

Abstract
Background
Cat eye syndrome (CES) is a rare congenital disease frequently caused by a partial tetrasomy of the proximal long (q) arm of chromosome 22, due to a small supernumerary marker chromosome (sSMC). CES patients show remarkable phenotypic variability. Despite the progress of molecular cytogenetic technology, the cause of phenotypic variability and the genotype–phenotype correlations remain unknown.
Methods
We analyzed clinical and genetic data of a new patient with CES together with 27 previously reported ones with a confirmed genomic gain in the PubMed database between 2012 and 2023.
Results
We reported a boy with CES carrying a 22q11.1-q11.21 duplication of 1.76 Mb tetrasomy (16888900_18644241, hg19) who presented currently rare or unreported clinical findings such as congenital aural atresia, hearing loss, PLSVC, and IVC. The results of the whole exome sequencing (WES) showed a heterozygous mutation of the GJB2 gene (NM_004004.6: exon2: c.109G > A). In addition, the results of our literature review showed that the presence of a classical sSMC was the most frequent cytogenetic abnormality in CES (82%). 63% of cases were in a homogenous state and 37% of cases were in a mosaic state. 72% of cases had a 1–2 Mb duplication. In the majority of CES patients the breakpoints in chromosome 22 are localized to a 50 kb region (18610000_18660000 bp). The CES critical region (CESCR) may be further delimited to a 0.3 Mb region (17799398_18111588 bp). Within this region CECR2, SLC25A18, ATP6V1E1, and BCL2L13 are strong candidate genes for causing the main CES phenotype. The ear anomalies are the most frequent features in CES patients (89%) and hearing loss was present in 36% of CES patients.
Conclusions
The phenotypic features in CES are highly variable. Our findings expand the symptom spectrum of CES and lay the foundation for better delineating the clinical phenotype, molecular cytogenetic features associated with CES and genotype–phenotype correlations. We recommend performing WES to rule out the involvement of other genetic factors in the patient’s phenotype. In addition, our findings also highlight the need for genetic counseling and recurrence risk assessment.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12887-024-05136-9.
Keywords: Cat eye syndrome, Small supernumerary marker chromosome (sSMC), 22q11.1q11.21 duplication, Hearing loss, Congenital aural atresia, Genetic counseling
Background
Cat eye syndrome (CES) (OMIM 115470), also known as Schmid-Fraccaro syndrome, is a rare congenital disease with a population incidence of about 1:50,000–1:150,000 [1]. CES is frequently caused by a partial tetrasomy of the proximal long (q) arm of chromosome 22, due to a small supernumerary marker chromosome (sSMC). The 22q11 region is highly susceptible to chromosomal rearrangements due to multiple highly homologous repetitive regions, known as low copy repeats (LCRs) [2, 3]. Duplications and deletions of proximal chromosome 22q have been associated with a few syndromes and developmental abnormalities such as CES and DiGeorge/velocardiofacial syndrome (DGS/VCFS) [4–7].
Almost all the published studies on CES have highlighted a widely variable phenotype. Coloboma of the iris, ear, and anal malformations are known as the classic clinical triad. However, only 41% of CES patients display all three classic features [1, 8]. CES patients can also present down-slanting palpebral fissures, hypertelorism, cleft palate, spina bifida, congenital heart, skeletal anomalies and renal malformations [1, 9, 10]. In addition, almost half of patients have mild or moderate developmental delay, but growth is not usually affected [11, 12]. However, congenital aural atresia and hearing loss are less discussed in published studies. In the last 10 years, single nucleotide polymorphism array (SNP-array), array comparative genomic hybridization (array-CGH) and copy number variation sequencing (CNV-seq) technologies have been widely used in the detection of genomic diseases. Some new molecular alternations have been described in CES patients. To better delineate the clinical and molecular cytogenetics findings associated with CES, we reviewed 26 CES patients reported since 2012 (including the current case) with confirmed molecular cytogenetic testing and found some meaningful results.
Methods
Case presentation
The patient was a 2-year-old boy who was referred for cytogenetic studies because of speech delay and hearing abnormality. He was born to a 32-year-old mother following an unremarkable pregnancy G4P1 at 39 weeks by caesarean section. His birth weight was 3.00 kg (10th centile), head circumference was 34 cm (< 50th centile), and birth length was 52 cm (> 75th centile). The parents were non-consanguineous and healthy. Newborn physical examination revealed congenital anal atresia and craniofacial dysmorphism, including micrognathia, left auricular malformations, atresia of the left external auditory meatus, bilateral preauricular pits and tags, fistula on the right cheek and auricle, sinus in the left cheek (suggestive of branchial cleft sinus tract), and ocular hypertelorism.
Fetal cardiac malformations were detected by fetal echocardiography at 26+ 5 weeks’ pregnant. The cardiac malformations included persistent left superior vena cava (PLSVC), interrupted inferior vena cava (IVC), right atrium enlargement (RAE) and right ventricle enlargement (RVE). Neonatal heart color ultrasound showed PLSVC, atrial septal defect (ASD), and anomalies of the coronary sinus. However, a non-invasive prenatal testing (NIPT) examination at another hospital yielded a negative result at 16+ 4 weeks. The patient had anoplasty at 1 day after birth, and recovered well. At 2 years old, physical examination showed that his height was 94 cm (97th centile), his weight was 12.5 kg (50th centile) and his head circumference was 49.0 cm (> 50th centile). His craniofacial characteristics were characterized by a broad and prominent forehead, micrognathia, and mild facial asymmetry. Ocular features included hypertelorism, downslanting palpebral fissures, bilateral eyelid ptosis, and epicanthus. The ophthalmological examination, which included visual acuity testing, evaluation of ocular motility, and retinal examination via ophthalmoscopy, indicated that the boy exhibited normal visual acuity and excluded the presence of iris coloboma, chorioretinal coloboma, strabismus, and any abnormalities in ocular motility. However, assessments of pupillary function, visual fields, and intraocular pressure were not performed. The otorhinolaryngological evaluation diagnosed normal hearing on the right and severe loss on the left. Craniofacial features of the patient at the age of 2 were showed in Fig. 1.
Fig. 1.
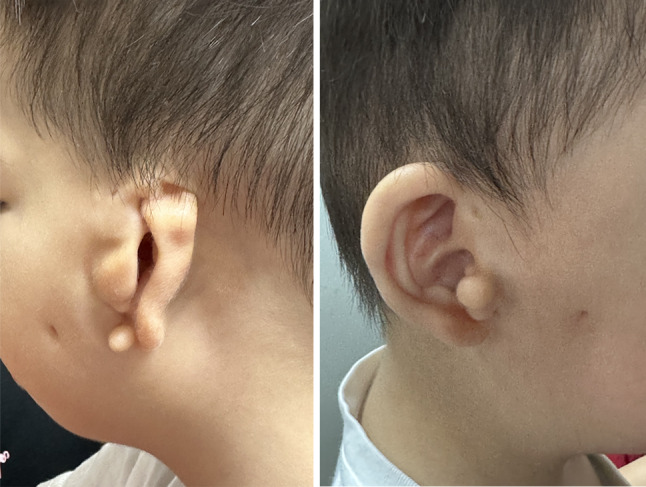
Craniofacial features of the patient at the age of 2. Severe left ear malformation, bilateral preauricular pits and tags, fistula on the right cheek, and sinus in the left cheek
Karyotyping
For cytogenetic analysis, metaphase chromosomes were obtained from peripheral blood lymphocytes after 72 h of incubation. Metaphase spreads were prepared for GTG banding and high-resolution staining according to standard procedures. Karyotypes were obtained from the patient and his parents. Twenty metaphases were analyzed from each subject by GTG banding. An Olympus microscope (BX41) was used for karyotyping and metaphase images were captured using VideoTesT-Karyo software (Meta systems, Altlussheim, Germany).
Chromosomal microarray analysis
Affymetrix CytoScan® 750 K (Affymetrix, Inc., Santa Clara, CA, USA) arrays were utilized to analyze genome-wide copy number aberrations. DNA amplification, tagging, and hybridization were conducted according to the manufacturer’s protocols. The original chip data was analyzed using Chromosome Analysis Suite software (ChAS, version 4.3) (Affymetrix, CA, USA). The pathogenicity of detected CNVs was assessed according to the technical standards issued by the American College of Medical Genetics and Genomics and the Clinical Genome Resource (ClinGen) [13].
Whole exome sequencing (WES)
WES was performed to investigate the potential pathogenic variant in this case. Sequencing was performed immediately after library synthesis. The paired-end WES was performed on an Illumina HiSeq X platform with an average coverage of 100x mean depth (WeHealth, Shanghai, China). Illumina Casava 1.8.2 software was used for base calling. Sequenced reads were mapped to the human reference genome sequence (hg38).
Literature review
The PubMed online database was searched for the period January 2012 through October 2023 to find all papers indexed for the subject ‘cat eye syndrome’ and/or ‘supernumerary marker chromosome 22’. Only patients with cytogenetic and a confirmed array-CGH, SNP-array, or CNV-seq results were included in the current study in order to determine the exact location and size of the duplication. Those cases only proven by traditional cytogenetics and/or typical symptoms were excluded. Prenatal cases that ended in abortion were also excluded from the current study because postnatal phenotyping could not be traced. After ultimate revision, twenty-five articles were included in the final analysis. We included our patient in the phenotype analysis.
Results
Chromosome karyotype analysis
The G-banding chromosomal analysis revealed an additional sSMC. The karyotype of the patient was defined as 47, XY, +psu idic(22)(q11.21), combined with the typical clinical features suggestive of CES (Fig. 2). The karyotypes of his parents are normal.
Fig. 2.

The G-banded karyotype of a sSMC(22) (red arrow)
Chromosomal microarray analysis
In order to further identify the size and position of the chromosomal aberration, we performed CMA on genomic DNA. The results disclosed a partial repeat of the 22q11 region with a size of approximately 1.76 Mb on q11.1-q11.21 (16888900_18644241) (Fig. 3). The final karyotype based on ISCN [2020] was 47,XY,+psu idic(22)(q11.21).arr[GRCh37] 22q11.1q11.21(16888900_18644241)×4 dn.
Fig. 3.

Result of the SNP-array genotyping (GRCh37/hg19) and mapping of the genomic gain originating from chromosome 22. Chromosomal microarray showing tetrasomy region (denoted by arrow) -arr[GRCh37] 22q11.1q11.21(16888900_18644241) x4
Our patient’s breakpoints correspond to the CESCR. There were four copies of each of the XKR3, CCT8L2, GAB4, IL17RA, TMEM121B, HDHD5(CECR5), ADA2(CECR1), CECR2, SLC25A18, ATP6V1E1, BCL2L13, BID, MICAL3, PEX26, TUBA8, and USP18 (partial) genes (Fig. 4). Seven genes can be disease-causing: IL17RA (OMIM genes – 605461), ADA2(CECR1) (OMIM genes – 607575), ATP6V1E1 (OMIM genes – 108746), PEX26 (OMIM genes – 608666), TUBA8 (OMIM genes − 605742), and USP18 (OMIM genes – 607057) (Fig. 4). ClinGen dosage sensitivity analysis showed that ISCA-37,393 (Dosage ID, chr22:17392953_18591860) within our repeating area was triplosensitive. The duplication did not overlap with the VCF/DiGeorge locus. Thus, our patient was classified as type I CES with breakpoints within LCR22A and between D22S427 and D22S1638 (Fig. 5).
Fig. 4.

Overview of the genes in the duplication region of our patient. The region extends to position 18,644,241 according to UCSC Genome Browser on Human (GRCh37/hg19), corresponding to a CES-SMC type I, including 16 genes
Fig. 5.

Diagram denoting the CESCR together with the location of the duplication region in the index patient. The blue blocks along with references under the map indicate the location of duplication in the index cases. The red block on the map represents the region of common breakpoints of CES. The red arrow indicates the DGS/VCFS region. The further defined CESCR is located between the red dotted lines
Whole exome sequencing (WES)
WES was performed to identify the potential pathogenic variant in this case. The results also revealed a chromosome duplication of 22q11.1-q11.21. And in addition to this, a heterozygous mutation of the GJB2 gene was found in this patient (GJB2: NM_004004.6: exon2:c.109G > A:p.V37I).
Clinical data from literature
Finally, 28 recorded cases met the inclusion criteria (including the present case). Cytogenetics data analyses were performed on these 28 cases. A complete list of references is available on request (Supplementary Tables S1). The results of the cytogenetic analysis of these patients are shown in Table 1. The results showed that the presence of a classical sSMC(22) was the most frequent cytogenetic abnormality in these patients (23/28 cases, 82%). In the five additional cases, the chromosome aberration consisted of triplication of the 22q11 region (14%) and an atypical ring sSMC(22) (4%) (Table 1). sSMC(22) was found in a homogenous state in 63% of cases and in a mosaic state in 37% of cases (the state of one case was not available) (Table 1). De novo cases accounted for 58% (11/19 cases) and inherited cases accounted for 42% (8/19 cases) (inheritance information was not available for nine cases).
Table 1.
Cytogenetic data from 28 CES cases
| Type of chromosome aberration (n = 28) | State (n = 27) | Inheritance (n = 19) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Classical sSMC | Ring sSMC | Trp (22) | Homogeneous | Mosaicism | de novo | Inherited | |||
| Number of cases | 23/28 | 1/28 | 4/28 | 17/27 | 10/27 | 11/19 | 8/19 | ||
| Frequency (%) | 82 | 4 | 14 | 63 | 37 | 58 | 42 | ||
sSMC: small supernumerary marker chromosome; Trp: triplication
The results of molecular cytogenetic analyses (Table 2) showed that the CESCR was tetrasomic in 89% (24/27), trisomic in 7% (2/27), and both trisomic and tetrasomic in 4% (1/27) (duplication copy number was not available for one case). A 1–2 Mb duplication was most frequent (18/25, 72%) in 25 cases and no information was available for three cases. Type I chromosome aberration is the main molecular basis of CES, as it was the cause of CES in 27/28 of our cases (96%). In addition, the results of the CMA showed that the breakpoints in chromosome 22 were located in 18610000_18660000 bp in the majority CES patients (13/17, 76%) (only the CMA results with reference to human GRCh37/hg19 were analyzed for convenience) (Fig. 5).
Table 2.
Molecular cytogenetic finding of 28 CES cases
| Duplication copy number(n = 27) | Size of duplication(n = 25) | Type of anomaly(n = 28) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Tetrasomy | Trisomy | Both | < 1 Mb | 1-2 Mb | > 2 Mb | type I | type II | |||
| Number of cases | 24/27 | 2/27 | 1/27 | 4/25 | 18/25 | 3/25 | 27/28 | 1/28 | ||
| Frequency (%) | 89 | 7 | 4 | 16 | 72 | 12 | 96 | 4 | ||
Table 3 presents the frequency of the main clinical features in the 28 CES patients. Among these, 89% (25/28) had ear anomalies. Ear malformation and hearing loss were present in 39% (11/28) and 36% (10/28) respectively. Another two signs of the classic triad were present in less than half of the cases. Abdominal malformations including anal atresia, fistula/sinus, and other abdominal malformations were present in 50% (14/28). However, total colobomas were only present in 25% (7/28).
Table 3.
Frequency of main signs in CES patients
| Number of patients (n = 28) | Frequency (%) | |
|---|---|---|
| Ear anomalies | 25/28 | 89 |
| Preauricular pits/ tags | 24 | 86 |
| Ear malformation | 11 | 39 |
| Hearing loss | 10 | 36 |
| Abdominal malformations | 14/28 | 50 |
| Anal atresia | 13 | 46 |
| Fistula/sinus | 5 | 18 |
| Other abdominal malformations | 4 | 14 |
| Ophthalmologic abnormalities | 14/28 | 50 |
| Total colobomas | 7 | 25 |
| Ocular motility defect | 8 | 29 |
| Eye abnormalities | 5 | 18 |
| Craniofacial anomalies | 16/28 | 57 |
| Micrognathia | 15 | 54 |
| Hemifacial hypoplasia | 9 | 32 |
| Cardiovascular anomalies | 15/28 | 54 |
| TAPVR | 4 | 14 |
| ASD/VSD | 9 | 32 |
| Other anomalies | 11 | 39 |
| Urogenital malformation | 6/28 | 21 |
| Musculoskeletal anomalies | 9/28 | 32 |
| Mental retardation | 11/28 | 39 |
| Growth retardation | 7/28 | 25 |
TAPVR: total anomalous pulmonary venous return. ASD: atrial septal defects. VSD: ventricular septal defects
Craniofacial anomalies were the second most frequent signs noted in 57% (16/28) of cases. Cardiovascular anomalies were the third most frequent signs noted in 54% (15/28) of cases. ASD and/or ventricular septal defects (VSD) was present in 32% of cases and total anomalous pulmonary venous return (TAPVR) was present in 14% of cases. There were also some less common heart malformations such as PLSVC, type B interruption of the aortic arch and a large patent ductus arteriosus, a left-sided aortic arch with an aberrant right subclavian artery, and discrete coarctation of the aorta. Urogenital malformations such as renal agenesis, small dysplastic kidney, abnormal male external genitalia, and cryptorchidism are relatively uncommon in CES patients, being present in 21% of cases. Musculoskeletal anomalies were present in 32% of cases. Growth retardation was present in 25% of cases and mental retardation was present in 39% of cases.
Discussion
CES is a rare malformation syndrome that was first reported in 1965 by Schachenmann [14]. About 300 patients with CES have been reported to date [11]. The male to female ratio was about 1:3 according to the reported cases. We report a 2-year-old boy carrying a 22q11.1-q11.21 duplication of 1.76 Mb tetrasomy with the basic characteristics of CES, but lacking iris colobomas.
In the current case, his mother underwent a non-invasive prenatal testing (NIPT) examination at 16+ 4 weeks’ pregnant. Unfortunately, this NIPT yielded a negative result. In recent years, NIPT has been widely promoted for prenatal screening for trisomy 21, trisomy 18, and trisomy 13. However, so far, it is difficult for NIPT to screen for small CNVs, especially CNVs less than 2 Mb in length [15, 16]. Our case carries a microduplication of the 22q11 region with a size of approximately 1.76 Mb. Poor sensitivity for CNVs less than 2 Mb cause a false-negative NIPT result. Our case also received a screening of prenatal fetal cardiac malformation at 26+ 5 weeks’ pregnant. Fetal echocardiography confirmed that the fetus suffered from congenital heart disease. Unfortunately, the pregnant woman did not receive invasive prenatal diagnostic testing by karyotyping and CNV analysis. The results of the present study indicated that pregnant women who meet clinical indications for invasive prenatal diagnosis should be offered interventional prenatal diagnostic tests and whole-genome chip testing.
Although congenital heart anomalies do not belong to the clinical presentation triad, they are one of the major signs of CES presented in about 50–60% of affected patients, with ASD/VSD and TAPVR being the most frequently identified [11, 17]. It was reported that VSD and TAPVR are two relatively common malformations with incidence rates of 23% (18/80) and 19% (15/80) respectively [9]. Our patient did not present TAPVR, but had two other congenital heart defects, IVC and PLSVC. To the best our knowledge, this is the first time, in the literature, that the two cardiovascular malformations have been reported in CES. Our results expand the spectrum of symptoms of CES.
Our patient presented severe left ear malformations, atresia of the left external auditory meatus, and hearing loss. It is generally acknowledged that hearing impairment is not a major feature in CES patients because it was reported in only 17% (9/54) of CES patient presenting hearing loss [1]. However, recently, Jedraszak et al. found that hearing loss was present in 28% of the patients in a large cohort of 43 patients [11]. Thereby, the incidence of hearing loss may have been underestimated in CES patients. Although the involvement of the CESCR is well established, the significance of genotype-phenotype correlations remains largely unknown. The CECR2 (CES chromosome region candidate 2, MIM 607576) in the CESCR is known as a chromatin remodeling gene involved in neural tube closure and inner ear development. The influence of overexpression of CECR2 on the development of the brain, eye and ear might be responsible for frequent abnormalities of these organs in CES patients [18]. A recent study showed that the Cecr2 mutant mouse effectively demonstrates many of the abnormal features present in human patients with CES [19].
Interestingly, the results of the WES of our patient showed a heterozygous mutation of the GJB2 gene (NM_004004.6: exon2: c.109G > A). The GJB2 gene is found on chromosome 13q11 and encodes for the protein connexin 26, a beta class gap junction protein expressed in the cochlea and in the epidermis. Pathogenic variants of GJB2 are the most common cause of autosomal recessive sensorineural hearing loss [20]. However, as for the pathogenicity of heterozygotes, there is a lack of genetic information about the GJB2 c.109G > A mono-allelic mutation, although a study by Lin et al. showed that GJB2 c.109G > A heterozygotes had poorer hearing than did homozygotes [20]. Therefore, we cannot rule out that GJB2 c.109G > A may play additional roles in the phenotype of hearing loss in the present case.
To better delineate the clinical and molecular cytogenetics findings associated with CES, we reviewed 28 patients (including the present case) with a confirmed duplication size and position of CES and obtained meaningful results. According to the literature, three types of CES have been described previously based on the localization of breakpoints in 22q [21]. Our patient has a de novo sSMC with a 22q11.1-q11.21 duplication of 1.76 Mb (16888900_18644241, hg19). The sSMC was classified as type I with breakpoints within LCR22A and between D22S427 (18.49–18.69 Mb, hg19) and D22S1638 (18.89–19.10 Mb, hg19) [21, 22]. The amplification did not overlap with the VCF/DiGeorge critical region (Fig. 5).
Our data from 28 patients indicated that a classical sSMC was the most frequent cytogenetic abnormality in CES patients (82%). The duplicated chromosome segments in most of these patients are tetramorphic (a ring sSMC including both trisomy and tetrasomy) and with a duplication size ranging from 0.2 to 2.96 Mb. Type I chromosome aberration is the main molecular basis of CES (27/28, 96%). The results are consistent with a study by Jedraszak et al. and highlight that type I CES chromosomes are the most common cause of CES [11].
It has been reported that the development of the majority of neocentric sSMCs is based on a U-type exchange during meiosis I [3, 23]. 22q11.1-q11.2 is a critical region for chromosomal rearrangements due to its low copy repeats (LCRs). There are eight LCRs termed LCR22A-LCR22H in 22q11.1-q11.2 region. Based on the findings of the literature review, we speculate that recombination events during meiosis are more likely to happen in LCR22A, the proximal region of the long arm of chromosome 22. Chromosome 22 is most likely to generate a tetrasomy of about 1.7 Mb on 22q11.1-q11.21 by a U-type exchange. It was found that more than two-thirds of cases had a 1–2 Mb duplication and that the breakpoints of chromosome 22 of the majority of cases are localized to a 50 kb region between 18,610,000 bp and 18,660,000 bp (Fig. 5). These results support our above speculation.
According to Mears et al., the smaller CESCR spans around 2 Mb, from the centromere to the locus of the ATP6V1E1 gene (Cen_18111588 bp, hg19) [24, 25]. Subsequently, Knijnenburg et al. reported a 0.6 Mb partial tetrasomy (chr22: 17799398_18397897 bp) in a three-generation family with all the cardinal features of CES [26]. Consequently, the CESCR should be further delimited to a 0.3 Mb region between 17799398_18111588 bp (Fig. 5). CECR2, SLC25A18, and ATP6V1E1 within this region are strong candidate genes for causing the main CES phenotype [26]. Based on these results, we speculate that CECR2 (but not CECR1) gene dosage and functional interactions among amplified genes are the determining factors expressing the CES phenotype.
Our patient has the main characteristic clinical symptoms, but no iris coloboma, urogenital malformation, or growth retardation. Our findings highlight the phenotypic variability of CES. It has previously been reported that iris coloboma is the most frequently missing typical feature [11]. In the present study, we found that the top three anomalies are ear anomalies (89%), craniofacial anomalies (57%), and cardiovascular anomalies (54%). However, total colobomas were only present in 25% (7/28). The origin of the phenotypic variability is still unknown, despite efforts to define the CESCR. The duplication of the CES region is not necessarily symmetrical. Some partial trisomy patients have similar clinical features to cases with the CESCR in partial tetrasomy [18, 27]. In addition, the size of the duplicated fragment is not associated with the phenotype [1, 28]. In fact, some patients with the sSMC (22) and almost the same size of approximately 1.76 Mb duplication and same amplified gene content have different clinical symptoms [5, 29–31].
It is difficult to define the genotype–phenotype correlations according to the present results because of the relatively modest number of patients and limitation to type I CES. A possible cause of phenotypic variability is mosaicism of sSMC [32]. It has been reported that there is a direct correlation between the phenotype and the degree of mosaicism in the carriers [33]. In the present study, we found an unexpectedly high frequency of patients with mosaicism for sSMC (37%). The results are consistent with the findings of a recent study by Jedraszak et al. (40% of patients with mosaicism) [11]. It was reported that there are some CES patients with low mosaicism levels which could be associated with a milder phenotype [11, 33]. The rate of mosaicism of sSMC varied greatly between individuals as well as between tissues, with twice as many cells with the SMC in epithelial cells compared to blood [33]. A low percentage of sSMC mosaicism is not easily detected by blood karyotype analysis. Thus, a patient who was thought to be a de novo case previously may be inherited from a low percentage of sSMC mosaicism. These results suggest that we should pay more attention to parent karyotype analysis and prenatal diagnosis, which is essential for genetic counseling on risk of recurrence.
Conclusions
We have reported a CES patient presenting with rare or not reported clinical findings such as congenital aural atresia, hearing loss, PLSVC, and IVC. The GJB2 c.109G > A mono-allelic mutation may play additional roles in the phenotype of hearing loss. Given the lessons arising from this case, we strongly recommend that pregnant women who meet clinical indications such as fetal cardiac malformations should accept invasive prenatal diagnosis because NIPT shows low sensitivity for CNVs of less than 2 Mb. In addition, our review of the literature indicates that the CESCR may be further delimited to a 0.3 Mb region between 17799398_18111588 bp. CECR2 genes within this region are strong candidate genes for causing the main CES phenotype. Given the high frequency of patients with mosaicism for sSMC and the low proportion of mosaicism, parents who have had a child with CES are strongly advised to routinely propose prenatal diagnosis to prevent recurrence. We also highlight that hearing loss is not uncommon in CES. We recommend that CES patients should receive early hearing loss detection and intervention.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We thank the patient and his family for participating in this study.
Abbreviations
- array-CGH
Array comparative genomic hybridization
- ASD
Atrial septal defect
- CES
Cat eye syndrome
- CESCR
CES critical region
- CMA
Chromosomal microarray analysis
- CNV-seq
Copy number variation sequencing
- DGS/VCFS
DiGeorge/velocardiofacial syndrome
- IVC
Interrupted inferior vena cava
- LCRs
Low copy repeats
- NIPT
Non-invasive prenatal testing
- PLSVC
Persistent left superior vena cava
- RAE
Right atrium enlargement
- RVE
Right ventricle enlargement
- sSMC
Small supernumerary marker chromosome
- TAPVR
Total anomalous pulmonary venous return
- Trp
Triplication
- WES
Whole exome sequencing
Author contributions
Y.L. guided this work and reviewed the article. L.X. and X.C. finished the article. L.T. and S.M. helped to finish the experiment. J.W and H.Z. collected relevant information. All authors read and approved the final manuscript.
Funding
This study was supported by grants from the Natural Science Research Project of Anhui Educational Committee, China (No. KJ2021A0704).
Data availability
All data generated or analysed during this study are included in this published article or are available from the corresponding author on reasonable request.
Declarations
Ethics approval and consent to participate
The patient’s parents have given their informed written consent for their son’s participation to this study. This research received ethics approval from the Ethics Committee of Bengbu Medical University.
Consent for publication
We obtained written informed consent from the subjects’ parents for publication of this case report and any accompanying images.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Liang Xu and Xia Cheng contributed equally to this work.
References
- 1.Berends MJ, Tan-Sindhunata G, Leegte B, van Essen AJ. Phenotypic variability of Cat-Eye syndrome. Genet Couns. 2001;12(1):23–34. [PubMed] [Google Scholar]
- 2.Emanuel BS. Molecular mechanisms and diagnosis of chromosome 22q11.2 rearrangements. Dev Disabil Res Rev. 2008;14(1):11–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liehr T, Claussen U, Starke H. Small supernumerary marker chromosomes (sSMC) in humans. Cytogenet Genome Res. 2004;107(1–2):55–67. [DOI] [PubMed] [Google Scholar]
- 4.Glaeser AB, Diniz BL, Santos AS, Guarana BB, Muniz VF, Carlotto BS, Everling EM, Noguchi PY, Garcia AR, Miola J, et al. A child with cat-eye syndrome and oculo-auriculo-vertebral spectrum phenotype: a discussion around molecular cytogenetic findings. Eur J Med Genet. 2021;64(11):104319. [DOI] [PubMed] [Google Scholar]
- 5.Lu Y, Shen L, Zheng Y, Zhang H, Liu Y, Qi M, Huang S, Shen B. A Chinese family with cat eye syndrome and abnormality of eye movement: first case report. Front Pediatr. 2023;11:1145183. [DOI] [PMC free article] [PubMed]
- 6.Portnoi MF. Microduplication 22q11.2: a new chromosomal syndrome. Eur J Med Genet. 2009;52(2–3):88–93. [DOI] [PubMed] [Google Scholar]
- 7.Katz B, Enright J, Couch S, Harocopos G, Lee AR. Atypical presentation of Cat Eye Syndrome in an infant with Peters anomaly and microphthalmia with cyst. Ophthalmic Genet. 2020;41(6):645–9. [DOI] [PubMed] [Google Scholar]
- 8.Quintero-Rivera F, Martinez-Agosto JA. Hemifacial microsomia in cat-eye syndrome: 22q11.1-q11.21 as candidate loci for facial symmetry. Am J Med Genet A. 2013;161A(8):1985–91. [DOI] [PubMed] [Google Scholar]
- 9.Rosias PR, Sijstermans JM, Theunissen PM, Pulles-Heintzberger CF, De Die-Smulders CE, Engelen JJ, Van Der Meer SB. Phenotypic variability of the cat eye syndrome. Case report and review of the literature. Genet Couns. 2001;12(3):273–82. [PubMed] [Google Scholar]
- 10.Spineli-Silva S, Monlleó IL, Félix TM, Gil-da-Silva-Lopes VL, Vieira TP. Overlapping spectrum of Craniofacial Microsomia phenotype in Cat-Eye Syndrome. Cleft Palate Craniofac J. 2024; 61(9):1578-85. [DOI] [PubMed]
- 11.Jedraszak G, Jobic F, Receveur A, Bilan F, Gilbert-Dussardier B, Tiffany B, Missirian C, Willems M, Odent S, Lucas J et al. Cat eye syndrome: clinical, cytogenetics and familial findings in a large cohort of 43 patients highlighting the importance of congenital heart disease and inherited cases. Am J Med Genet A. 2024; 4:e63807. [DOI] [PubMed]
- 12.Gaspar NS, Rocha G, Grangeia A, Soares HC. Cat-Eye Syndrome: a report of two cases and literature review. Cureus. 2022;14(6):e26316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, Raca G, Ritter DI, South ST, Thorland EC, et al. Correction: technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med. 2021;23(11):2230. [DOI] [PubMed] [Google Scholar]
- 14.Schachenmann G, Schmid W, Fraccaro M, Mannini A, Tiepolo L, Perona GP, Sartori E. Chromosomes in Coloboma and Anal Atresia. Lancet. 1965;2(7406):290. [DOI] [PubMed] [Google Scholar]
- 15.Qian YQ, Wang XQ, Chen M, Luo YQ, Yan K, Yang YM, Liu B, Wang LY, Huang YZ, Li HG, et al. Detection of fetal subchromosomal aberration with cell-free DNA screening led to diagnosis of parental translocation: review of 11344 consecutive cases in a university hospital. Eur J Med Genet. 2019;62(2):115–23. [DOI] [PubMed] [Google Scholar]
- 16.Ye X, Lin S, Song X, Tan M, Li J, Wang J, Yan H, Zhang H, Li S, Chen D, et al. Identification of copy number variants by NGS-based NIPT at low sequencing depth. Eur J Obstet Gynecol Reproductive Biology. 2021;256:297–301. [DOI] [PubMed] [Google Scholar]
- 17.Williams JL, McDonald MT, Seifert BA, Deak KL, Rehder CW, Campbell MJ. An Unusual Association: total anomalous pulmonary venous return and aortic arch obstruction in patients with Cat Eye Syndrome. J Pediatr Genet. 2021;10(1):35–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haltrich I, Pikó H, Kiss E, Tóth Z, Karcagi V, Fekete G. A de novo atypical ring sSMC(22) characterized by array CGH in a boy with cat-eye syndrome. Mol Cytogenet 2014, 7(1). [DOI] [PMC free article] [PubMed]
- 19.Dicipulo R, Norton KA, Fairbridge NA, Kibalnyk Y, Fox SC, Hornberger LK, McDermid HE. Cecr2 mutant mice as a model for human cat eye syndrome. Sci Rep. 2021;11(1):3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin Y-F, Lin H-C, Tsai C-L, Hsu Y-C. GJB2 mutation spectrum in the Taiwanese population and genotype–phenotype comparisons in patients with hearing loss carrying GJB2 c.109G > A and c.235delC mutations. Hear Res. 2022; 413:108135. [DOI] [PubMed]
- 21.McTaggart KE, Budarf ML, Driscoll DA, Emanuel BS, Ferreira P, McDermid HE. Cat eye syndrome chromosome breakpoint clustering: identification of two intervals also associated with 22q11 deletion syndrome breakpoints. Cytogenet Cell Genet. 1998;81(3–4):222–8. [DOI] [PubMed] [Google Scholar]
- 22.Bartsch O, Rasi S, Hoffmann K, Blin N. FISH of supernumerary marker chromosomes (SMCs) identifies six diagnostically relevant intervals on chromosome 22q and a novel type of bisatellited SMC(22). Eur J Hum Genet. 2005;13(5):592–8. [DOI] [PubMed] [Google Scholar]
- 23.Voullaire L, Saffery R, Earle E, Irvine DV, Slater H, Dale S, du Sart D, Fleming T, Choo KH. Mosaic inv dup(8p) marker chromosome with stable neocentromere suggests neocentromerization is a post-zygotic event. Am J Med Genet. 2001;102(1):86–94. [DOI] [PubMed] [Google Scholar]
- 24.Mears AJ, Duncan AM, Budarf ML, Emanuel BS, Sellinger B, Siegel-Bartelt J, Greenberg CR, McDermid HE. Molecular characterization of the marker chromosome associated with cat eye syndrome. Am J Hum Genet. 1994;55(1):134–42. [PMC free article] [PubMed] [Google Scholar]
- 25.Mears AJ, el-Shanti H, Murray JC, McDermid HE, Patil SR. Minute supernumerary ring chromosome 22 associated with cat eye syndrome: further delineation of the critical region. Am J Hum Genet. 1995;57(3):667–73. [PMC free article] [PubMed] [Google Scholar]
- 26.Knijnenburg J, van Bever Y, Hulsman LO, van Kempen CA, Bolman GM, van Loon RL, Beverloo HB, van Zutven LJ. A 600 kb triplication in the cat eye syndrome critical region causes anorectal, renal and preauricular anomalies in a three-generation family. Eur J Hum Genet. 2012;20(9):986–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsai M-C, Chou Y-Y, Wang J-N, Wu J-M, Huang C-C, Kuo P-L, Tsai Y-S. Type B interrupted aortic Arch and Hydrocephalus Associated with Mosaicism of a 1.37 mb amplified Cat Eye syndrome critical region. Pediatr Neonatology. 2015;56(4):277–9. [DOI] [PubMed] [Google Scholar]
- 28.Serra G, Giambrone C, Antona V, Cardella F, Carta M, Cimador M, Corsello G, Giuffrè M, Insinga V, Maggio MC et al. Congenital hypopituitarism and multiple midline defects in a newborn with non-familial Cat Eye syndrome. Ital J Pediatr 2022, 48(1). [DOI] [PMC free article] [PubMed]
- 29.AlSubaihin A, VanderMeulen J, Harris K, Duck J, McCready E. Müllerian Agenesis in Cat Eye Syndrome and 22q11 chromosome abnormalities: a Case Report and Literature Review. J Pediatr Adolesc Gynecol. 2018;31(2):158–61. [DOI] [PubMed] [Google Scholar]
- 30.Li J, Zhang Y, Diao Y, Li R, Jiang L, Zhou L, Liu J, Duan W, Yang L, Mittal B. A De Novo sSMC (22) characterized by high-resolution chromosome microarray analysis in a Chinese boy with Cat-Eye Syndrome. Case Rep Genet. 2021;2021:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y, Zhang P, Chai Y, Zang W. Cat eye syndrome caused by 22q11.1q11.21 duplication: case report in a Chinese family. Mol Cytogenet. 2023;16(1):28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Córdova-Fletes C, Domínguez MG, Vázquez-Cárdenas A, Figuera LE, Neira VA, Rojas-Martínez A, Ortiz-López R. A de novo sSMC(22) characterized by high-resolution arrays in a girl with Cat-Eye Syndrome without Coloboma. Mol Syndromol. 2012;3(3):131–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kvarnung M, Lindstrand A, Malmgren H, Thåström A, Jacobson L, Dahl N, Lundin J, Blennow E. Inherited mosaicism for the supernumerary marker chromosome in cat eye syndrome: Inter- and intra‐individual variation and correlation to the phenotype. Am J Med Genet Part A. 2012;158A(5):1111–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analysed during this study are included in this published article or are available from the corresponding author on reasonable request.