

ABSTRACT
Aim
Periodontal disease is driven by oral pathogens, including Porphyromonas gingivalis, and the release of inflammatory cytokines. These cytokines (e.g., TNF) or their receptors (e.g., IL‐1R) are substrates of a disintegrin and metalloproteinases (ADAMs). In this study, we aimed to determine the effects of ADAMs on periodontal disease phenotypes.
Materials and Methods
Western blot and FRET‐based activity measurements of the gingival crevicular fluid (GCF) of patients were compared with those of infected (P. gingivalis) or cytokine‐stimulated oral keratinocytes and primary human neutrophils, respectively. This was accompanied by an analysis of the released extracellular vesicles and MMP9 activity.
Results
In the GCF of patients, ADAM8 protein expression and activity were correlated with disease stage, whereas ADAM10 protein expression was inversely correlated with disease stage. Infection and the resulting cytokine release orchestrated the release of soluble ADAM8 by oral keratinocytes and primary neutrophils as soluble ectodomain and on exosomes, respectively. Furthermore, ADAM8 regulated the release of ADAM10 and MMP9.
Conclusion
Dysregulation of cell‐associated and extracellular ADAM proteolytic activity may be an essential regulatory element in the progression of periodontal disease driven by ADAM8. The influence of ADAM8 on disease onset and the evaluation of targeting ADAM8 as a potential and novel local treatment option should be addressed in future translational in vivo studies.
Keywords: cell–matrix interactions, infectious disease(s), matrix metalloproteinases, periodontal disease, proteases/proteinases
Abbreviations
- ADAM
a disintegrin and metalloproteinase
- BHIB
brain heart infusion broth
- ELISA
enzyme‐linked immunosorbent assay
- EVs
extracellular vesicles
- FCS
foetal calf serum
- FRET
fluorescence resonance energy transfer
- GAPDH
glyceraldehyde 3‐phosphate dehydrogenase
- IFN‐γ
interferon gamma
- IL‐1β
interleukin 1 beta
- McF
Macfarland
- MMP‐9
matrix metalloproteinase 9
- P. gingivalis
Porphyromonas gingivalis
- RT
room temperature
- TNF‐α
tumour necrosis factor alpha
1. Introduction
Periodontal diseases are among the most common pathologies with an estimated global prevalence of 19% and more than 1 billion cases worldwide (Jain et al. 2024). Chronic periodontitis may develop into systemic responses based on hyperinflammation and disruption of the immune response, with the risk of endomyocarditis or changes in the gut microbiome (Martinez‐Garcia and Hernandez‐Lemus 2021). Periodontitis is initiated and driven by dysbiosis of the oral microbiome, resulting in overactivation of the host immune response and changes in the cytokine network (Pan, Wang, and Chen 2019). Poor oral hygiene and a lack of frequent biofilm disruption lead to the accumulation of bacteria, such as Fusobacterium nucleatum, eliciting gingival inflammation and the proliferation of Porphyromonas gingivalis (P. gingivalis) (Meyle and Chapple 2015). These oral bacteria manipulate innate and adaptive immune responses, specifically targeting the antimicrobial mechanisms of neutrophils to evade killing and promote inflammation (Miralda and Uriarte 2021). Excess of reactive oxygen species, released matrix metalloproteinases (MMPs) and cytokines lead to collateral periodontal tissue loss and osteoclast activation, resulting in bone loss (Meyle and Chapple 2015; Pan, Wang, and Chen 2019). Cytokines central to periodontitis are interleukin‐1β (IL‐1β), IL‐6 and tumour necrosis factor alpha (TNF‐α). Upon secretion by periodontal cells and innate immune cells, they elicit the release of other cytokines from T cells, such as interferon gamma (IFN‐γ) (Pan, Wang, and Chen 2019). These cytokines (TNF‐α) or their corresponding receptors (IL‐1R and IL‐6R) are either substrates of a disintegrin and metalloproteinases (ADAMs) or induce their expression or activation (for review, see Dreymueller, Uhlig, and Ludwig 2015). ADAMs are multidomain transmembrane proteins that are activated upon cleavage of the inhibitory prodomain. In addition to their proteolytic activity, ADAMs perform several non‐proteolytic functions, such as interaction with and regulation of integrins or the extracellular matrix. In various infectious diseases, ADAMs are involved in each step of the infection process, starting with initial pathogen recognition and entrance, phagocytosis and leukocyte recruitment, up to regeneration (for review, see Aljohmani and Yildiz 2020). ADAM proteases may not only act on the expressing cells themselves but may also be released as soluble variants or on exosomes, performing proteolytic cleavage in the extracellular matrix and at distinct cellular sites. Earlier studies had shown that ADAM8 is up‐regulated in the saliva of patients with periodontitis, displaying proteolytic activity (Feng et al. 2019; Hiyoshi et al. 2022), and its expression is enhanced by stimulation with Fusobacterium nucleatum in epithelial cells (Aung et al. 2017). To date, few studies have addressed the relevance of ADAM10 and ADAM17 in periodontal disease. For ADAM17, the up‐regulation of mRNA expression in gingival tissue has been observed (Tomita et al. 2013), and an increase in receptor activator of nuclear factor‐κB ligand (RANKL) expression in osteoblast has been linked to ADAM17 protein levels (Lee et al. 2011). Inhibition experiments have revealed that ADAM10 and ADAM17 contribute to the release of chemokines from human gingival fibroblasts, thereby controlling the migration of leukocytes into inflamed tissues (Hosokawa et al. 2007). However, comparative and mechanistic studies on ADAM‐based proteolytic activity and its functional impact are lacking.
We hypothesised that the proteolytic activity of ADAMs released into the extracellular space plays an essential role in the initiation, progression and severity of periodontitis. In this study, we observed an increase in ADAM8 protein expression and proteolytic activity in the gingival crevicular fluid (GCF) of patients suffering from periodontitis, which was correlated with MMP9 release and the disease stage. Especially, the release of ADAM8 on exosomes disturbed the oral keratinocyte regeneration, further fostered by its influence on MMP9 and ADAM10. Our observation suggests that ADAM8 and its interaction partners might represent novel treatment targets for the prevention of periodontal disease progression, which should be—together with a potential impact on disease initiation—addressed in future translational in vivo studies.
2. Materials and Methods
2.1. Patient Study
This study was approved by the ethics committee of the Landesärztekammer des Saarlandes (345/20 by D.Y. and M.H.) and was performed in accordance with the Declaration of Helsinki. Written informed consent was obtained from all individuals visiting either the Clinic for Dental Preservation, Periodontology, and Preventive Dentistry of Saarland University Hospital, or the dental office of Reinstädtler and Hektor. The samples were pseudo‐anonymized during their transfer from the clinic to the research area. The exclusion criteria, patient characteristics (Table S1) and classifications are given in the Supporting Information.
2.2. Sample Preparation
Paper points were inserted into the periodontal pockets and left for 30 s. The same procedure was followed for up to three further sampling sites on the same tooth, and all paper points were transferred together into a reaction vessel. The samples were stored at 4°C for a maximum of 7 days until further processing. Detailed sample preparation methods for western blotting and activity assays are given in the Supporting Information.
2.3. Buffers and Solutions
Buffers and solutions used in this study are shown in Table S2.
2.3.1. Anaerobic Bacteria Preparation
P. gingivalis strain DSM 20709 (syn. ATCC 33277), originally isolated from human gingival sulcus, was obtained from the German Collection of Microorganisms and Cell Cultures GmbH (DSMZ) and used throughout the study. Bacterial culture and cell preparation are described in the Supporting Information.
2.3.2. Cell Culture and Cell Stimulation
Human keratinocytes (K2; a gift from Dr. Miosge, University of Göttingen) were cultured in Keratinocyte Growth Medium 2 (PromoCell, Heidelberg, Germany). Primary human neutrophils were cultured in RPMI1640 (with 0.2% foetal calf serum [FCS]). Neutrophil isolation, culture conditions, inhibitors and stimulant treatments are described in the Supporting Information.
2.3.3. Western Blot Analysis
K2 cells or human neutrophils were lysed with 150–200 μL of lysis buffer (Table S2), and proteins were separated according to their molecular weight by SDS‐PAGE as described in the Supporting Information (Aljohmani, Andres, and Yildiz 2022; Aljohmani, Opitz, et al. 2022).
2.3.4. Activity Measurement
To assess the activity of soluble ADAM8 in patient samples, as well as its release from K2 cells and human neutrophils, fluorescence resonance energy transfer (FRET)‐based substrates containing specific substrate cleavage sites were used, as described in the Supporting Information.
2.3.5. Gelatin Zymography
Equal protein amounts isolated from the GCF were loaded onto an SDS‐polyacrylamide (5%) gel supplemented with 0.1% gelatin (from porcine skin, Sigma‐Aldrich, Taufkirchen, Germany). Enzyme activity was evaluated as described in the Supporting Information.
2.3.6. Enzyme‐Linked Immunosorbent Assay
Cell lysates, cell culture supernatants and sulcus fluid from patients at different disease stages were evaluated for MMP‐9 content using the ELISA Duo Set (R&D System, Wiesbaden, Germany, Cat No: DY911) following the manufacturer's protocol.
2.3.7. Exosome Preparation and Wound Closure (Scratch) Assay
Isolation of exosomes from the cell culture medium of K2 cells or human neutrophils and the scratch assay, an in vitro wound closure and tissue layer regeneration assay, are described in detail in the Supporting Information.
2.4. Statistical Analysis
Quantitative data are shown as mean ±SD. Statistical analyses were performed using GraphPad PRISM 9.0 (GraphPad Software, La Jolla, CA, USA). The numbers (n) and statistical tests are provided in the figure legends. Statistical significance was set at p < 0.05.
3. Results
3.1. Presence of ADAM8 and Its Proteolytic Activity in Gingival Fluid Increases With Disease Stage
To determine the potential contribution of ADAMs to the severity of periodontal disease, the protein expression levels of ADAM8, 10 and 17 were investigated in the sulcus fluid of healthy patients and those with periodontitis. We observed a stage‐dependent increase in ADAM8 total protein expression, shown by an increase in both the pro‐form (120 kDa, only seen in 72% of the patients) and mature form (90 kDa), as well as an increase in maturation (the transformation of the pro‐form to the mature form; Figures 1A–C and S1A). The remnant form (the part of ADAM8 remaining in the membrane after the autocatalytic release of the extracellular domain) was slightly, but not significantly, increased at all disease stages (Figure S1B). Interestingly, the protein expression of ADAM10, a substrate of ADAM8, was strongly increased upon disease onset (Stages I–II) but reduced at later stages of the disease (Figure 1D,E and Figure S1C; pro‐form 100 kDa, mature form 70 kDa). This was further reflected in the maturation level (Figure 1F), which was measured as the ratio of the mature form to the pro‐form. In contrast, the expression of ADAM17 did not change (Figure S1D–G; pro‐form 130 kDa, mature form 100 kDa). Notably, the healthy patients did not show the presence of these forms of ADAM8, 10 or 17. Since maturation does not directly indicate activation, we analysed the activity of ADAM8 using a specific FRET substrate, including the cleavage site of CD23. At the early onset of disease (Stages I–II), no changes were observed in patients with periodontal disease compared with healthy patients, and activity massively increased in highly diseased patients (Figure 1G,H).
FIGURE 1.
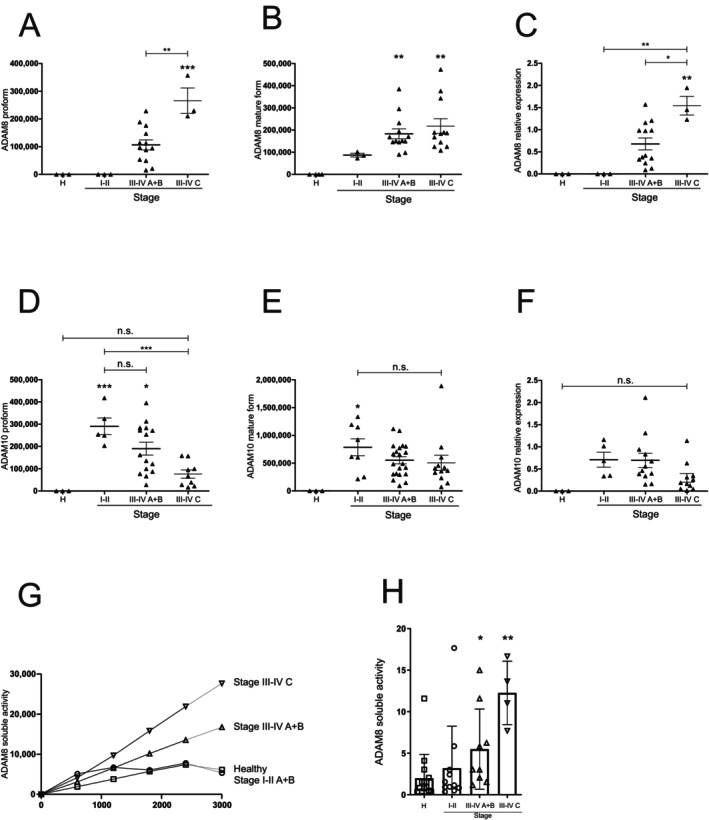
ADAM8 proteolytic activity in gingival fluid is associated with disease severity. (A–F) Sulcus fluid from patients with different disease stages were investigated by Western blotting to analyse the protein expression and maturation of ADAM8 and ADAM10 using antibodies against the C‐terminal domains. Band intensity was quantified by densitometry (A, pro‐form ADAM8; 120 kDa; B, mature form ADAM8, 90 kDa; D, pro‐form ADAM10; 100 kDa; E, mature form ADAM10, 70 kDa). Maturation was determined as ratio of the mature form and pro‐form of the respective protease (C and F). (G, H) Sulcus fluid was resuspended in activity buffer and subjected to ADAM8 activity measurement using a FRET‐based substrate (n = 3–12 per group). Activity over time is shown in (G) and was used for the calculation of the slope shown in (H). Data are shown as mean ± SD in (A–F) and (H), and activity over time in (G). Statistical analysis was performed with one‐way ANOVA followed by Tukey's post hoc test. Asterisks without line represent statistically significant differences to healthy patients; asterisks with line between groups (*p < 0.05, **p < 0.01, ***p < 0.001).
3.2. P. gingivalis Induces ADAM8 Gene Expression and Release of ADAM8 to the Extracellular Matrix
The development of periodontitis is driven by initial dysbiosis at the local site, which includes P. gingivalis as a central player in the red complex causing gingivitis. Therefore, we analysed ADAM8 and ADAM10 expression in primary human keratinocytes infected with P. gingivalis. We observed an increase in ADAM8 pro‐form and mature form, and a decrease in the pro‐form of ADAM10 (Figures 2A–D and S2A,B). Since the intracellular marker protein glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) can be detected only in the sulcus fluid of a few patients, we hypothesised that the observed expression and activity of ADAMs in patient samples may be mostly derived from cell‐free structures. As mentioned previously, part of ADAM8 is released as a soluble ectodomain. We observed increased levels of the remaining ADAM8 remnant form upon infection (Figure 2E). The second major source of external activity is the ADAM8‐carrying exosomes. However, during the period monitored in our study, only higher amounts of the remnant form, not the mature form, were released on exosomes, (Figure 2F). However, infection with P. gingivalis induced a clear release of mature ADAM10 on exosomes (Figure 2G). Furthermore, FRET‐based activity measurements revealed a strong increase in ADAM8 activity in P. gingivalis‐challenged cells, which was significantly inhibited by the ADAM8‐specific inhibitor BK‐1361 (Schlomann et al. 2015) (Figure 2H). Furthermore, we observed an increase in ADAM10 activity, which was significantly inhibited by the ADAM10‐specific inhibitor GI254023X but only slightly affected by BK‐1361 (Figure 2I). Thus, the release of ADAM8 as a soluble ectodomain by oral keratinocytes may contribute to the proteolytic pool observed in patient samples, whereas the membrane‐associated forms may be derived from different cellular sources.
FIGURE 2.

P. gingivalis induces ADAM8 expression and activity in human oral keratinocytes. Human primary keratinocytes (K2 cells) were grown until confluency and infected with P. gingivalis (2 h, 0.1 McF). (A–E) The cells were lysed, and the samples were analysed by Western blotting using antibodies against the C‐terminal domains of ADAM8 and ADAM10, respectively. GAPDH served as loading control. Band intensity was quantified by densitometry and normalized to uninfected cells. (F, G) The medium of the infected and non‐infected cells was subjected to differential centrifugation and density fractionation for exosome purification. The resulting pellets of different densities were analysed by Western blotting for the expression of ADAM8 and ADAM10 as well as Flottilin‐1 and CD9 as exosomal markers. Representative blots of three independent experiments are shown. (H, I) K2 cells were pre‐incubated with 10 μM BK‐1361, 10 μM GI254023X, or 0.01% DMSO (vehicle control) for 30 min and infected with P. gingivalis (2 h, 0.1 McF). The medium was analysed for ADAM8 or ADAM10 activity using a FRET‐based assay, and the slope of the activity over time was calculated. Quantitative data are shown as mean + SD of three independent experiments. Statistical analysis was performed with one‐sample t‐test (A–E) and one‐way ANOVA (H, I). Asterisks represent statistical differences (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, n.s. not significant).
3.3. P. gingivalis‐Induced ADAM8 Activation Correlates With Tissue Destructive Activity
One characteristic of periodontal disease development is over‐activation of the immune response, in which neutrophils dominate the activated lesion, bridging both the innate and acquired immune systems (Meyle and Chapple 2015). Infection of primary human neutrophils with P. gingivalis resulted in the release of exosomes carrying both the pro‐form and the remnant form of ADAM8 (Figure 3A). In contrast, neutrophils showed a moderate constitutive release of ADAM10 in exosomes under these culture conditions, which did not increase upon infection (Figure 3B). Key factors for periodontal connective tissue destruction are MMPs such as MMP9, which have been shown to be regulated by ADAM8 (Cook et al. 2022). The zymography of patient samples revealed an increase in gelatinase activity at higher disease stages (Figure S3A), as confirmed by the MMP9 ELISA (Figure 3C). Infection of primary human neutrophils with P. gingivalis resulted in an increase in cell‐associated MMP9 expression (Figure 3D), whereas the released content decreased (Figure 3E). However, in both cases, MMP9 expression was significantly inhibited by BK‐1361, whereas no effect on ADAM10 inhibition was observed. In contrast to neutrophils, MMP9 expression or release by keratinocytes was unaffected by infection and inhibition, respectively (Figure S3B,C). To address the tissue‐destructive action of the extracellular vesicles (EVs) and soluble factors (vesicle‐free supernatant) in more detail, wound healing assays (scratch assays) were performed. Exosomes released by P. gingivalis‐infected neutrophils strongly reduced keratinocyte wound closure, which was reversed by co‐treatment with BK‐1361. Inhibition of ADAM10 by GI254023X had no beneficial effect on the migration capacity of exosome‐challenged keratinocytes but displayed general migration inhibition even in control‐treated cells (Figures 4A and S4A), as observed earlier (Pruessmeyer et al. 2014). Similar to exosomes, soluble factors released by neutrophils inhibited scratch closure, which was not affected by BK‐1361 because this compound does not inhibit the soluble form of ADAM8 (Figure 4B) (Conrad et al. 2022). We next investigated whether the release of damaging exosomes and soluble factors could be prevented by ADAM8 inhibition. Indeed, pre‐incubation of neutrophils with BK‐1316 before P. gingivalis challenge reduced the tissue‐damaging effects of the released exosomes (Figure 4C) and soluble factors (Figure 4D). Thus, ADAM8 in neutrophils may regulate tissue destruction in periodontal disease.
FIGURE 3.

P. gingivalis‐induced ADAM8 activation correlates with proteolytic disbalance. (A, B) Primary human neutrophils were infected with P. gingivalis (2 h, 0.1 McF) or left uninfected. The medium of the infected and non‐infected cells was subjected to differential centrifugation and density fractionation for exosome purification. The resulting pellets of different density were analysed by Western blotting for the expression of ADAM8 and ADAM10 as well as Flottilin‐1 and CD9 as exosomal markers. Representative images of three independent experiments are shown. (C) Sulcus fluid from patients with different disease stages was analysed for MMP‐9 expression by ELISA. (D, E) Primary human neutrophils were pre‐incubated with 10 μM BK‐1361, 10 μM GI254023X or 0.01% DMSO (vehicle control) for 30 min and infected with P. gingivalis (2 h, 0.1 McF). Subsequently, the cells were lysed, and the cell lysate and the medium were analysed for MMP‐9 expression and release by ELISA. Neutrophil data are shown as mean ± SD of three independent experiments. Statistical analysis was performed using one‐way ANOVA followed by Tukey's post hoc test. Asterisks without line represent statistically significant differences with healthy patients and asterisks with line between groups (*p < 0.05, ***p < 0.001, ****p < 0.0001, n.s. not significant).
FIGURE 4.

P. gingivalis‐induced ADAM8 activation correlates with tissue destruction. (A–D) K2 cells were grown until forming a monolayer, incubated with mitomycin (1 μg/mL) for 2 h followed by wound induction using an automatic scratcher. (A, B) Primary human neutrophils were infected with P. gingivalis (2 h, 0.1 McF). The medium of the infected cells was subjected to differential centrifugation and density fractionation for exosome purification and collection of soluble factors (supernatant). Subsequently, K2 cells were incubated with (A) exosomes or (B) soluble factors in the presence or absence of 10 μM BK‐1361 or 10 μM GI254023X and evaluated for wound closure. (C, D) Primary human neutrophils were pre‐incubated with 10 μM BK‐1361 or 0.01% DMSO (vehicle control) for 30 min and infected with P. gingivalis (2 h, 0.1 McF) followed by exosomes purification and collection of soluble factors from the medium. Subsequently, K2 cells were incubated with (C) exosomes or (D) soluble factors and evaluated for wound closure. Data are shown as a percentage of wound closure relative to closed wound. Quantitative data are shown as means ± SD of eight independent experiments. Asterisks indicate significance among treated cells calculated using one‐way ANOVA and Tukey's post‐test (*p < 0.05, **p < 0.01, ***p < 0.001).
3.4. Cytokines May Orchestrate the ADAM Response in Periodontal Disease
Pathogens in the oral cavity lead to the release of cytokines that not only regulate the host immune response and osteolytic activity but also affect the tissue damage (Pan, Wang, and Chen 2019). The cytokine pool consists of the IL‐1 and TNF families, which are released by periodontal cells, myeloid cells and lymphocytes. To mimic this process, we stimulated oral keratinocytes with a combination of TNFα, IL‐1β and IFNγ. After 24 h, stimulated keratinocytes showed an increase in ADAM8 pro‐form, mature form and remnant form (Figure 5A–C). Interestingly, upon long‐term stimulation (48 h), only the pro‐form remained elevated, whereas the other forms returned to their unstimulated levels. For ADAM10, we observed a similar time‐dependent increase in protein expression; however, this did not reach significance due to the high standard deviations (Figure 5D,E). In contrast to the expression of ADAM8, that of the pro‐form of ADAM10 was reduced after 48 h of stimulation. Stimulated keratinocytes released an increased number of exosomes 48 h post stimulation, as indicated by an increase in flotillin‐1 and CD9 intensity (Figure 6A,B). This was accompanied by moderate expression of ADAM8 on these exosomes but a strong increase in mature ADAM10 and a potential cleavage fragment (50 kDa). Notably, almost no ADAM8 signal was detected in the exosome fraction of unstimulated K2 cells. Activity measurements of ADAM8 and ADAM10 released from human primary keratinocytes and neutrophils indicated a strong increase in ADAM8 activity in stimulated cells, which was inhibited by BK‐1361 (Figure 6C,E). The increase in ADAM10 activity caused by stimulation with the cytokine mixture was inhibited by GI254023X in both keratinocytes and neutrophils. However, ADAM8 inhibition by BK‐1361 affected ADAM10 activity in cytokine‐stimulated neutrophils but not in K2 cells (Figure 6D,F).
FIGURE 5.

Cytokines regulate ADAM8 expression in human oral keratinocytes. (A–E) K2 cells were grown until confluency and stimulated with a combination of TNFα, INF‐γ and IL‐1β (10 μM each) or sterile water (vehicle control) for 24 and 48 h. Subsequently, the cells were lysed, and the samples were analysed by Western blotting using antibodies against the C‐terminus of ADAM8 and ADAM10, respectively, with β‐actin as loading control. Band intensity was quantified by densitometry and normalized to unstimulated cells. Data are shown as mean + SD of three independent experiments. Statistical analysis was performed by one‐sample t‐test. Asterisks represent statistical differences (*p < 0.05, **p < 0.01, ***p < 0.001).
FIGURE 6.

Regulation of extracellular release of ADAM8 and ADAM10 by cytokines. (A, B) K2 cells were grown until confluency and stimulated with a combination of TNFα, INF‐γ and IL‐1β (10 μM each) or sterile water (vehicle control) for 48 h. The medium of the stimulated and non‐stimulated cells was subjected to differential centrifugation and density fractionation for exosome purification. The resulting pellets of different density were analysed by Western blotting for the expression of ADAM8 and ADAM10 as well as Flottilin‐1 and CD9 as exosomal markers. Representative blots of three independent experiments are shown. (C–F) K2 cells (C, D) or primary human neutrophils (E, F) were pre‐incubated with 10 μM BK‐1361, 10 μM GI254023X or 0.01% DMSO (vehicle control) for 30 min followed by stimulation with a combination of TNFα, INF‐γ and IL‐1β (10 μM each) or sterile water (vehicle control) for 48 h. The medium was analysed for ADAM8 (C, E) or ADAM10 (D, F) activity using a FRET‐based activity assay. Data are shown as mean + SD of three independent experiments. Statistical analysis was performed using one‐sample t‐test. Asterisks without line represent statistically significant differences to healthy patients, and asterisks with line between groups (*p < 0.05, **p < 0.01, ***p < 0.001).
Thus, bacterial infection and induced cytokine release stimulate a cell‐specific release of ADAM8, orchestrating tissue‐destructive activity and resulting in novel proteolytic effector molecules such as ADAM10.
4. Discussion
In the present study, we observed an increase in ADAM8 protein expression and activity in the sulcus fluid, correlating with the periodontal disease stage. Our data suggest that both P. gingivalis infection and the resulting cytokine release orchestrate the release of soluble ADAM8 by keratinocytes and neutrophils as soluble ectodomains or on exosomes, respectively. Furthermore, we observed ADAM8 regulated the release of ADAM10 and MMP9, which influenced wound healing and tissue destruction at least in vitro.
In previous studies, only total ADAM8 protein expression was examined (Aung et al. 2017; Feng et al. 2019), which was similarly increased in our patient cohort and correlated with the disease stage. ADAMs are strongly regulated at the post‐transcriptional level, resulting in the activation and generation of different forms of the protein. In our study, an increase in ADAM8 maturation and proteolytic activity was observed for the first time, again correlating with disease stage. It has been recently reported that ADAM8 exerts proteolytic activity on ADAM10 and ADAM17, thereby generating soluble ectodomains (Scharfenberg et al. 2020). Following this, we observed a decrease in the expression of membrane‐associated forms of ADAM10 protein in the sulcus fluid. In vitro, we confirmed that ADAM8 activation was responsible for the increase in ADAM10 proteolytic activity in both oral keratinocytes and neutrophils.
The observed soluble activity may result from the release of ADAM‐carrying EVs (Cook et al. 2022; Groth et al. 2016) or soluble ectodomains that still exert proteolytic activity. ADAM8 requires trimerization for activation, resulting in the autocatalytic removal of the inactivating pro domain and the release of its soluble ectodomain (Naus et al. 2006). This trimerization is inhibited by the cyclic peptide inhibitor BK‐1361 (Schlomann et al. 2015). While we observed strong inhibition of ADAM8 activity by BK‐1361 in cytokine‐stimulated oral keratinocytes, the inhibition of ADAM8 activity was weaker in P. gingivalis‐infected cells. This was further reflected in the membrane‐associated release of the mature form of ADAM8 in EVs, which was absent upon infection; only the remnant form was observed. Furthermore, ADAM8 inhibition did not affect, or only slightly affected, the increase of ADAM10 activity in cytokine‐stimulated keratinocytes. In contrast, the neutrophil‐derived ADAM8 activity was strongly inhibited by BK‐1361, which further inhibited ADAM8 release. Thus, keratinocytes appear to release ADAM8 as a soluble ectodomain, whereas neutrophils are the primary cellular source of ADAM8‐carrying exosomes.
Periodontal pathogens have developed strategies to evade the immune defences of the host (Miralda and Uriarte 2021). It was recently shown that ADAM8‐deficient mice are protected against bacterial lung infections, accompanied by a higher clearance capacity (Conrad et al. 2022). Similarly, increased ADAM8 protein expression in periodontal disease may be associated with reduced bacterial clearance and increased bacterial burden. Furthermore, ADAM8 is important for MMP‐9 release in the inflammatory tumour environment (Cook et al. 2022). Another study demonstrated that MMP‐9 is released during infection with P. gingivalis, thereby affecting the breakdown of inflamed human pulp tissue (Ding et al. 1997), and MMP inhibition protects against alveolar bone loss (Ramamurthy et al. 2002). In our study, neutrophils were identified as the major source of MMP9 release, which was inhibited by ADAM8. However, ADAM8 may modulate tissue destruction and progression not only through MMP9 but also through the release of ADAM10. The soluble form of ADAM10 can cleave fibronectin (Scharfenberg et al. 2020), and fibronectin fragments have been associated with periodontal disease status (Huynh et al. 2002). In addition to the release of soluble ADAM10 variants, we detected a decrease in membrane‐associated expression with increasing disease stage, which was also observed in our cell culture–based experiments. Cell‐associated ADAM10 is required for the cleavage of growth factors such as epidermal growth factor, thereby ensuring proper wound healing (Shin et al. 2023). Cell‐associated ADAM10 also contributes to keratinocyte adhesion and migration (Maretzky et al. 2005). Thus, ADAM8‐dependent modulation of ADAM10 expression and activity may promote tissue degradation while inhibiting repair processes. Which factors and mechanisms drive the transition from gingivitis as inflammatory lesion to periodontitis is still poorly understood. Dysbiosis and inflammation are involved in both the onset and progression of periodontitis, which led to the ‘inflammation‐mediated polymicrobial emergence and disease exacerbation’ (IMPEDE) model (Abdulkareem et al. 2023). Our recent observations indicate an ADAM8‐dependent hyperactivity of neutrophils, leading to reduced regeneration and increased tissue destruction. Thus, it seems feasible that the proteolytic imbalance driven by ADAM8 may influence the shift from controlled inflammation in gingivitis to dysbiosis, increased recruitment of neutrophils and the transition to chronic periodontitis. However, further translational studies are required to identify the relevance of ADAM8 and its interaction with other proteases such as ADAM10 and MMP9 in the transition to periodontitis.
5. Conclusion
The dysregulation of cell‐associated and extracellular ADAM proteolytic activity, mainly driven by ADAM8, may be an essential regulatory element in periodontal disease severity, potentially fostering disease progression. In particular, ADAM8, which seems to play a minor role under physiological conditions and is mostly up‐regulated under pathophysiological conditions (Chen et al. 2016; Conrad et al. 2022; Cook et al. 2022; Dreymueller et al. 2017; Schlomann et al. 2015), may be a potential target for local treatment or used as a diagnostic/prognostic marker, which should be addressed in future translational studies.
Author Contributions
Ahmad Aljohmani contributed to the conception, data analysis and interpretation; drafted the original manuscript; and critically revised the manuscript. Hakon Heinze contributed to the conception, data analysis, and interpretation and drafted the original manuscript. Federico Guillermo Gharzia contributed to data analysis and interpretation, drafted the original manuscript and critically revised the manuscript. Ahmed Mohamed Mostafa Abdrabou and Bashar Reda contributed to the acquisition and data analysis and critically revised the manuscript. Sören L. Becker and Markus Bischoff contributed to the conception, acquisition of data, and critically revised the manuscript. Matthias Hannig contributed to the conception and design of the study, acquisition and data analysis and critically revised the manuscript. Daniela Yildiz contributed to the conception and design of the study; acquisition, data analysis and interpretation; drafted the original manuscript and critically revised it. All authors provided their final approval and agreed to be accountable for all aspects of this study.
Consent
Written informed consent was obtained from all participants.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Data S1.
Acknowledgements
We thank Nina Schnellbach and Maria Rieseweber for technical support, and Jörg‐Walter Bartsch and Nicolai Miosge for providing the BK‐1361 and K2 cells, respectively. We thank the employees of the Clinic for Dental Preservation, Periodontology, and Preventive Dentistry of Saarland University Hospital as well as those of the dental office Reinstädtler and Hektor for their support in acquiring the samples. We also acknowledge the German Academic Exchange Service (DAAD) for providing financial support. Open Access funding enabled and organized by Projekt DEAL.
Funding: This study was supported by the German Research Foundation (DR1013/1‐1 by DY) and the HIPS‐UdS TANDEM initiative (Saarland University by DY, MB, and SLB). We also acknowledge the German Academic Exchange Service (DAAD) for providing financial support.
The first three authors share first authorship.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- Abdulkareem, A. A. , Al‐Taweel F. B., Al‐Sharqi A. J. B., Gul S. S., Sha A., and Chapple I. L. C.. 2023. “Current Concepts in the Pathogenesis of Periodontitis: From Symbiosis to Dysbiosis.” Journal of Oral Microbiology 15, no. 1: 2197779. 10.1080/20002297.2023.2197779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aljohmani, A. , Andres N. N., and Yildiz D.. 2022. “ Pseudomonas aeruginosa Alters Critical Lung Epithelial Cell Functions Through Activation of ADAM17.” Cells 11, no. 15: 2303. 10.3390/cells11152303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aljohmani, A. , Opitz B., Bischoff M., and Yildiz D.. 2022. “ Pseudomonas aeruginosa Triggered Exosomal Release of ADAM10 Mediates Proteolytic Cleavage in Trans.” International Journal of Molecular Sciences 23, no. 3: 1259. 10.3390/ijms23031259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aljohmani, A. , and Yildiz D.. 2020. “A Disintegrin and Metalloproteinase‐Control Elements in Infectious Diseases.” Frontiers in Cardiovascular Medicine 7: 608281. 10.3389/fcvm.2020.608281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aung, W. P. P. , Chotjumlong P., Pata S., et al. 2017. “Inducible Expression of a Disintegrin and Metalloproteinase 8 in Chronic Periodontitis and Gingival Epithelial Cells.” Journal of Periodontal Research 52, no. 3: 582–593. 10.1111/jre.12426. [DOI] [PubMed] [Google Scholar]
- Chen, J. , Deng L., Dreymuller D., et al. 2016. “A Novel Peptide ADAM8 Inhibitor Attenuates Bronchial Hyperresponsiveness and Th2 Cytokine Mediated Inflammation of Murine Asthmatic Models.” Scientific Reports 6: 30451. 10.1038/srep30451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad, C. , Yildiz D., Cleary S. J., et al. 2022. “ADAM8 Signaling Drives Neutrophil Migration and ARDS Severity.” JCI Insight 7, no. 3: e149870. 10.1172/jci.insight.149870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook, L. , Sengelmann M., Winkler B., et al. 2022. “ADAM8‐Dependent Extracellular Signaling in the Tumor Microenvironment Involves Regulated Release of Lipocalin 2 and MMP‐9.” International Journal of Molecular Sciences 23, no. 4: 1976. 10.3390/ijms23041976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, Y. , Haapasalo M., Kerosuo E., Lounatmaa K., Kotiranta A., and Sorsa T.. 1997. “Release and Activation of Human Neutrophil Matrix Metallo‐ and Serine Proteinases During Phagocytosis of Fusobacterium nucleatum, Porphyromonas gingivalis and Treponema denticola .” Journal of Clinical Periodontology 24, no. 4: 237–248. 10.1111/j.1600-051x.1997.tb01837.x. [DOI] [PubMed] [Google Scholar]
- Dreymueller, D. , Pruessmeyer J., Schumacher J., et al. 2017. “The Metalloproteinase ADAM8 Promotes Leukocyte Recruitment in Vitro and in Acute Lung Inflammation.” American Journal of Physiology. Lung Cellular and Molecular Physiology 313, no. 3: L602–L614. 10.1152/ajplung.00444.2016. [DOI] [PubMed] [Google Scholar]
- Dreymueller, D. , Uhlig S., and Ludwig A.. 2015. “ADAM‐Family Metalloproteinases in Lung Inflammation: Potential Therapeutic Targets.” American Journal of Physiology. Lung Cellular and Molecular Physiology 308, no. 4: L325–L343. 10.1152/ajplung.00294.2014. [DOI] [PubMed] [Google Scholar]
- Feng, Y. , Li Q., Chen J., et al. 2019. “Salivary Protease Spectrum Biomarkers of Oral Cancer.” International Journal of Oral Science 11, no. 1: 7. 10.1038/s41368-018-0032-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groth, E. , Pruessmeyer J., Babendreyer A., et al. 2016. “Stimulated Release and Functional Activity of Surface Expressed Metalloproteinase ADAM17 in Exosomes.” Biochimica et Biophysica Acta 1863, no. 11: 2795–2808. 10.1016/j.bbamcr.2016.09.002. [DOI] [PubMed] [Google Scholar]
- Hiyoshi, T. , Domon H., Maekawa T., et al. 2022. “Neutrophil Elastase Aggravates Periodontitis by Disrupting Gingival Epithelial Barrier via Cleaving Cell Adhesion Molecules.” Scientific Reports 12, no. 1: 8159. 10.1038/s41598-022-12358-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa, Y. , Hosokawa I., Ozaki K., Nakae H., and Matsuo T.. 2007. “CXC Chemokine Ligand 16 in Periodontal Diseases: Expression in Diseased Tissues and Production by Cytokine‐Stimulated Human Gingival Fibroblasts.” Clinical and Experimental Immunology 149, no. 1: 146–154. 10.1111/j.1365-2249.2007.03398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh, Q. N. , Wang S., Tafolla E., et al. 2002. “Specific Fibronectin Fragments as Markers of Periodontal Disease Status.” Journal of Periodontology 73, no. 10: 1101–1110. 10.1902/jop.2002.73.10.1101. [DOI] [PubMed] [Google Scholar]
- Jain, N. , Dutt U., Radenkov I., and Jain S.. 2024. “WHO's Global Oral Health Status Report 2022: Actions, Discussion and Implementation.” Oral Diseases 30: 73–79. 10.1111/odi.14516. [DOI] [PubMed] [Google Scholar]
- Lee, J. H. , Choi Y. J., Heo S. H., Lee J. M., and Cho J. Y.. 2011. “Tumor Necrosis Factor‐Alpha Converting Enzyme (TACE) Increases RANKL Expression in Osteoblasts and Serves as a Potential Biomarker of Periodontitis.” BMB Reports 44, no. 7: 473–477. 10.5483/BMBRep.2011.44.7.473. [DOI] [PubMed] [Google Scholar]
- Maretzky, T. , Reiss K., Ludwig A., et al. 2005. “ADAM10 Mediates E‐Cadherin Shedding and Regulates Epithelial Cell‐Cell Adhesion, Migration, and Beta‐Catenin Translocation.” Proceedings of the National Academy of Sciences of the United States of America 102, no. 26: 9182–9187. 10.1073/pnas.0500918102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez‐Garcia, M. , and Hernandez‐Lemus E.. 2021. “Periodontal Inflammation and Systemic Diseases: An Overview.” Frontiers in Physiology 12: 709438. 10.3389/fphys.2021.709438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyle, J. , and Chapple I.. 2015. “Molecular Aspects of the Pathogenesis of Periodontitis.” Periodontology 2000 69, no. 1: 7–17. 10.1111/prd.12104. [DOI] [PubMed] [Google Scholar]
- Miralda, I. , and Uriarte S. M.. 2021. “Periodontal Pathogens' Strategies Disarm Neutrophils to Promote Dysregulated Inflammation.” Molecular Oral Microbiology 36, no. 2: 103–120. 10.1111/omi.12321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naus, S. , Reipschlager S., Wildeboer D., et al. 2006. “Identification of Candidate Substrates for Ectodomain Shedding by the Metalloprotease‐Disintegrin ADAM8.” Biological Chemistry 387, no. 3: 337–346. 10.1515/BC.2006.045. [DOI] [PubMed] [Google Scholar]
- Pan, W. , Wang Q., and Chen Q.. 2019. “The Cytokine Network Involved in the Host Immune Response to Periodontitis.” International Journal of Oral Science 11, no. 3: 30. 10.1038/s41368-019-0064-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruessmeyer, J. , Hess F. M., Alert H., et al. 2014. “Leukocytes Require ADAM10 but Not ADAM17 for Their Migration and Inflammatory Recruitment Into the Alveolar Space.” Blood 123, no. 26: 4077–4088. 10.1182/blood-2013-09-511543. [DOI] [PubMed] [Google Scholar]
- Ramamurthy, N. S. , Xu J. W., Bird J., et al. 2002. “Inhibition of Alveolar Bone Loss by Matrix Metalloproteinase Inhibitors in Experimental Periodontal Disease.” Journal of Periodontal Research 37, no. 1: 1–7. 10.1034/j.1600-0765.2002.00342.x. [DOI] [PubMed] [Google Scholar]
- Scharfenberg, F. , Helbig A., Sammel M., et al. 2020. “Degradome of Soluble ADAM10 and ADAM17 Metalloproteases.” Cellular and Molecular Life Sciences 77, no. 2: 331–350. 10.1007/s00018-019-03184-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlomann, U. , Koller G., Conrad C., et al. 2015. “ADAM8 as a Drug Target in Pancreatic Cancer.” Nature Communications 6: 6175. 10.1038/ncomms7175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, S. H. , Koh Y. G., Lee W. G., Seok J., and Park K. Y.. 2023. “The Use of Epidermal Growth Factor in Dermatological Practice.” International Wound Journal 20, no. 6: 2414–2423. 10.1111/iwj.14075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita, T. , Kubota T., Nakasone N., et al. 2013. “Gene and Protein Localisation of Tumour Necrosis Factor (TNF)‐alpha Converting Enzyme in Gingival Tissues From Periodontitis Patients With Drug‐Induced Gingival Overgrowth.” Archives of Oral Biology 58, no. 8: 1014–1020. 10.1016/j.archoralbio.2013.02.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.