

Abstract
The relative reactivity of a systematic series of simple aliphatic acetate esters has been measured. Exposure of pairs of esters of increasing remote steric hindrance (by altering the degree of branching of the ester alkyl group) to a methanolic solution of Cs2CO3 proved to be a reliable (and general) method for quantitating the rate differences in these base-catalyzed transesterification reactions. The trends in relative rates are in accordance with the qualitative “Rule of Six” put forward by Melvin S. Newman in 1950, as deduced then from interpretation of earlier reports of ease of Fischer esterification reactions.
Graphical Abstract
INTRODUCTION
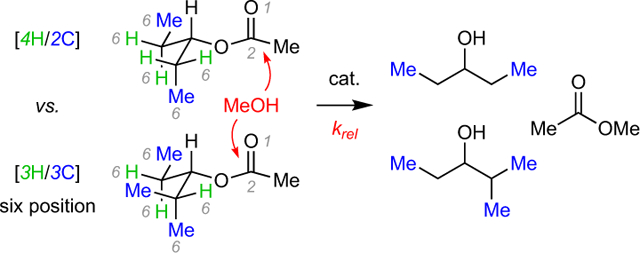
In 1950 M. S. Newman proposed an empirical rule for analyzing reactions that involved additions to carbon-containing unsaturated functional groups (Csp2=X).1 As stated there:
In reactions involving addition to an unsaturated function, the greater number of atoms in the six position the greater will be the steric effect. For convenience this will be referred to as the rule of six and the number of atoms in the six position will be called the six-number (cf. [nH/mC] in 1, Figure 1a).
Newman presented this in the specific context of analysis of three earlier literature reports2 of the rates of acid-catalyzed esterification reactions of ca. two dozen aliphatic carboxylic acids that differed in the types and number of alkyl groups near the carboxyl functionality (cf., e.g., 2a-c). These reactions proceed, of course, through a tetrahedral intermediate in which the carbonyl carbon has become sp3-hydridized. Several specific examples that serve to demonstrate the categorization of some of these simple aliphatic acids are shown in Figure 1b. The number of hydrogen atoms and/or carbon atoms (here, always methyl groups) in the six-position are indicated by the convention [nH/mC]. (A methyl carbon atom in the six-position necessarily, of course, carries with it three hydrogen atoms in the seven-position.) The relative rates of Fischer esterification (with methanol) are shown below each of these structures.
Figure 1.

(a) A generic structure in which the rule of six can be considered for the reactivity of addition reactions to the C=X moiety. (b) Three (of the dozens) of Fischer esterification reactions that Newman drew upon in developing his empirical rule of six. (c) An example showing the benefit of understanding the role of remote steric hindrance.
Newman followed his initial report with a more extensive discussion in a chapter titled “Additions to Unsaturated Functions” in his book on steric effects in organic chemistry.3 He limited his analyses to the identification of qualitative (rather than quantitative) trends because of the added complexity of the conformational landscape as well as the fact that substituents in the 5-postion also contribute to differential reactivities, albeit to a lesser extent.4 Most of the (relatively few) subsequent citations to the rule of six refer to it in only a passing comment,5 likely a reflection of its qualitative nature. Nonetheless, we felt that gaining a sense for the magnitude of this kind of remote steric effect would still be of value.
In a previous study in our laboratory of a natural product synthesis,6 we observed the efficient conversion of the triacetate 3 to the monoacetate-diol 4 upon exposure to aqueous methanolic potassium carbonate. No trace of another product was detected. This was very much in accord with Newman’s empirical rule of six. We decided to more systematically study the impact of substitution of related transesterification reactions, a type of transformation that also typically proceeds through a tetrahedral intermediate derived from the ester carbonyl in the substrate. The results of these studies are presented here.
RESULTS AND DISCUSSION
Cs2CO3 Catalyzed Transesterifications.
Using competition experiments, we have measured the relative rates of transesterification of the ten acetate esters 5–11 (Figure 2) in a homogeneous solution of methanol using cesium carbonate as a basic catalyst. Temperatures of 20, 50, and 120 °C were used in order to achieve comparable time scales for analysis. These homologous substrates differ by the orderly change arising from replacement of a six-position hydrogen atom by a six-position methyl group. The systematic nature of structural change across the series is easily seen in the bracketed designations where the sum of the six position atoms is constant (i.e., n + m = 6).
Figure 2.

Ten model acetates ranging from zero [6H/0C] to six [0H/6C] carbons in the six-position were studied.
Although we initially used a gas-chromatographic method to assess the relative rates of many of these,5 we have more recently used a series of pairwise competition studies using 1H NMR analysis. This latter approach ensures that the temperature and catalyst concentration was identical for any one experiment and benefits from the fact that the detector response is more straightforward (1H integration). Mesitylene (ca. 0.5 equiv) was added as an internal standard. Following the addition of a stock solution of Cs2CO3 (ca. 0.5 equiv), small aliquots (ca. 20 mL) of the reaction mixture were removed over time and directly added to C6D6 (ca. 0.5 mL). The 1H NMR spectrum was recorded soon after; controls showed that there was no significant further change in the ratio of components in the C6D6 samples, even days later. The use of C6D6 rather than CDCl3 provided superior resolution of the methyl group resonances of the two acetate substrates. Their disappearance could then be monitored over time relative to the aromatic proton resonances of the mesitylene standard. Each of the pairwise competition experiments was performed in duplicate, and the agreement between the relative rates measure in both runs was good.
As an example of this methodology, analysis of the pair of acetates 6 vs. 7b is shown in Figure 3. The spectrum in panel (b) is for the zero time point. The starting ratio of acetates in this particular experiment is measured to be 1.12 from the relative integrations of each acetate methyl group singlet at ca. 1.7 ppm. The stack plot in panel (c) shows the disappearance of each of the two substrates (blue vs. red) as well as the appearance of the methyl acetate product (d 1.60 ppm, green) over time. These data are plotted in panel (d) (as ln[acetate]/ln[mesitylene] vs. time). The near linearity is consistent with an expected pseudo first-order reaction mechanism for the transesterification. The ratio of the slopes for the rate of disappearance for this trial was 3.2 (and 3.0 for a second trial). We judged that the data obtained using this approach were sufficiently good to provide the type of semi-quantitative, relative reactivities we were seeking.
Figure 3.

An example of the data collection for one trial (a) of 6 vs. 7b. (b) Time zero spectrum from which the initial ratios of each acetate vs. the mesitylene standard were measured. (c) Spectra of aliquots taken over time. (d) Plot of the consumption of each of acetates 6 and 7b over time.
Table 1 presents the relative rate comparisons for the entire complement of acetates. All of the fastest reacting subset (5–8b) were measured at ambient temperature. The 3-pentyl acetate (7b) was used as the common substrate (i.e., the benchmark compound) in each of these five pairwise competition experiments. As shown in the first column of relative rate data in Table 1, the transesterification rapidity for these six acetates ranged over a factor of 22. The more hindered substrates 9a–9b reacted more slowly, so their methanolyses were carried out at 50 °C (in comparison to 8b as the benchmark). Similarly, for acetate 10, we found a reaction temperature of 120 °C to be convenient; it was measured in competition with 9b. The most hindered substrate, 11, not surprisingly, reacted extremely slowly. At 150 °C it was only partially cleaved to its corresponding alcohol after 20 h, making it challenging to perform a pairwise comparison under the same conditions. Moreover, there is the possibility that 11 may be giving rise to the alcohol 11’ (di-tert-butyl carbinol) by an alternative mechanism – ketene formation from the ester enolate.7 Consequently, we have not offered any quantitative comparison for this slowest substrate of all.
Table 1.
The full set of acetate ester substrates and their observed relative rates of methanolysis.
| ester | # of | relative ratesa | ||||
|---|---|---|---|---|---|---|
| # | structure | Six-Hs | Six-Cs | 20 °C | 50 °C | 120 °C |
| 5 |

|
6 | 0 | 22 | - | - |
| 6 |
|
5 | 1 | 10 | - | - |
| 7a |

|
4 | 2 | 3.7 | - | - |
| 7b |

|
4 | 2 | 3.3b | - | - |
| 8a |

|
3 | 3 | 1.1 | - | - |
| 8b |

|
3 | 3 | 1.0 | 8.3b | - |
| 9a |

|
2 | 4 | - | 1.2 | - |
| 9b |

|
2 | 4 | - | 1.0 | 2.2b |
| 10 |
|
1 | 5 | - | - | 1.0 |
| 11 |

|
0 | 6 | - | - | see text |
The relative rates at each individual temperature were normalized by assigning the rate of the slowest reacting substate a krel of 1.0.
The benchmark compound used for all experiments at this temperature (i.e., to determine the krels within any one column). This benchmark was present in each of the pairwise competition experiments used at this temperature.
Several trends can be seen from the information in Table 1. Within the set of the first six acetates, as each additional methyl group is added to the previous homolog (i.e., progressing from [6H/0C] to [5H/1C] to [4H/2C] to [3H/3C]) the transesterification rate slows by a factor of 2–3 with the addition of each new methyl group. Isomers 7a and 7b differ only by which side of the ester alkyl moiety has gained the extra methyl group (each is [4H/2C]). They have nearly the same rate (krel(7a/7b) = 1.1). The same is true for the pair 8a and 8b (as well as 9a and 9b). Moreover, for each of these pairs the ester with the more balanced degree of branching in the ester alkyl group (i.e., the b-isomer) reacted slightly slower than the a-isomer. Interestingly, portions of this trend can be gleaned from the experimental data for Fischer esterification (in both MeOH2c and in EtOH2a) that Newman used when first formulating the rule of six.
Upon adding the fourth methyl group and moving from 8a/b to 9a/b (i.e., progressing from [3H/3C] to [2H/4C]), there is more substantial rate retardation that deserves some special consideration. More specifically, the isomers of 9 react ca. 8x slower than the acetate 8b.8 Why is this not more like the factor of 2–3 seen for all of the earlier homologs? To explore this we carried out a multiconformational search and full set of density functional theory (DFT) calculations to find the conformational preferences for each acetate (see Supporting Information for details).
For 9a and 9b, the lowest energy conformer for each is shown in Figure 4.9 Notice that each has an extended, anti-anti arrangement of its parent pentane backbone. The terminal methyl groups (i.e., C1 and C5 of the pentane) serve to dissuade tetrahedral intermediate formation arising from any nucleophilic addition to the acetate carbonyl group. Regardless of whether the rate limiting step in the mechanism for base-catalyzed transesterification under the conditions used in this study is formation of the tetrahedral intermediate or its subsequent collapse to release methyl acetate, steric congestion near that tetrahedral intermediate would both slow its rate of formation as well as diminish its equilibrium concentration. The conformer for each of 9a and 9b that allows access for methanol to engage the acetate carbonyl was computed to be 3.0 and 3.5 kcal mol−1 higher in energy, respectively. Each is created by rotation about the C3–C4 pentane bond (i.e., is an anti-gauche conformer) but is greatly destabilized by the syn-pentane interaction that accompanies that geometric change. Thus, each of these conformers, which are more conducive to participating in the transesterification, is populated to only <1% of the equilibrium ensemble of conformers. In contrast, the same analysis of the [3H/3C] homolog 8b, shows, again, its lowest energy conformer to have the anti-anti pentane geometry; however, an anti-gauche conformer with freer access for tetrahedral intermediate formation is only 1.0 kcal mol−1 higher in energy than the global minimum and comprises 8% of the full conformational ensemble for 8b. Said another way, a reasonably large portion of the 8b molecules can adopt a conformation that avoids a syn-pentane interaction, yet still allows access for attack at the carbonyl carbon. The larger rate retardation for 9a/b relative to all the earlier progressions from 5 through 8b can be attributed to the significantly reduced population of the more reactive conformers 9a/b.
Figure 4.

Conformational analysis of the lowest energy conformers of 9a (2,2-dimethyl-3-pentyl acetate) and 9b (2,4-dimethyl-3-pentyl acetate).
The relative rate ratio of 9b vs. 10 was measured to be 2.2 (at 120 °C, last column in Table 1). The acetate in the latter is flanked by one tert-butyl and one iso-propyl group. As with 9b, the most reactive (accessible) conformer of 10, which has its iso-propyl methine proton oriented toward the acetate carbonyl group, is ca. 3 kcal mol−1 higher in energy than the most stable conformer. A simple argument that can account for the reactivity difference (of 2.2) in this 9b/10 pair is that the former has two iso-propyl groups that can orient so as to more readily accommodate the tetrahedral intermediate while 10 has only one. The mole fractions of those conformations that have a Me2C–H oriented toward the carbonyl group in 9b vs. 10 are computed to be 0.43% vs. 0.28%, respectively.
The overall range of rate differences between the most and least reactive acetates, i.e., 5 vs. 10, was estimated in the following way. The steric factor: Acetate 5 reacts 22 times faster than 8b, which reacts 8.3 times faster than 9b, which reacts 2.2 times faster than 10. If these were all being measured at the same temperature, the steric factor alone would then account for a factor of 400 in relative rates. The temperature correction: Using the half-life of the transesterification at 20 °C for 7b of 21 h and i) assuming a pseudo first-order reaction (the methanol solvent was in large excess in all reactions), ii) assuming a pre-exponential factor of 1012, and iii) ignoring the concentration of the Cs2CO3 catalyst (which was similar in all experiments), we deduce an activation energy of 22.8 kcal mol−1. The impact of the temperature change from 20 to 120 °C is a factor of 21,500 (k = Ae(–Eact/RT)). Assuming that all of the transesterification reactions are subject to the same temperature dependence, we estimate an overall difference in reactivity of krel(5/10) = 8,600,000.
Related transesterification experiments.
Transesterifications of 7g with different alcohols.
We compared the rates of alcoholysis in the series of increasing sterically hindered (and less polar) solvents MeOH, EtOH, and iPrOH for the single acetate 7b (Table 2). We used 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 2.0 equiv) as the basic catalyst because the solubility of Cs2CO3 was limited in the higher alcohols. In MeOH more than half of 7b was consumed over 24 h at 20 °C. The bulkier EtOH required heating the reaction mixture to 50 °C and iPrOH necessitated extended heating at 120 °C to achieve convenient rates of reaction. From the observed precipitous drop in reactivity, one can see that the hindrance of the nucleophilic agent forming the tetrahedral intermediate impacts the transesterification rate significantly. Thus (and not surprisingly), the choice of the solvent contemplated for use in a deprotection of a complex substrate is a factor to be considered.
Table 2.
Transesterification reaction of 7b with alcohols of increasing steric bulk, catalyzed by DBU (2.0 equiv).

| |||
|---|---|---|---|
| ROH solvent | Temp | Time | Consumption of 7b |
| MeOH | 20 °C | 24 h | 62% |
| EtOH | 50 °C | 48 h | 25% |
| iPrOH | 120 °C | 168 h | 23% |
Cs2CO3 vs. DBU as the (base) catalyst.
The acetates 6, 7b, and 8b (ranging from [5H/1C] to [4H/2C] to [3H/3C]) displayed a three-fold decrease in reactivity for each successive carbon in the six-position when using 0.5 equiv of Cs2CO3 as the base (Table 3, first column of relative rates). Use of DBU (1.0 equiv) led to essentially the same relative rates of acetate cleavage among 6, 7b, and 8b, suggestive of a similar mechanism (e.g., both general base catalysis) for each of the two different bases.
Table 3.
Relative rates of transesterification for acetates 6, 7b, and 8b using Cs2CO3 vs. DBU in MeOH.

Normalized to that of 7b for each set of conditions.
From Table 1.
MeOH, 20 °C, Cs2CO3 (0.5 equiv).
MeOH, 20 °C, DBU (1.0 equiv, which gave similar absolute rates of the methanolysis as did use of 0.5 equiv of Cs2CO3, thus allowing for the same array of timepoints to be taken during the NMR monitoring).
H2SO4 as the (acid) catalyst.
We briefly examined a Brønsted acid-catalyzed variant of the methanolysis using the same set of acetate esters 6, 7b, and 8b. In MeOH containing ~0.5 equiv of H2SO4 the relative rate ratios were 2.0:1.0:0.43, respectively (Figure 5). Thus, the acid-catalyzed methanolyses showed a slightly lower level of selectivity compared to the analogous base-catalyzed reactions (i.e., difference of ca. 2x vs. 3x). This suggests that a base-catalyzed ester cleavage would likely be more selective in a structurally complex molecule containing more than one ester (cf. 3 to 4, Figure 1).
Figure 5.
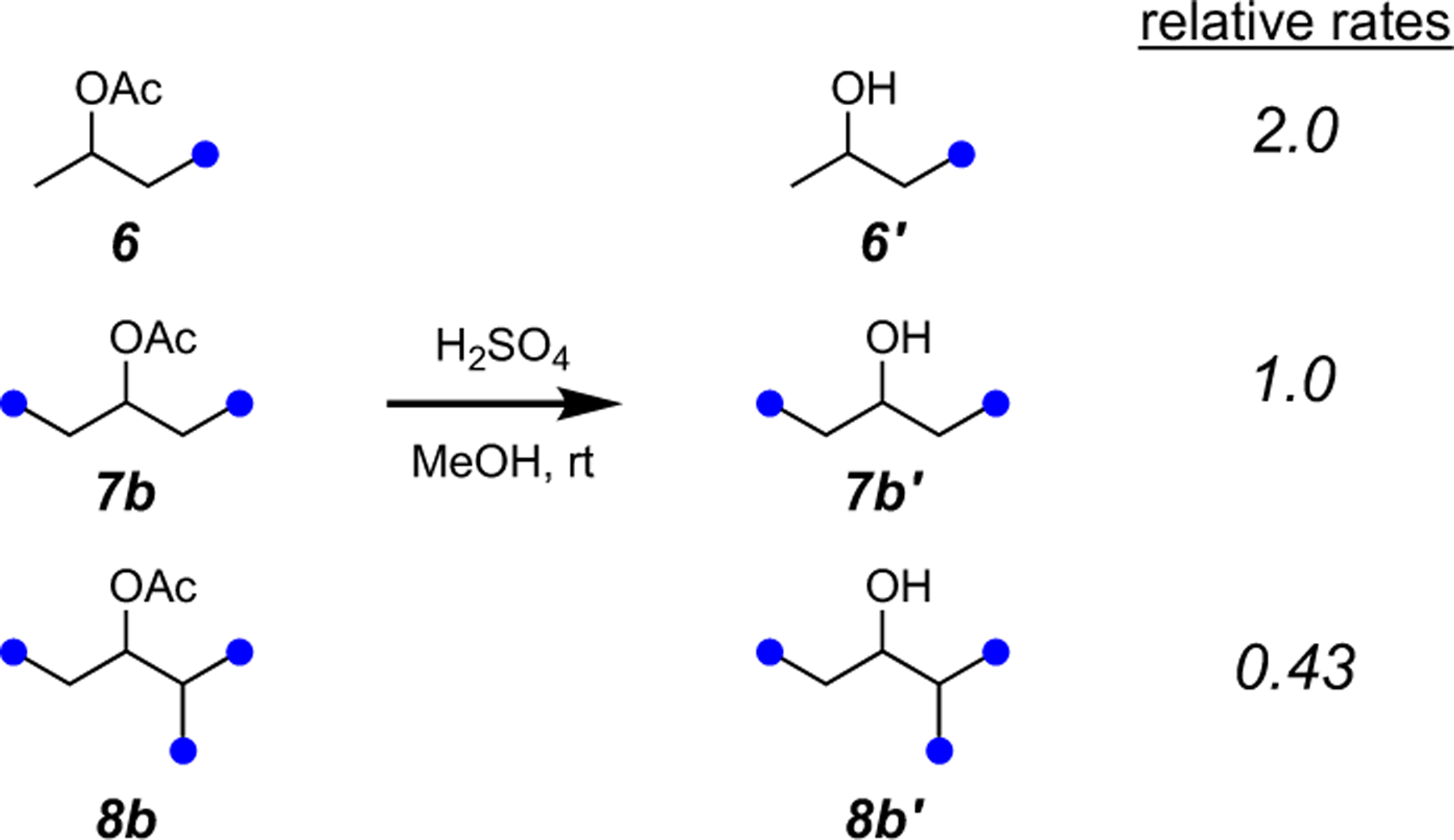
Relative rates of Brønsted acid-catalyzed methanolysis for the acetates 6, 7b, and 8b.
CONCLUSIONS
We have systematically examined the relative rates of base-catalyzed transesterification for ten model acetates containing, in total, a combination of six hydrogen and/or methyl groups in the six position relative to the acetate carbonyl oxygen atom. The observed drop in reactivity for each successive carbon in the six position reaffirms Newman’s qualitative, empirical rule of six. Computation (DFT) of the ground state conformational populations of the acetates was used to rationalize an observed discontinuity (8a/b vs. 9a/b) in the rate depression resulting from systematic replacement of six-Hs by six-Cs. Of the two bookend acetates studied, [6H/0C] (5) vs. [1H/5C] (10), an overall difference in the rate of transesterification of seven orders of magnitude was approximated. Increasing the steric bulk of the solvent was shown to significantly slow the rate of base-catalyzed transesterification. Finally, the rates of analogous Brønsted acid-catalyzed methanolysis was shown to be slightly less sensitive to the degree of hindrance in the acetate relative to the base-catalyzed reaction.
EXPERIMENTAL SECTION
General Procedures for Kinetic Experiments.
a). Cs2CO3 or H2SO4 or DBU as catalyst in MeOH.
To a screw cap culture tube was added i) 0.2 mL each of a 0.5 M stock solution in methanol of each of the two acetates being compared, ii) 0.1 mL of an ca. 0.5 M solution of mesitylene in methanol, and iii) 0.5 mL of a 0.1 M stock solution of either Cs2CO3 or H2SO4 or 0.5 mL of a 0.2 M stock solution of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU). An aliquot (20 mL) was taken and immediately diluted into ca. 0.5 mL of C6D6; the NMR spectrum was recorded to determine the initial (t = 0) ratio of the two acetates relative to the mesitylene internal standard. Additional aliquots were removed and analyzed at multiple timepoints over two days. The reactions were performed at ambient temperature or in an oil bath equilibrated to either 50, 120, or 150 °C.
b). DBU as catalyst in MeOH, EtOH, or iPrOH.
To a screw-cap culture tube was added 7b (0.1 mmol, 0.013 g), mesitylene (0.05 mmol, 0.006 g), DBU (0.2 mmol, 0.0304 g), and alcohol solvent (1 mL). An aliquot (20 mL) was taken and immediately diluted into ca. 0.5 mL of C6D6 or CDCl3 to record the initial amount of 7b in the mixture relative to the mesitylene standard. Additional aliquots were removed and analyzed at multiple timepoints over two days. The reactions were performed at ambient temperature or in an oil bath equilibrated to either 50 or 120 °C.
Spectral Data for Acetates 3, 4, and 6–11.
(±)-(2R,4R,6R,8R)-2,4,6,8-Tetramethylnonane-1,5,9-triyl Triacetate (3) and (±)-(2R,4R,6R,8R)-1,9-Dihydroxy-2,4,6,8-tetramethylnonan-5-yl Acetate (4).
To a suspension of potassium carbonate (1.49 g, 10.8 mmol) in aq MeOH (MeOH:H2O = 3 : 1, v/v, 100 mL) was added the triacetate 36 (1.76 g, 4.9 mmol). After 10 h of stirring at RT, the complete consumption of the starting material was confirmed by TLC analysis. The mixture was extracted (CH2Cl2 × 4), washed (saturated NH4Cl), and dried (MgSO4). Concentration followed by MPLC (1 : 1.5 hexane/EtOAc, containing 5% isopropanol) yielded the diol monoacetate 4 (1.20 g, 4.3 mmol, 89 %). For triacetate 3. 1H NMR (CDCl3, 200 MHz): δ 4.64 (dd, J = 7.0, 7.0 Hz, CH3CO2CH), 3.92–3.74 (m, 2 × CH3CO2CH2), 2.03 (s, CH3CO2CH), 2.01 (s, 2 × CH3CO2CH2), 1.94–1.68 (m, 4 × CH3CH), 1.21–1.03 (m, 2 × CHCH2CH), 0.85 (d, J = 6.5 Hz, CH3CH), 0.84 (d, J = 7.0 Hz, CH3CH), 0.82 (d, J = 6.4 Hz, CH3CH), and 0.80 (d, J = 7.2 Hz, CH3CH). 13C{1H} NMR (CDCl3, 50.3 MHz): δ 170.5 (2x), 170.5, 81.3, 69.5, 69.2, 36.8, 34.5, 31.0, 30.7, 29.4, 29.3, 20.3 (2x), 20.3, 16.0, 15.5, 15.4, and 13.1. IR (CDCl3) 3000, 2850, 1710, 1250, and 1030 cm−1. HRMS (Cl) Calcd for C19H34O6 + NH4+: 376.2694. Found: 376.2685. For acetate 4. 1H NMR (CDCl3, 300 MHz): δ 4.67 (dd, J = 7.0, 4.9 Hz, CHOAc), 3.47 (dd, J = 11.1, 4.0 Hz, CHaHbOH), 3.46 (dd, J = 10.2, 5.6 Hz, CHbHaOH), 3.42 (dd, J = 10.6, 6.6 Hz, CHaHbOH), 3.41 (dd, J = 11.1, 6.2 Hz, CHbHaOH), 2.08 (s, OAc), 1.95–1.80 (m, CH3CHCHOAc), 1.75–1.60 (m, CH3CHCH2OH), 1.21 (ddd, J = 13.5, 9.9, 4.5 Hz, HCH), 1.20 (ddd, J = 13.2, 10.9, 3.3 Hz, HCH’), 1.06 (ddd, J = ~13.0, 10.9, 3.0 Hz, HCH”), 1.05 (ddd, J = 13.7, 9.5, 4.6 Hz, HCH””), 0.89 (d, J = 6.6 Hz, CHCH3), 0.88 (two overlapping d’s, J = 6.6 Hz, CHCH3’/”), and 0.86 (d, J = 6.6 Hz, CHCH3”’). 13C{1H} NMR (CDCl3, 50.3 MHz): δ 170.7, 81.2, 68.6, 68.2, 36.7, 34.5, 32.6, 32.4, 31.1, 30.7, 20.4, 15.6, 15.5, 15.3, and 13.0. IR (neat) 3400, 2980, 2960, 2890, 1730, 1460, 1380, 1240, 1040, and 960 cm−1. Anal. Calcd for C15H30O4: C, 65.66; H, 11.02. Found: C, 65.49; H, 10.88.
NMR Spectral Data for Acetates 6–11.
Because the NMR spectral data in the literature for 6–10 are of variable quality, the line listings for the 1H and 13C NMR spectra of those acetates are provided here. Full characterization data for 11 are provided here for this reported10 but uncharacterized acetate.
6: (0.36 g, 62%, pale yellow oil) 1H NMR (CDCl3, 400 Hz): δ 4.83 (nfom, 1H), 2.03 (s, 3H), 1.59 (dqd, J = 13.7, 7.5, 6.9 Hz, 1H), 1.53 (dqd, J = 13.7, 7.5, 5.9 Hz, 1H), 1.20 (d, J = 6.5 Hz, 3H), and 0.90 (dd, J = 7.5, 7.5 Hz, 3H). 13C{1H} NMR (CDCl3, 100.6 Hz): δ 171.1, 72.4, 28.9, 21.5, 19.6, and 9.8.
7a: (0.43 g, 66%, pale yellow oil) 1H NMR (CDCl3, 400 Hz): δ 4.72 (dq, J = 6.7, 6.3 Hz, 1H), 2.04 (s, 3H), 1.77 (qqd, J = 6.8, 6.8, 5.8 Hz, 1H), 1.16 (d, J = 6.1 Hz, 3H), 0.902 (d, J = 6.8 Hz, 3H), and 0.901 (d, J = 6.8 Hz, 3H). 13C{1H} NMR (CDCl3, 100.6 Hz): δ 171.0, 75.4, 32.8, 21.5, 18.2, 18.1, and 16.8.
7b: (0.43 g, 66%, pale yellow oil) 1H NMR (CDCl3, 400 Hz): δ 4.75 (tt, J = 6.8, 5.5 Hz, 1H), 2.05 (s, 3H), 1.57 (dqd, J = 14.0, 7.5, 5.5 Hz, 2H, CH3CHaHb), 1.55 (dqd, J = 14.0, 7.1, 7.1 Hz, 2H, CH3CHaHb), and 0.88 (dd, J = 7.3, 7.3 Hz, 6H). 13C{1H} NMR (CDCl3, 100.6 Hz): δ 171.3, 76.9, 26.6, 21.4, and 9.7.
8a: (0.54 g, 75%, pale yellow oil) 1H NMR (CDCl3, 400 Hz): δ 4.68 (q, J = 6.5 Hz, 1H), 2.04 (s, 3H), 1.13 (d, J = 6.5 Hz, 3H), and 0.90 (s, 9H). 13C{1H} NMR (CDCl3, 100.6 Hz): δ 171.0, 77.8, 34.2, 25.8, 21.4, and 15.0.
8b: (0.47 g, 65%, pale yellow oil) 1H NMR (CDCl3, 400 Hz): δ 4.67 (ddd, J = 8.2, 5.8, 4.6 Hz, 1H), 2.06 (s, 3H), 1.83 (qqd, J = 6.8, 6.8, 5.8 Hz, 1H), 1.58 (dqd, J = 13.9, 7.4, 4.4 Hz, 1H, CHaHb), 1.52 (ddq, J = 14.3, 8.2, 7.2 Hz, 1H, CHaHb), 0.894 (dd, J = 6.8 Hz, 3H), 0.888 (d, J = 6.9 Hz, 3H, HCCH3), and 0.87 (t, J = 7.5, 3H). 13C{1H} NMR (CDCl3, 100.6 Hz): δ 171.3, 80.0, 31.1, 24.2, 21.2, 18.7, 17.7, and 10.0.
9a: (0.67 g, 84%, pale yellow oil) 1H NMR (CDCl3, 400 Hz): δ 4.66 (dd, J = 2.5, 10.6 Hz, 1H), 2.08 (s, 3H), 1.62 (dqd, J = 14.0, 7.4, 2.5 Hz, 1H, CH3CHaHb), 1.44 (ddq, J = 14.2, 10.7, 7.1 Hz, 1H, CH3CHaHb), 0.89 (s, 9H), and 0.86 (dd, J = 7.4, 7.4 Hz, 3H). 13C{1H} NMR (CDCl3, 100.6 Hz): δ 171.4, 82.5, 34.7, 26.1, 22.6, 21.1, and 11.1.
9b: (0.62 g, 79%, pale yellow oil) 1H NMR (CDCl3, 400 Hz): δ 4.58 (t, J = 6.0 Hz, 1H), 2.07 (s, 3H), 1.89 (qqd, J = 6.9, 6.9, 6.1 Hz, 2H), 0.90 (d, J = 6.9 Hz, 6H), and 0.90 (d, J = 6.9 Hz, 6H). 13C{1H} NMR (CDCl3, 100.6 Hz): δ 171.4, 82.8, 29.5, 21.0, 19.7, and 17.3.
10: (0.76 g, 85%, pale yellow oil) 1H NMR (CDCl3, 400 Hz): δ 4.55 (d, J = 3.3 Hz, 1H), 2.08 (s, 3H), 2.01 (qqd, J = 6.8, 6.8, 3.3 Hz, 1H), 0.96 (d, J = 6.9 Hz, 3H), 0.92 (s, 9H), and 0.88 (d, J = 6.8 Hz, 3H). 13C{1H} NMR (CDCl3, 100.6 Hz): δ 171.3, 84.2, 35.5, 28.6, 26.8, 23.3, 21.0, and 18.1.
11: (0.56 g, 60%, clear crystalline solid) 1H NMR (CDCl3, 400 Hz): δ 4.58 (s, 1H), 2.08 (s, 3H), and 1.00 (s, 18 H). 13C{1H} NMR (CDCl3, 100.6 Hz): 171.1, 85.9, 37.2, 28.8, and 21.1. IR (neat): 2959 (Csp3-H), 1733 (C=O), and 1240 (C-O st as) cm−1. HRMS (EI-TOF) m/z: [M+-tBu•] Calcd for C7H13O2+ 129.0910, Found 129.0913 (10 %); [tBuC(H)=O+H] Calcd for C5H11O+ 87.0804, Found 87.0808 (100%); [tBu+] Calcd for C4H9+ 57.0699, Found 57.0705 (50%). mp: 27.6–30.3 °C.
Supplementary Material
The Supporting Information is available free of charge on the ACS Publications website.
PDF containing the experimental procedure for preparing compound 4, graphs of the data from all pairwise competition reactions between acetates, description of DFT computational methods and energies and co-ordinates for all conformers of acetates 5–11, and the static copies of the proton and carbon spectra for each of acetates 6–11.
A .zip file of the .xyz files of all computed conformers. A .zip file of the folders of raw NMR data for 6–11 titles FID for Publication.
ACKNOWLEDGMENT
This work was supported by a grant from the National Science Foundation (NSF, CHE-2155042). A.M.H.W. was supported by a Heisig-Gleysteen Summer Fellowship. Partial support for NMR instrumentation came from an NIH Shared Instrumentation Grant (NIH S10 OD011952). Mass spectrometry data were obtained with instrumentation in the University of Minnesota Department of Chemistry Mass Spectrometry Laboratory (MSL), obtained with an NSF instrumentation grant (CHE-1336940). The DFT computational studies were made possible by the Minnesota Supercomputing Institute (MSI).
Footnotes
None of the authors have a competing financial interest in this work.
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.
REFERENCES
- 1.Newman MS Some observations concerning steric factors. J. Am. Chem. Soc 1950, 72, 4783–4786. [Google Scholar]
- 2.(a) Bhide BV; Sudborough JJ Studies in transesterification. J. Ind. Inst. Sci 1925, 8, 89–128. [Google Scholar]; (b) von Braun J; Fischer F Beiträge zur Kenntnis der sterischen Hinderung, VII. Mitteil.: Veresterung und Verseifung vom Standpunkt der elektronischen Theorie der Bindung. Chem. Ber 1933, 66, 101–104. [Google Scholar]; (c) Smith HA; Burn J Kinetics of the acid-catalyzed esterification of phenyl- and cyclohexyl-substituted aliphatic acids in methanol. J. Am. Chem. Soc 1944, 66, 1494–1497. [Google Scholar]
- 3.Newman MS Additions to unsaturated functions. In Steric Effects in Organic Chemistry; John Wiley & Sons, 1956; pp 201–248. [Google Scholar]
-
4.(a) This type of longer-range steric effect (or wrap-around effect) is similar to what is learned from the A-values in the monoalkylcyclohexanes (i.e., 1.7 Me to 1.8 Et to 2.2 iPr to ~5 tBu kcal mol−1); Anslyn EV; Dougherty DA
Strain and stability. In Modern Physical Organic Chemistry; University Science, 2005; pp 65–143. [Google Scholar]; (b) The change of iPr to tBu introduces, perforce, a methyl carbon in place of a hydrogen that is six atoms removed from the axial hydrogen atoms at C3 and C5 of the cyclohexane ring.

- 5.A Google Scholar search (6–27-24) for “rule of six” within the 196 articles that cite reference #1 resulted in 61 hits, of which approximately half are in the context of polymer chemistry.
- 6.Ahn S-C Studies toward the total synthesis of the venturicidin aglycone. Ph.D. Thesis, University of Minnesota, Minneapolis, MN, 1991. [Google Scholar]
- 7.(a) Tidwell TT Preparation of ketenes. In Ketenes, 2nd ed.; Wiley-Interscience, 2006; pp 76–86. [Google Scholar]; (b) Pratt RF; Bruice TC The carbanion mechanism (E1cB) of ester hydrolysis. III. Some structure–reactivity studies and the ketene intermediate. J. Am. Chem. Soc 1970, 92, 5956–5964. [Google Scholar]; (c) Inoue M; Bruice TC Influence of steric effects upon the rate constants for competing BAC2 and E1cB ester hydrolyses. J. Org. Chem 1983, 48, 3559–3561. [Google Scholar]
- 8.This trend of a non-linear increase in the relative energetics of a set of substrates containing substituents changing in the steric bulk of substituents (e.g., Me to Et to iPr to tBu) is, again, reminiscent of the change in A-values for the alkylcyclohexanes.4a
- 9.The conformers were visualized using CYLView. Legault CY CYLView20; Université de Sherbrooke, 2020. (http://www.cylview.org; accessed 6-20-24). [Google Scholar]
- 10.Newman MS; Hishida S Alkaline hydrolysis of normal and pseudo methyl esters of o-benzoylbenzoates and of hindered alkyl acetates. J. Am. Chem. Soc 1962, 84, 3582–3584. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.