Adv. Exp. Med. Biol. 2009;665:17–29. doi: 10.1007/978-1-4419-1599-3_2. [DOI] [PubMed] [Google Scholar]
- 55.Zheng C., Yu X., Liang Y., Zhu Y., He Y., Liao L., Wang D., Yang Y., Yin X., Li A., He Q., Li B. Targeting PFKL with penfluridol inhibits glycolysis and suppresses esophageal cancer tumorigenesis in an AMPK/FOXO3a/BIM-dependent manner. Acta Pharm. Sin. B. 2022;12:1271–1287. doi: 10.1016/J.APSB.2021.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Castilla L.H. NRAS palmitoylation and oncogenic fitness. Blood. 2020;135:1725–1726. doi: 10.1182/BLOOD.2020005720. [DOI] [PubMed] [Google Scholar]
- 57.Adam K., Lesperance J., Hunter T., Zage P.E. The potential functional roles of NME1 histidine kinase activity in neuroblastoma pathogenesis. Int. J. Mol. Sci. 2020;21 doi: 10.3390/IJMS21093319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alsanafi M., Brown R.D.R., Oh J., Adams D.R., Torta F., Pyne N.J., Pyne S. Dihydroceramide desaturase functions as an inducer and rectifier of apoptosis: effect of retinol derivatives, antioxidants and phenolic compounds. Cell Biochem. Biophys. 2021;79:461. doi: 10.1007/S12013-021-00990-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang W., Cui X., Sun D., Sun G., Yan Z., Wei M., Wang Z., Yu W. POU5F1 promotes the proliferation, migration, and invasion of gastric cancer cells by reducing the ubiquitination level of TRAF6. Cell Death Dis. 2023;14 doi: 10.1038/S41419-023-06332-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fadda A., Syed N., Mackeh R., Papadopoulou A., Suzuki S., Jithesh P.V., Kino T. Genome-wide regulatory roles of the C2H2-type zinc finger protein ZNF764 on the glucocorticoid receptor. Sci. Rep. 2017;7 doi: 10.1038/srep41598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin M., Li Y., Qin S., Jiao Y., Hua F. Ubiquitin-like modifier-activating enzyme 7 as a marker for the diagnosis and prognosis of breast cancer. Oncol. Lett. 2020;19:2773–2784. doi: 10.3892/OL.2020.11406/DOWNLOAD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mackenzie A.E., Milligan G. The emerging pharmacology and function of GPR35 in the nervous system. Neuropharmacology. 2017;113:661–671. doi: 10.1016/J.NEUROPHARM.2015.07.035. [DOI] [PubMed] [Google Scholar]
- 63.Bao K.C., Wang F.F. The role of SPDEF in cancer: promoter or suppressor. Neoplasma. 2022;69:1270–1276. doi: 10.4149/NEO_2022_220529N571. [DOI] [PubMed] [Google Scholar]
- 64.Rudd J.C., Maity S., Grunkemeyer J.A., Snyder J.C., Lovas S., Hansen L.A. Membrane structure and internalization dynamics of human Flower isoforms hFWE3 and hFWE4 indicate a conserved endocytic role for hFWE4. J. Biol. Chem. 2023;299 doi: 10.1016/j.jbc.2023.104945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Spiegler S., Kirchmaier B., Rath M., Korenke G.C., Tetzlaff F., Van De Vorst M., Neveling K., Acker-Palmer A., Kuss A.W., Gilissen C., Fischer A., Schulte-Merker S., Felbor U. FAM222B is not a likely novel candidate gene for cerebral cavernous malformations. Mol. Syndromol. 2016;7:144–152. doi: 10.1159/000446884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Miller K.M., Tjeertes J.V., Coates J., Legube G., Polo S.E., Britton S., Jackson S.P. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nature Structural & Molecular Biology 2010. 2010;17(9 17):1144–1151. doi: 10.1038/nsmb.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Islam M.N., Rabby M.G., Hossen M.M., Kamal M.M., Zahid M.A., Syduzzaman M., Hasan M.M. In silico functional and pathway analysis of risk genes and SNPs for type 2 diabetes in Asian population. PLoS One. 2022;17 doi: 10.1371/JOURNAL.PONE.0268826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.George R.M., Firulli A.B. Hand factors in cardiac development. Anat. Rec. 2019;302:101–107. doi: 10.1002/AR.23910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang S., Wu J., Wang Z., Gong Z., Liu Y., Wang Z. Emerging roles of Circ-ZNF609 in multiple human diseases. Front. Genet. 2022;13 doi: 10.3389/FGENE.2022.837343/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vijayalingam S., Ezekiel U.R., Xu F., Subramanian T., Geerling E., Hoelscher B., San K.K., Ganapathy A., Pemberton K., Tycksen E., Pinto A.K., Brien J.D., Beck D.B., Chung W.K., Gurnett C.A., Chinnadurai G. Human iPSC-derived neuronal cells from CTBP1-mutated patients reveal altered expression of neurodevelopmental gene networks. Front. Neurosci. 2020;14 doi: 10.3389/fnins.2020.562292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huang J., Gou H., Yao J., Yi K., Jin Z., Matsuoka M., Zhao T. The noncanonical role of EZH2 in cancer. Cancer Sci. 2021;112:1376. doi: 10.1111/CAS.14840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Alam R., Rahman Imon R., Enamul M., Talukder K., Akhter S., Hossain M.A., Ahammad F., Rahman M.M. GC-MS analysis of phytoconstituents from Ruellia prostrata and Senna tora and identification of potential anti-viral activity against SARS-CoV-2. 2021. [DOI] [PMC free article] [PubMed]
- 73.Long Q.Z., Du Y.F., Ding X.Y., Li X., Bin Song W., Yang Y., Zhang P., Zhou J.P., Liu X.G. Replication and fine mapping for association of the C2orf43, FOXP4, GPRC6A and RFX6 genes with prostate cancer in the Chinese population. PLoS One. 2012;7 doi: 10.1371/JOURNAL.PONE.0037866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Manichaikul A., Wang X.Q., Sun L., Dupuis J., Borczuk A.C., Nguyen J.N., Raghu G., Hoffman E.A., Onengut-Gumuscu S., Farber E.A., Kaufman J.D., Rabinowitz D., Stukovsky K.D.H., Kawut S.M., Hunninghake G.M., Washko G.R., O'Connor G.T., Rich S.S., Barr R.G., Lederer D.J. Genome-wide association study of subclinical interstitial lung disease in MESA. Respir. Res. 2017;18:1–11. doi: 10.1186/S12931-017-0581-2/FIGURES/3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shiraishi K., Okada Y., Takahashi A., Kamatani Y., Momozawa Y., Ashikawa K., Kunitoh H., Matsumoto S., Takano A., Shimizu K., Goto A., Tsuta K., Watanabe S.I., Ohe Y., Watanabe Y., Goto Y., Nokihara H., Furuta K., Yoshida A., Goto K., Hishida T., Tsuboi M., Tsuchihara K., Miyagi Y., Nakayama H., Yokose T., Tanaka K., Nagashima T., Ohtaki Y., Maeda D., Imai K., Minamiya Y., Sakamoto H., Saito A., Shimada Y., Sunami K., Saito M., Inazawa J., Nakamura Y., Yoshida T., Yokota J., Matsuda F., Matsuo K., Daigo Y., Kubo M., Kohno T. Association of variations in HLA class II and other loci with susceptibility to EGFR-mutated lung adenocarcinoma. Nature Communications 2016. 2016;7(1 7):1–7. doi: 10.1038/ncomms12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang Z., Seow W.J., Shiraishi K., Hsiung C.A., Matsuo K., Liu J., Chen K., Yamji T., Yang Y., Chang I.S., Wu C., Hong Y.C., Burdett L., Wyatt K., Chung C.C., Li S.A., Yeager M., Hutchinson A., Hu W., Caporaso N., Landi M.T., Chatterjee N., Song M., Fraumeni J.F., Kohno T., Yokota J., Kunitoh H., Ashikawa K., Momozawa Y., Daigo Y., Mitsudomi T., Yatabe Y., Hida T., Hu Z., Dai J., Ma H., Jin G., Song B., Wang Z., Cheng S., Yin Z., Li X., Ren Y., Guan P., Chang J., Tan W., Chen C.J., Chang G.C., Tsai Y.H., Su W.C., Chen K.Y., Huang M.S., Chen Y.M., Zheng H., Li H., Cui P., Guo H., Xu P., Liu L., Iwasaki M., Shimazu T., Tsugane S., Zhu J., Jiang G., Fei K., Park J.Y., Kim Y.H., Sung J.S., Park K.H., Kim Y.T., Jung Y.J., Kang C.H., Park I.K., Kim H.N., Jeon H.S., Choi J.E., Choi Y.Y., Kim J.H., Oh I.J., Kim Y.C., Sung S.W., Kim J.S., Il Yoon H., Kweon S.S., Shin M.H., Seow A., Chen Y., Lim W.Y., Liu J., Wong M.P., Lee V.H.F., Bassig B.A., Tucker M., Berndt S.I., Chow W.H., Ji B.T., Wang J., Xu J., Sihoe A.D.L., Ho J.C.M., Chan J.K.C., Wang J.C., Lu D., Zhao X., Zhao Z., Wu J., Chen H., Jin L., Wei F., Wu G., An S.J., Zhang X.C., Su J., Wu Y.L., Gao Y.T., Xiang Y.B., He X., Li J., Zheng W., Shu X.O., Cai Q., Klein R., Pao W., Lawrence C., Hosgood H.D., Hsiao C.F., Chien L.H., Chen Y.H., Chen C.H., Wang W.C., Chen C.Y., Wang C.L., Yu C.J., Chen H.L., Su Y.C., Tsai F.Y., Chen Y.S., Li Y.J., Yang T.Y., Lin C.C., Yang P.C., Wu T., Lin D., Zhou B., Yu J., Shen H., Kubo M., Chanock S.J., Rothman N., Lan Q. Meta-analysis of genome-wide association studies identifies multiple lung cancer susceptibility loci in never-smoking Asian women. Hum. Mol. Genet. 2016;25:620–629. doi: 10.1093/HMG/DDV494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jin G., Ma H., Wu C., Dai J., Zhang R., Shi Y., Lu J., Miao X., Wang M., Zhou Y., Chen J., Li H., Pan S., Chu M., Lu F., Yu D., Jiang Y., Dong J., Hu L., Chen Y., Xu L., Shu Y., Pan S., Tan W., Zhou B., Lu D., Wu T., Zhang Z., Chen F., Wang X., Hu Z., Lin D., Shen H. Genetic variants at 6p21.1 and 7p15.3 are associated with risk of multiple cancers in Han Chinese. 2012. [DOI] [PMC free article] [PubMed]
- 78.Wu C., Hu Z., He Z., Jia W., Wang F., Zhou Y., Liu Z., Zhan Q., Liu Y., Yu D., Zhai K., Chang J., Qiao Y., Jin G., Liu Z., Shen Y., Guo C., Fu J., Miao X., Tan W., Shen H., Ke Y., Zeng Y., Wu T., Lin D. Genome-wide association study identifies three new susceptibility loci for esophageal squamous-cell carcinoma in Chinese populations. Nature Genetics 2011. 2011;43(7 43):679–684. doi: 10.1038/ng.849. [DOI] [PubMed] [Google Scholar]
- 79.Takata R., Akamatsu S., Kubo M., Takahashi A., Hosono N., Kawaguchi T., Tsunoda T., Inazawa J., Kamatani N., Ogawa O., Fujioka T., Nakamura Y., Nakagawa H. Genome-wide association study identifies five new susceptibility loci for prostate cancer in the Japanese population. Nature Genetics 2010. 2010;42(9 42):751–754. doi: 10.1038/ng.635. [DOI] [PubMed] [Google Scholar]
- 80.Luo Y.S., Zhang K., Cheng Z.S. Absence of association between a long COVID and severe COVID-19 risk variant of FOXP4 and lung cancer. Front. Genet. 2023;14 doi: 10.3389/FGENE.2023.1258829/PDF. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data included in article/supplementary material/referenced in article.