

Significance
There are many hurdles in developing biological drugs against neurodegenerative disorders, such as Alzheimer’s disease (AD). One is the blood–brain barrier (BBB), which strictly limits the entering of exogenous molecules, including drugs. This has been overcome by various strategies with huge efforts and advances during the last few decades. Another significant barrier is the inefficient accumulation of drugs to the desired target site in the brain, in particular, the hippocampus, which is damaged in neurodegenerative disorders, including AD. Here, we genetically engineered insulin-fusion proteins [called hippocampal neuron–targeting (Ht) proteins] and provided evidence that Ht proteins could be targeted to hippocampal neurons in vitro and in vivo. Our method will help in the development of protein drugs for neurodegenerative disorders.
Keywords: hippocampal neuron, insulin, drug delivery, fusion protein, Alzheimer’s disease
Abstract
Hippocampal neurons can be the first to be impaired with neurodegenerative disorders, including Alzheimer’s disease (AD). Most drug candidates for causal therapy of AD cannot either enter the brain or accumulate around hippocampal neurons. Here, we genetically engineered insulin-fusion proteins, called hippocampal neuron–targeting (Ht) proteins, for targeting protein drugs to hippocampal neurons because insulin tends to accumulate in the neuronal cell layers of the hippocampus. In vitro examinations clarified that insulin and Ht proteins were internalized into the cultured hippocampal neurons through insulin receptor–mediated macropinocytosis. Cysteines were key determinants of the delivery of Ht proteins to hippocampal neurons, and insulin B chain mutant was most potent in delivering cargo proteins. In vivo accumulation of Ht proteins to hippocampal neuronal layers occurred after intracerebroventricular administration. Thus, hippocampal neuron–targeting technology can provide great help for developing protein drugs against neurodegenerative disorders.
Neurodegenerative disorders increase with age. In particular, dementia, such as Alzheimer’s disease (AD), not only affects the patients but also their family and caregivers. To extend healthy life expectancy, effective preventive and therapeutic strategies against neurodegenerative disorders must be developed. Clinically, a limited number of medications are available for AD, including donepezil, memantine, and galantamine, which symptomatically prevent decrease in neuronal activity and communication (1). Recently, an antibody drug (lecanemab) for removing pathogenic amyloid β (Aβ) was approved in the United States and Japan (2, 3). However, the use of antibody drugs is limited for mild cognitive impairment and early stages of AD because there is no evidence that they are effective on the progressive stage of dementia, with severe pathological conditions and memory loss. Furthermore, their effectiveness is controversial between specialists and researchers.
Most candidates are currently proposed as drugs for treating AD (Fig. 1A). Although antibodies are thought to remove soluble Aβ oligomers and protofibrils from the brain and blood, inhibitors for Aβ-producing enzymes from amyloid precursor protein, such as β-secretase (BACE1), can prevent the generation of Aβ. Nucleic acids, including antisense oligos and small interfering RNA (siRNA), can reduce Aβ production by silencing BACE1 gene expression. Recently, nucleic acids were tested for silencing the gene encoding microtubule-associated protein tau, another pathogen in AD (4, 5). Neurotrophins, such as brain-derived neurotrophic factor, may be used to recover loss of neuronal function and activity under pathological conditions (6–9). It is important that the drugs should work in or around neuronal cells at the “hippocampus” because hippocampal neurons are initial points for the production and secretion of Aβ, as well as the phosphorylation or propagation of tau.
Fig. 1.

Schematic conceptual illustration of the study. (A) Need for targeted delivery of drugs, including antibodies, neurotrophic factors, and nucleic acids, to hippocampal neurons for protection and repair in neurodegenerative disorders, such as dementia (AD). (B) Obstacles for transport of drugs to the brain. The BBB prevents entry of exogenous molecules, even therapeutic drugs. Drugs are required to reach deep target regions of the brain, such as the hippocampus, through the brain parenchyma. (C) Unique strategy to deliver protein-based drugs to the hippocampus via fusion with insulin, which potentially accumulates on hippocampal neurons. Our goal is to effectively deliver insulin-fusion protein therapeutics from administration sites (nose or systemic circulation) to hippocampal neurons. Illustrations were created with BioRender.com.
In recent decades, pharmaceutical companies have been struggling to develop treatments, including antibody drugs, for the causal treatment of AD. Most potential candidates have been dropped out from clinical trials, and development was switched for next drug candidates, without considering their pharmacokinetic problems. Because only few molecules (transporter substrates like glucose and hydrophobic small molecules) can permeate through the blood–brainbarrier (BBB), most drugs cannot penetrate the brain parenchyma (Fig. 1B). Pharmaceutical research has little addressed the transport of antibody drugs from systemic circulation to the brain. Various promising techniques for overcoming and bypassing the BBB have been established (10–12). The most well-known strategy is based on antibodies against transferrin receptor (TfR), which is abundantly expressed on the surface of brain microvascular endothelial cells. Cargo drugs fused to the TfR antibody can be delivered to the brain (13–18). Another useful option is nose-to-brain delivery, in which drugs intranasally administered can directly move to the brain without systemic absorption and subsequent transport through BBB (19–22). These techniques will accelerate drug development for AD and other neurodegenerative disorders. However, as described above, one of the major therapeutic targets for these diseases related to memory loss and cognitive dysfunction is the hippocampus. Therefore, the technology for specifically accumulating them to hippocampal neuronal cells is necessary with or after brain delivery to maximize the efficacy of the potential drugs for dementia; however, such approaches have not been studied yet.
Insulin is a bioactive peptide that performs many functions in the body via receptor stimulation, which triggers phosphorylation of downstream signaling molecules, such as insulin receptor substrate (IRS), phosphoinositide 3-kinase (PI3K), and protein kinase B (Akt) (23). Over several decades, the IR on the BBB was referred to as a potential target for drug delivery to the brain, but transcytosis via the IR on the brain endothelial cells might not be mediated by normal IR signaling (24). On the other hand, recent studies have clarified that insulin can enhance memory and learning after intranasal administration (25, 26). We recently found that insulin could be delivered efficiently to the brain via intranasal coadministration with cell-penetrating peptide (penetratin), as the epithelial permeation enhancer, and eventually distributed specifically around the neuronal cell layers in the mouse hippocampus (27). We hypothesized that the disposition of insulin in the brain makes it eligible for efficient drug delivery to the hippocampus and neuronal cells (Fig. 1C). Therefore, we developed targeting technology to deliver protein-based drugs, such as antibodies and neurotrophic factors, to hippocampal neurons, based on genetic engineering of fusion proteins. First, we evaluated the characteristics of insulin in the brain to estimate its potential as a drug carrier. Second, we generated plasmid vectors for bacterial expression of proteins fused to insulin (insulin A chain, B chain, and full-length) and tested the ability of insulin-fusion proteins to accumulate in hippocampal neurons. The fusion proteins were named “hippocampal neuron–targeting (Ht) proteins.” Third, we studied the mechanisms associated with specific localization of Ht proteins in the brain under various experimental conditions and with insulin mutants. Furthermore, we demonstrated the in vivo performance of Ht proteins in effective and specific delivery to hippocampal neurons after intracerebroventricular (ICV) administration in mice and tested the brain delivery potential through noninvasive intranasal administration. This study provides evidence that a specific targeting approach with insulin-inspired fusion proteins can enhance accumulation of protein drugs and other molecules in hippocampal neurons. Our findings have great application in brain neurodegenerative disorder therapy.
Results
In Vivo and In Vitro Disposition of Insulin around the Hippocampus after Efficient Brain Delivery.
In our previous study, insulin seemed to be localized on hippocampal neuronal layers after enhanced nose-to-brain delivery (27). To discuss the potential of insulin in delivering other drugs to hippocampal neurons, we first evaluated the in vivo distribution property of insulin after intranasal administration, based on immunohistological examinations (Fig. 2A). The results reproduced our results that insulin (green) could accumulate around hippocampal neuronal layers (Fig. 2B). Multiple staining results showed that insulin did not colocalize with marker proteins for astrocytes and neuronal stem cells glial fibrillary acidic protein (GFAP) and nestin, respectively). By contrast, colocalization with the neuronal cell body marker microtubule-associated protein 2 (MAP2) clarified that insulin was internalized by neuronal cells, suggesting that insulin was not only bound to the cell surface but was also taken up by neuronal cells. Microglial marker Iba1 was not included in this study because insulin disposition was clearly different from typical microglial localization.
Fig. 2.

In vitro and in vivo characterization of insulin as a carrier to hippocampal neurons. (A) Brief illustration of the in vivo study for evaluating the specificity of insulin accumulation in the brain. For microscopic observation, brain specimens were isolated from mice receiving intranasal insulin (10 IU/kg) with mucosal permeation enhancer (L-penetratin, 2 mM). Frozen brain sections were treated with primary antibodies to insulin and various cell markers (MAP2, neuronal cell body; GFAP, astrocytes; nestin, neuronal stem cells) and stained with secondary antibodies conjugated with fluorescent dyes. (B) Representative images (from three mice) of the hippocampus after intranasal delivery of insulin. (C) Four types of cells were used for the in vitro study (NIH3T3 cells as unrelated control, bEnd.3 cells as BBB model, C6 cells as glial model, and HT22 cells as hippocampal neuron model). (D) Confocal micrographs of cells incubated with FITC-insulin (10 μg/mL) for 120 min at 37 °C. Prior to observation, the cells were washed thrice with glycine–HCl buffer and once with phosphate-buffered saline (PBS) for optimal detection of intracellular fluorescence. The bars indicate 20 μm. (E) Histograms generated from flow cytometry after incubation with FITC-insulin (10 μg/mL) for 120 min at 37 °C. (F) Geometric mean fluorescence intensity (MFI) calculated from the results of flow cytometry as the quantitative parameter. (G) Comparison of cellular uptake of FITC-insulin by HT22 and N2a cells. (H and I) Flow cytometry for examining energy dependence of cellular uptake of FITC-insulin by comparing them at biological (37 °C) and energy-abolishing (4 °C) conditions. (J) Western blot analysis to compare the expression of IR between cells. Samples were loaded at 6 μg of total protein. β-Actin was used as housekeeping control. (K) Relative expression levels of IR to β-actin between cells based on western blotting.
We further investigated the in vitro property of insulin by testing its cellular uptake by various cell types (Fig. 2C). Insulin accumulation was compared mainly between C6 (glial cell model) and HT22 (neuronal cell model) cells. The brain microvascular model (bEnd.3 cells) was used as insulin might have the potential to transport through BBB (28). Fibroblasts NIH3T3 cells were used as brain-unrelated control cells. Peripheral tissue-derived cells, such as HEK293T cells, with more abundant expression of IR than neuronal cells (SI Appendix, Fig. S1 A and B), were excluded from comparison of cellular uptake of Ht-fusion proteins. Fig. 2D shows confocal micrographs of cells treated with fluorescein isothiocyanate (FITC)-insulin for 2 h. Fig. 2 E and F shows more quantitative data with histograms and geometric mean fluorescence intensity, based on flow cytometry, respectively. Among brain cells, neuronal HT22 cells are highly capable of FITC-insulin uptake compared with glial C6 cells. In contrast to the known property of insulin passing through BBB, FITC-insulin was less taken up by bEnd.3 cells. Because IR is widely expressed in the body, FITC-insulin was effectively taken up by 3T3 cells. As shown in Fig. 2G, FITC-insulin was more effectively taken up by HT22 cells than neuroblastoma Neuro2a (N2a) cells, suggesting specific accumulation of insulin to the hippocampal neurons. Examination of temperature sensitivity (Fig. 2 H and I) demonstrated that internalization of FITC-insulin was completely abolished at low temperature (4 °C), suggesting that insulin internalization involves energy-dependent pathways, including endocytosis. Punctate cellular fluorescence (Fig. 2D) indicated internalization of FITC-insulin via endocytosis.
We further examined IR expression on the cells to clarify the relationship with cellular uptake of insulin. IR was most highly expressed in the 3T3 cells, and the order of expression level in the brain-related cells was HT22, C6, and bEnd.3 (Fig. 2 J and K). According to the company’s datasheet of primary antibody, the main band for IR should appear around 95 kDa, but in mouse tissues, the band at 72 kDa seemed correct. This trend was similar with that of cellular internalization capacity (Fig. 2F), implying that insulin accumulated in hippocampal neurons via receptor binding. Because receptor binding of some growth factors may have cross-effect with insulin, we examined insulin-like growth factor 1 receptor (IGF-1R) (29, 30). However, IGF-1R was not detected in any cell lysate used in this study (SI Appendix, Fig. S1C).
Thus, insulin can deliver other molecules into hippocampal neurons. We tested the ability of insulin to deliver the low molecular fluorescent dye, FITC, via ICV administration of FITC-insulin. Fluorescence was observed in the hippocampal neuronal layer after ICV administration of FITC-insulin, whereas FITC-dextran (4.4 kDa, FD-4) tagged with FITC could not be localized in neuronal cells (SI Appendix, Fig. S2). The results suggest that insulin can deliver other molecules, possibly protein drugs, through conjugation.
Genetic Engineering, Expression, and Characterization of Ht Proteins.
We engineered fusion proteins of insulin and target proteins for delivery of protein drugs to hippocampal neurons. Luminescent and fluorescent proteins—NanoLuc (Nluc) and mNeonGreen (mNG), respectively—were used to detect biodistribution, brain penetration, and neuronal accumulation in the hippocampus (31, 32). pET vectors with genes encoding insulin (A chain, B chain, and full-length proinsulin, including A and B chains and c-peptide) and probe proteins (Nluc and mNG) were designed and transformed into bacteria for protein expression (Fig. 3A). Insulin A– and B chain–fused proteins were named as HtA- and HtB proteins, respectively. Plasmids encoding full-length proinsulin-fused proteins were tested being transformed into different bacteria; the fusion proteins expressed in BL21(DE3) and SHuffle were named HtC and HtD proteins, respectively. Here, SHuffle Escherichia coli was used to enhance the correct folding of disulfide-bonded proteins (33). All proteins tagged with a histidine–asparagine repeat (6× HN) were purified with nickel-based gravity columns. The proteins were confirmed eluted in fraction No. 2, and expression yields were partially reduced due to fusion with insulin (Fig. 3 B and C). Luminescence activity and fluorescence intensity were considerably decreased with addition of insulin to the N terminus of the proteins (Fig. 3 D and E). We used western blotting to confirm that insulin was fused to Nluc (Fig. 3F) and mNG (Fig. 3G). Antibodies specific to insulin A or B chain could react to HtA or HtB protein, respectively, and HtC- and HtD proteins were detected with both anti-insulin antibodies. Because multiple bands were observed under nonreducing condition, fusion proteins might be complexed intermolecularly. Analysis with high-performance liquid chromatography confirmed purified Ht-fusion proteins without any contamination (SI Appendix, Fig. S3).
Fig. 3.

Production and characterization of Ht proteins. (A) Design of plasmid vectors for generating fusion proteins with insulin. The constructs were designed for fusing target proteins with A chain (21 amino acids), B chain (30 amino acids), or full-length insulin (B chain/c-peptide/A chain, proinsulin form). Nluc luciferase and mNG were chosen as experimental model proteins for detection in several assays. (B and C) The Ht proteins expressed in bacteria [BL21(DE3) or SHuffle T7 E. coli] were purified with nickel affinity columns through the HN-repeat tag at the C terminus of fusion proteins. Ht proteins were mostly eluted in fraction #2. (D and E) Luminescence or fluorescence intensity of Ht proteins (Nluc/mNG) was measured with a microplate reader at 100 pg/mL or 20 μg/mL, respectively. (F and G) Western blotting of Ht-Nluc and mNG, respectively. Samples were loaded at 75 ng/mL of purified protein. Poly vinylidene difluoride (PVDF) membranes were treated with antibodies for Nluc, mNG, insulin A chain, and B chain. Arrows indicate the molecular weight of original Nluc or mNG.
Comparison of Cellular Accumulation and Internalization Properties of Ht Proteins.
To examine the hippocampal neuronal specificity of Ht proteins, we compared qualitatively and quantitatively their accumulation and internalization into different cell types with confocal microscopy and flow cytometry (Fig. 4A). As described for examination with FITC-insulin, NIH3T3, bEnd.3, C6, and HT22 cells were used in this examination (Fig. 2C). First, we compared the internalization of all Ht-fusion mNG in bEnd.3 (BBB model) and HT22 cells (hippocampal neuron model) and found that insulin fusion effectively enhanced the cellular uptake of mNG, particularly by HT22 cells (Fig. 4B). Intriguingly, even incomplete insulin (only A and B chains) could intensively increase cellular internalization of mNG. The specificity of most effective HtB- and complete HtD-mNG was further tested with various cell types (Fig. 4C). Consistent with the results using FITC-insulin (Fig. 2D), punctate fluorescence was observed in 3T3, C6, and HT22 cells after treatment with HtB- and HtD-mNG, but these proteins were slightly internalized into bEnd.3 cells. Flow cytometry–based quantification revealed that insulin fusion could potentially enhance the internalization of mNG into various cell types (3T3, C6, and HT22 cells) (Fig. 4 D and E); importantly, mNG could be predominantly taken up by HT22 cells, especially when fused to insulin B chain (HtB-mNG). These in vitro estimations suggest that insulin, even fragments such as the B chain, can effectively deliver the proteins to hippocampal neurons.
Fig. 4.

In vitro cellular uptake and in vivo brain distribution of Ht-mNG. (A) Brief illustration of in vitro evaluation with confocal scanning microscopy and flow cytometry. NIH3T3 cells (unrelated control), bEnd.3 cells (BBB model), C6 cells (glial model), and HT22 cells (hippocampal neuron model) were used in this study. (B and C) Confocal micrographs of cells treated with Ht-mNG (25 μg/mL) for 120 min at 37 °C. Prior to observation, the cells were washed thrice with glycine–HCl buffer and once with PBS for optimal fluorescence investigation. Panel (B) compares cellular uptake of all Ht-mNG by bEnd.3 and HT22 cells. Panel (C) compares uptake of HtB- and HtD-mNG between all four cell types. (D and E) Histograms and geometric MFI, respectively, generated from flow cytometry after treatment with Ht-mNG at 25 μg/mL at 37 °C. (F) Brief illustration of the SPR assay. Purified IR with α and β chains was immobilized on the CM5 sensorchip, and various concentrations of Ht-mNG were injected into the flow as analytes. Intermolecular binding was detected as mass change on the sensorchip. (G) Binding sensorgrams after Ht-mNG was injected to the IR –immobilized flow cell, where specific binding was expressed by subtracting nonspecific binding to blank flow cell from the total binding. (H) Comparison of cellular uptake of Ht-mNG by HT22 and N2a cells. (I) Brief illustration of ICV administration to mice. Mice brains were isolated 30 min after ICV administration of Ht-mNG (250 μg/mL, 10 μL/30 g mouse). Frozen tissue slices were treated with primary antibodies to neuronal nucleic marker (NeuN) and stained with secondary antibodies conjugated to fluorescent dyes. (J) Representative images (from three mice for each group) of hippocampal regions after ICV administration of Ht-mNG. The bars indicate 500 μm.
The higher specificity of HtB- and HtD-mNG to HT22 cells was confirmed in HT22 and C6 cells cocultured Transwell assay. To avoid direction vias of Transwell (between upper and lower chambers), one experiment was conducted with HT22 and C6 cultured in upper and lower chambers, respectively, and another with the reverse order of cultures (SI Appendix, Fig. S4A). HtB- and HtD-mNG were more preferentially taken up by HT22 cells than C6 cells in both conditions (SI Appendix, Fig. S4B) The uptake pattern of Ht-mNG was similar to that of FITC-insulin (SI Appendix, Fig. S4C). To elucidate the involvement of the receptor binding of Ht-mNG in their cell accumulation specificity, intermolecular interactions between Ht-mNG and IR were analyzed with surface plasmon resonance (SPR) (Fig. 4F). In this experiment, purified IR, consisting of α and β subunits, which was cloned from HEK293T cells and expressed in BL21(DE3) E. coli, was immobilized on the CM5 sensorchip [706.4 response unit (RU)], and then, Ht-mNG were injected as analyte. A negligible change was observed in the sensorgram after injection of mNG, confirming that mNG without insulin could not specifically bind to IR (Fig. 4G). By contrast, a concentration-dependent increase in the sensorgram was observed after all Ht-mNG was injected. Unexpectedly, fusion with incomplete forms of insulin (A and B chains of insulin) provided mNG the ability to bind IR. Based on affinity analysis, the dissociation constant (Kd) and maximal RU (Rmax) were calculated. The lowest Kd for HtB-mNG (3.330 μM) among fusion proteins suggested possible specificity to IR –expressing cells such as hippocampal neurons, and the highest Rmax (152.4 RU) could be associated with the enhanced cellular uptake of HtB-mNG.
Because HT22 cells had the largest capacity to internalize insulin and Ht-mNG in the cells (Figs. 2F and 4E), these in vitro data strongly reinforced the potential of insulin to deliver protein drugs to hippocampal neurons. However, HT22 cells are immortalized and may not completely reflect the in vivo properties of hippocampal neurons. Therefore, we characterized rat hippocampal primary neurons and examined the accumulation of Ht proteins in these primary cells. The expression of IR was similar between HT22 cells and hippocampal primary neurons (SI Appendix, Fig. S5 A and B). In addition, IGF-1R was not detected in both cell lysates. Despite similar receptor expression, Ht-mNG (HtB- and HtD-mNG) were more effectively merged with hippocampal primary neurons than HT22 cells (SI Appendix, Fig. S5C). Because primary neurons are nonproliferative, enough cells for flow cytometry were not obtained. Instead, the effect of Ht-fusion on the uptake by hippocampal primary neurons was quantitatively analyzed with Ht-Nluc. HtB-fusion effectively enhanced Nluc uptake by primary neurons, but the quantitative comparison between the HT22 cell and hippocampal primary neuron might not be precise because of the low content and limited detection of nonproliferating primary neurons (SI Appendix, Fig. S5D). The specificity of Ht-mNG accumulation to hippocampal neurons was further investigated by comparing their uptake by HT22 and N2a cells. HtB- and HtD-mNG were efficiently taken up by HT22 cells, but not by N2a cells (Fig. 4H), suggesting the potential of Ht proteins to specifically target to the hippocampal neurons.
Hippocampal Distribution of Ht Proteins after ICV Administration.
Based on the results of in vitro examinations (Fig. 4 B, C, and E), we tested the capacity of Ht proteins in animals. Mice received Ht-mNG via ICV injection, and their distribution in the brain was evaluated with confocal microscopy of brain sections (Fig. 4I). The molecules administered via ICV injection could rapidly reach neighboring hippocampal regions; however, normally, most molecules cannot penetrate the neuronal cell layer. Although mNG without insulin was located outside the neuronal cell layer, HtA- and HtB-tagged mNG could accumulate in neuronal cells, including axonal parts (Fig. 4I). Contrary to expectation, full-length insulin (HtD-mNG) was less effective in delivering mNG to neuronal cells, possibly because its large molecular weight reduced hippocampal diffusion.
Mechanisms for Accumulation of Ht Proteins in the Hippocampal Neurons.
Although the expression of IR on hippocampal neurons may be associated with the specificity of cell accumulation of insulin and Ht proteins (Figs. 2K and 4G), the detailed mechanisms must be understood. To examine the involvement of energy-dependent endocytosis in the accumulation and internalization of Ht-mNG by HT22 cells, we compared uptake between 37 °C and 4 °C (Fig. 5A). The cells were washed with acidic glycine–HCl buffer or neutral PBS to separate the fraction attached on the cell surface from the fraction internalized by the cells. We have reported that glycine–HCl buffer is most effective for detaching fractions from the cell surface and maximizing fluorescence intensity inside the cells (34). Strong fluorescence of HtB-mNG and HtD-mNG in HT22 cells was abrogated by incubation at 4 °C and washing with glycine–HCl buffer (Fig. 5 B, Upper). Flow cytometry confirmed the reduced fluorescence of cells treated with Ht-mNG at 4 °C (Fig. 5C). Even after washing with PBS, the internalization of Ht-mNG was almost abolished at 4 °C (Fig. 5 B, Lower), suggesting that Ht-mNG is mainly internalized by fluid-phase endocytosis and not cell surface receptor-mediated endocytosis. To examine the involvement of IR as a possible determinant, HT22 cells were incubated with high concentration of insulin (100 μg/mL) and then treated with Ht-mNG, where receptor should be occupied with pretreated insulin. Contrary to expectation, in the presence of excess insulin, Ht-mNG uptake by HT22 cells was partially elevated (Fig. 5C), suggesting that uptake of Ht-mNG cannot be competed with insulin and that Ht-mNG can be transferred into cells via receptor-independent or indirect receptor-dependent pathways. We confirmed that insulin itself (FITC-insulin) is internalized in a receptor-independent manner (Fig. 5D), suggesting that insulin signaling (IRS/PI3K/Akt) for glucose metabolism and insulin internalization, possibly for memory and learning, in the brain occur separately (24, 35). This possibility was confirmed by testing the hypoglycemic effect of insulin and Ht-mNG and phosphorylation of IR via insulin stimulation of HEK293T cells, which abundantly express IR (SI Appendix, Fig. S1 A and B). Although subcutaneous human insulin injection (1 IU/kg body weight) in mice showed strong hypoglycemic effects, HtB- and HtD-mNG did not affect blood glucose levels (SI Appendix, Fig. S6A). The gradual elevation of blood glucose levels after injection was normal reaction of mice due to the stress during experimental operation (22). Western blotting demonstrated that IR in HEK293T cells was phosphorylated by adding human insulin consisting of both A and B chains, but no effect of only A or B chain and Ht-fusion proteins was observed (SI Appendix, Fig. S6 B and C).
Fig. 5.
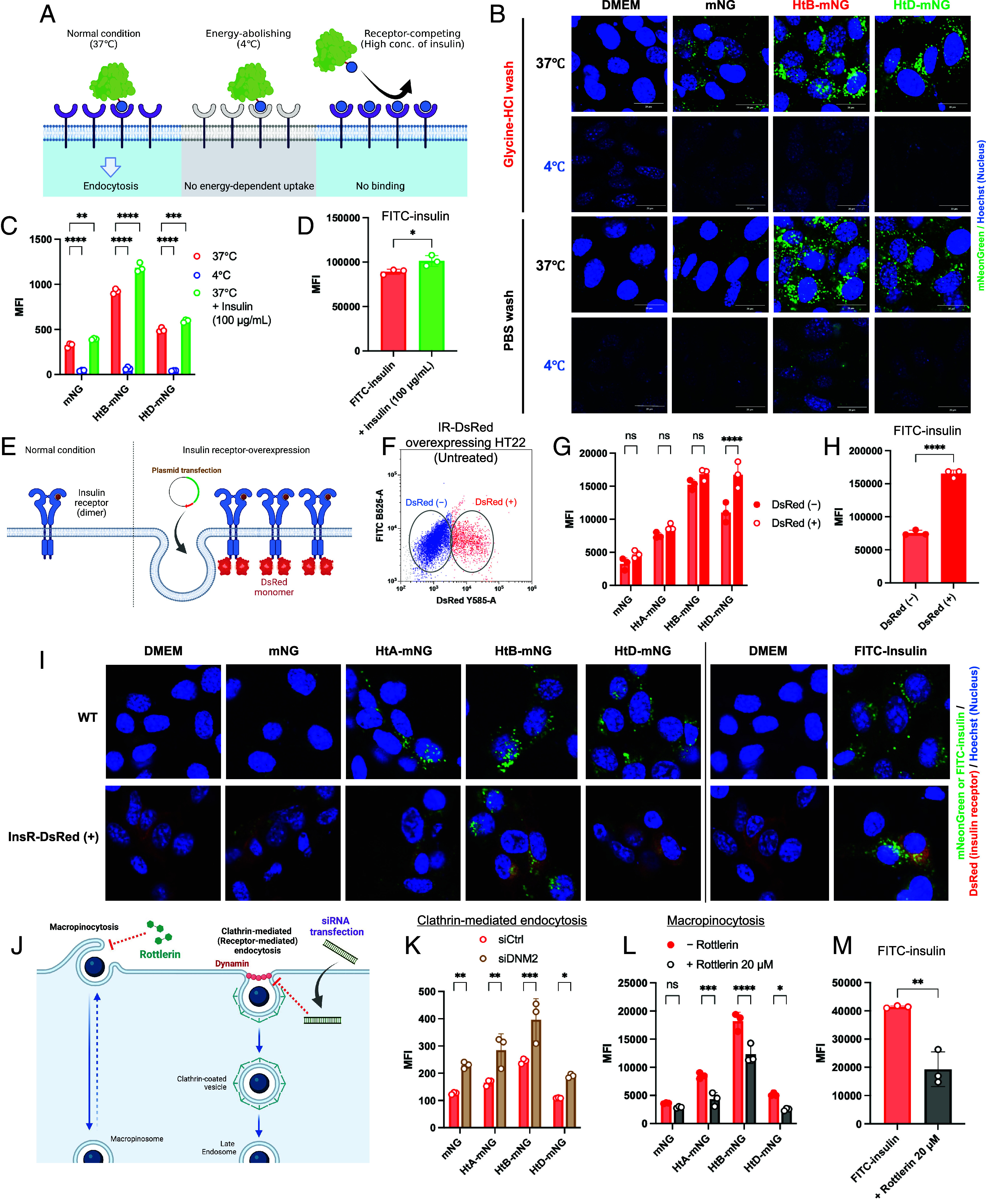
Mechanistic evaluation of the cellular accumulation and uptake of Ht-mNG and FITC-insulin. (A) Schematic illustration of the experimental conditions for examining involvement of energy-dependent endocytosis and specific cell surface receptor. (B) Confocal micrographs of HT22 cells after incubation with Ht-mNG (25 μg/mL) at 37 °C or 4 °C followed by washing with glycine–HCl buffer or PBS. (C) MFI of HT22 cells calculated from flow cytometry after incubation with Ht-mNG at 37 °C or 4 °C or in the presence of excessive concentration of unlabeled insulin (100 μg/mL). (D) MFI of HT22 cells after incubation with FITC-insulin at 37 °C in the presence or absence of unlabeled insulin. (E) Illustration of IR-overexpressing cells for determining the contribution of IR to Ht-fusion proteins. IR was transiently expressed in HT22 cells transfected with the IR-encoding plasmid. In this plasmid, DsRed is tagged at the 3′ end (C terminus) of the IR for detecting IR-overexpressing cells. (F) Dot plot from flow cytometry for gating IR-DsRed-positive and -negative cells. (G and H) MFI from flow cytometry after incubation with Ht-mNG and FITC-insulin, respectively. Fluorescence intensity was compared between DsRed-positive and -negative cells. (I) Confocal micrographs of IR-DsRed-transfected HT22 cells and wild-type (WT) HT22 cells after incubation with Ht-mNG or FITC-insulin. Two micrographs shown for untreated control cells Dulbecco’s modified Eagle medium (DMEM) were taken from the same samples, but detection settings were differently optimized for comparison with mNG and FITC-insulin samples. (J) Schematic illustration of the experiment for evaluating involvement of endocytosis pathways. siRNA was used for knocking down the major protein (dynamin) associated with clathrin-mediated endocytosis, and rottlerin (20 μM) was used for reducing macropinocytosis. (K) Flow cytometric analysis of dynamin-deleted HT22 cells after incubation with Ht-mNG. (L and M) Flow cytometric analysis of rottlerin-treated HT22 cells after incubation with Ht-mNG or FITC-insulin, respectively.
The contribution of IR to the specific accumulation of Ht proteins was further investigated with IR-overexpressing HT22 cells (Fig. 5E). We cloned the plasmid for expressing IR linked to a fluorescent tag (DsRed) in HT22 cells. The cells expressed IR-DsRed after transfection with reagents (TransIT-X2 and TransIT 293) for 24 or 48 h, with low cytotoxicity (SI Appendix, Fig. S7 A and B). We separated DsRed-positive (IR-overexpressing) and DsRed-negative (wild-type) cells by flow cytometry and compared the cellular internalization of Ht-mNG (Fig. 5F). Only HtD-mNG internalized more effectively into DsRed-positive cells than DsRed-negative cells, but no statistically significant increase was observed for other Ht-mNG (Fig. 5G). Because FITC-insulin was more efficiently taken up by DsRed-positive cells (Fig. 5H), the contribution of IR could be assessed with this system. These results suggest that accumulation of HtA- and HtB-mNG is IR independent, although the SPR assay demonstrated the binding of Ht-mNG—even incomplete insulin forms A- and B-chains—to IR. Confocal microscopy could not reveal the colocalization of Ht-mNG with IR-DsRed in cells (Fig. 5I), strengthening the hypothesis that IR does not contribute to the cellular accumulation of Ht-mNG, in particular HtA- and HtB-mNG.
To find the mechanisms for Ht-fusion-mediated neuronal cell accumulation of proteins, we investigated the involvement of endocytosis types using siRNA for knocking down dynamin-2, which is the main protein for clathrin-, caveolae-, and receptor-mediated endocytosis, and the macropinocytosis inhibitor, rottlerin (Fig. 5J) (36–41). The expression of dynamin-2 in HT22 cells was reduced by treatment with siRNA for dynamin-2 (siDNM2) (SI Appendix, Fig. S7 C and D). Because the transfection reagent or siRNA treatment was potentially cytotoxic, Ht-mNG uptake was compared between control siRNA and siDNM2-transfected cells (SI Appendix, Fig. S7E). No reduction in the internalization of Ht-mNG was observed in siDNM2-transfected HT22 cells (Fig. 5K). The treatment of HT22 cells with rottlerin (20 μM) drastically decreased the internalization of Ht-mNG (Fig. 5L). FITC-insulin was less efficiently internalized into rottlerin-treated HT22 cells, compared with untreated cells (Fig. 5M), which is consistent with our recent publication (37). By contrast, treatment of cells with a macropinocytosis marker (FD70) clarified that the activity of macropinocytosis in HT22 cells was not intrinsically high compared with other cells (3T3, bEnd.3, and C6 cells) (SI Appendix, Fig. S8 A and B). The uptake of FD70 by HT22 cells was reduced at 4 °C and in the presence of rottlerin (SI Appendix, Fig. S8 C and D). Interestingly, FD70 uptake was facilitated the by addition of excessive insulin in concentration-dependent and saturable manner (SI Appendix, Fig. S8 D and E), but not increased in IR-DsRed-positive cells without receptor stimulation (SI Appendix, Fig. S8F). These findings suggest that insulin and Ht-mNG were internalized into HT22 cells via enhanced macropinocytosis triggered by receptor stimulation. Insulin is often supplemented as a growth factor for cell culture and can potentially activate macropinocytosis as biological response, like other growth factors (42). HtB-mNG might unlock the maximal potential of macropinocytosis in HT22 cells; therefore, the additional internalization of HtB-mNG might be difficult to detect even when overexpressing IR (Fig. 5G). These in vitro data suggest that enhanced macropinocytosis of neuronal cells through exposure to insulin or Ht-fusion proteins contributes to accumulation in hippocampal neurons in vivo.
Dominant Amino Acid Sequences in Insulin for Hippocampal Neuronal Delivery.
We determined the amino acid sequence in insulin necessary for hippocampal neuronal targeting. We generated various insulin mutants (M1 to M6) from native insulin A and B chains (Fig. 6A). M7 and M8 were prepared as nonspecific cell-penetrating peptide (penetratin) and random sequence peptide with 30 amino acids (two cysteines remained), respectively. Both one-third reversed (M1) and scrambled (M2) mutants of insulin A chain effectively increased mNG uptake by HT22 cells (Fig. 6B). Similarly, the effect of fusion with one-third reversed (M3) and scrambled (M4) mutants of insulin B chain did not decrease compared with native HtB-fusion, considering interfered fluorescence intensity (Fig. 6 B and C). Surprisingly, M3-fusion was approximately five times effective in enhancing mNG uptake compared with native HtB-fusion. Comparison with HT22 and N2a cells showed the potential of M3-fusion to deliver proteins specifically to the hippocampal neurons (Fig. 6D). Random peptide M8 was efficiently taken up by HT22 cells, thus the complete sequence of insulin might not be essential as a critical determinant. Unlike one-third reversed and scrambled mutants, the cysteine-substitution mutants (with alanine [M5] or serine [M6]) dramatically reduced HtB-mNG uptake (Fig. 6B), suggesting that the intermolecular disulfide bond is important.
Fig. 6.

Determination of important amino acid sequences in insulin and identifying more powerful mutants for delivering protein drugs to hippocampal neurons. (A) List of insulin mutants and their amino acid sequences. M1 and M2 are insulin A mutants, and M3 to M6 are insulin B mutants. M7 and M8 are typical strong CPP (penetratin) and random peptides with 30 amino acids, respectively. M9, M3v2, and M8v2 are additional mutants from original insulin B, M3, and M8, respectively. (B) MFI from flow cytometry of HT22 cells incubated with insulin mutant-mNG for 2 h at 37 °C. (C) Variation in fluorescence intensity of insulin mutant-mNG at the same concentration (10 μg/mL). (D) Comparison of insulin mutant-mNG uptake by NIH3T3, bEnd.3, C6, and HT22 cells with flow cytometry. (E) Comparison of cellular uptake of M3-mNG by HT22 and N2a cells. (F) Western blotting of insulin mutant-mNG. Samples were loaded at 75 ng/mL of purified protein. PVDF membranes were treated with antibodies for mNG. Arrows indicate the molecular weights of original mNG. (G) Binding sensorgrams after insulin mutant-mNG was injected to IR –immobilized flow cell, where specific binding was expressed by subtracting nonspecific binding to blank flow cell from total binding. (H) Uptake by HT22 cells overexpressing IR-DsRed. (I) Concentration dependence of HtB- and M3-mNG on HT22 cell uptake. (J) Uptake of insulin mutant-mNG by HT22 cells at 37 °C and 4 °C. (K) Uptake by HT22 cells treated with rottlerin (20 μM) or unlabeled insulin (100 μg/mL). (L) Representative images (from three mice for each time point) around hippocampal regions at 30 and 60 min after ICV administration of the insulin mutant (M3)-mNG in mice. The slices prepared from frozen brain specimens were stained with primary antibodies against NeuN and secondary antibodies with fluorescent dye (AF647). The bars indicate 500 μm.
To examine the necessity of cysteine in effective internalization into hippocampal neurons, additional mutants (M9, M3v2, and M8v2) were prepared. Cysteine-substitution mutants (M3v2 and M8v2) were less effective in enhancing the internalization of mNG (Fig. 6E). By contrast, changing the position of cysteine in insulin B chain (M9) did not affect the ability of insulin B chain to enhance cellular internalization. The importance of the intermolecular disulfide bond was further investigated with western blotting. As shown in Fig. 6F, western blotting without reducing agent clarified that HtB-, M3-, M8-, and M9-fusion mNG, which are highly sensitive to cellular internalization (Fig. 6 B and E), made an intermolecular complex. Western blotting with reduced samples revealed single bands of each fusion mNG, suggesting that the complex dissociated by reducing intermolecular disulfide bonds (Fig. 6F). Thus, inclusion of cysteine in carrier peptides and the resultant intermolecular disulfide bonds can promote the delivery of Ht proteins to hippocampal neurons. A recent study also reported that multivalent insulin nanocomplex showed higher activation of IR compared to monovalent insulin (43).
In experiments with single culture, the accumulation of these mutants was not specific to HT22 cells compared with other cells (3T3, bEnd.3, and C6 cells) (Fig. 6E). By contrast, uptake by HT22/C6 cells in coculture Transwell systems showed a tendency of specific accumulation of M3-mNG to HT22 cells (SI Appendix, Fig. S4D). SPR analysis clarified that M3- and M6-mNG could bind IR, although the amino acid sequences within these peptides were mostly modified from original human insulin (Fig. 6G). The binding of M3- and M6-mNG did not contribute to IR phosphorylation (SI Appendix, Fig. S6D). Interestingly, M8 mutant-mNG, which is unrelated to insulin, could bind IR (Fig. 6G), indicating its effect on enhanced uptake of M8-mNG by HT22 cells. The uptake study with IR-DsRed-overexpressing HT22 cells showed that HtB-mNG and M8-mNG were similarly taken up by cells, and no differences between DsRed-positive and -negative cells suggest that their addition led to maximal macropinocytosis (Fig. 6H). By contrast, internalization of M3-mNG was much higher than HtB- and M8-mNG and not enhanced in DsRed-positive cells, indicating the involvement of additional or other mechanisms than IR-mediated macropinocytosis. Compared with HtB-mNG uptake, M3-mNG uptake was highly saturable with increase in concentration (Fig. 6I). This suggests that other potential receptors or shuttle proteins were involved in the neuronal internalization of M3-mNG. The examination of internalization pathways clarified that the uptake of M3- and other mutant-mNG were temperature dependent and rottlerin sensitive (Fig. 6 J and K). Treatment with excessive insulin did not affect the uptake of M3- and M6-mNG (Fig. 6K), suggesting that IR-mediated macropinocytosis was fully activated with M3-mNG even without extra stimulation and contributed less to M6-mNG.
Thus, insulin mutant M3 was most efficient in delivering proteins to hippocampal neurons. Therefore, we tested the in vivo performance of M3-mNG for hippocampal neuronal targeting after ICV administration. Fig. 6L shows the distribution of M3-mNG around the hippocampal region at 30 and 60 min after ICV administration. M3-mNG-derived fluorescence accumulated in the neuronal layers, suggesting that M3 can specifically deliver proteins to hippocampal neurons.
Noninvasive and Direct Nose-to-Brain Delivery and Hippocampal Neuronal Targeting of Ht Proteins.
The in vivo studies with Ht protein (mNG) showed their potential in delivering therapeutic proteins effectively and specifically to hippocampal neurons after ICV administration (Figs. 4 J and 6L). Non- or less-invasive routes must be developed for targeting protein drugs to hippocampal neurons for the future clinical application. Nose-to-brain delivery has been well studied and is considered as an approach for the direct and effective transport of drugs, including peptides and proteins (44). Nasal mucosal transport of macromolecules, such as peptides, proteins, and nucleic acids, prior to brain distribution is intrinsically low. Therefore, we examined the transport efficiency of Ht-fusion proteins from the nasal cavity to the brain with or without epithelial permeation enhancers (SI Appendix, Fig. S9A). Nluc was used as cargo for considering detectability after intranasal administration. We found that the cell-penetrating peptide penetratin could be a useful carrier for therapeutic peptides and proteins crossing the nasal epithelial barrier (21, 27, 45, 46). We mixed HtB-Nluc with penetratin and administered both intranasally to mice. Coadministration with penetratin significantly enhanced the nose-to-brain transfer of HtB-Nluc (SI Appendix, Fig. S9B). The highest luminescence activity of HtB-Nluc was detected in the olfactory bulb, suggesting direct nose-to-brain transport via the space surrounding olfactory neurons. By contrast, HtB-Nluc was efficiently absorbed into the blood because the lamina propria has abundant microvessels. Importantly, HtB-Nluc could be distributed throughout the brain, including the deepest part––hippocampus (SI Appendix, Fig. S9B). Thus, protein drugs can be noninvasively targeted to hippocampal neurons with the Ht-fusion strategy.
Discussion
The hippocampus is a functionally and positionally central part of the brain, contributing to memory and learning. The hippocampus should be a therapeutic target when neurodegenerative disorders, including AD, impair memory and learning. Delivering drugs that modify Aβ and tau, as well as repair neurons, to appropriate parts of the brain is important for effective causal AD therapy. In this study, insulin could be localized in hippocampal neurons after effective nose-to-brain delivery (Fig. 2B), consistent with our recent findings (27). The specificity of insulin accumulation in the brain was related to the level of IR expression on the cells (Fig. 2 F and K). Therefore, we designed Ht proteins that target the hippocampus. In vitro and in vivo data demonstrated that Ht proteins could be delivered preferentially to hippocampal neurons by binding the IR (Fig. 4). Importantly, even incomplete forms of insulin (only B chain) could bind the IR and deliver cargo. In neuronal cells, insulin and Ht proteins induced macropinocytosis through IR binding (Fig. 5). Increased macropinocytosis, possibly actin-dependent neuronal ultrafast endocytosis (47, 48), contributed to effective delivery to hippocampal neurons in vivo. Although the substitution of cysteines in Ht proteins decreased their neuronal uptake, the partial reversal of the amino acid sequence within the insulin B chain unexpectedly boosted cellular uptake by approximately fivefold, showing the in vivo performance of hippocampal neuronal targeting (Fig. 6).
Thus, fusion or conjugation with insulin (even a single chain) or its mutants can be a revolutionary approach for delivering therapeutic proteins to hippocampal neurons. This is potentially applicable to antibodies and neurotrophic factors, as well as low molecular weight medicines. In recent years, neuronal targeting with Rabies virus glycoprotein derived short peptide (RVG29) has been most studied for effective neuronal delivery of proteins and nucleic acids (49, 50). However, RVG29 is not specific for delivering to hippocampal neurons (51), and virus-derived carriers are unfavorable considering their clinical use. Although studies have shown efficient genome editing in hippocampal neurons with nonviral targeted axonal import peptide carrier, which was identified by phage display screening (52), this peptide was originally studied as a carrier to spinal cord motor neurons (53). Another study suggested that lipid nanoparticles (LNP) could deliver siRNAs into cultured neurons to achieve targeted gene silencing (54). However, the silencing effect was based on the specific action of siRNAs against neuronal genes, and the LNP themselves were located widely in the brain. Unlike other techniques, our hippocampal neuron–targeting technology (or HiNT) can deliver drugs more safely and specifically to hippocampal neurons. Importantly, HiNT will contribute to maximizing the therapeutic potential of various biological drugs for AD and other neurodegenerative disorders. Because intranasal coadministration with the membrane permeation enhancer, penetratin, might be capable of delivering Ht proteins (Nluc) to the brain, less- or noninvasive and convenient administration will be more optimized for clinical application of HiNT in the future.
Materials and Methods
Detailed materials and methods are provided in SI Appendix, Materials and Methods, including all materials and instruments, genetic engineering, protein expression, and all other in vitro and in vivo biological analyses. Key protocols used in this study and ethical issues are summarized below, with fully detailed descriptions present in SI Appendix.
Cell Culture.
Human embryonic kidney HEK293T (CTL-3216), mouse cerebral cortex endothelial bEnd.3 (CRL-2299), and rat glial fibroblast C6 (CCL-107) cells were purchased from the American Type Culture Collection (Rockville, MD, USA). Mouse hippocampal neuronal HT22 cells (SCC129) were purchased from Merck KGaA (Darmstadt, Germany). NIH3T3 and N2a cells were purchased from the RIKEN Bioresource Research Center (Tsukuba, Japan). For detail, see SI Appendix.
Generation of Constructs.
The genetic engineering experiments were approved by the Institutional Review Board of Kobe Gakuin University (protocol codes: 22-01, 22-07, and 23-03). Original vectors containing mNG (pNCS-mNG) and Nluc (N1361, pNLF1) were purchased from Allele Biotechnology (San Diego, CA, USA) and Promega Corp. (Madison, WI, USA), respectively. The DNA fragments of preproinsulin including B chain, c-peptide, and A chain, were synthesized by Integrated DNA Technologies, Inc. (Coralville, IA, USA). PCR and sequencing primers were synthesized by Eurofins Genomics, Inc. (Tokyo, Japan). Fragments encoding insulin mutants were synthesized by Integrated DNA Technologies or Eurofins. For detail, see SI Appendix.
Protein Expression.
The constructs encoding insulin or mutated insulin-Nluc or mNG fusion proteins were transformed into BL21(DE3) competent cells (#69450, Merck KGaA). HtD-Nluc and HtD-mNG were transformed into SHuffle T7 Express competent cells (C3029J, New England Biolabs). For detail, see SI Appendix.
Animals.
All animal studies were performed at Kobe Gakuin University and complied with the regulations of the Committee on Ethics in the Care and Use of Laboratory Animals. The animal experiments were approved by the Institutional Review Board of Kobe Gakuin University (protocol codes: A22-02, A22-03, A23-27, and A23-28). Male ddY mice (6 wk old, 30 g body weight) were purchased from Japan SLC (Shizuoka, Japan). For detail, see SI Appendix.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
This study was supported in part by JSPS KAKENHI for Scientific Research B (grant numbers 23H03751 and 23K28439), Mochida Memorial Foundation for Medical and Pharmaceutical Research, and Takeda Science Foundation. We are grateful to Ms. Mai Itagaki and Mr. Yoshinori Nasu (DDS Laboratory, Kobe Gakuin University, Japan) for their technical help in this study.
Author contributions
N.K., K.I., Y. Ohmoto, S.F., R.S., M. Maki, M. Miyata, Y.M., N.N., M.Y., and Y. Ohigashi designed research; N.K., K.I., Y. Ohmoto, S.F., R.S., M. Maki, M. Miyata, Y.M., N.N., M.Y., and Y. Ohigashi performed research; N.K., K.I., Y. Ohmoto, S.F., R.S., M. Maki, M. Miyata, Y.M., N.N., M.Y., and Y. Ohigashi analyzed data; M.T.-M. gave suggestions; and N.K., K.I., Y. Ohmoto, S.F., R.S., M. Maki, M. Miyata, Y.M., N.N., M.Y., Y. Ohigashi, and M.T.-M. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Boxer A. L., Sperling R., Accelerating Alzheimer’s therapeutic development: The past and future of clinical trials. Cell 186, 4757–4772 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Dyck C. H., et al. , Lecanemab in early Alzheimer’s disease. N. Engl. J. Med. 388, 9–21 (2023). [DOI] [PubMed] [Google Scholar]
- 3.Honig L. S., et al. , ARIA in patients treated with lecanemab (BAN2401) in a phase 2 study in early Alzheimer’s disease. Alzheimers Dement. N9, e12377 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mummery C. J., et al. , Tau-targeting antisense oligonucleotide MAPT(Rx) in mild Alzheimer’s disease: A phase 1b, randomized, placebo-controlled trial. Nat. Med. 29, 1437–1447 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vogel J. W., et al. , Spread of pathological tau proteins through communicating neurons in human Alzheimer’s disease. Nat. Commun. 11, 2612 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu Z., et al. , Neurotrophic signaling deficiency exacerbates environmental risks for Alzheimer’s disease pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 118, e2100986118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fukushima Y., et al. , Treatment of ischemic neuronal death by introducing brain-derived neurotrophic factor mRNA using polyplex nanomicelle. Biomaterials 270, 120681 (2021). [DOI] [PubMed] [Google Scholar]
- 8.Nagahara A. H., et al. , Early BDNF treatment ameliorates cell loss in the entorhinal cortex of APP transgenic mice. J. Neurosci. 33, 15596–15602 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nagahara A. H., et al. , Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat. Med. 15, 331–337 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Terstappen G. C., Meyer A. H., Bell R. D., Zhang W., Strategies for delivering therapeutics across the blood-brain barrier. Nat. Rev. Drug Discov. 20, 362–383 (2021). [DOI] [PubMed] [Google Scholar]
- 11.Pardridge W. M., A historical review of brain drug delivery. Pharmaceutics 14, 1283 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zuchero Y. J., et al. , Discovery of novel blood-brain barrier targets to enhance brain uptake of therapeutic antibodies. Neuron 89, 70–82 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Okuyama T., et al. , Iduronate-2-sulfatase with anti-human transferrin receptor antibody for neuropathic mucopolysaccharidosis II: A phase 1/2 trial. Mol. Ther. 27, 456–464 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arguello A., et al. , Molecular architecture determines brain delivery of a transferrin receptor-targeted lysosomal enzyme. J. Exp. Med. 219, e20211057 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Okuyama T., et al. , A phase 2/3 trial of Pabinafusp Alfa, IDS fused with anti-human transferrin receptor antibody, targeting neurodegeneration in MPS-II. Mol. Ther. 29, 671–679 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao P., et al. , A tetravalent TREM2 agonistic antibody reduced amyloid pathology in a mouse model of Alzheimer’s disease. Sci. Transl. Med. 14, eabq0095 (2022). [DOI] [PubMed] [Google Scholar]
- 17.van Lengerich B., et al. , A TREM2-activating antibody with a blood-brain barrier transport vehicle enhances microglial metabolism in Alzheimer’s disease models. Nat. Neurosci. 26, 416–429 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niewoehner J., et al. , Increased brain penetration and potency of a therapeutic antibody using a monovalent molecular shuttle. Neuron 81, 49–60 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Correa D., et al. , Intranasal delivery of full-length anti-Nogo-A antibody: A potential alternative route for therapeutic antibodies to central nervous system targets. Proc. Natl. Acad. Sci. U.S.A. 120, e2200057120 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aly A. E., et al. , Delivery of mutant huntingtin-lowering antisense oligonucleotides to the brain by intranasally administered apolipoprotein A-I nanodisks. J. Control Release 360, 913–927 (2023). [DOI] [PubMed] [Google Scholar]
- 21.Kamei N., Takeda-Morishita M., Brain delivery of insulin boosted by intranasal coadministration with cell-penetrating peptides. J. Control Release 197, 105–110 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Kamei N., et al. , Effective nose-to-brain delivery of exendin-4 via coadministration with cell-penetrating peptides for improving progressive cognitive dysfunction. Sci. Rep. 8, 17641 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Talbot K., et al. , Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Invest. 122, 1316–1338 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gray S. M., Aylor K. W., Barrett E. J., Unravelling the regulation of insulin transport across the brain endothelial cell. Diabetologia 60, 1512–1521 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Craft S., et al. , Safety, efficacy, and feasibility of intranasal insulin for the treatment of mild cognitive impairment and alzheimer disease dementia: A randomized clinical trial. JAMA Neurol. 77, 1099–1109 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kamei N., et al. , Effect of an enhanced nose-to-brain delivery of insulin on mild and progressive memory loss in the senescence-accelerated mouse. Mol. Pharm. 14, 916–927 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Kamei N., et al. , Investigation of the transport pathways associated with enhanced brain delivery of peptide drugs by intranasal coadministration with penetratin. Pharmaceutics 13, 1745 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu D., Yang J., Pardridge W. M., Drug targeting of a peptide radiopharmaceutical through the primate blood-brain barrier in vivo with a monoclonal antibody to the human insulin receptor. J. Clin. Invest. 100, 1804–1812 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kleinridders A., Ferris H. A., Cai W., Kahn C. R., Insulin action in brain regulates systemic metabolism and brain function. Diabetes 63, 2232–2243 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soto M., Cai W., Konishi M., Kahn C. R., Insulin signaling in the hippocampus and amygdala regulates metabolism and neurobehavior. Proc. Natl. Acad. Sci. U.S.A. 116, 6379–6384 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hall M. P., et al. , Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem. Biol. 7, 1848–1857 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shaner N. C., et al. , A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat. Methods 10, 407–409 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lobstein J., et al. , SHuffle, a novel Escherichia coli protein expression strain capable of correctly folding disulfide bonded proteins in its cytoplasm. Microb. Cell Fact. 11, 56 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kamei N., et al. , Optimization of the method for analyzing endocytosis of fluorescently tagged molecules: Impact of incubation in the cell culture medium and cell surface wash with glycine-hydrochloric acid buffer. J. Control Release 310, 127–140 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Konishi M., et al. , Endothelial insulin receptors differentially control insulin signaling kinetics in peripheral tissues and brain of mice. Proc. Natl. Acad. Sci. U.S.A. 114, E8478–E8487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rennick J. J., Johnston A. P. R., Parton R. G., Key principles and methods for studying the endocytosis of biological and nanoparticle therapeutics. Nat. Nanotechnol. 16, 266–276 (2021). [DOI] [PubMed] [Google Scholar]
- 37.Itagaki M., Nasu Y., Sugiyama C., Nakase I., Kamei N., A universal method to analyze cellular internalization mechanisms via endocytosis without non-specific cross-effects. FASEB J. 37, e22764 (2023). [DOI] [PubMed] [Google Scholar]
- 38.Sarkar K., Kruhlak M. J., Erlandsen S. L., Shaw S., Selective inhibition by rottlerin of macropinocytosis in monocyte-derived dendritic cells. Immunology 116, 513–524 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oh P., McIntosh D. P., Schnitzer J. E., Dynamin at the neck of caveolae mediates their budding to form transport vesicles by GTP-driven fission from the plasma membrane of endothelium. J. Cell Biol. 141, 101–114 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cocucci E., Gaudin R., Kirchhausen T., Dynamin recruitment and membrane scission at the neck of a clathrin-coated pit. Mol. Biol. Cell 25, 3595–3609 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaksonen M., Roux A., Mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 19, 313–326 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Nakase I., Kobayashi N. B., Takatani-Nakase T., Yoshida T., Active macropinocytosis induction by stimulation of epidermal growth factor receptor and oncogenic ras expression potentiates cellular uptake efficacy of exosomes. Sci. Rep. 5, 10300 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spratt J., et al. , Multivalent insulin receptor activation using insulin-DNA origami nanostructures. Nat. Nanotechnol. 19, 237–245 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Borrajo M. L., Alonso M. J., Using nanotechnology to deliver biomolecules from nose to brain–Peptides, proteins, monoclonal antibodies and RNA. Drug Deliv. Transl. Res. 12, 862–880 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khafagy E. S., et al. , Systemic and brain delivery of leptin via intranasal coadministration with cell-penetrating peptides and its therapeutic potential for obesity. J. Control Release 319, 397–406 (2020). [DOI] [PubMed] [Google Scholar]
- 46.Kamei N., et al. , Therapeutic effects of anti-amyloid beta antibody after intravenous injection and efficient nose-to-brain delivery in Alzheimer’s disease mouse model. Drug Deliv. Transl. Res. 12, 2667–2677 (2022). [DOI] [PubMed] [Google Scholar]
- 47.Watanabe S., et al. , Ultrafast endocytosis at mouse hippocampal synapses. Nature 504, 242–247 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ogunmowo T. H., et al. , Membrane compression by synaptic vesicle exocytosis triggers ultrafast endocytosis. Nat. Commun. 14, 2888 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kumar P., et al. , Transvascular delivery of small interfering RNA to the central nervous system. Nature 448, 39–43 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Alvarez-Erviti L., et al. , Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 29, 341–345 (2011). [DOI] [PubMed] [Google Scholar]
- 51.Kim S. S., et al. , Targeted delivery of siRNA to macrophages for anti-inflammatory treatment. Mol. Ther. 18, 993–1001 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sellers D. L., Lee K., Murthy N., Pun S. H., TAxI-peptide targeted Cas12a ribonuclease protein nanoformulations increase genome editing in hippocampal neurons. J. Control Release 354, 188–195 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sellers D. L., et al. , Targeted axonal import (TAxI) peptide delivers functional proteins into spinal cord motor neurons after peripheral administration. Proc. Natl. Acad. Sci. U.S.A. 113, 2514–2519 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rungta R. L., et al. , Lipid nanoparticle delivery of sirna to silence neuronal gene expression in the brain. Mol. Ther. Nucleic Acids 2, e136 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or SI Appendix.