

Abstract
Background:
Neuropsychiatric symptoms (NPS) can be an early manifestation of Alzheimer’s disease (AD). However, the associations among NPS, cognition, and AD biomarkers across the disease spectrum are unclear.
Objectives:
We analyzed cross-sectional mediation pathways between cerebrospinal fluid (CSF) biomarkers of AD (Aβ1–42, p-tau181), cognitive function, and NPS.
Methods:
Primary models included 781 participants from the National Alzheimer’s Coordinating Center (NACC) data set who had CSF analyzed for AD biomarkers using Lumipulse. NPS were assessed with the Neuropsychiatric Inventory Questionnaire (NPI-Q). We assessed cognition with the harmonized MMSE/MoCA, as well as neuropsychological tests sensitive to AD pathology: story recall, naming, animal fluency, and Trails B. The Clinical Dementia Rating (CDR®) scale assessed dementia severity. Mediation models were estimated with Kemeny metric covariance in a structural equation model framework, controlling for age, education, sex, and APOE ε4.
Results:
The sample was older adults (M=73.85, SD=6.68; 49.9% male, 390; 27.9% dementia, 218) who were predominantly white (n=688, 88.1%). Higher p-tau181/Aβ1–42 ratio predicted higher NPI-Q, which was partially mediated by the MMSE/MoCA and, in a second model, story recall. No other pathway was statistically significant. Both the MMSE/MoCA and NPI-Q independently mediated the association between p-tau181/Aβ1–42 ratio and CDR global impairment. With dementia excluded, p-tau181/Aβ1–42 ratio was no longer associated with the NPI-Q.
Conclusion:
NPS may be secondary to cognitive impairment and AD pathology through direct and indirect pathways. NPS independently predict dementia severity in AD. However, AD pathology likely plays less of a role in NPS in samples without dementia. Table 1,2,3,4,5,6
Keywords: Alzheimer’s disease, Neuropsychiatric Symptoms, Cognition, Cerebrospinal Fluid, Biomarkers, Amyloid, P-tau
INTRODUCTION
Neuropsychiatric symptoms (NPS) are a common feature experienced by patients diagnosed with dementia and mild cognitive impairment (MCI) due to Alzheimer’s disease (AD)[1,2]. NPS are associated with early pathological changes, including disruption in various neurotransmitter systems [3], and brainstem involvement may be instrumental in AD pathogenesis [4]. NPS are also predictors of cognitive decline, and thus are of growing interest to clinicians and caregivers [2,5–7]. NPS in AD include changes to personality as well as mood and behavioral disturbances with the most prevalent symptoms including apathy [8,9], anxiety [10,11], and depression [12]. Some authors have proposed mild behavioral impairment as a distinct syndrome of prodromal dementia [5,13,14]. However, the etiology of NPS in dementia remains unclear. A number of studies have suggested that NPS are driven by AD pathology including the aggregation of tau neurofibrillary tangles and amyloid plaques [15–17]. There may even be region specific mechanisms for NPS profiles in AD. For example, apathy appears related to AD pathology impacting the anterior cingulate-subcortical circuit, whereas depression and anxiety appear related to impact on frontal-limbic circuits, hallucinations appear related to impact on frontal and temporal lobes, and delusions appear related to impact on anterior-posterior networks and anterior insular regions (see [18]). Other studies have shown that NPS are a prodrome for incident cognitive impairment among healthy community-dwelling participants [19–21]. However, the etiology of NPS in individuals across disease stages is not entirely understood (e.g., preclinical, MCI, and AD dementia) [22,23].
NPS in dementia have been linked to AD pathology in numerous studies of fluid biomarkers [11,17,24–26]. For example, a study with a large AD sample showed patients with a slightly elevated and subsequently increasing score on the Cornell Scale for Depression in Dementia (CSDD) had significantly lower cerebrospinal fluid amyloid beta (CSF Aβ1–42) level compared to patients who scored consistently low on the CSDD, suggesting that symptoms of depression may be linked to higher disease burden [25]. Further, a study of patients with MCI from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort showed that the presence of anxiety was associated with abnormal CSF Aβ1–42 and total tau (t-tau) concentrations [11]. A recent review study found a large degree of heterogeneity in findings related to CSF correlates of NPS in patients with AD and MCI, emphasizing the need for further exploration into these relationships [27]. Most of the research done in this area has focused on or included patients with cognitive impairment, leading to a gap in knowledge regarding the etiology of NPS in individuals without substantial disease burden. It remains undetermined if AD pathology causes NPS directly or if NPS are indirectly mediated by a decline in cognitive function. That is, NPS could be exacerbated by AD-related cognitive decline without a direct relationship to the underlying pathology (e.g., through secondary effects such as diminished ability to engage in hobbies and social interaction).
A small number of studies have considered cognition when examining the potential relationship between NPS and AD pathology [24,28,29]. Krell-Roesch et al. found that lower CSF Aβ1–42 and higher t-tau/ Aβ1–42 and phosphorylated tau (p-tau)/ Aβ1–42 ratios were associated with clinical symptoms of depression and anxiety in older non-demented adults, suggesting that NPS expression may result from AD pathology and potentially independent of cognitive status [28]. Analyses did not control for cognitive function; however, a stronger association between lower CSF Aβ1–42 and clinical symptoms of anxiety and depression was found in individuals with MCI (versus normal cognition). This stresses the need for further investigation into the role of cognitive status in preclinical AD. Babulal et al. [29] examined the presence of NPS and mood changes in a mixed cohort from the WU Knight ADRC (cognitively normal patients, some with positive AD biomarkers). Results showed that participants with higher CSF tau/ Aβ1–42 ratio had a greater increase in overall mood disturbance across a one year follow up compared to participants with lower values. Higher values also predicted specific increases in symptoms of anxiety and depression. However, there was no significant correlation between change in scores on the Mini-Mental State Examination (MMSE) and total mood disturbance, NPS, or depression. In contrast, Banning et al. [24] found that lower levels of CSF Aβ1–42 and higher levels of CSF p-tau and t-tau were associated with the presence of NPS, and that these associations were mediated by MMSE scores. The results suggest NPS in dementia are partially mediated by cognitive functioning. However, the study failed to assess psychometric properties of NPS in the sample and combined different measures of CSF biomarker analysis. Although an agreement between methods has been established, this relationship is not absolute and could lead to skewed results [30]. Likewise, it is unclear whether these findings will only hold for AD dementia, versus earlier stages in the disease spectrum.
In this study, we investigated the associations between CSF biomarkers (Aβ1–42, p-tau181), NPS, and cognitive function across the disease spectrum. We leveraged a large cross-sectional cohort from the National Alzheimer’s Coordinating Center (NACC) Uniform Data Set (UDS) but restricted our primary analyses to participants with a single CSF assay method (Lumipulse). This study includes only cross-sectional data because of the complexity of the models and the lack of consistent data across timepoints. We included multiple neuropsychological tests that are sensitive to AD pathology and conducted factor analyses to assess the structure of the NPS measure. Along with models examining NPS as the outcome, we assessed a secondary model to see whether cognition and NPS independently mediate the relationship between CSF biomarkers and dementia severity.
METHODS
Participants and Design
The sample included data from 2009 participants from the NACC who completed UDS visits between 2005 and 2020, and who had baseline CSF biomarker data from within a year of their UDS visit. Of these, 781 had CSF analyzed by Lumipulse, which was the primary analytic sample used for this study. This was chosen because it was the most common CSF collection method, and originated from a single site (the Knight Alzheimer Disease Research Center (ADRC) at Washington University in St. Louis, MO). Of note, all participants from the Knight ADRC are offered biomarker testing, including lumbar puncture (LP) with CSF analysis. Approximately 76.8% of all Knight ADRC participants receive a baseline LP in place of or along with another biomarker method (e.g., amyloid positron emission tomography). Participants agree to LP in principle, excluding participants with advanced dementia. LP participation is also optional for participants from under-represented groups. Further, many participants who agree to LP are later excluded due to physician objection or known contraindication (e.g., ongoing anticoagulant therapy or space-occupying cerebral lesion). Despite this, the majority of eligible participants receive at least a baseline LP (see [31]).
Since 2005, >30 ADRCs have participated in the NACC-UDS, a database of standardized cognitive, behavioral, and functional evaluations (full description can be found elsewhere [32,33]). Of these, nine sites had available CSF biomarker data. We included a larger sample from all nine sites in secondary models (to increase the generalizability of the findings; see analytic plan). Supplemental Figure 1 provides a CONSORT flow diagram of participants included.
Standard protocol approvals, registrations, and patient consents
The NACC database was approved by the University of Washington Institutional Review Board. Informed consent was obtained at each individual ADC. Involvement of human subjects was done in accord with the Helsinki Declaration of 1975.
CSF Biomarkers
LP is not routine for all ADRCs that contribute to NACC and sharing of CSF data to NACC is voluntary. Nine sites in the current data had available CSF data. In the present sample, we analyzed participants whose CSF biomarker data was analyzed using Lumipulse (n=781). All Lumipulse-based samples were collected at a single ADRC, the Knight ADRC. Primary models included p-tau181/Aβ1–42 ratio as the assumed best predictor of underlying AD pathology (i.e., given widespread clinical use and superior concordance with amyloid PET scans compared to individual biomarkers) [34–36]. As a sensitivity analysis, models were recomputed with p-tau181 and Aβ1–42 as individual predictors. Of note, t-tau was not a significant predictor in any model (alone or in combination with other biomarkers), and was not included in the analysis to minimize model complexity. Samples were collected and processed as previously described [37]. LP was done after overnight fasting. Samples were collected in a 50 mL polypropylene tube using an atraumatic Sprotte 22-gauge spinal needle. CSF was placed on ice and centrifuged at low speed within two hours. CSF was transferred to another 50 mL tube and aliquoted at 500 uL into polypropylene tubes and stored at −80°C [38]. For biomarker analysis, samples were brought to room temperature, vortexed, and transferred to polystyrene cuvettes. Concentrations of each biomarker were measured by chemiluminescent enzyme immunoassay using a fully automated platform (LUMIPULSE G1200, Fujirebio, Ghent, Belgium). Reagents were derived from a single lot.
Of the other eight sites, 704 samples were analyzed using Luminex, 512 were analyzed by enzyme-linked immunosorbent assay (ELISA), and 12 were analyzed using Athena ADMark. These last 12 participants were excluded due to the small numbers in the group. These participants were only analyzed in secondary models due to differences in analyte concentrations by assay methods (see Methods).
Neuropsychiatric Symptoms
The Neuropsychiatric Inventory Questionnaire (NPI-Q) was used as a measure of NPS [39]. In the NPI-Q, study partners (collateral sources) are asked to evaluate the patient on 12 symptoms over the past month. Each symptom is rated on a scale from 0 (absent) to 3 (severe). The NPI-Q was intended to produce a composite or summary score. Numerous factor analyses have been conducted in various samples with the NPI-Q, with diverse structures retained across the samples (see [40]). However, several of these studies (e.g., [41–43]) used principal component analysis, a factor analytic method which hypothesizes the absence of measurement error, and is therefore inappropriate for a Likert scale such as the NPI-Q (see [44]). In this study, we employed a factor analytic method without this limitation (see Measurement and Latent Constructs). We used factor models given the original intention of the NPI-Q [39], and to reduce multiple comparisons.
Neuropsychological Tests and Dementia Severity
Participants receive the UDS neuropsychological battery at approximately annual visits to each ADRC [45–47]. The UDS has had three versions, with the most recent (UDS version 3.0) released in March 2015 [45]. At this most recent update, several tests from the neuropsychological battery were replaced with non-proprietary alternatives. We used results from the Crosswalk study to harmonize scores between versions [46]. From the full UDS battery, we included tests that are sensitive to AD pathology, including tests of episodic memory (i.e., story recall; Logical Memory Delayed Recall and Craft Story Delayed Recall) [48,49], confrontation naming (Boston Naming Test and Multilingual Naming Test) [50–52], semantic fluency (Animals) [53,54], and set shifting (i.e., switching between instructional sets; Trail Making Test Part B [TMT-B]) [55]. We also included a global cognitive screening measure (Mini-Mental State Examination [MMSE] or Montreal Cognitive Assessment [MoCA]) [56,57]. Raw scores were used for these measures. Of note, the full battery of tests did not fit into a latent measurement model (see Results).
Lower scores indicate worse performance for all but one test, TMT-B, where slower speed in seconds indicates worse performance. As noted, for participants who received earlier versions of the NACC UDS, neuropsychological test scores were harmonized with the Crosswalk study [46]. This included scores from Logical Memory, the Boston Naming Test, and the MoCA. Final harmonized cognitive test variables included the MMSE/MoCA, Story Recall, Naming, Animal Fluency, and TMT-B.
We assessed dementia severity using global rating scores from the Clinical Dementia Rating® (CDR®) Dementia Staging Instrument [58]. The CDR stages severity of dementia using informant report of orientation, judgment/problem solving, memory, home and hobbies, personal care, and community affairs. Higher scores reflect worse severity of dementia. Participants were excluded from respective models if data on mediators or outcomes were missing, including the MMSE/CDR (n=21) and other neuropsychological tests (n=68). The same participants were missing data from the MMSE/MoCA and CDR.
Measurement and Latent Constructs
Prior to inferential models, we examined the factor structure of the NPI-Q to better understand our primary outcome in this sample. Given diverse factor structures previously documented with the NPI-Q [40], we initially conducted parallel analysis (see [59]). We next assessed the recommended number of retained factors with confirmatory factor analyses (CFAs). For all factor analyses and subsequent structural models, we used the Kemeny metric space to construct a non-parametric covariance matrix with a generalized linear rank framework. These associations are affine-linearly invariant over all monotone transformations, enabling stable linear decomposition and analysis [60]. Loadings were estimated with generalized least squares (GLS). Higher factor scores suggest increased NPS. Confirmatory fit was assessed with the comparative fit index (CFI), the root mean square error of approximation (RMSEA), and the standardized root-mean-square residual (SRMR).[61–63] Satisfactory fit criteria were as follows: CFI > 0.95, RMSEA < 0.06, and SRMR < 0.08 [63]. When relevant, models were compared with the Bayesian information criterion (BIC), with lower values suggesting a better model [64,65].
Missing NPI-Q data were rare in the sample (≤ 1%) and no NPI-Q data were missing in samples included in structural models (i.e., participants without missing relevant moderators). Mean values for NPI-Q items ranged from 0.02 (hallucinations) to 0.37 (irritability/lability). Skewness was high for several items including delusions (5.99), hallucinations (11.97), elation/euphoria (9.49), disinhibition (3.89), motor disturbance (4.91), and appetite/eating (3.21); kurtosis was > 4 for all items, further supporting a nonparametric approach. While many of the factor loadings were low and cannot be validly interpreted as part of the construct, we did not remove these items from structural models because to do so would remove the ability to account for measurement error from these items, and because they were considered an important part of the overall construct when the scale was designed (i.e., items to be interpreted with a summary score; see [39]). In addition, exploratory factor analyses were run to compare unidimensional and multidimensional models. In all cases, the unidimensional model was the preferred model (e.g., BICfactor1= −255.7, BICfactor2= 38.18, BICfactor3 = 286.12).
As a final step, we assessed whether the full battery of neuropsychological tests would fit into a latent measurement model following the same procedure.
Statistical Analyses
Analyses were performed with the lavaan package in the statistical programming language R (version 4.2.3) [66]. Primary models assessed whether cognition, as measured by neuropsychological tests, mediated the relationship between CSF biomarkers and the NPI-Q. As described, harmonized raw scores were used for neuropsychological tests. In addition, p-tau181/ Aβ1–42 ratio was used in primary models and individual CSF biomarkers in sensitivity models. To replicate the most similar past study, and due to discrepancies in missing data, we initially used the MMSE/MoCA as a stand-alone mediator. We then conducted a second model with Story Recall, Naming, Animal Fluency, and TMT-B as independent mediators. Mediation models were assessed in an SEM framework. Unlike the Baron & Kenny approach [67], SEM simultaneously estimates latent variables and mediation pathways while correcting for measurement error; the schema for and definition of mediation analyses can be found in Figure 1. Based on these findings, an exploratory model included NPI-Q factor scores and MMSE/MoCA scores as independent mediators of the relationship between CSF biomarkers and CDR global impairment scores. We did not include additional neuropsychological tests in this model given the sample size differences and the complexity of the model. Age, biological sex, years of education, and Apolipoprotein E (APOE) ε4 allele genotyping (ε4 carriers vs. non-carriers) were included as covariates in all models. When multiple mediation pathways were statistically significant, contrasts were used to compare statistical differences between pathways. Statistics of variance are presented as R2. The absolute value of standardized weights can be directly compared to assess the relative importance of a predictor in a particular model.
Figure 1.

Schema for Mediation Analyses. An initial structural equation model estimated the c path, which is the total direct effect of the predictors on the outcome. Next, the same direct path was estimated (c’) while simultaneously estimating mediation effects (indirect pathway a × b). The total effect is the sum of direct and indirect pathways for a predictor. Mediators included 1) harmonized MMSE/MoCA scores, or 2) harmonized neuropsychological tests: Story Recall, Naming, Animal Fluency, and Trail Making Test Part B. Note: 42-Residue-Long Amyloid β Isoform (Aβ1–42); Neuropsychiatric Inventory Questionnaire (NPI-Q); Tau Phosphorylated at Threonine-181 (p-tau181).
To examine the impact of early versus late-stage disease, we conducted sensitivity analyses in which we removed all participants with dementia from the models (as diagnosed by the local ADRC). These models included smaller samples with complete data on the MMSE/MoCA (n=546) and complete data on other neuropsychological tests (n=539). To ensure that the findings from the sample with Lumipulse CSF collection were not due to undocumented confounds compared to other participants, we replicated primary models in the full sample with any specified CSF collection method (N=2009), including participants with complete data on the MMSE/MoCA (n=1665) and complete data on other neuropsychological tests (n=1478). We added covariates to account for CSF collection method to these models. These variables were converted into binary indicators (i.e., dummy coding) to permit analysis with our generalized linear rank framework. We recognize that adding collection method as a covariate does not fully and properly account for discrepancies in method. For this reason, we only included these models as a sensitivity analysis and focused on a single biomarker method (Lumipulse) for primary models.
RESULTS
Descriptive Characteristics
The primary sample included 781 NACC participants with Lumipulse CSF biomarker collection. Of these, 688 identified as non-Hispanic White (88.1%) and 390 identified as male (49.9%). The sample were older adults (Median = 74, Interquartile range = (69, 78)), highly educated (Myears = 15.67; SD = 2.81), primarily English speaking (n=776; 99.4%), and right-hand dominant (n=686; 87.8%). Descriptive characteristics stratified by dementia status can be found in Table 1. In the sample with dementia (n = 218), most received a clinical diagnosis of Alzheimer’s disease (94.5%). There were two individuals diagnosed with traumatic brain injury (0.9%), one participant was diagnosed with frontotemporal lobar degeneration (0.5%), and there were nine participants with an unspecified or unclear diagnosis (4.1%).
Table 1.
Descriptive Characteristics Stratified by Dementia Status in the Primary Sample (n = 781).
| Non-Dementia | Dementia | SMD | |
|---|---|---|---|
| (n = 563) | (n = 218) | ||
| Age, yrs, Median (IQR) | 74 [70, 78] | 74 [68, 79] | 0.151 |
| Sex (female), n (%) | 297 (52.8) | 94 (43.1) | 0.194 |
| Education, yrs | 15.92 (2.74) | 15.01 (2.91) | 0.322 |
| Race, n (%) | 0.183 | ||
| White | 490 (87.0) | 198 (90.8) | |
| Black or African American | 49 (8.7) | 10 (4.6) | |
| Native Hawaiian or Pacific Islander | 1 (0.2) | 0 (0.0) | |
| Asian | 4 (0.7) | 1 (0.5) | |
| Multiracial | 19 (3.4) | 9 (4.1) | |
| Primary Language, n (%) | 0.086 | ||
| English | 559 (99.3) | 217 (99.5) | |
| Japanese | 1 (0.2) | 0 (0.0) | |
| Other | 3 (0.6) | 1 (0.5) | |
| Handedness, n (%) | 0.132 | ||
| Left | 51 (9.1) | 27 (12.4) | |
| Right | 498 (88.5) | 188 (86.2) | |
| Mixed | 14 (2.5) | 3 (1.4) | |
| APOE ε4 status, n (%) | 0.578 | ||
| Νο ε4 allele | 356 (63.3) | 84 (38.5) | |
| One ε4 allele copy | 187 (33.3) | 102 (46.8) | |
| Two ε4 allele copies | 19 (3.4) | 32 (14.7) | |
| History of Hypertension, n (%) | 0.325 | ||
| Absent | 137 (24.3) | 70 (32.1) | |
| Present | 143 (25.4) | 73 (33.5) | |
| Unknown | 283 (50.3) | 75 (34.4) | |
| History of Diabetes, n (%) | 0.103 | ||
| Absent | 499 (88.6) | 198 (90.8) | |
| Type-2 | 33 (5.9) | 8 (3.7) | |
| Unknown | 31 (5.5) | 12 (5.5) | |
| History of Hypercholesterolemia, n (%) | 0.478 | ||
| Absent | 172 (30.6) | 91 (41.7) | |
| Present | 221 (39.3) | 102 (46.8) | |
| Unknown | 170 (30.2) | 25 (11.5) | |
| History of TBI, n (%) | 0.428 | ||
| Absent | 357 (63.4) | 173 (79.4) | |
| Present | 37 (6.6) | 17 (7.8) | |
| Unknown | 169 (30.0) | 28 (12.8) | |
| CSF Aβ1–42 (pg/mL) | 795.77 (360.70) | 513.51 (251.95) | 0.907 |
| CSF p-tau181 (pg/mL) | 50.68 (32.30) | 89.65 (50.48) | 0.920 |
| CSF t-tau (pg/mL) | 382.40 (240.33) | 634.33 (381.48) | 0.790 |
| CSF p-tau181/Aβ1–42 (pg/mL) | 0.08 (0.08) | 0.21 (0.15) | 1.063 |
| MMSE/MoCA | 28.81 (1.52) | 24.42 (4.48) | 1.312 |
| Naming | 27.68 (2.37) | 23.56 (6.06) | 0.896 |
| Story Recall | 12.20 (4.71) | 4.04 (4.38) | 1.794 |
| Animal Fluency | 19.83 (5.33) | 13.75 (5.73) | 1.10 |
| TMT-B | 91.71 (44.91) | 155.82 (85.37) | 0.94 |
| CDR Global n (%) | 3.769 | ||
| 0.0 | 489 (86.9) | 0 (0.0) | |
| 0.5 | 74 (13.1) | 146 (67.0) | |
| 1.0 | 0 (0.0) | 63 (28.9) | |
| 2.0 | 0 (0.0) | 9 (4.1) |
Values reflect mean with SD in parentheses, unless otherwise indicated. Sample includes NACC participants with Lumipulse-generated biomarker results. Harmonized neuropsychological variables include the MMSE/MoCA (Mini-Mental State Examination or Montreal Cognitive Assessment), Naming (Boston Naming Test or Multilingual Naming Test), Story Recall (Logical Memory Delayed Recall or Craft Story Recall), Animal Fluency, and TMT-B (Trail Making Test Part B). Amyloid-β protein (Aβ); Apolipoprotein E (APOE); Cerebrospinal fluid (CSF); Clinical Dementia Rating (CDR®) Global Impairment Scores; Phosphorylated-tau (p-tau); Standardized Mean Difference (SMD); Total tau (t-tau).
Overall, moderate to severe NPS were relatively rare in the sample, but this varied by NPI-Q item (see Table 2). Of note, 376 (48.1%) had at least one positive value for at least one symptom (NPI-Q > 0). In the sample without dementia, 199 (35.3%) had at least one positive value.
Table 2.
Rates of Neuropsychiatric Symptoms for the Primary Sample (n=781).
| NPI-Q Item | Absent | Mild | Moderate | Severe | Sum |
|---|---|---|---|---|---|
| Delusions | 752 (96.41%) | 15 (1.92%) | 10 (1.28%) | 3 (0.38%) | 780 (100.00%) |
| Hallucinations | 773 (98.98%) | 4 (0.51%) | 3 (0.38%) | 1 (0.13%) | 781 (100.00%) |
| Agitation/Aggression | 659 (84.38%) | 67 (8.58%) | 46 (5.89%) | 9 (1.15%) | 781 (100.00%) |
| Depression/Dysphoria | 645 (82.69%) | 81 (10.38%) | 46 (5.90%) | 8 (1.03%) | 780 (100.00%) |
| Anxiety | 667 (85.40%) | 60 (7.68%) | 46 (5.89%) | 8 (1.02%) | 781 (100.00%) |
| Elation/Euphoria | 768 (98.34%) | 9 (1.15%) | 3 (0.38%) | 1 (0.13%) | 781 (100.00%) |
| Apathy/Indifference | 669 (85.66%) | 65 (8.32%) | 42 (5.38%) | 5 (0.64%) | 781 (100.00%) |
| Disinhibition | 705 (90.27%) | 53 (6.79%) | 18 (2.30%) | 5 (0.64%) | 781 (100.00%) |
| Irritability/Lability | 600 (76.82%) | 94 (12.04%) | 73 (9.35%) | 14 (1.79%) | 781 (100.00%) |
| Motor Disturbance | 734 (94.10%) | 26 (3.33%) | 15 (1.92%) | 5 (0.64%) | 780 (100.00%) |
| Nighttime Behaviors | 667 (86.40%) | 53 (6.87%) | 37 (4.79%) | 15 (1.94%) | 772 (100.00%) |
| Appetite/Eating Problems | 680 (87.29%) | 67 (8.60%) | 27 (3.47%) | 5 (0.64%) | 779 (100.00%) |
Sample includes NACC participants with Lumipulse-generated biomarker results. Neuropsychiatric Inventory Questionnaire (NPI-Q).
Factor analyses
For samples with Lumipulse-generated CSF biomarker results and complete data on the MMSE/MoCA/CDR (n=760), as well as complete data on neuropsychological tests (n=713), parallel analysis suggested that only one factor should be retained from the NPI-Q. A specified unidimensional CFA demonstrated excellent model fit in both samples (χ2 = 4.27, df = 54, p = 1.00, CFI = 1.00, RMSEA = 0.00, SRMR = 0.01, and, χ2 = 3.61, df = 54, p = 1.00, CFI = 1.00, RMSEA = 0.00, SRMR = 0.01, respectively). While most factor loadings were significantly different from zero in both samples (p < 0.05), several had minimal contribution to the factor (standardized loadings below 0.20; see Table 3). Overall item contribution was similar between both samples. Items were not removed from the model as described in Methods (Measurement and Latent Constructs). All correlation residuals were below |0.10|.
Table 3.
Item Fit in Confirmatory Factor Analysis of the Neuropsychiatric Inventory Questionnaire.
| NPI-Q Item | CFA 1 (n=760) | CFA 2 (n=713) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Loadings | β | SE | z | p | Loadings | β | SE | z | p | |
| Irritability/Lability* | 0.44 | 1 | . | . | . | 0.44 | 1 | . | . | . |
| Hallucinations | 0.02 | 0.01 | 0.03 | 0.31 | 0.759 | 0.01 | 0.00 | 0.02 | 0.14 | 0.886 |
| Delusions | 0.11 | 0.11 | 0.06 | 1.98 | 0.048 | 0.10 | 0.08 | 0.05 | 1.61 | 0.108 |
| Agitation/Aggression | 0.36 | 0.70 | 0.16 | 4.48 | 0.000 | 0.35 | 0.68 | 0.16 | 4.16 | 0.000 |
| Depression/Dysphoria | 0.30 | 0.61 | 0.15 | 4.13 | 0.000 | 0.29 | 0.58 | 0.15 | 3.84 | 0.000 |
| Anxiety | 0.26 | 0.49 | 0.13 | 3.84 | 0.000 | 0.26 | 0.47 | 0.13 | 3.57 | 0.000 |
| Elation/Euphoria | 0.05 | 0.04 | 0.04 | 0.98 | 0.328 | 0.05 | 0.03 | 0.04 | 0.87 | 0.383 |
| Apathy/Indifference | 0.28 | 0.53 | 0.13 | 3.98 | 0.000 | 0.28 | 0.50 | 0.13 | 3.74 | 0.000 |
| Disinhibition | 0.20 | 0.32 | 0.10 | 3.23 | 0.001 | 0.20 | 0.30 | 0.10 | 2.99 | 0.003 |
| Motor Disturbance | 0.14 | 0.17 | 0.07 | 2.39 | 0.017 | 0.12 | 0.13 | 0.07 | 1.92 | 0.055 |
| Nighttime Behaviors | 0.21 | 0.37 | 0.12 | 3.23 | 0.001 | 0.19 | 0.32 | 0.11 | 2.82 | 0.005 |
| Appetite/Eating Problems | 0.13 | 0.24 | 0.10 | 2.29 | 0.022 | 0.14 | 0.23 | 0.11 | 2.20 | 0.028 |
Note: Table reflects item fit for sample with Lumipulse-generated biomarker results and complete data on the MMSE/MoCA/CDR (n=760) or complete data on other neuropsychological tests (n=713). Fit reflects standardized factor loadings and non-standardized fit relative to the reference item and Wald test.
The item with the highest standardized loading was chosen as the reference item. Confirmatory Factor Analysis (CFA); Neuropsychiatric Inventory Questionnaire (NPI-Q); Standard Error (SE).
In the sample with Lumipulse-generated CSF biomarker results and complete data on neuropsychological tests (n=713), parallel analysis suggested that five factors should be retained for the full battery of neuropsychological tests. A specified unidimensional CFA demonstrated poor fit (χ2 = 618.82, df = 54, p < 0.002, CFI = 0.34, RMSEA = 0.12, SRMR = 0.15). No model with fewer than four factors demonstrated adequate fit with any of these fit indices and there were not sufficient indicators (i.e., neuropsychological tests) to construct a multi-dimensional latent model. As a result, only raw scores for a priori selected tests were used.
Mediation Models
In the sample with Lumipulse-generated CSF biomarker results and complete data on the MMSE/MoCA (n=760), we assessed the MMSE/MoCA as mediator for the association between CSF biomarkers and the NPI-Q. The initial (c-path) model had excellent fit, χ2 = 12.13, df = 109, p = 1.00, CFI = 1.00, RMSEA = 0.00, SRMR = 0.01, R2 = 0.09, as did the full mediation model, χ2 = 14.38, df = 120, p = 1.00, CFI = 1.00, RMSEA = 0.00, SRMR = 0.01, R2 = 0.15. In the initial model, higher p-tau181/Aβ1–42 ratio predicted higher NPI-Q. These effects were present but reduced in the full mediation model, suggesting partial mediation (i.e., the mediator only partially accounts for the initial association). Specifically, the association between p-tau181/ Aβ1–42 ratio and the NPI-Q was partially mediated by the MMSE/MoCA, standardized indirect effect (IEz) = 0.066, 95% CI [0.034, 0.099]. See Table 4a and Figure 2a.
Table 4a.
Mediation Model for Cerebrospinal Fluid Biomarkers, MMSE/MoCA scores, and Neuropsychiatric Symptoms
| Path | Predictor | Dependent/Mediator | βz | SE | z | p | 95% CI |
|---|---|---|---|---|---|---|---|
| Initial: c | p-tau181/Aβ1–42 ratio | NPI-Q | 0.262 | 0.055 | 4.753 | 0.000 | (0.154, 0.371) |
| . | Age, yrs | . | −0.086 | 0.054 | −1.581 | 0.114 | (−0.192, 0.021) |
| . | Sex (female) | . | −0.059 | 0.054 | −1.087 | 0.277 | (−0.165, 0.047) |
| . | Education, yrs | . | −0.084 | 0.054 | −1.550 | 0.121 | (−0.190, 0.022) |
| . | APOE ε4 status | . | 0.029 | 0.056 | 0.516 | 0.606 | (−0.081, 0.138) |
| Mediation: c’ | p-tau181/Aβ1–42 ratio | NPI-Q | 0.198 | 0.057 | 3.487 | 0.000 | (0.087, 0.309) |
| . | Age, yrs | . | −0.095 | 0.054 | −1.771 | 0.077 | (−0.200, 0.010) |
| . | Sex (female) | . | −0.043 | 0.054 | −0.798 | 0.425 | (−0.148, 0.062) |
| . | Education, yrs | . | −0.054 | 0.054 | −0.991 | 0.321 | (−0.159, 0.052) |
| . | APOE ε4 status | . | 0.017 | 0.055 | 0.307 | 0.759 | (−0.091, 0.125) |
| a | p-tau181/Aβ1–42 ratio | MMSE/MoCA | −0.252 | 0.035 | −7.187 | 0.000 | (−0.321, −0.183) |
| . | Age, yrs | . | −0.034 | 0.035 | −0.965 | 0.335 | (−0.102, 0.035) |
| . | Sex (female) | . | 0.058 | 0.035 | 1.657 | 0.097 | (−0.011, 0.126) |
| . | Education, yrs | . | 0.124 | 0.035 | 3.577 | 0.000 | (0.056, 0.192) |
| . | APOE ε4 status | . | −0.054 | 0.036 | −1.515 | 0.130 | (−0.124, 0.016) |
| b | MMSE/MoCA | NPI-Q | −0.264 | 0.055 | −4.785 | 0.000 | (−0.372, −0.156) |
| indirect (a×b) | p-tau181/Aβ1–42 ratio | . | 0.066 | 0.017 | 3.966 | 0.000 | (0.034, 0.099) |
| total (c + a×b) | p-tau181/Aβ1–42 ratio | . | 0.264 | 0.055 | 4.778 | 0.000 | (0.156, 0.373) |
Sample included NACC participants with Lumipulse-generated biomarker results and complete data on the MMSE/MoCA (n=760). Amyloid-β protein (Aβ); Apolipoprotein E (APOE); Cerebrospinal fluid (CSF); Mini-Mental State Examination or Montreal Cognitive Assessment (MMSE/MoCA); Neuropsychiatric Inventory Questionnaire (NPI-Q); Phosphorylated-tau (p-tau); Standard Error (SE).
Figure 2a.

Mediation Model for Cerebrospinal Fluid Biomarkers, MMSE/MoCA scores, and Neuropsychiatric Symptoms. Note: Dashed lines represent indirect (mediation) pathways. Sample included NACC participants with Lumipulse-generated biomarker results and complete data on the MMSE/MoCA (n=760). Amyloid-β protein (Aβ); Mini-Mental State Examination or Montreal Cognitive Assessment (MMSE/MoCA); Neuropsychiatric Inventory Questionnaire (NPI-Q); Phosphorylated-tau (p-tau).
In the sample with Lumipulse-generated CSF biomarker results and complete data on neuropsychological tests (n=713), we assessed Story Recall, Naming, Animal Fluency, and TMT-B scores as mediators for the association between CSF biomarkers and the NPI-Q. The initial (c-path) model had excellent fit, χ2 = 9.59, df = 109, p = 1.00, CFI = 1.00, RMSEA = 0.00, SRMR = 0.01, R2 = 0.08, as did the full mediation model, χ2 = 112.55, df = 159, p = 0.99, CFI = 1.00, RMSEA = 0.00, SRMR = 0.04, R2 = 0.13. In the initial model, higher p-tau181/ Aβ1–42 ratio predicted higher NPI-Q. These effects were present but reduced in the full mediation model, suggesting partial mediation. The association between p-tau181/ Aβ1–42 ratio and the NPI-Q was significantly mediated by Story Recall, IEz = 0.049, 95% CI [0.011, 0.088]. None of the other mediation pathways were statistically significant. However, the mediation effect of TMT-B was near statistical significance, IEz = 0.024, 95% CI [−0.003, 0.051]. See Table 4b and Figure 2b.
Table 4b.
Mediation Model for Cerebrospinal Fluid Biomarkers, Neuropsychological Tests, and Neuropsychiatric Symptoms.
| Path | Predictor | Dependent/Mediator | βz | SE | z | p | 95% CI |
|---|---|---|---|---|---|---|---|
| Initial: c | p-tau181/Aβ1–42 ratio | NPI-Q | 0.242 | 0.058 | 4.157 | 0.000 | (0.128, 0.356) |
| . | Age, yrs | . | −0.082 | 0.057 | −1.445 | 0.148 | (−0.194, 0.029) |
| . | Sex (female) | . | −0.061 | 0.057 | −1.078 | 0.281 | (−0.173, 0.050) |
| . | Education, yrs | . | −0.086 | 0.057 | −1.509 | 0.131 | (−0.197, 0.026) |
| . | APOE ε4 status | . | 0.032 | 0.058 | 0.550 | 0.583 | (−0.082, 0.147) |
| Mediation: c’ | p-tau181/Aβ1–42 ratio | NPI-Q | 0.152 | 0.069 | 2.213 | 0.027 | (0.017, 0.287) |
| . | Age, yrs | . | −0.107 | 0.058 | −1.846 | 0.065 | (−0.221, 0.007) |
| . | Sex (female) | . | −0.046 | 0.058 | −0.796 | 0.426 | (−0.159, 0.067) |
| . | Education, yrs | . | −0.030 | 0.062 | −0.489 | 0.625 | (−0.151, 0.091) |
| . | APOE ε4 status | . | 0.016 | 0.059 | 0.270 | 0.787 | (−0.100, 0.131) |
| a | p-tau181/Aβ1–42 ratio | Story Recall | −0.286 | 0.038 | −7.539 | 0.000 | (−0.360, −0.212) |
| . | Age, yrs | . | −0.015 | 0.038 | −0.382 | 0.703 | (−0.090, 0.060) |
| . | Sex (female) | . | 0.108 | 0.038 | 2.834 | 0.005 | (0.033, 0.182) |
| . | Education, yrs | . | 0.114 | 0.038 | 3.002 | 0.003 | (0.039, 0.188) |
| . | APOE ε4 status | . | −0.064 | 0.039 | −1.649 | 0.099 | (−0.141, 0.012) |
| b | Story Recall | NPI-Q | −0.173 | 0.064 | −2.709 | 0.007 | (−0.298, −0.048) |
| indirect (a×b) | p-tau181/Aβ1–42 ratio → Story Recall | . | 0.049 | 0.019 | 2.542 | 0.011 | (0.011, 0.088) |
| total (c + a×b) | p-tau181/Aβ1–42 ratio → Story Recall | . | 0.221 | 0.065 | 3.408 | 0.001 | (0.094, 0.349) |
| a | p-tau181/Aβ1–42 ratio | Naming | −0.136 | 0.041 | −3.275 | 0.001 | (−0.217, −0.055) |
| . | Age, yrs | . | −0.053 | 0.040 | −1.321 | 0.187 | (−0.133, 0.026) |
| . | Sex (female) | . | −0.085 | 0.040 | −2.099 | 0.036 | (−0.163, −0.006) |
| . | Education, yrs | . | 0.139 | 0.040 | 3.474 | 0.001 | (0.061, 0.217) |
| . | APOE ε4 status | . | −0.067 | 0.041 | −1.608 | 0.108 | (−0.148, 0.015) |
| b | Naming | NPI-Q | −0.077 | 0.064 | −1.214 | 0.225 | (−0.202, 0.048) |
| indirect (a×b) | p-tau181/Aβ1–42 ratio → Naming | . | 0.010 | 0.009 | 1.138 | 0.255 | (−0.008, 0.029) |
| total (c + a×b) | p-tau181/Aβ1–42 ratio → Naming | . | 0.183 | 0.067 | 2.752 | 0.006 | (0.053, 0.313) |
| a | p-tau181/Aβ1–42 ratio | Animal Fluency | −0.182 | 0.042 | −4.327 | 0.000 | (−0.264, −0.099) |
| . | Age, yrs | . | −0.075 | 0.041 | −1.814 | 0.070 | (−0.156, 0.006) |
| . | Sex (female) | . | 0.027 | 0.041 | 0.660 | 0.509 | (−0.054, 0.108) |
| . | Education, yrs | . | 0.162 | 0.041 | 3.980 | 0.000 | (0.082, 0.241) |
| . | APOE ε4 status | . | −0.013 | 0.042 | −0.309 | 0.758 | (−0.096, 0.070) |
| b | Animal Fluency | NPI-Q | −0.064 | 0.066 | −0.976 | 0.329 | (−0.193, 0.065) |
| indirect (a×b) | p-tau181/Aβ1–42 ratio → Animal Fluency | . | 0.012 | 0.012 | 0.952 | 0.341 | (−0.012, 0.036) |
| total (c + a×b) | p-tau181/Aβ1–42 ratio → Animal Fluency | . | 0.178 | 0.065 | 2.734 | 0.006 | (0.050, 0.305) |
| a | p-tau181/Aβ1–42 ratio | TMT-B | 0.203 | 0.040 | 5.072 | 0.000 | (0.125, 0.282) |
| . | Age, yrs | . | 0.103 | 0.039 | 2.621 | 0.009 | (0.026, 0.181) |
| . | Sex (female) | . | −0.023 | 0.040 | −0.575 | 0.565 | (−0.100, 0.055) |
| . | Education, yrs | . | −0.139 | 0.039 | −3.545 | 0.000 | (−0.215, −0.062) |
| . | APOE ε4 status | . | −0.004 | 0.041 | −0.107 | 0.915 | (−0.084, 0.075) |
| b | TMT-B | NPI-Q | 0.119 | 0.063 | 1.891 | 0.059 | (−0.004, 0.243) |
| indirect (a×b) | p-tau181/Aβ1–42 ratio → TMT-B | . | 0.024 | 0.014 | 1.770 | 0.077 | (−0.003, 0.051) |
| total (c + a×b) | p-tau181/Aβ1–42 ratio → TMT-B | . | 0.104 | 0.081 | 1.284 | 0.199 | (−0.055, 0.263) |
Sample included NACC participants with Lumipulse-generated biomarker results and complete data on neuropsychological tests (n=713). Amyloid-β protein (Aβ); Apolipoprotein E (APOE); Boston Naming Test or Multilingual Naming Test (Naming); Cerebrospinal fluid (CSF); Logical Memory Delayed Recall or Craft Story Recall (Story Recall); Neuropsychiatric Inventory Questionnaire (NPI-Q); Phosphorylated-tau (p-tau); Standard Error (SE); Trail Making Test Part B (TMT-B).
Fig 3 Figure 2b.
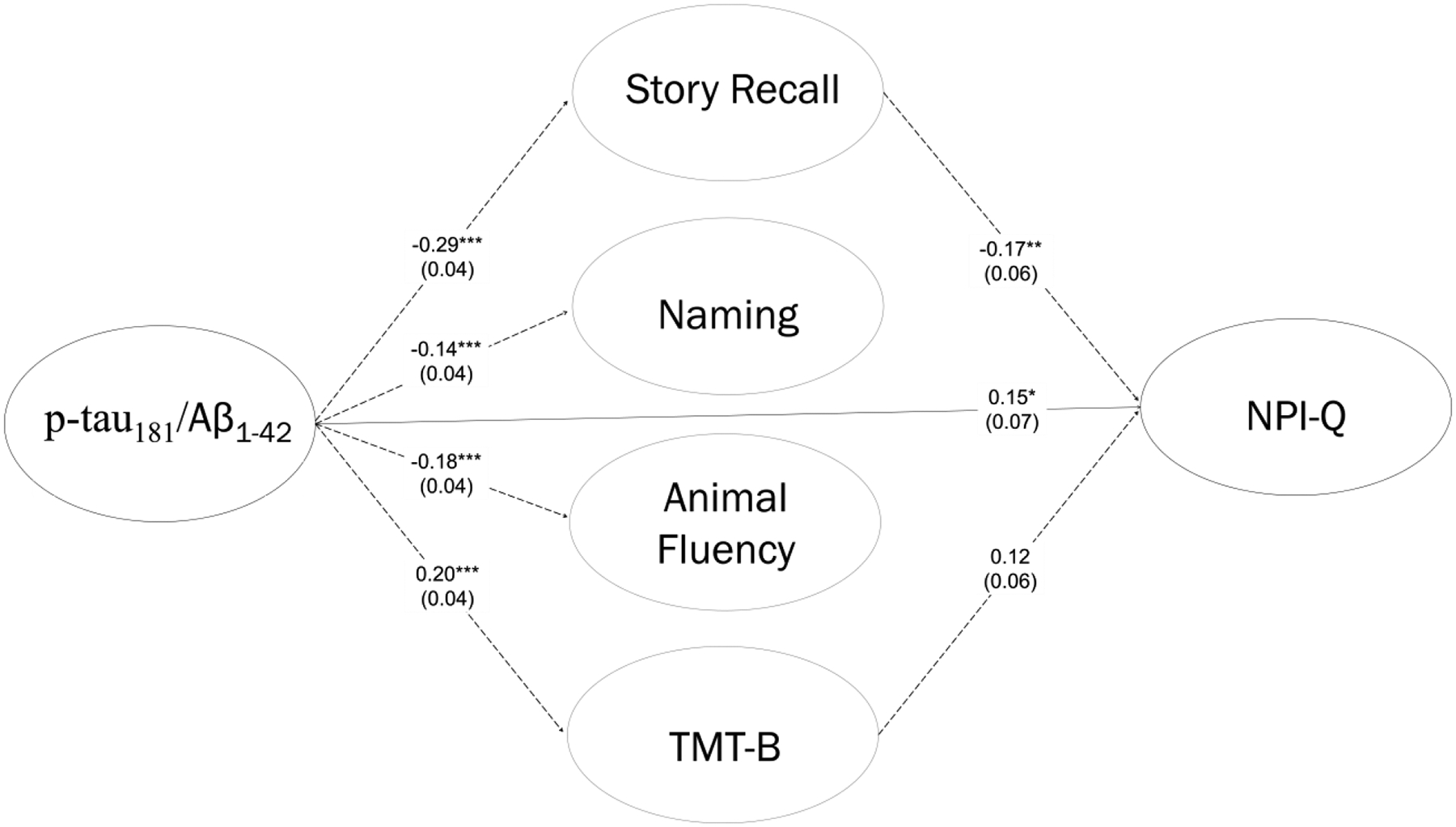
Mediation Model for Cerebrospinal Fluid Biomarkers, Neuropsychological Tests, and Neuropsychiatric Symptoms. Note: Dashed lines represent indirect (mediation) pathways. For simplicity, indirect pathways that are not statistically significant are omitted. Sample included NACC participants with Lumipulse-generated biomarker results and complete data on neuropsychological tests (n=713). Amyloid-β protein (Aβ); Boston Naming Test or Multilingual Naming Test (Naming); Logical Memory Delayed Recall or Craft Story Recall (Story Recall); Neuropsychiatric Inventory Questionnaire (NPI-Q); Phosphorylated-tau (p-tau); Trail Making Test Part B (TMT-B).
In the sample with complete data on the MMSE/MoCA and CDR (n=760), we assessed the MMSE/MoCA and NPI-Q as mediators for the association between CSF biomarkers and CDR global impairment scores. The initial (c-path) model had excellent fit, χ2 < 0.01, df = 5, p = 1.00, CFI = 1.00, RMSEA = 0.00, SRMR = 0.00, R2 = 0.11, as did the full mediation model, χ2 = 36.08, df = 132, p = 1.00, CFI = 1.00, RMSEA = 0.00, SRMR = 0.02, R2 = 0.25. In the initial model, higher p-tau181/ Aβ1–42 ratio predicted higher CDR global impairment. These effects were present but reduced in the full mediation model, suggesting partial mediation. The association between p-tau181/ Aβ1–42 ratio and CDR global impairment was partially mediated by the MMSE/MoCA, IEz = 0.058, 95% CI [0.034, 0.082], as well as the NPI-Q, IEz = 0.091, 95% CI [0.040, 0.143]. The difference between these pathways was not statistically significant (suggesting neither is the more complete mediator), βz = 0.034, 95% CI [−0.021, 0.088]. See Table 4c and Figure 2c.
Table 4c.
Mediation Model for Cerebrospinal Fluid Biomarkers, MMSE/MoCA scores, Neuropsychiatric Symptoms, and Dementia Severity.
| Path | Predictor | Dependent/Mediator | βz | SE | z | p | 95% CI |
|---|---|---|---|---|---|---|---|
| Initial: c | p-tau181/Aβ1–42 ratio | CDR | 0.299 | 0.034 | 8.717 | 0.000 | (0.232, 0.366) |
| . | Age, yrs | . | −0.032 | 0.035 | −0.937 | 0.349 | (−0.100, 0.035) |
| . | Sex (female) | . | −0.040 | 0.035 | −1.156 | 0.248 | (−0.108, 0.028) |
| . | Education, yrs | . | −0.051 | 0.035 | −1.480 | 0.139 | (−0.119, 0.017) |
| . | APOE ε4 status | . | 0.055 | 0.036 | 1.557 | 0.120 | (−0.014, 0.125) |
| Mediation: c’ | p-tau181/Aβ1–42 ratio | CDR | 0.152 | 0.042 | 3.605 | 0.000 | (0.069, 0.234) |
| . | Age, yrs | . | −0.010 | 0.036 | −0.290 | 0.772 | (−0.080, 0.059) |
| . | Sex (female) | . | −0.006 | 0.035 | −0.174 | 0.862 | (−0.076, 0.063) |
| . | Education, yrs | . | 0.009 | 0.036 | 0.246 | 0.806 | (−0.061, 0.079) |
| . | APOE ε4 status | . | 0.031 | 0.036 | 0.850 | 0.395 | (−0.040, 0.102) |
| a | p-tau181/Aβ1–42 ratio | NPI-Q | 0.279 | 0.059 | 4.744 | 0.000 | (0.164, 0.395) |
| Age, yrs | . | −0.091 | 0.058 | −1.579 | 0.114 | (−0.205, 0.022) | |
| Sex (female) | . | −0.064 | 0.058 | −1.115 | 0.265 | (−0.178, 0.049) | |
| Education, yrs | . | −0.090 | 0.058 | −1.551 | 0.121 | (−0.203, 0.024) | |
| APOE ε4 status | . | 0.030 | 0.059 | 0.506 | 0.613 | (−0.086, 0.147) | |
| b | NPI-Q | CDR | 0.327 | 0.057 | 5.767 | 0.000 | (0.216, 0.438) |
| indirect (a×b) | p-tau181/Aβ1–42 ratio → NPI-Q | . | 0.091 | 0.026 | 3.467 | 0.001 | (0.040, 0.143) |
| total (c + a×b) | p-tau181/Aβ1–42 ratio → NPI-Q | . | 0.159 | 0.041 | 3.903 | 0.000 | (0.079, 0.239) |
| a | p-tau181/Aβ1–42 ratio | MMSE/MoCA | −0.258 | 0.036 | −7.183 | 0.000 | (−0.328, −0.187) |
| Age, yrs | . | −0.035 | 0.036 | −0.973 | 0.331 | (−0.105, 0.035) | |
| Sex (female) | . | 0.059 | 0.036 | 1.666 | 0.096 | (−0.010, 0.129) | |
| Education, yrs | . | 0.126 | 0.035 | 3.565 | 0.000 | (0.057, 0.196) | |
| APOE ε4 status | . | −0.055 | 0.037 | −1.502 | 0.133 | (−0.127, 0.017) | |
| b | MMSE | CDR | −0.225 | 0.035 | −6.507 | 0.000 | (−0.293, −0.157) |
| indirect (a×b) | p-tau181/Aβ1–42 ratio → MMSE/MoCA | . | 0.058 | 0.012 | 4.796 | 0.000 | (0.034, 0.082) |
| total (c + a×b) | p-tau181/Aβ1–42 ratio → MMSE/MoCA | . | 0.147 | 0.048 | 3.036 | 0.002 | (0.052, 0.242) |
| contrast (a×b – a×b) | p-tau181/Aβ1–42 ratio → NPI-Q vs p-tau181/Aβ1–42 ratio → MMSE/MoCA | . | 0.034 | 0.028 | 1.214 | 0.225 | (−0.021, 0.088) |
Sample included NACC participants with Lumipulse-generated biomarker results and complete data on the MMSE/MoCA (n=760). Amyloid-β protein (Aβ); Apolipoprotein E (APOE); Cerebrospinal fluid (CSF); Clinical Dementia Rating (CDR®) Global Impairment Scores; Mini-Mental State Examination or Montreal Cognitive Assessment (MMSE/MoCA); Neuropsychiatric Inventory Questionnaire (NPI-Q); Phosphorylated-tau (p-tau); Standard Error (SE).
Fig 4 Figure 2c.

Mediation Model for Cerebrospinal Fluid Biomarkers, MMSE/MoCA scores, Neuropsychiatric Symptoms, and Dementia Severity. Note: Dashed lines represent indirect (mediation) pathways. Sample included NACC participants with Lumipulse-generated biomarker results and complete data on the MMSE/MoCA (n=760). Amyloid-β protein (Aβ); Clinical Dementia Rating (CDR®) Global Impairment Scores; Mini-Mental State Examination or Montreal Cognitive Assessment (MMSE/MoCA); Neuropsychiatric Inventory Questionnaire (NPI-Q); Phosphorylated-tau (p-tau).
Sensitivity Models
For all models, including CSF Aβ1–42 and CSF p-tau181 as independent predictors replicated findings from models using p-tau181/Aβ1–42 ratio. That is, lower CSF Aβ1–42 and higher CSF p-tau181 predicted higher NPI-Q. These effects reduced and/or were no longer statistically significant in full mediation models, suggesting partial to more complete mediation. The same mediation pathways were and were not statistically significant (see Supplemental Table 1a,b). Statistics of variance were slightly higher (MMSE/MoCA: R2 = 0.16; neuropsychological tests: R2 = 0.14). Likewise, in the sample with complete data on the MMSE/MoCA and CDR (n=760), lower CSF Aβ1–42 and higher CSF p-tau181 predicted higher CDR global impairment. These effects were present but reduced in the full mediation model, suggesting partial mediation. Statistics of variance were again slightly higher (baseline: R2 = 0.12, mediation: R2 = 0.26). The same mediation pathways were statistically significant (see Supplemental Table 2). Likewise, the difference between these pathways was not statistically significant.
To examine the impact of early versus late-stage disease, we assessed whether the MMSE/MoCA continued to mediate the association between CSF biomarkers and the NPI-Q in participants without dementia (n=546). The initial (c-path) model had excellent fit, χ2 = 4.16, df = 109, p = 1.00, CFI = 1.00, RMSEA = 0.00, SRMR = 0.01. However, CSF biomarkers were not significant predictors of the NPI-Q in the sample. As a result, a full mediation model was not conducted. We assessed whether Story Recall, Naming, Animal Fluency, or TMT-B mediated the association between CSF biomarkers and the NPI-Q in participants without dementia (n=539). The initial (c-path) model had excellent fit, χ2 = 4.09, df = 109, p = 1.00, CFI = 1.00, RMSEA = 0.00, SRMR = 0.01. However, CSF biomarkers were again not significant predictors of the NPI-Q in the sample. As a result, a full mediation model was not conducted.
To ensure generalizability, we assessed whether the MMSE/MoCA continued to mediate the association between CSF biomarkers and the NPI-Q in the sample with relevant complete data and any CSF collection method (n=1665). The initial (c-path) model had excellent fit, χ2 = 35.45, df = 142, p = 1.00, CFI = 1.00, RMSEA = 0.00, SRMR = 0.01, R2 = 0.10, as did the full mediation model, χ2 = 41.30, df = 153, p = 1.00, CFI = 1.00, RMSEA = 0.00, SRMR = 0.01, R2 = 0.19. Findings were largely the same compared to the sample with Lumipulse-generated CSF biomarker results. Next, we assessed whether Story Recall, Naming, Animal Fluency, or TMT-B scores mediated the association between CSF biomarkers and the NPI-Q in the sample with relevant complete data and any CSF collection method (n=1478). The initial (c-path) model had excellent fit, χ2 = 25.61, df = 142, p = 1.00, CFI = 1.00, RMSEA = 0.00, SRMR = 0.01, R2 = 0.08. However, there was evidence of poor fit in the full mediation model, χ2 = 318.62, df = 192, p < 0.001, CFI = 0.64, RMSEA = 0.02, SRMR = 0.05, R2 = 0.14. There were also a few differences in findings compared to the sample with Lumipulse-generated CSF biomarker results. Along with story recall, which remained a significant mediator, TMT-B was a statistically significant mediator in the larger sample, IEz = 0.029, 95% CI [0.007, 0.051]. See Supplemental Tables 3a,b for full model results from these analyses.
DISCUSSION
The purpose of this study was to investigate whether cognition plays a mediating role in the relationship between CSF biomarkers (Aβ1–42, p-tau181) and NPS across the AD disease spectrum. In addition to analyzing models with NPS as the primary outcome, we also explored whether cognition and NPS act as independent mediators in the association between CSF biomarkers and dementia severity. Findings suggest that higher p-tau181/ Aβ1–42 ratio, the assumed best predictor of underlying AD pathology [34–36], was associated with worse cognitive performance, consequently resulting in more pronounced NPS. Estimates of variance explained were modest and relatively consistent between models, likely due to the previously stated measurement error within the NPI-Q (see Measurement and Latent Constructs). CSF biomarkers and mediators were relatively stronger predictors than other covariates based on standardized weights. However, cognitive measures only partially mediated the relationship between CSF biomarkers and NPS, suggesting an independent, direct relationship between AD pathology and NPS (i.e., independent of cognitive effects in an AD sample). Among cognitive tests included in this study, story recall emerged as the only retained mediator, with the highest standardized effect. This is consistent with past research that poor recall is a prominent cognitive deficit expected in AD throughout the disease course [68,69]. Global cognition and NPS independently mediated the relationship between CSF biomarkers and dementia severity, with minimal difference between the pathways. As such, AD pathology may disrupt both cognition and mental health, which independently contribute to functional disturbance.
Patients with AD have higher frequency of NPS than general populations [70,71], which can precede cognitive impairment [7,72], and impacts quality of life, daily function, caregiver burden, and institutionalization [13,73,74]. However, the independent and shared pathways between cognition and NPS in AD may lead to different expressions throughout the disease course. Studies have shown that NPS tend to fluctuate within and between patients over time, compared to the relatively steady cognitive decline seen in AD [72,75,76]. Likewise, some studies have found that baseline NPS predicts longitudinal cognitive decline [77–79], but this is not consistent [72,80]. Since psychiatric symptoms impact cognitive test performance (see [81]), it is possible that cognitive functions and NPS create a feedback loop in AD patients over time [82]. That is, cognitive deficits exacerbate NPS and elevated NPS may worsen cognitive deficits.
In our study, when participants with dementia were excluded from models, CSF biomarkers no longer predicted NPS. In contrast to past research ([23] provides a review), this suggests that NPS may not necessarily serve as a better early indicator of AD compared to cognitive measures. However, this observation also supports the directional assumptions made in our primary analyses. In early stages of the disease with less AD pathology, NPS might stem from a variety of factors, including pre-existing psychiatric conditions and psychosocial factors. When pathology builds and disperses in the brain, we observe increased cognitive consequences, potentially exacerbating NPS. It may be that only stable, late onset NPS stemming specifically from AD correlates with worsened cognitive decline (versus transient psychiatric symptoms resulting from factors other than AD; see [83]).
A difference between transient and stable NPS would explain inconsistent associations between AD pathology and NPS across the disease spectrum, particularly in studies that combine groups [16,24,84]. Prevalence of NPS at a given disease stage may vary by symptom, with each symptom having a distinct atrophy, neuropathologic, and regional cerebral blood activation profile [85–87]. There may also be instrumental limitations impacting findings. For example, the NPI-Q was validated to assess psychopathology in dementia patients, rather than earlier in the disease course [39]. Longitudinal worsening of NPS may be a better indicator of AD pathology [17,29,81,88], which we did not examine in this cross-sectional study. Regardless, our findings are relatively consistent with and expand upon the most similar past study described above [24]. Likewise, these findings have implications for management of NPS in AD patients.
The cognitive sequelae of AD may exacerbate NPS, making it crucial to address cognitive symptoms in treatment of these patients. Memantine, particularly in combination with acetylcholinesterase inhibitors (AChEI), demonstrates improved efficacy for NPS, compared to AChEI alone [89,90]. However, monotherapy with AchEI can also provide relief [91]. Conversely, psychotropic medications in this context have varying support and/or more severe side-effect profiles [92–94]. One meta-analysis found that risperidone and galantamine had the best evidence for treatment of NPS across dementia syndromes [95]. Current investigations explore glycogen synthase kinase-3 (GSK3) inhibitors and other kinase inhibitors [96,97], while monoclonal antibody medications targeting Ab (like lecanemab) await evaluation for NPS impact [98]. For non-responsive or contraindicated cases, emerging evidence supports behavioral, interpersonal, and environmental interventions [99–102]. However, additional studies are needed to identify at-risk AD patients and tailor treatment to specific symptom profiles.
This study has several strengths which contribute to the reliability and significance of findings. First, we addressed non-standardized CSF assay methods by focusing primary analyses on participants with CSF analyzed by Lumipulse, and conducting sensitivity analyses to confirm the consistency of results. Second, our study employed appropriate psychometric analyses of the NPI-Q, which improves upon the existing literature. Third, utilizing innovative nonparametric methods presents a practical alternative for studies employing the NPI-Q (versus assessing items individually or combining items without consideration for measurement error). Fourth, we extended past research by incorporating several neuropsychological tests sensitive to AD pathology. This permits interrogation of the specific aspects of cognition linked to the proposed mediation pathways (e.g., memory). Finally, leveraging data from NACC provided a large, methodologically consistent cohort, further fortifying the credibility of the research.
Our study has several limitations, particularly stemming from analysis of the NPI-Q. We observed that NPI-Q item loadings varied. In some instances, items cannot be reliably interpreted as integral to the construct. In our sample, these items do not operate as intended by the scale’s design, which was meant for interpretation with a summary score (see [39]). Despite this, we opted to include these items in analyses. Doing so permits the opportunity to account for measurement error, and these items were considered a crucial part of the construct when the scale was formulated. The limited number of tests administered as part of the UDS neuropsychological battery prevented the estimation of a multidimensional model for these variables. Analyses are further complicated by reliance on the Crosswalk study [46] to harmonize across UDS versions, which could increase noise/error. In primary models, we focused on a single CSF biomarker method, which limits the generalizability of the sample. Likewise, our sensitivity models are limited in addressing variations in CSF assay method. Partialling out variance likely does not fully account for these differences. While mediation suggests potential causality in a cross-sectional study, this represents only one possible explanation. For example, these models do not include all possible covariates; unmeasured variables could be either primary or secondary confounds to the outcome or association being studied (see [103] for a discussion). As noted, we included cross-sectional data for this study because of the complexity of our models and the lack of consistent data across timepoints. Future studies should investigate longitudinal models with NACC samples while perhaps sacrificing psychometric complexity. Finally, the NACC is comprised of individuals from ADRCs across the U.S. and is not representative of the general population. As with any CSF biomarker study, samples from the NACC that choose to undergo LP are not necessarily reflective of all AD patients. For example, participants from the Knight ADRC with advanced dementia do not receive LP. Regardless, examining NPS in this sample provides value, and ongoing replication will address concerns from any particular study.
Our findings suggest that higher p-tau181/ Aβ1–42 ratio, a strong indicator of AD pathology, was associated with worse cognitive performance and more pronounced NPS. However, cognitive measures only partially explained the relationship, indicating an independent link between AD pathology and NPS. Global cognition and NPS independently mediated the relationship between CSF biomarkers and dementia severity, suggesting the independent impact of AD pathology on functional disturbance through both cognitive decline and NPS. Early in the disease course, NPS may result from various factors. As AD pathology progresses, cognitive effects may exacerbate NPS, creating complex and varied symptom expressions in patients with AD.
Supplementary Material
Acknowledgments:
The authors have no acknowledgments to report. The NACC database is funded by NIA/NIH Grant U24 AG072122. NACC data are contributed by the NIA-funded ADRCs: P30 AG062429 (PI James Brewer, MD, PhD), P30 AG066468 (PI Oscar Lopez, MD), P30 AG062421 (PI Bradley Hyman, MD, PhD), P30 AG066509 (PI Thomas Grabowski, MD), P30 AG066514 (PI Mary Sano, PhD), P30 AG066530 (PI Helena Chui, MD), P30 AG066507 (PI Marilyn Albert, PhD), P30 AG066444 (PI John Morris, MD), P30 AG066518 (PI Jeffrey Kaye, MD), P30 AG066512 (PI Thomas Wisniewski, MD), P30 AG066462 (PI Scott Small, MD), P30 AG072979 (PI David Wolk, MD), P30 AG072972 (PI Charles DeCarli, MD), P30 AG072976 (PI Andrew Saykin, PsyD), P30 AG072975 (PI David Bennett, MD), P30 AG072978 (PI Neil Kowall, MD), P30 AG072977 (PI Robert Vassar, PhD), P30 AG066519 (PI Frank LaFerla, PhD), P30 AG062677 (PI Ronald Petersen, MD, PhD), P30 AG079280 (PI Eric Reiman, MD), P30 AG062422 (PI Gil Rabinovici, MD), P30 AG066511 (PI Allan Levey, MD, PhD), P30 AG072946 (PI Linda Van Eldik, PhD), P30 AG062715 (PI Sanjay Asthana, MD, FRCP), P30 AG072973 (PI Russell Swerdlow, MD), P30 AG066506 (PI Todd Golde, MD, PhD), P30 AG066508 (PI Stephen Strittmatter, MD, PhD), P30 AG066515 (PI Victor Henderson, MD, MS), P30 AG072947 (PI Suzanne Craft, PhD), P30 AG072931 (PI Henry Paulson, MD, PhD), P30 AG066546 (PI Sudha Seshadri, MD), P20 AG068024 (PI Erik Roberson, MD, PhD), P20 AG068053 (PI Justin Miller, PhD), P20 AG068077 (PI Gary Rosenberg, MD), P20 AG068082 (PI Angela Jefferson, PhD), P30 AG072958 (PI Heather Whitson, MD), P30 AG072959 (PI James Leverenz, MD).
Funding:
This work was supported by grants from the National Institutes of Health (P30AG072978) and the Department of Veterans’ Affairs (I01BX005933). This publication was also supported through BU-CTSI Grant Number 1UL1TR001430. HZ is a Wallenberg Scholar and a Distinguished Professor at the Swedish Research Council supported by grants from the Swedish Research Council (#2023-00356; #2022-01018 and #2019-02397), the European Union’s Horizon Europe research and innovation programme under grant agreement No 101053962, Swedish State Support for Clinical Research (#ALFGBG-71320), the Alzheimer Drug Discovery Foundation (ADDF), USA (#201809-2016862), the AD Strategic Fund and the Alzheimer’s Association (#ADSF-21-831376-C, #ADSF-21-831381-C, #ADSF-21-831377-C, and #ADSF-24-1284328-C), the Bluefield Project, Cure Alzheimer’s Fund, the Olav Thon Foundation, the Erling-Persson Family Foundation, Familjen Rönströms Stiftelse, Stiftelsen för Gamla Tjänarinnor, Hjärnfonden, Sweden (#FO2022-0270), the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 860197 (MIRIADE), the European Union Joint Programme - Neurodegenerative Disease Research (JPND2021-00694), the National Institute for Health and Care Research University College London Hospitals Biomedical Research Centre, and the UK Dementia Research Institute at UCL (UKDRI-1003). KB is supported by the Swedish Research Council (#2017-00915), the Alzheimer Drug Discovery Foundation (ADDF), USA (#RDAPB-201809-2016615), the Swedish Alzheimer Foundation (#AF-742881), Hjärnfonden, Sweden (#FO2017-0243), the Swedish state under the agreement between the Swedish government and the County Councils, the ALF-agreement (#ALFGBG-715986), the European Union Joint Program for Neurodegenerative Disorders (JPND2019-466-236), and the National Institute of Health (NIH), USA, (grant #1R01AG068398-01). TKK was funded by the Brightfocus Foundation (#A2020812F), the Swedish Alzheimer Foundation (Alzheimerfonden; #AF-930627), the Swedish Brain Foundation (Hjärnfonden; #FO2020-0240), the Swedish Parkinson Foundation (Parkinsonfonden), the Swedish Dementia Foundation (Demensförbundet), the Agneta Prytz-Folkes & Gösta Folkes Foundation (#2020-00124), the Aina (Ann) Wallströms and Mary-Ann Sjöbloms Foundation, the Anna Lisa and Brother Björnsson’s Foundation, Gamla Tjänarinnor, and the Gun and Bertil Stohnes Foundation. KWT is supported by the US Department of Veterans Affairs, (# IK2 CX002065). JCM is funded by grants from the National Institutes of Health (P30 AG066444; P01AG003991; P01AG026276). GMB is supported by the National Institute on Aging (R01AG080469, R01AG068183, R01AG067428, R01AG074302, R01AG074302) and Bright Focus Foundation (A2021142S).
Footnotes
CRediT Author Statement:
Brandon Frank (Conceptualization, Data Curation, Formal Analysis, Methodology, Project Administration, Visualization, Writing – original draft), Michael Walsh (Conceptualization, Methodology, Project Administration, Writing – original draft), Landon Hurley (Conceptualization, Formal Analysis, Methodology, Software, Validation, Writing – original draft), Jenna Groh (Conceptualization, Writing – original draft), Kaj Blennow (Methodology, Supervision, Writing – review & editing), Henrik Zetterberg (Methodology, Supervision, Writing – review & editing), Yorghos Tripodis (Funding Acquisition, Supervision, Writing – review & editing), Andrew E. Budson (Funding Acquisition, Supervision, Writing – review & editing), Maureen O’Connor (Funding Acquisition, Supervision, Writing – review & editing), Brett Martin (Data Curation, Methodology), Jason Weller (Funding Acquisition, Supervision, Writing – review & editing), Ann McKee (Funding Acquisition, Supervision, Writing – review & editing), Wendy Qiu (Funding Acquisition, Supervision, Writing – review & editing), Thor D. Stein (Funding Acquisition, Supervision, Writing – review & editing), Robert A. Stern (Funding Acquisition, Supervision, Writing – review & editing), Jesse Mez (Funding Acquisition, Supervision, Writing – review & editing), Rachel Henson (Data Curation, Funding Acquisition, Investigation, Resources, Writing – review & editing), Justin Long (Data Curation, Funding Acquisition, Investigation, Resources, Writing – review & editing), Andrew J. Aschenbrenner (Investigation, Writing – review & editing), Ganesh M. Babulal (Data Curation, Funding Acquisition, Investigation, Resources, Writing – review & editing), John C. Morris (Data Curation, Funding Acquisition, Investigation, Resources, Supervision, Writing – review & editing), Suzanne Schindler (Data Curation, Funding Acquisition, Investigation, Resources, Supervision, Writing – review & editing), Michael L Alosco (Conceptualization, Funding Acquisition, Methodology, Project Administration, Supervision, Writing – review & editing).
Conflict of Interest: Drs. Henrik Zetterberg and Ganesh Babulal are Editorial Board Members of this journal but were not involved in the peer-review process of this article nor had access to any information regarding its peer-review. MLA receives research support from Life Molecular Imaging Inc and Rainwater Charitable Foundation Inc. He has also received a single time honorarium from the Michael J Fox Foundation for services unrelated to this study. He received royalties from Oxford University Press Inc. SES served on advisory boards for Eisai. AEB receives research support from Bristol-Myers Squibb and Vox Neuro. He receives consulting monies from Eli Lilly and AbbVie. He receives royalties from Elsevier and Oxford University Press. HZ has served at scientific advisory boards and/or as a consultant for Abbvie, Acumen, Alector, Alzinova, ALZPath, Amylyx, Annexon, Apellis, Artery Therapeutics, AZTherapies, Cognito Therapeutics, CogRx, Denali, Eisai, Merry Life, Nervgen, Novo Nordisk, Optoceutics, Passage Bio, Pinteon Therapeutics, Prothena, Red Abbey Labs, reMYND, Roche, Samumed, Siemens Healthineers, Triplet Therapeutics, and Wave, has given lectures in symposia sponsored by Alzecure, Biogen, Cellectricon, Fujirebio, Lilly, Novo Nordisk, and Roche, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program (outside submitted work). None of this research, consulting, or royalties are related to this study.
Data Availability:
The data supporting the findings of this study are available upon request from the National Alzheimer’s Coordinating Center (https://naccdata.org/requesting-data/data-request-process).
REFERENCES
- [1].Lyketsos CG, Carrillo MC, Ryan JM, Khachaturian AS, Trzepacz P, Amatniek J, Cedarbaum J, Brashear R, Miller DS (2011) Neuropsychiatric symptoms in Alzheimer’s disease. Alzheimers Dement J Alzheimers Assoc 7, 532–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Masters MC, Morris JC, Roe CM (2015) “Noncognitive” symptoms of early Alzheimer disease. Neurology 84, 617–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Šimić G, Babić Leko M, Wray S, Harrington CR, Delalle I, Jovanov-Milošević N, Bažadona D, Buée L, de Silva R, Di Giovanni G, Wischik CM, Hof PR (2017) Monoaminergic neuropathology in Alzheimer’s disease. Prog Neurobiol 151, 101–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Simic G, Stanic G, Mladinov M, Jovanov-Milosevic N, Kostovic I, Hof PR (2009) Does Alzheimer’s disease begin in the brainstem? Neuropathol Appl Neurobiol 35, 532–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ismail Z, Smith EE, Geda Y, Sultzer D, Brodaty H, Smith G, Agüera-Ortiz L, Sweet R, Miller D, Lyketsos CG, ISTAART Neuropsychiatric Symptoms Professional Interest Area (2016) Neuropsychiatric symptoms as early manifestations of emergent dementia: Provisional diagnostic criteria for mild behavioral impairment. Alzheimers Dement J Alzheimers Assoc 12, 195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Vik-Mo AO, Giil LM, Ballard C, Aarsland D (2018) Course of neuropsychiatric symptoms in dementia: 5-year longitudinal study. Int J Geriatr Psychiatry 33, 1361–1369. [DOI] [PubMed] [Google Scholar]
- [7].Wise EA, Rosenberg PB, Lyketsos CG, Leoutsakos J-M (2019) Time course of neuropsychiatric symptoms and cognitive diagnosis in National Alzheimer’s Coordinating Centers volunteers. Alzheimers Dement Diagn Assess Dis Monit 11, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nobis L, Husain M (2018) Apathy in Alzheimer’s disease. Curr Opin Behav Sci 22, 7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dolphin H, Dyer AH, McHale C, O’Dowd S, Kennelly SP (2023) An Update on Apathy in Alzheimer’s Disease. Geriatr Basel Switz 8, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mendez MF The Relationship Between Anxiety and Alzheimer’s Disease1. J Alzheimers Dis Rep 5, 171–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ramakers IHGB, Verhey FRJ, Scheltens P, Hampel H, Soininen H, Aalten P, Rikkert MO, Verbeek MM, Spiru L, Blennow K, Trojanowski JQ, Shaw LM, Visser PJ, Alzheimer’s Disease Neuroimaging Initiative and DESCRIPA Investigators (2013) Anxiety is related to Alzheimer cerebrospinal fluid markers in subjects with mild cognitive impairment. Psychol Med 43, 911–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chi S, Wang C, Jiang T, Zhu X-C, Yu J-T, Tan L (2015) The prevalence of depression in Alzheimer’s disease: a systematic review and meta-analysis. Curr Alzheimer Res 12, 189–198. [DOI] [PubMed] [Google Scholar]
- [13].Sheikh F, Ismail Z, Mortby ME, Barber P, Cieslak A, Fischer K, Granger R, Hogan DB, Mackie A, Maxwell CJ, Menon B, Mueller P, Patry D, Pearson D, Quickfall J, Sajobi T, Tse E, Wang M, Smith EE, Investigators for the P registry (2018) Prevalence of mild behavioral impairment in mild cognitive impairment and subjective cognitive decline, and its association with caregiver burden. Int Psychogeriatr 30, 233–244. [DOI] [PubMed] [Google Scholar]
- [14].Ismail Z, McGirr A, Gill S, Hu S, Forkert ND, Smith EE (2021) Mild Behavioral Impairment and Subjective Cognitive Decline Predict Cognitive and Functional Decline. J Alzheimers Dis 80, 459–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Krell-Roesch J, Zaniletti I, Syrjanen JA, Kremers WK, Algeciras-Schimnich A, Dage JL, van Harten AC, Fields JA, Knopman DS, Jack CR, Petersen RC, Vassilaki M, Geda YE (2023) Plasma-derived biomarkers of Alzheimer’s disease and neuropsychiatric symptoms: A community-based study. Alzheimers Dement Amst Neth 15, e12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Krell-Roesch J, Vassilaki M, Mielke MM, Kremers WK, Lowe VJ, Vemuri P, Machulda MM, Christianson TJ, Syrjanen JA, Stokin GB, Butler LM, Traber M, Jack CR, Knopman DS, Roberts RO, Petersen RC, Geda YE (2019) Cortical β-amyloid burden, neuropsychiatric symptoms, and cognitive status: the Mayo Clinic Study of Aging. Transl Psychiatry 9, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Donovan NJ, Locascio JJ, Marshall GA, Gatchel J, Hanseeuw BJ, Rentz DM, Johnson KA, Sperling RA (2018) Longitudinal Association of Amyloid Beta and Anxious-Depressive Symptoms in Cognitively Normal Older Adults. Am J Psychiatry 175, 530–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Chen Y, Dang M, Zhang Z (2021) Brain mechanisms underlying neuropsychiatric symptoms in Alzheimer’s disease: a systematic review of symptom-general and –specific lesion patterns. Mol Neurodegener 16, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Krell-Roesch J, Syrjanen JA, Machulda MM, Christianson TJ, Kremers WK, Mielke MM, Knopman DS, Petersen RC, Vassilaki M, Geda YE (2021) Neuropsychiatric symptoms and the outcome of cognitive trajectories in older adults free of dementia: The Mayo Clinic Study of Aging. Int J Geriatr Psychiatry 36, 1362–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Xu X, Ang SL, Hilal S, Chan QL, Wong TY, Venketasubramanian N, Ikram MK, Chen CL-H (2015) Association of neuropsychiatric symptoms and sub-syndromes with cognitive impairment in community-dwelling Asian elderly. Int Psychogeriatr 27, 1839–1847. [DOI] [PubMed] [Google Scholar]
- [21].Kociolek AJ, Fernandez KK, Hernandez M, Jin Z, Cosentino S, Zhu CW, Gu Y, Devanand DP, Stern Y (2023) Neuropsychiatric Symptoms and Trajectories of Dependence and Cognition in a Sample of Community-dwelling Older Adults with Dementia. Curr Alzheimer Res 20, 409–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Geda YE, Krell-Roesch J, Sambuchi N, Michel BF (2017) Neuropsychiatric Symptoms and Neuroimaging Biomarkers in Alzheimer Disease: “Which is the Cart and Which is the Horse?” Am J Geriatr Psychiatry Off J Am Assoc Geriatr Psychiatry 25, 694–696. [DOI] [PubMed] [Google Scholar]
- [23].Ng KP, Chiew H, Rosa-Neto P, Kandiah N, Ismail Z, Gauthier S (2021) Associations of AT(N) biomarkers with neuropsychiatric symptoms in preclinical Alzheimer’s disease and cognitively unimpaired individuals. Transl Neurodegener 10, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Banning LCP, Ramakers IHGB, Köhler S, Bron EE, Verhey FRJ, de Deyn PP, Claassen JAHR, Koek HL, Middelkoop HAM, van der Flier WM, van der Lugt A, Aalten P, Alzheimer’s Disease Neuroimaging Initiative, Parelsnoer Institute Neurodegenerative Diseases study group (2020) The Association Between Biomarkers and Neuropsychiatric Symptoms Across the Alzheimer’s Disease Spectrum. Am J Geriatr Psychiatry Off J Am Assoc Geriatr Psychiatry 28, 735–744. [DOI] [PubMed] [Google Scholar]
- [25].Barca ML, Persson K, Eldholm R, Benth JŠ, Kersten H, Knapskog A-B, Saltvedt I, Selbaek G, Engedal K (2017) Trajectories of depressive symptoms and their relationship to the progression of dementia. J Affect Disord 222, 146–152. [DOI] [PubMed] [Google Scholar]
- [26].Goukasian N, Hwang KS, Romero T, Grotts J, Do TM, Groh JR, Bateman DR, Apostolova LG (2019) Association of brain amyloidosis with the incidence and frequency of neuropsychiatric symptoms in ADNI: a multisite observational cohort study. BMJ Open 9, e031947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Showraki A, Murari G, Ismail Z, Barfett JJ, Fornazzari L, Munoz DG, Schweizer TA, Fischer CE (2019) Cerebrospinal Fluid Correlates of Neuropsychiatric Symptoms in Patients with Alzheimer’s Disease/Mild Cognitive Impairment: A Systematic Review. J Alzheimers Dis JAD 71, 477–501. [DOI] [PubMed] [Google Scholar]
- [28].Krell-Roesch J, Rakusa M, Syrjanen JA, van Harten AC, Lowe VJ, Jack CR Jr., Kremers WK, Knopman DS, Stokin GB, Petersen RC, Vassilaki M, Geda YE Association between CSF biomarkers of Alzheimer’s disease and neuropsychiatric symptoms: Mayo Clinic Study of Aging. Alzheimers Dement n/a,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Babulal GM, Ghoshal N, Head D, Vernon EK, Holtzman DM, Benzinger TLS, Fagan AM, Morris JC, Roe CM (2016) Mood Changes in Cognitively Normal Older Adults are Linked to Alzheimer Disease Biomarker Levels. Am J Geriatr Psychiatry 24, 1095–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Leitão MJ, Silva-Spínola A, Santana I, Olmedo V, Nadal A, Le Bastard N, Baldeiras I (2019) Clinical validation of the Lumipulse G cerebrospinal fluid assays for routine diagnosis of Alzheimer’s disease. Alzheimers Res Ther 11, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Day GS, Rappai T, Sathyan S, Morris JC (2020) Deciphering the factors that influence participation in studies requiring serial lumbar punctures. Alzheimers Dement Diagn Assess Dis Monit 12, e12003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Beekly DL, Ramos EM, Lee WW, Deitrich WD, Jacka ME, Wu J, Hubbard JL, Koepsell TD, Morris JC, Kukull WA, Centers TNAD (2007) The National Alzheimer’s Coordinating Center (NACC) Database: The Uniform Data Set. Alzheimer Dis Assoc Disord 21, 249–258. [DOI] [PubMed] [Google Scholar]
- [33].Weintraub S, Salmon D, Mercaldo N, Ferris S, Graff-Radford NR, Chui H, Cummings J, DeCarli C, Foster NL, Galasko D (2009) The Alzheimer’s Disease Centers’ Uniform Data Set (UDS): the neuropsychologic test battery. Alzheimer Dis Assoc Disord 23, 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hansson O, Seibyl J, Stomrud E, Zetterberg H, Trojanowski JQ, Bittner T, Lifke V, Corradini V, Eichenlaub U, Batrla R, Buck K, Zink K, Rabe C, Blennow K, Shaw LM (2018) CSF biomarkers of Alzheimer’s disease concord with amyloid-β PET and predict clinical progression: A study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimers Dement 14, 1470–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Leuzy A, Mattsson-Carlgren N, Cullen NC, Stomrud E, Palmqvist S, La Joie R, Iaccarino L, Zetterberg H, Rabinovici G, Blennow K, Janelidze S, Hansson O (2023) Robustness of CSF Aβ42/40 and Aβ42/P-tau181 measured using fully automated immunoassays to detect AD-related outcomes. Alzheimers Dement 19, 2994–3004. [DOI] [PubMed] [Google Scholar]
- [36].Campbell MR, Ashrafzadeh-Kian S, Petersen RC, Mielke MM, Syrjanen JA, van Harten AC, Lowe VJ, Jack CR Jr, Bornhorst JA, Algeciras-Schimnich A (2021) P-tau/Aβ42 and Aβ42/40 ratios in CSF are equally predictive of amyloid PET status. Alzheimers Dement Diagn Assess Dis Monit 13, e12190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Barthélemy NR, Saef B, Li Y, Gordon BA, He Y, Horie K, Stomrud E, Salvadó G, Janelidze S, Sato C, Ovod V, Henson RL, Fagan AM, Benzinger TLS, Xiong C, Morris JC, Hansson O, Bateman RJ, Schindler SE (2023) CSF tau phosphorylation occupancies at T217 and T205 represent improved biomarkers of amyloid and tau pathology in Alzheimer’s disease. Nat Aging 3, 391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Fagan AM, Mintun MA, Mach RH, Lee S-Y, Dence CS, Shah AR, LaRossa GN, Spinner ML, Klunk WE, Mathis CA, DeKosky ST, Morris JC, Holtzman DM (2006) Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Aβ42 in humans. Ann Neurol 59, 512–519. [DOI] [PubMed] [Google Scholar]
- [39].Cummings JL (1997) The Neuropsychiatric Inventory: Assessing psychopathology in dementia patients. Neurology 48, 10S–16S. [DOI] [PubMed] [Google Scholar]
- [40].Lai CK (2014) The merits and problems of Neuropsychiatric Inventory as an assessment tool in people with dementia and other neurological disorders. Clin Interv Aging 9, 1051–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Aalten P, Verhey FRJ, Boziki M, Brugnolo A, Bullock R, Byrne EJ, Camus V, Caputo M, Collins D, De Deyn PP, Elina K, Frisoni G, Holmes C, Hurt C, Marriott A, Mecocci P, Nobili F, Ousset PJ, Reynish E, Salmon E, Tsolaki M, Vellas B, Robert PH (2008) Consistency of neuropsychiatric syndromes across dementias: results from the European Alzheimer Disease Consortium. Part II. Dement Geriatr Cogn Disord 25, 1–8. [DOI] [PubMed] [Google Scholar]
- [42].Frisoni GB, Frisoni GB, Rozzini L, Gozzetti A, Binetti G, Zanetti O, Bianchetti A, Trabucchi M, Cummings JL (1999) Behavioral syndromes in Alzheimer’s disease: description and correlates. Dement Geriatr Cogn Disord 10, 130–138. [DOI] [PubMed] [Google Scholar]
- [43].Lange RT, Hopp GA, Kang N (2004) Psychometric properties and factor structure of the Neuropsychiatric Inventory Nursing Home version in an elderly neuropsychiatric population. Int J Geriatr Psychiatry 19, 440–448. [DOI] [PubMed] [Google Scholar]
- [44].Göb R, McCollin C, Ramalhoto MF (2007) Ordinal methodology in the analysis of Likert scales. Qual Quant Int J Methodol 41, 601–626. [Google Scholar]
- [45].Besser L, Kukull W, Knopman DS, Chui H, Galasko D, Weintraub S, Jicha G, Carlsson C, Burns J, Quinn J, Sweet RA, Rascovsky K, Teylan M, Beekly D, Thomas G, Bollenbeck M, Monsell S, Mock C, Zhou XH, Thomas N, Robichaud E, Dean M, Hubbard J, Jacka M, Schwabe-Fry K, Wu J, Phelps C, Morris JC (2018) Version 3 of the National Alzheimer’s Coordinating Center’s Uniform Data Set. Alzheimer Dis Assoc Disord 32, 351–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Monsell SE, Dodge HH, Zhou X-H, Bu Y, Besser LM, Mock C, Hawes SE, Kukull WA, Weintraub S (2016) Results from the NACC Uniform Data Set neuropsychological battery Crosswalk Study Running head: Neuropsychological Battery Crosswalk Study Results. Alzheimer Dis Assoc Disord 30, 134–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Morris JC, Weintraub S, Chui HC, Cummings J, DeCarli C, Ferris S, Foster NL, Galasko D, Graff-Radford N, Peskind ER, Beekly D, Ramos EM, Kukull WA (2006) The Uniform Data Set (UDS): Clinical and Cognitive Variables and Descriptive Data From Alzheimer Disease Centers. Alzheimer Dis Assoc Disord 20, 210–216. [DOI] [PubMed] [Google Scholar]
- [48].Wechsler D (1945) A Standardized Memory Scale for Clinical Use. J Psychol 19, 87–95. [Google Scholar]
- [49].Craft S, Newcomer J, Kanne S, Dagogo-Jack S, Cryer P, Sheline Y, Luby J, Dagogo-Jack A, Alderson A (1996) Memory improvement following induced hyperinsulinemia in alzheimer’s disease. Neurobiol Aging 17, 123–130. [DOI] [PubMed] [Google Scholar]
- [50].Gollan TH, Weissberger GH, Runnqvist E, Montoya RI, Cera CM (2012) Self-ratings of Spoken Language Dominance: A Multi-Lingual Naming Test (MINT) and Preliminary Norms for Young and Aging Spanish-English Bilinguals. Biling Camb Engl 15, 594–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Jefferson AL, Wong S, Gracer TS, Ozonoff A, Green RC, Stern RA (2007) Geriatric Performance on an Abbreviated Version of the Boston Naming Test. Appl Neuropsychol 14, 215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Mack WJ, Freed DM, Williams BW, Henderson VW (1992) Boston Naming Test: Shortened Versions for Use in Alzheimer’s Disease. J Gerontol 47, P154–P158. [DOI] [PubMed] [Google Scholar]
- [53].Henry JD, Crawford JR, Phillips LH (2004) Verbal fluency performance in dementia of the Alzheimer’s type: a meta-analysis. Neuropsychologia 42, 1212–1222. [DOI] [PubMed] [Google Scholar]
- [54].Lucas JA, Ivnik RJ, Smith GE, Bohac DL, Tangalos EG, Graff-Radford NR, Petersen RC (1998) Mayo’s Older Americans Normative Studies: Category Fluency Norms. J Clin Exp Neuropsychol 20, 194–200. [DOI] [PubMed] [Google Scholar]
- [55].Tombaugh TN (2004) Trail Making Test A and B: Normative data stratified by age and education. Arch Clin Neuropsychol 19, 203–214. [DOI] [PubMed] [Google Scholar]
- [56].Smith T, Gildeh N, Holmes C (2007) The Montreal Cognitive Assessment: Validity and Utility in a Memory Clinic Setting. Can J Psychiatry 52, 329–332. [DOI] [PubMed] [Google Scholar]
- [57].Sabe L, Jason L, Juejati M, Leiguarda R, Starkstein S (1993) Sensitivity and specificity of the Mini-Mental State Exam in the diagnosis of dementia. Behav Neurol 6, 207–210. [DOI] [PubMed] [Google Scholar]
- [58].Morris JC (1993) The Clinical Dementia Rating (CDR): Current version and scoring rules. Neurology 43, 2412–2414. [DOI] [PubMed] [Google Scholar]
- [59].Lim S, Jahng S (2019) Determining the number of factors using parallel analysis and its recent variants. Psychol Methods 24, 452–467. [DOI] [PubMed] [Google Scholar]
- [60].Emond EJ, Mason DW (2002) A new rank correlation coefficient with application to the consensus ranking problem. J Multi-Criteria Decis Anal 11, 17–28. [Google Scholar]
- [61].Bentler PM (1990) Comparative fit indexes in structural models. Psychol Bull 107, 238–246. [DOI] [PubMed] [Google Scholar]
- [62].Browne MW, Cudeck R (1993) Alternative ways of assessing model fit. In Bollen KA & Long JS (Eds.), Testing structural equation models. Sage, Newbury Park, CA, pp. 136–162. [Google Scholar]
- [63].Hu L, Bentler PM (1999) Cutoff criteria for fit indexes in covariance structure analysis: Conventional criteria versus new alternatives. Struct Equ Model Multidiscip J 6, 1–55. [Google Scholar]
- [64].Neath AA, Cavanaugh JE (2012) The Bayesian information criterion: background, derivation, and applications. WIREs Comput Stat 4, 199–203. [Google Scholar]
- [65].Schwarz G (1978) Estimating the Dimension of a Model. Ann Stat 6, 461–464. [Google Scholar]
- [66].Rosseel Y, Oberski DL, Byrnes J, Vanbrabant L, Savalei V, Merkle E, Hallquist M, Rhemtulla M, Katsikatsou M, Barendse M (2014) lavaan: Latent variable analysis. [Google Scholar]
- [67].Baron RM, Kenny DA (1986) The moderator-mediator variable distinction in social psychological research: conceptual, strategic, and statistical considerations. J Pers Soc Psychol 51, 1173–1182. [DOI] [PubMed] [Google Scholar]
- [68].Ally BA, Hussey EP, Ko PC, Molitor RJ (2013) Pattern separation and pattern completion in Alzheimer’s disease: Evidence of rapid forgetting in amnestic mild cognitive impairment. Hippocampus 23, 1246–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Yassa MA, Stark SM, Bakker A, Albert MS, Gallagher M, Stark CEL (2010) High-resolution structural and functional MRI of hippocampal CA3 and dentate gyrus in patients with amnestic Mild Cognitive Impairment. NeuroImage 51, 1242–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Lyketsos CG, Lopez O, Jones B, Fitzpatrick AL, Breitner J, DeKosky S (2002) Prevalence of Neuropsychiatric Symptoms in Dementia and Mild Cognitive ImpairmentResults From the Cardiovascular Health Study. JAMA 288, 1475–1483. [DOI] [PubMed] [Google Scholar]
- [71].Siafarikas N, Selbaek G, Fladby T, Benth JŠ, Auning E, Aarsland D (2018) Frequency and subgroups of neuropsychiatric symptoms in mild cognitive impairment and different stages of dementia in Alzheimer’s disease. Int Psychogeriatr 30, 103–113. [DOI] [PubMed] [Google Scholar]
- [72].Eikelboom WS, van den Berg E, Singleton EH, Baart SJ, Coesmans M, Leeuwis AE, Teunissen CE, van Berckel BNM, Pijnenburg YAL, Scheltens P, van der Flier WM, Ossenkoppele R, Papma JM (2021) Neuropsychiatric and Cognitive Symptoms Across the Alzheimer Disease Clinical Spectrum: Cross-sectional and Longitudinal Associations. Neurology 97, e1276–e1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Travis Seidl JN, Massman PJ (2016) Cognitive and Functional Correlates of NPI-Q Scores and Symptom Clusters in Mildly Demented Alzheimer Patients. Alzheimer Dis Assoc Disord 30, 145. [DOI] [PubMed] [Google Scholar]
- [74].van der Linde RM, Dening T, Stephan BCM, Prina AM, Evans E, Brayne C (2016) Longitudinal course of behavioural and psychological symptoms of dementia: systematic review. Br J Psychiatry 209, 366–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Brodaty H, Connors MH, Xu J, Woodward M, Ames D (2015) The Course of Neuropsychiatric Symptoms in Dementia: A 3-Year Longitudinal Study. J Am Med Dir Assoc 16, 380–387. [DOI] [PubMed] [Google Scholar]
- [76].Vik-Mo AO, Giil LM, Borda MG, Ballard C, Aarsland D (2020) The individual course of neuropsychiatric symptoms in people with Alzheimer’s and Lewy body dementia: 12-year longitudinal cohort study. Br J Psychiatry 216, 43–48. [DOI] [PubMed] [Google Scholar]
- [77].Burhanullah MH, Tschanz JT, Peters ME, Leoutsakos J-M, Matyi J, Lyketsos CG, Nowrangi MA, Rosenberg PB (2020) Neuropsychiatric Symptoms as Risk Factors for Cognitive Decline in Clinically Normal Older Adults: The Cache County Study. Am J Geriatr Psychiatry 28, 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].David ND, Lin F, Porsteinsson AP (2016) Trajectories of Neuropsychiatric Symptoms and Cognitive Decline in Mild Cognitive Impairment. Am J Geriatr Psychiatry 24, 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Defrancesco M, Marksteiner J, Kemmler G, Dal-Bianco P, Ransmayr G, Benke T, Mosbacher J, Höller Y, Schmidt R (2020) Specific Neuropsychiatric Symptoms Are Associated with Faster Progression in Alzheimer’s Disease: Results of the Prospective Dementia Registry (PRODEM-Austria). J Alzheimers Dis 73, 125–133. [DOI] [PubMed] [Google Scholar]
- [80].Canevelli M, Adali N, Cantet C, Andrieu S, Bruno G, Cesari M, Vellas B, The ICTUS/DSA Group (2013) Impact of behavioral subsyndromes on cognitive decline in Alzheimer’s disease: data from the ICTUS study. J Neurol 260, 1859–1865. [DOI] [PubMed] [Google Scholar]
- [81].Gatchel JR, Rabin JS, Buckley RF, Locascio JJ, Quiroz YT, Yang H-S, Vannini P, Amariglio RE, Rentz DM, Properzi M, Donovan NJ, Blacker D, Johnson KA, Sperling RA, Marshall GA, Harvard Aging Brain Study (2019) Longitudinal Association of Depression Symptoms With Cognition and Cortical Amyloid Among Community-Dwelling Older Adults. JAMA Netw Open 2, e198964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Babulal GM, Chen L, Murphy SA, Doherty JM, Johnson AM, Morris JC (2023) Neuropsychiatric Symptoms and Alzheimer Disease Biomarkers Independently Predict Progression to Incident Cognitive Impairment. Am J Geriatr Psychiatry 31, 1190–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].McGirr A, Nathan S, Ghahremani M, Gill S, Smith EE, Ismail Z (2022) Progression to Dementia or Reversion to Normal Cognition in Mild Cognitive Impairment as a Function of Late-Onset Neuropsychiatric Symptoms. Neurology 98, e2132–e2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Malpas CB, Sharmin S, Kalincik T (2021) The histopathological staging of tau, but not amyloid, corresponds to antemortem cognitive status, dementia stage, functional abilities and neuropsychiatric symptoms. Int J Neurosci 131, 800–809. [DOI] [PubMed] [Google Scholar]
- [85].Bruen PD, McGeown WJ, Shanks MF, Venneri A (2008) Neuroanatomical correlates of neuropsychiatric symptoms in Alzheimer’s disease. Brain 131, 2455–2463. [DOI] [PubMed] [Google Scholar]
- [86].Ng KP, Chiew HJ, Rosa-Neto P, Kandiah N, Ismail Z, Gauthier S (2019) Brain Metabolic Dysfunction in Early Neuropsychiatric Symptoms of Dementia. Front Pharmacol 10,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Ehrenberg AJ, Suemoto CK, de França Resende EP, Petersen C, Leite REP, Rodriguez RD, de Ferretti-Rebustini REL, You M, Oh J, Nitrini R, Pasqualucci CA, Jacob-Filho W, Kramer JH, Gatchel JR, Grinberg LT (2018) Neuropathologic Correlates of Psychiatric Symptoms in Alzheimer’s Disease. J Alzheimers Dis 66, 115–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Babulal GM, Zhu Y, Trani J-F (2023) Racial and ethnic differences in neuropsychiatric symptoms and progression to incident cognitive impairment among community-dwelling participants. Alzheimers Dement 19, 3635–3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Kishi T, Matsunaga S, Iwata N (2017) The effects of memantine on behavioral disturbances in patients with Alzheimer’s disease: a meta-analysis. Neuropsychiatr Dis Treat 13, 1909–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Chen R, Chan P-T, Chu H, Lin Y-C, Chang P-C, Chen C-Y, Chou K-R (2017) Treatment effects between monotherapy of donepezil versus combination with memantine for Alzheimer disease: A meta-analysis. PLOS ONE 12, e0183586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Tan C-C, Yu J-T, Wang H-F, Tan M-S, Meng X-F, Wang C, Jiang T, Zhu X-C, Tan L (2014) Efficacy and safety of donepezil, galantamine, rivastigmine, and memantine for the treatment of Alzheimer’s disease: a systematic review and meta-analysis. J Alzheimers Dis JAD 41, 615–631. [DOI] [PubMed] [Google Scholar]
- [92].Gerlach LB, Kales HC (2020) Pharmacological Management of Neuropsychiatric Symptoms of Dementia. Curr Treat Options Psychiatry 7, 489–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Calsolaro V, Antognoli R, Okoye C, Monzani F (2019) The Use of Antipsychotic Drugs for Treating Behavioral Symptoms in Alzheimer’s Disease. Front Pharmacol 10,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Masopust J, Protopopová D, Vališ M, Pavelek Z, Klímová B (2018) Treatment of behavioral and psychological symptoms of dementias with psychopharmaceuticals: a review. Neuropsychiatr Dis Treat 14, 1211–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Huang Y-Y, Teng T, Giovane CD, Wang R-Z, Suckling J, Shen X-N, Chen S-D, Huang S-Y, Kuo K, Cai W-J, Chen K-L, Feng L, Zhang C, Liu C-Y, Li C-B, Zhao Q-H, Dong Q, Zhou X-Y, Yu J-T (2023) Pharmacological treatment of neuropsychiatric symptoms of dementia: a network meta-analysis. Age Ageing 52, afad091. [DOI] [PubMed] [Google Scholar]
- [96].Griebel G, Stemmelin J, Lopez-Grancha M, Boulay D, Boquet G, Slowinski F, Pichat P, Beeské S, Tanaka S, Mori A, Fujimura M, Eguchi J (2019) The selective GSK3 inhibitor, SAR502250, displays neuroprotective activity and attenuates behavioral impairments in models of neuropsychiatric symptoms of Alzheimer’s disease in rodents. Sci Rep 9, 18045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].D’Mello SR (2021) When Good Kinases Go Rogue: GSK3, p38 MAPK and CDKs as Therapeutic Targets for Alzheimer’s and Huntington’s Disease. Int J Mol Sci 22, 5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Swanson CJ, Zhang Y, Dhadda S, Wang J, Kaplow J, Lai RYK, Lannfelt L, Bradley H, Rabe M, Koyama A, Reyderman L, Berry DA, Berry S, Gordon R, Kramer LD, Cummings JL (2021) A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimers Res Ther 13, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Kouloutbani K, Venetsanou F, Markati A, Karteroliotis KE, Politis A (2022) The effectiveness of physical exercise interventions in the management of neuropsychiatric symptoms in dementia patients: a systematic review. Int Psychogeriatr 34, 177–190. [DOI] [PubMed] [Google Scholar]
- [100].Wolinsky D, Drake K, Bostwick J (2018) Diagnosis and Management of Neuropsychiatric Symptoms in Alzheimer’s Disease. Curr Psychiatry Rep 20, 117. [DOI] [PubMed] [Google Scholar]
- [101].Nagata T, Shinagawa S, Inamura K, Shigeta M (2022) Pathogenesis and Personalized Interventions for Pharmacological Treatment-Resistant Neuropsychiatric Symptoms in Alzheimer’s Disease. J Pers Med 12, 1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Koch J, Amos JG, Beattie E, Lautenschlager NT, Doyle C, Anstey KJ, Mortby ME (2022) Non-pharmacological interventions for neuropsychiatric symptoms of dementia in residential aged care settings: An umbrella review. Int J Nurs Stud 128, 104187. [DOI] [PubMed] [Google Scholar]
- [103].Pearce N, Vandenbroucke JP (2016) Causation, mediation and explanation. Int J Epidemiol 45, 1915–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of this study are available upon request from the National Alzheimer’s Coordinating Center (https://naccdata.org/requesting-data/data-request-process).