

Abstract
Inhibiting angiotensin-converting enzyme 1 (ACE1) is a key strategy for managing hypertension as it prevents the formation of angiotensin II, a potent vasoconstrictor. Given the adverse effects associated with synthetic inhibitors, there is an increasing focus on exploring natural bioactive peptides as potential ACE1 inhibitors. Hemopressins (Hp) are peptides derived from hemoglobin. The present study investigated the ACE1 inhibitory activity of two Hp variants, Hp bearing phenylalaine (Hp-F) and Hp bearing leucine (Hp-L), using a combination of in vitro and in silico methodologies. In enzyme inhibition assays, Hp-L variants exhibited better inhibition when compared to Hp-F variants. Furthermore, in molecular docking and molecular dynamics simulations, Hp-L variants displayed favorable binding characteristics, in terms of binding energy and interactions, supporting their potential to be effective ACE1 inhibitors. The peptides were observed to interact with key residues involved in binding widely used ACE1 inhibitors. Notably, peptide RVD-Hp-L (RVDPVNFKLLSH) showed the lowest IC50 value, higher binding affinity and sustained interactions while binding to the catalytic site of ACE1. Finally, the substitution of phenylalanine with leucine in hemopressins significantly enhances their binding affinity and inhibitory potency.
Keywords: Hemopressin, ACE1, Molecular docking, Molecular simulation, Hypertension
Subject terms: Molecular modelling, Screening
Introduction
Cardiovascular disease (CVD) and hypertension are significant global health concerns1. The key hormonal system that maintains cardiovascular homeostasis in humans is the renin-angiotensin-aldosterone system (RAAS) where the angiotensin-converting enzyme 1 (ACE1) plays a significant role2. ACE1 is a chloride-dependent zinc metallopeptidase responsible for the conversion of decapeptide angiotensin I (Ang I) to octapeptide angiotensin II (Ang II), a potent vasoconstrictor, by cleaving two amino acids from the carboxy-terminal. In addition, it inactivates the vasodilator bradykinin by sequentially cleaving a dipeptide from its C-terminal. Thus, ACE1 regulates the balance between the vasodilatory properties of bradykinin and the vasoconstrictive properties of Ang II. This multifaceted role of ACE1 has garnered significant attention in the area of hypertension, CVDs and renal diseases3.
Dysregulation of the RAAS is critical in the etiology of hypertension, and the relationship between ACE1 and hypertension has been widely explored. Hence, human ACE1 has been identified as an excellent target for the treatment of hypertension and other cardiovascular problems. Two ACE1 isoforms have been identified in humans, encoded by a gene located on chromosome 17 (cytogenic location: 17q23.3) with 26 exons. Somatic ACE1 is transcribed from exons 1–12 and 13-26 whereas testicular ACE1 is transcribed from exons 13–26. Somatic ACE1 has two homologous enzymatic domains (N and C), whereas testicular ACE1 is a single-domain protein that is identical to somatic ACE’s C domain4. Even though both domains cleave AngI, it has demonstrated that C-ACE1 is adequate to sustain blood pressure regulation in vivo5,6. Hence, it is considered the predominant site for Ang II generation. Within the C domain, a Zn2+ ion is coordinated by His383, His387 and Glu411. Three subsites have been identified surrounding this catalytic site – S1 subsite (Ala354, Glu384 and Tyr523), S2 subsite (Asn281, His353, Lys511, His513 and Tyr520) and S1′subsite (Glu162). Molecules that are capable of binding to these sites could inhibit ACE1 activity7.
ACE1 inhibitors competitively bind to ACE1 and block Ang II generation and breakdown of bradykinin5. A number of antihypertensive medications are currently on the market, with lisinopril and captopril being the most popular. These drugs protect not only against hypertension but also associated disorders including diabetes, chronic kidney diseases and CVDs by reducing the synthesis of Ang II8. Despite remarkable growth in the use of ACE1 inhibitors worldwide, there exists an increased risk of adverse consequences such as persistent dry cough, loss of taste, hypotension, dizziness, hyperkalemia and angioedema9,10. Additionally, ACE1 inhibition can boost chymase activity in cardiac interstitial fluid, creating an additional channel for Ang II production11. As a result, for a significant number of patients receiving this therapy as well as those receiving other antihypertensive medication, blood pressure management is still suboptimal. Furthermore, chronic usage of ACE1 inhibitors might result in ACE1 inhibitor escape, a condition in which Ang II levels do not return to normal despite significant ACE suppression. This makes a compelling justification for finding more effective strategies for treating patients with hypertension, a major risk factor for cardiovascular problems12.
Peptides have been extensively studied for their antihypertensive property for regulating blood pressure13. These peptides could be derived from numerous sources and could act as an alternative to synthetic drugs due to their potential therapeutic benefits14,15. For instance, peptides derived from plants, microalgae, fruits, mushrooms, and fish have been found to exhibit ACE1 inhibitory activity16–23.
Hemoglobin, a vital protein involved in oxygen transport has been studied for the generation of bioactive peptides24. Bioactive peptides derived from the proteolysis of hemoglobin chains exhibit diverse physiological activities25. For example, hemorphins derived from the β-chain of hemoglobin exhibit opioid activity along with antiproliferative, immunomodulatory, and antihypertensive properties26–28. Hemopressin (Hp) peptides, a group of endogenous polypeptides derived from the proteolytic degradation of α-chain of hemoglobin, have garnered attention due to their interactions with cannabinoid receptors29. Hemopressin peptides act as modulators of the cannabinoid receptors and could function as agonists or antagonists depending on their structure. A nonapeptide hemopressin PVNFKFLSH (Hp-F), isolated from the rat and other related organisms, acts as an inverse agonist at the cannabinoid receptor 1 (CB1R) and modulates its signalling30,31. Furthermore, N-terminal extended hemopressin peptides such as RVDPVNFKFLSH (RVD-Hp-F) and VDPVNFKFLSH (VD-Hp-F) were the first peptides to exhibit agonist activity at CB1R, making them a promising possibility for stimulating brain CB1R without producing addictive adverse effects. Later RVD-Hp-F was identified as a CB1R negative allosteric modulator and a positive cannabinoid receptor 2 (CB2) allosteric modulator32,33.
Interestingly, Hp-F peptide PVNFKFLSH was conserved in rats and other mammals including lions and jaguars. A single amino acid variation of this peptide PVNFKLLSH (F > L) was observed in humans and other mammals including mice and pigs29 (Fig. 1).
Fig. 1.

Variation of hemopressin peptides. Multiple sequence alignment of the α-chain of hemoglobin from Mus musculus (mouse), Sus scrofa (pig), Homo sapiens (human), Rattus norvegicus (rat), Panthera leo (lion) and Panthera onca (jaguar). The alignment was produced using Clustal Omega. Hemopressin peptide is boxed in red.
Studies have reported the dose-dependent hypotensive action of Hp-F peptide in anaesthetized rats34. Even though the inhibitory activity of this Hp-F has been documented, the inhibitory potential of other hemopressin peptides on ACE1 remains unexplored. Understanding the mechanisms through which these peptides interact with the ACE1, could lead to new opportunities for targeted interventions. This study aimed to provide a deeper understanding of the molecular mechanism behind how hemopressins interact with ACE1 by utilizing computational tools and enzyme inhibition assays. The peptides used for the present analysis are shown in Table 1. Molecular docking and molecular dynamics simulations were used to predict peptide interaction and stability in the active site of ACE1 by assessing binding energetics, dynamics, and protein-peptide contact stability. Additionally, in vitro absorbance assays were conducted to compare the inhibitory effects of hemopressin variants.
Table 1.
Hemopressin peptides used for the analysis.
| No. | Name | Short name | Amino acid sequence |
|---|---|---|---|
| 1 | Hemopressin-F | Hp-F | PVNFKFLSH |
| 2 | Hemopressin-L | Hp-L | PVNFKLLSH |
| 3 | VD-Hemopressin-F | VD-Hp-F | VDPVNFKFLSH |
| 4 | VD-Hemopressin-L | VD-Hp-L | VDPVNFKLLSH |
| 5 | RVD-Hemopressin-F | RVD-Hp-F | RVDPVNFKFLSH |
| 6 | RVD-Hemopressin-L | RVD-Hp-L | RVDPVNFKLLSH |
| 7 | Neokyotorphin | Hpα138−142 | TSKYR |
| 8 | Kyotorphin | Hpα141−142 | YR |
Results
In vitro inhibition assay
In vitro ACE1 inhibition assays were performed using the eight hemopressin peptides listed in Table 1. The six peptides with the highest ACE1 inhibitory activity were further tested in triplicate. The IC50 values of these peptides, shown in Table 2, indicate varying inhibitory activities. Among the peptides tested, RVD-Hp-L exhibited the best inhibition with an IC50 value of 2.52 ± 0.11 µM. The other peptides also showed moderate to good inhibition profile. The dose-response curve depicts the varying inhibitory potencies of hemopressin variants against ACE1 (Fig. 2). Comparing Hp-F and Hp-L, it was observed that Hp-L had higher inhibition levels at lower concentrations, indicating a more potent inhibitory action (Fig. 2). A similar pattern was found in VD-Hp-F and VD-Hp-L, where inhibition levels increased progressively as the VD-Hp-F concentration was raised (Fig. 2). In contrast, VD-Hp-L was inhibited more effectively at lower concentrations. RVD-Hp-L had a stronger inhibitory effect, reaching a high degree of inhibition at around 20 µM compared to RVD-Hp-F (Fig. 2). These results highlight the improvement in inhibitory potency achieved by substituting phenylalanine with leucine in the peptide sequence.
Table 2.
IC50 (mean ± SD) of the peptides tested against ACE1.
| Peptide | IC50 ± SD (µM) |
|---|---|
| PVNFKFLSH (Hp-F) | 6.22 ± 0.44 |
| PVNFKLLSH (Hp-L) | 3.86 ± 0.87 |
| VDPVNFKFLSH (VD-Hp-F) | 10.75 ± 0.79 |
| VDPVNFKLLSH (VD-Hp-L) | 4.54 ± 0.83 |
| RVDPVNFKFLSH (RVD-Hp-F) | 12.44 ± 2.92 |
| RVDPVNFKLLSH (RVD-Hp-L) | 2.52 ± 0.11 |
Fig. 2.
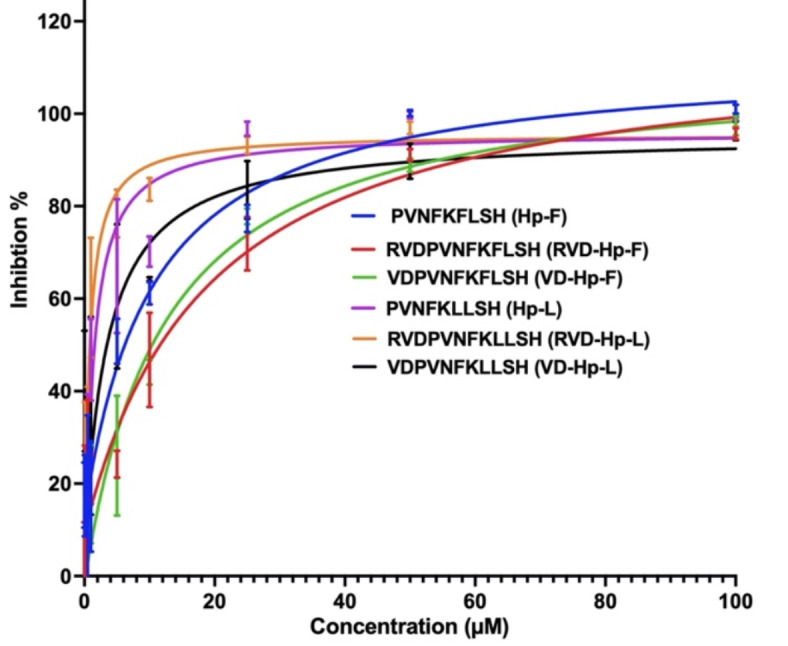
Dose response curves of hemopressin variants. ACE1 inhibition is shown in percentage for each dose.
Molecular docking
Six hemopressins, were docked flexibly to the catalytic site of ACE1 protein. Table 3 provides the docking score, molecular mechanics-generalized Born surface area (MM-GBSA) based binding free energy and amino acid residues of ACE1 that interacted with the hemopressins. Based on the GlideScore (GScore), binding energy and peptide binding orientation, it was observed that all six peptides docked well in the active site of the protein. Therefore, these peptides were analyzed further.
Table 3.
Interactions of the best binding pose of hemopressin variants with angiotensin-I converting enzyme (ACE1).
| Peptide | GlideScore (kcal/mol) |
MM-GBSA binding free energy (kcal/mol) |
Hydrogen bonds | Hydrophobic Interactions | π-π or cation-π Interactions | Salt bridge |
|---|---|---|---|---|---|---|
|
PVNFKFLSH (Hp-F) |
-14.13 | -99.51 | Asn66, Asn70, Glu143, Glu384, Asp415, Ser516, Ser517 | Ala63, Ala207, Ala208, Ala354, Ala356, Ile88, Ile204, Leu139, Phe359, Phe391, Phe457, Phe512, Phe527, Pro519, Trp220, Trp358, Tyr62, Tyr 135, Tyr360, Try523, Val351, Val379, Val380, Val518 | Arg124 | |
|
PVNFKLLSH (Hp-L) |
-13.97 | -111.29 | Asn66, Asn70, Glu143,Trp220,Glu384,Asp415, Ser517 | Ala63, Tyr62, Ile88, Met223, Trp220, Leu139, Tyr135, Ala354, Ala356, Trp357, Tyr360, Val351, Phe391, Val379, Val380, Pro407, Pro519, Phe457, Phe512, Val528, Tyr523, Phe527 | His387 | Glu143 |
| VDPVNFKFLSH (VD Hp-F) | -13.29 | -98.78 | Asp453, Ala354, Ala356, Tyr360, Tyr523, Arg522 | Trp59, Ile88, Met223, Trp220, Ala354, Ala356, Tyr360, Trp279, Trp357, Phe391, Phe457, Leu375, Pro407, Phe512, Val379, Val380, Val518, Pro519, Tyr520, Tyr523, Phe527 | Phe512, Lys118 |
Glu376, Glu403, Asp453 |
| VDPVNFKLLSH (VD Hp-L) | -12.53 | -104.65 | Asn66, Glu143, Gln281, Thr282, Asp453,Glu384, Ser516, | Trp59, Tyr62, Ala63, Ile88, Tyr135, Leu139, Met223, Trp220, Ala354, Ala356, Trp279, Trp357, Tyr360, Val379, Val380, Phe391, Phe457, Phe512, Phe527, Val518, Pro519, Tyr520, Tyr523 | His387 | Arg124 |
| RVDPVNFKFLSH (RVD Hp-F) | -12.40 | -99.10 |
Ala354, Ala356 Asn277, Gln281 Met450, Glu403, Glu411, Tyr 523 |
Trp59, Tyr62, Ala63, Leu139, Leu140, Met278, Trp279, Ala354, Ala356, Cys352, Tyr360, Val379, Val380, Cys370, Phe391, Tyr394, Pro407, Met450, Phe457, Pro515, Phe512, Val518, Tyr520, Tyr523, Phe527 | Glu403, Asp453 | |
| RVDPVNFKLLSH (RVD Hp-F | -13.73 | -118.34 | Glu403, Asn277, Gln281, Asp453, His513, His353 | Trp59, Tyr62, Ala63, Leu139, Leu140, Leu375, Val379, Val380, Tyr360, Ala354, Ala356, Trp357, Val351, Met278, Trp279, Phe391, Pro507, Phe457, Phe512, Pro515, Val518, Tyr523, Phe527 | Glu403, Asp453, Glu376 |
Docking of Hp-F and Hp-L with ACE1
The molecular docking results of Hp-F and Hp-L with ACE1, were analysed by focusing on their GScore and MM-GBSA binding free energy values. The optimal binding orientation of Hp-F exhibited a GScore of -14.13 kcal/mol and an MM-GBSA binding energy of -99.51 kcal/mol. Several intermolecular interactions were observed in the Hp-F peptide, such as the carbonyl group of Pro1 formed water-coordinated hydrogen bonds with Asp415 of the protein, Asn3 of the peptide interacted with the Zn2+ ion and formed a hydrogen bond with Glu384. Additionally, Lys5 formed hydrogen bonds with Glu143 and Ser516 of ACE1. Notably, Asn66 of ACE1 established hydrogen bonds with Phe6 of the peptide and a salt bridge formed between Arg124 of ACE1 and the backbone of His9 of the peptide. (Fig. 3A)
Fig. 3.

Angiotensin-I converting enzyme (ACE1) with hemopressin variant peptides docked in the active site. (A) ACE1-Hp-F (B) ACE1- Hp-L (C) ACE1-VD-Hp-F (D) ACE1-VD-Hp-L (E) RVD-Hp-F (F) ACE1-RVD-Hp-L. The ACE1 protein is shown in brown cartoon representation and its interacting residues are shown in pink stick representation; the docked ligand is represented as green sticks, and hydrogen bonds are shown as black dashed lines.
The binding orientation of Hp-L was found to closely resemble that of Hp-F. Specifically, the Hp-L had a GScore of -13.97 kcal/mol and an MM-GBSA binding energy of -111.29 kcal/mol. Similar to Hp-F, the carbonyl group of Pro1 formed hydrogen bonds with Asp415 of the protein, while Asn3 interacted with the Zn2+ ion and formed a hydrogen bond with Glu384. Furthermore, Phe4 exhibited π-π interactions with His387. Lys5 was found to engage in a salt bridge and hydrogen bond interaction with Glu143, along with another hydrogen bond with Asn70. Leu6 adopted a similar orientation to that of Phe6 in Hp-F, establishing a hydrogen bond interaction with Asn66 of the ACE1 protein. Additionally, His9 interacted with Trp220 and Ser517 through hydrogen bond interactions. Furthermore, various hydrophobic, polar, and electrostatic interactions between the peptide and ACE1 active site residues were identified, contributing to the peptide’s binding affinity within the active site of ACE1 (Fig. 3B).
Docking of VD-Hp-F and VD-Hp-L with ACE1
The optimal binding configuration of VD-Hp-F with ACE1 yielded a GScore of -13.29 kcal/mol and an MM-GBSA binding energy of − 98.78 kcal/mol. The N-terminal amino group of Val1 engaged in electrostatic interactions and hydrogen bonding with Glu376 and Asp453 of the protein. Following this, Asp2 and Asn3 interacted with Trp279 and Tyr523 by forming aromatic hydrogen bonds. Furthermore, the carbonyl group of Asn5 established a hydrogen bond with Tyr523. Phe6 exhibited π-π interactions with Phe512 and hydrogen bonding with Ala354 and Ala356. The amino group of Lys7 formed a salt bridge with Glu403, while Leu9 and Ser10 interacted via hydrogen bonds with Tyr360 and Arg522, respectively. His11’s imidazole ring engaged in π-cation interaction with Lys118 and a hydrogen bond with Glu123 of the protein. (Fig. 3C).
The best binding orientation of VD-Hp-L exhibited a Gscore of -12.53 kcal/mol and an MMGBSA energy of -104.65 kcal/mol. While the binding orientations of both peptides exhibited similarities, slight variations were observed in certain amino acid residue positions. Similar to VD-Hp-F, the N-terminal Val1 of VD-Hp-L established a hydrogen bond and salt bridge with Asp453. Asp2 interacted with ACE1 by forming hydrogen bonds with Thr282 and Gln281. Additionally, Val4 was observed to form hydrogen bonds with His353 and Asn5 formed hydrogen bonds with Glu384 and coordinated with the Zn2+ ion coordinated by the protein. His387 engaged in π-π interactions with Phe6 of the peptide and the amino group of Lys7 formed two hydrogen bonds with Ser516 and Glu143. Leu8 of the peptide formed hydrogen bonds with Asn66. Apart from these interactions, His11 established two hydrogen bonds with Arg124 of the protein. These interactions are complemented by various hydrophobic and polar interactions, which contribute to the stability and specificity of the protein-peptide complex (Fig. 3D).
Docking of RVD-Hp-F and RVD-Hp-L with ACE1
The molecular docking results of RVD-Hp-F exhibited a GScore of -12.40 kcal/mol and MM-GBSA binding energy of -99.10 kcal/mol. The guanidino group of Arg1 interacted with Met450 and Gln281 by forming hydrogen bonds. The Asp3 formed a hydrogen bond with Asn277 and Asn6 engaged in a hydrogen bond with Tyr523. The amino group of Phe7 formed a hydrogen bond with Ala354, whereas carbonyl oxygen of Phe7 interacted with Ala356 via hydrogen bonding. Furthermore, the amino group of Lys8 formed a hydrogen bond with Glu411, due to the donor-acceptor relationship between the amine and carboxylate groups. Furthermore, Lys8 formed a salt bridge with Glu403 driven by the strong electrostatic attraction between the positively charged ammonium group of Lys8 and the negatively charged carboxylate group of Glu403, which is crucial for maintaining the structural integrity and binding affinity of the peptide to ACE1 (Fig. 3E).
The binding of RVD-Hp-L exhibited a GScore of -13.73 kcal/mol and MM-GBSA binding free energy of -118.34 kcal/mol. The guanidino group of Arg1 interacted with Asp453 through hydrogen bonds and salt bridges. Additionally, the amino group of Arg1 formed a salt bridge with Glu376 and hydrogen bonded with Gln281. The carbonyl oxygen of Asp3 formed a hydrogen bond with Asn277, while Val5 establishes hydrogen bonds with His513 and His353. Notably, Phe7 engaged in metal coordination with the Zn2+ ion bound to ACE1. Finally, Lys8 formed both a salt bridge and a hydrogen bond with Glu403. (Fig. 3F).
Protein-peptide molecular dynamics simulations
Molecular dynamics (MD) simulations were performed in triplicate to evaluate the stability and dynamics of the interactions between ACE1 and hemopressins. MD results showed that all six complexes retained structural stability throughout the 500 ns simulations, as illustrated by the Cα root mean square deviation (RMSD) and the root mean square fluctuation (RMSF) plots (Figs. 4 and 5).
Fig. 4.

Root mean square deviation (RMSD) of protein Cα atoms obtained from triplicate 500 ns simulations of (A) ACE1-Hp-F (B) ACE1-Hp-L (C) ACE1-VD-Hp-F (D) ACE1-VD-Hp-L (E) RVD-Hp-F (F) ACE1-RVD-Hp-L.
Fig. 5.

Root mean square fluctuations (RMSF) of protein Cα atoms obtained from triplicate 500 ns simulations of (A) ACE1-Hp-F (B) ACE1-Hp-L (C) ACE1-VD-Hp-F (D) ACE1-VD-Hp-L (E) RVD-Hp-F (F) ACE1-RVD-Hp-L.
The RMSD of protein C-alpha (Cα) atoms were evaluated to study the difference in protein conformation in each frame of the MD trajectory when compared to the initial structure. This provides insights into the ACE1 flexibility and conformational changes induced upon peptide binding.
The RMSD data for Hp-F across three runs showed that, following initial fluctuations, the peptide achieved a stable conformation with a deviation of 2 Å (Fig. 4A). Among the Hp-L variants, initial variations were noted, but both VD-Hp-L and RVD-Hp-L reached stable conformations faster than the Hp-L variant (Fig. 4B and D, and 4F). The observed variations in RMSD values across the three runs are expected due to the inherent stochastic nature of the simulations. This randomness can lead to different trajectories and conformational ensembles, contributing to the variability in RMSD values across the runs. Overall, RMSD data suggested that the N-terminal VD and RVD extensions may contribute to more stable peptide structures.
Protein RMSF was examined for the three independent 500 ns simulation runs. RMSF data offers valuable insights into the flexibility and stability of protein residues in the protein-peptide complex, which are crucial in elucidating binding dynamics. The peaks in the RMSF graphs indicate regions of higher flexibility and corresponded to the loop regions of the protein. Furthermore, the RMSF patterns for these peptides are highly consistent across the three runs, highlighting the stability and consistency of the peptide-ACE1 interactions (Fig. 5).
Protein–peptide interaction plots were used to identify the stability of residue-level polar and hydrophobic interactions generated by hemopressin varaints with ACE1. Interactions that persisted more than 50% of the simulation time were analyzed. In Hp-F and Hp-L peptides, it was observed that interactions formed during the docking analysis were retained and new interactions were formed confirming their stability in the active site region. In Hp-F, polar interactions involved ACE1 residues Ala354, Arg124, Asp415, Glu143, and Tyr520 with Hp residues Asn3, Lys5, His9, Pro1, and Phe4 (Fig. 6A). Similarly, in Hp-L, polar interactions were observed between ACE1 residues Arg124, Asp415, and Glu143 with peptide residues His9, Pro1, and Lys5 (Fig. 7A). Extensive hydrophobic interactions formed between ACE1 residues Ala354, Phe359, Phe391, Phe457, Trp357, and Tyr520 with Hp-F residues Phe4, Phe6, Leu7, and Val2 (Fig. 8A). In Hp-L, hydrophobic interactions formed between Ala354, Phe457, Pro519, Trp357, Tyr520, and Val518 with peptide residues Phe4, Val2, Leu6, and Leu7 (Fig. 9A). The persistence of these interactions across multiple simulation runs underscores their importance in maintaining the structural integrity and binding affinity of the hemopressin-ACE1 complex.
Fig. 6.

Percentage of simulation time during which intermolecular polar contacts were retained between ACE1 and Hp-F variant peptides in triplicate 500 ns MD simulations. (A) polar interaction of Hp-F with ACE1, (B) polar interaction of VD-Hp-F with ACE1, (C) polar interaction of RVD-Hp-F with ACE1. s signifies salt bridge.
Fig. 7.

Percentage of simulation time during which intermolecular polar contacts were retained between ACE1 and Hp-L variant peptides in the triplicate 500 ns MD simulations. (A) polar interaction of Hp-Lwith ACE1, (B) polar interaction of VD-Hp-L with ACE1, (C) polar interaction of RVD-Hp-L with ACE1. s signifies salt bridge.
Fig. 8.

Percentage of simulation time during which intermolecular hydrophobic contacts were retained between ACE1 and Hp-F variant peptides in triplicate 500 ns MD simulations. (A) hydrophobic interaction of Hp-F with ACE1, (B) hydrophobic interaction of VD-Hp-F with ACE1, (C) hydrophobic interaction of RVD-Hp-F with ACE1.
Fig. 9.

Percentage of simulation time during which intermolecular hydrophobic contacts were retained between ACE1 and and Hp-L variant peptides in the triplicate in 500 ns MD simulations. (A) hydrophobic interaction of Hp-L with ACE1, (B) hydrophobic interaction of VD-Hp-L with ACE1, (C) hydrophobic interaction of RVD-Hp-L with ACE1.
For VD-Hp-F and VD-Hp-L peptides, distinct and consistent interaction patterns were observed. In VD-Hp-F, key polar interactions included hydrogen bonds and electrostatic interactions between ACE1 residues Ala356, Arg522, Asp358, Glu403, Lys511, Ser219, Trp220, and Tyr360 with peptide residues Phe6, His11, Phe8, Val1, Lys7, and Asp2 (Fig. 6B). In VD-Hp-L, polar interactions are found between ACE1 residues Ala356, Arg124, Asp415, Asp453, Gln369, Glu143, and Tyr523 with peptide residues Asn5, His11, Val1, Lys7, and Asp2, indicating stable hydrogen bonds and electrostatic interactions throughout the simulation (Fig. 7B). Predominant hydrophobic interactions in both peptides involved ACE1 residues Ala354, Ala356, Phe457, Phe527, Tyr520, Tyr523, Val379, Val380, and Val518, highlighting the strong binding affinity and stability of the peptides in the active site of the protein (Figs. 8B and 9B).
In RVD-Hp-F, polar interactions were formed between ACE1 residues Ala354, Asn136, Asn85, Asp358, Asp415, Asp453, Gln281, Glu403, Lys511, Tyr360, and Tyr523 interacting with peptide residues Phe7, His12, Lys8, Arg1, Asp3, and Asn6 (Fig. 6C). Apart from this, numerous residues of ACE1 formed hydrophobic contacts with RVD-Hp-F. These residues include Ala354, Ala356, Ala63, Ile88, Phe359, Phe457, Phe512, Phe527, Pro519, Trp357, Trp59, Tyr360, Tyr520, Tyr523, Tyr62, Val379, and Val518. These residues interacted with Phe7, Pro4, Val2, Val5, Phe9, and Leu10 of the peptide. RVD-Hp-L showed similar interactions where ACE1 residues Ala354, Asp453, Gln281, Glu143, Glu376, Glu403, Lys511, and Tyr523 interacted with peptide residues Pro4, Arg1, Asp3, His12, Lys8, Val5, and Asn6 (Fig. 7C). Residues that formed hydrophobic interactions included Ala354, Ala356, Ala63, Phe359, Phe391, Phe457, Phe512, Phe527, Trp357, Trp59, Tyr360 and Tyr523 (Fig. 8C). These residues interacted with hydrophobic residues of the RVD-Hp-L peptide including Phe7, Pro4, Val2, Val5, Leu9, and Leu10 (Fig. 9C). These consistent interactions across multiple runs underscore their importance in maintaining the structural integrity and binding affinity of these peptides.
While comparing the binding profiles of hemopressins with clinically relevant inhibitors such as captopril and lisinopril, it was observed that both the peptides and the known inhibitors bound in a similar manner in the active site of ACE1. Captopril, the first orally active ACE1 inhibitor, has been widely used for the treatment of hypertension and cardiovascular diseases since the 1980s35. However, due to its side effects, non-sulfhydryl ACE1 inhibitors like enalaprilat and lisinopril were developed with improved potency36. Molecular docking studies revealed that inhibitors such as captopril and lisinopril interacted with ACE1 by making polar and hydrophobic contacts with key residues, including Glu162, His353, Ala354, Ala356, Trp357, His383, Glu 384, His387, His410, Glu411, Lys511, Phe512, His513, Val518, Tyr520, Arg522 and Tyr523.These critical interactions contribute to the effective inhibition of ACE1 by these drugs and hemopressin peptides (Fig. 10).
Fig. 10.
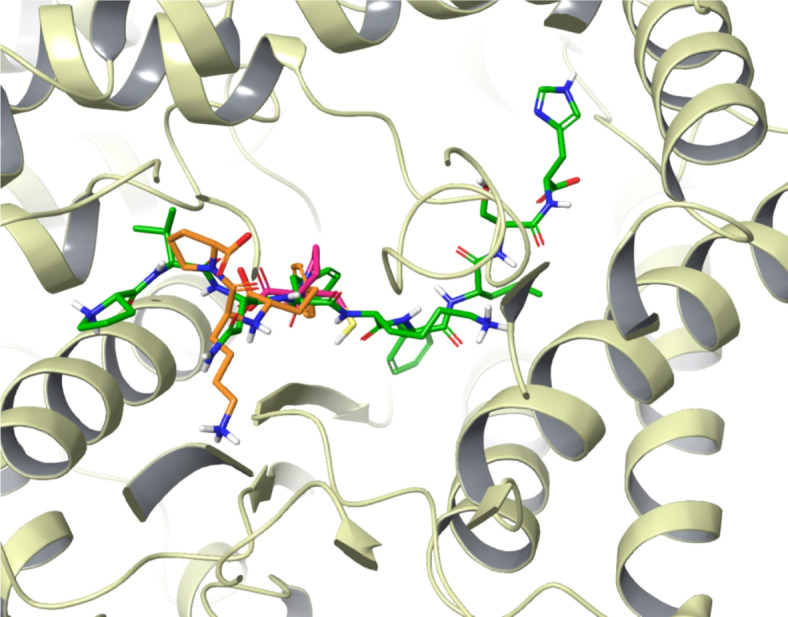
Binding comparison of Hp-F with drugs lisinopril and captopril. Hp-F (green), lisinopril (orange) and captopril (pink).
Discussion
Inhibiting ACE1 prevents the formation of Ang II, thereby reducing vasoconstriction and blood pressure. Therefore, ACE1 is a major drug target for hypertension. Owing to the potential side effects associated with chemically synthesized inhibitors, natural bioactive peptides exhibiting ACE1 inhibitory activities from diverse sources have been identified and reported37,38.
ACE1 inhibitory activity of hemopressin peptides, derived from hemoglobin, and their underlying molecular mechanisms have not been fully elucidated. To address this, the present study employed bioactive hemopressin peptides of two variations, to evaluate their potential inhibitory effects on ACE1. This study utilized a combination of in vitro and in silico methodologies, including inhibition assays, molecular docking, and MD simulations, to investigate the interactions between these peptides and ACE1. Among the eight peptides tested, six exhibited good ACE1 inhibitory activity. Notably, Hp-L variants demonstrated better inhibition profiles compared to Hp-F peptides. Following this, computational studies including molecular docking and simulations were performed to explore the binding orientation, free energy of binding and interacting residues. The findings revealed that Hp-L exhibited more favorable binding characteristics in terms of binding energy, GScore and intermolecular interactions, supporting their potential as effective ACE1 inhibitors.
In the in vitro inhibition assay, RVD-Hp-L had the lowest IC50 value among the peptides tested. The substitution of leucine in hemopressin peptides improves their binding affinity and inhibitory potency across peptide sequences. This may be due to several factors including being an aliphatic amino acid with a smaller side chain, which may reduce the steric hindrance compared to the bulky aromatic side chain of phenylalanine. This smaller side chain may allow better accommodation within the ACE1 catalytic site, enhancing peptide interactions with key residues and improving inhibitory activity.
Additionally, several structural and physicochemical characteristics of bioactive peptides have been shown to influence their ability to inhibit ACE139. The amino acid composition plays a crucial role in determining the inhibitory activity of peptides. Several studies have indicated that the ACE1-inhibitory peptides are generally short-chain peptides with 2–12 amino acids and crystallography studies show that large peptides cannot bind to the active site of ACE140. Typically, small oligopeptides are readily absorbed by the human body and can directly engage in tissue protein synthesis and metabolism41. Previous studies have indicated that the presence of cyclic, aromatic residues such as Phe, Tyr, His, Pro, and Tyr and hydrophobic amino acids such as Ala, Val, Leu, Pro, and Glu is associated with ACE1 inhibitory activity42. Here the reported peptides possess these amino acids which may assist in their inhibitory potential.
Moreover, it was noted that all the identified peptides appeared to bind strongly to the S1 and S2 subsites of ACE1, which is likely to contribute to their inhibitory activity. The presence of acidic amino acids Asp and Glu helps chelate the Zn2+ion bound to ACE1, which is essential for enzyme activity38,43,44. Zn2+is pivotal in the active site, forming a tetrahedral coordination with critical residues and playing a vital role in interactions between ACE1 and inhibitory peptides45. From molecular docking, it was observed that all Hp-L variants interacted with the Zn2+ ion using an asparagine residue of the peptide. This suggests that these peptides could inhibit ACE1 activity by disrupting the catalytically essential Zn2+coordination complex46.
The presence of hydrophobic N-terminus, such as Val, Leu, Ile, and basic amino acids, including Arg, Lys, and His, exhibit stronger affinity to ACE1 by interacting with protein hot spot residues47. In addition, the presence of Pro residue increases ACE1 inhibitory activity of a peptide. The distinctive cyclic structure of proline can induce kinks, which significantly aids their binding in the active site, thereby further hindering substrate binding48,49. These characteristics align with the structural features of peptides identified in the analysis that could boost their inhibitory activity against ACE1.
Additionally, when interaction profiles of hemopressins were compared with clinically relevant inhibitors such as captopril and lisinopril, it was observed that these peptides bound in a similar binding pattern occupying key regions of the ACE1 active site. These molecules interacted with critical residues such as Glu162, His353, Ala354, Ala356, Trp357, His383, Glu 384, His387, His410, Glu411, Lys511, Phe512, His513, Val518, Tyr520, Arg522, Tyr523 as well as the Zn2+ion. Notably, the docked pose of lisinopril was found to be identical to the observed pose in the crystal structure PDB ID 1O86, ensuring the reliability of the computational approaches40.
In conclusion, this study elucidated the inhibitory activity and underlying molecular interactions of hemopressin variants against ACE1. Results revealed that hemopressin variants bearing Leu, including Hp-L, VD-Hp-L, and RVD-Hp-L, exhibit better inhibition profiles compared to hemopressin variants bearing Phe amino acid. It was observed that substitution of leucine for phenylalanine in hemopressins greatly improves the binding affinity and inhibitory activity of these peptides. This highlights the role of unique amino acid residues in altering the inhibitory property of hemopressin peptides against ACE1. Future research should focus on the structural and physicochemical properties of these peptides for improving their inhibitory activity and optimizing their potential for hypertension management.
Materials and methods
In vitro ACE inhibition assay
ACE1 inhibition was assessed using the absorbance-based colorimetric ACE1 Kit-WST (Dojindo Laboratories, Japan), following manufacturer’s guidelines. Custom-synthesized peptides, listed in Table 1, were procured from Watson Biosciences, USA. Initially, the eight peptides were screened in a single run at varying concentrations of 0.01, 0.1, 0.5, 1, 5, 10, 25, 50, and 100 µM. The top six peptides showing notable ACE1 inhibition, namely Hp-F, Hp-L, VD-Hp-F, VD-Hp-L, RVD-Hp-F, and RVD-Hp-L were then repeated in triplicate. Dilutions of these peptides at concentrations of 0.01, 0.1, 0.5, 1, 5, 10, 25, 50, and 100 µM were prepared using phosphate saline buffer (PBS). In a 96-well microplate, 20 µL of each peptide concentration was added to the respective wells. Additionally, 20 µL and 40 µL of deionized water were pipetted into blank 1 and blank 2 wells, respectively. Subsequently, 20 µL of substrate buffer and 20 µL of enzyme working solution were added to each sample well and blank 1 well. The microplate was then incubated at 37 °C for 1 h.
Measurement of absorbance
Following the incubation period, 200 µL of indicator working solution was added to each well and incubated for 5 min at room temperature. The absorbance at 450 nm was subsequently determined using a Glomax Discover Microplate Reader (Promega, USA). ACE1 inhibition was quantified using the following formula:
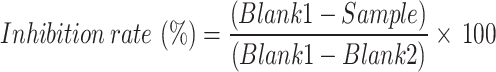 |
Where Blank1 (without a sample but with enzyme working solution) and Blank2 (without a sample and no enzyme working solution) are 20 µL and 40 µL deionized water, respectively. The dose-response curve and the half-maximal inhibitory concentration (IC50) were calculated using GraphPad Prism version 10 (GraphPad, San Diego, CA) by fitting a nonlinear regression inhibition plot against the peptide concentration.
Pre-processing of protein structure
The three-dimensional structure of ACE1 (PDB ID: 2XY9) was downloaded from the Protein Data Bank (PDB) and preprocessed using Schrödinger Suite 2022-4 50. Protein preparation involved accurately assigning bond orders, modifying the ionization state, reorienting groups, introducing disulfide bonds, removing unnecessary water molecules, capping amide termini, assigning partial charges, and including missing atoms and side chains. Missing hydrogen atoms were added, and hydrogen bonding networks were optimized to ensure an appropriate representation of protein interactions51. Finally, energy minimization was performed to optimize and refine the protein structure to preserve the structural stability and relieve steric clashes52.
Active site identification and grid generation
A receptor grid was generated around ACE1’s active site using the receptor grid generation panel of the Schrödinger Maestro53. For this, the default parameters and the OPLS 2001 force field were used, with a van der Waal radius scaling factor of 1.0 to minimize penalties for near encounters and a partial charge cut-off of 0.25. A cubic region appropriate for peptide docking was created around the centroid of the protein’s active site residues.
Peptide docking and binding energy calculation
Peptide docking was performed to determine the most likely binding orientation of hemopressins with ACE1. The extended conformations of the peptides were generated using the peptide docking panel of the Schrödinger suite. The peptide docking panel of Schrödinger Glide version 2022-4 was employed to perform the docking with increased sampling and several docking runs to improve the accuracy54. The GScore scoring method, along with free energy of binding, was used to rank the docked poses55. The top docked poses based on GScore and binding energy were shortlisted for further analysis. After docking, Schrödinger Maestro was used for visualization and analysis of the various types of contacts, including hydrogen bonds, salt bridges, π–π and π-cation contacts, and hydrophobic interactions56. The binding free energy of the best-docked poses was computed using the MM-GBSA approach with Schrödinger Prime using the OPLS 2005 force field and the VSGB 2.0 implicit solvent model54. For comparison, clinically approved drugs lisinopril and captopril were also docked to ACE1.
Protein–peptide molecular dynamics simulations
To assess the strength of intermolecular contacts and the stability of the ACE1-hemopressin complexes, molecular dynamics (MD) simulations of shortlisted peptides docked to ACE1 were performed. MD simulations were performed using Desmond version 6.9 57. Each protein-peptide complex was subjected to 500 ns of MD simulations in triplicate58. All complexes were placed in an orthorhombic box of size 89 Å × 89 Å × 89 Å and solvated with single-point charge water molecules using the Desmond System Builder57. Subsequently, the simulation system was neutralized with the required number of counterions, and the salt concentration was set at 0.15 M NaCl. Before running the MD simulations, all systems were subjected to the steepest descent minimization and Desmond’s default eight-stage relaxation protocol. The electrostatic interactions were calculated using the Particle Mesh Ewald (PME) method with 1.0 nm short-range electrostatic and van der Waals cutoffs59. An NPT ensemble with the temperature at 300 K and the pressure at 1 atm was applied in all runs. The Nosé–Hoover thermostat and the isotropic Martyna–Tobias–Klein barostat were used to maintain a temperature of 300 K and a pressure of 1 atm, respectively60,61. A time-reversible reference system propagator algorithm (RESPA) integrator was used with an inner time step of 2.0 fs and an outer time step of 6.0 fs62. Following the simulations, the number of protein-peptide hydrogen bonds, intermolecular interactions, root-mean-square deviation (RMSD) of the peptide and the root-mean-square fluctuation (RMSF) of both the protein and the peptide were computed. All bonds involving hydrogen atoms were constrained using the M-SHAKE algorithm implemented in Desmond63. Packaged and custom scripts were used to analyze the simulation data. Protein-ligand contacts, RMSF, and RMSD of the complexes were calculated from the trajectories and plotted using R version 4.2.3 (http://www.r-project.org).
Acknowledgements
This research was funded by a center-based research grant (Fund number: 12R107) from the United Arab Emirates University to R.V and a United Arab Emirates University National Faculty Research Program grant (Fund number: 31S468) to Y.A.D.
Author contributions
R.V conceived experiments. P.A, B.B, A.R, S.A.S and Y.A.D performed experiments. P.A and B.B analyzed data. P.A and R.V wrote the manuscript. All authors reviewed the manuscript.
Data availability
The datasets generated during and /or analyzed during the current study are available from the corresponding author on reasonable request.
Declarations
Conflict of interest
R.V. is an editorial board member of Scientific Reports. The other authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Fuchs, F. D. & Whelton, P. K. High blood pressure and cardiovascular disease. Hypertension. 75, 285–292 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Te Riet, L., van Esch, J. H. M., Roks, A. J. M., van den Meiracker, A. H. & Danser, A. H. J. Hypertension: renin-angiotensin-aldosterone system alterations. Circ. Res.116, 960–975 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Khurana, V. & Goswami, B. Angiotensin converting enzyme (ACE). Clin. Chim. Acta. 524, 113–122 (2022). [DOI] [PubMed] [Google Scholar]
- 4.Hubert, C., Houot, A. M., Corvol, P. & Soubrier, F. Structure of the angiotensin I-converting enzyme gene. Two alternate promoters correspond to evolutionary steps of a duplicated gene. J. Biol. Chem.266, 15377–15383 (1991). [PubMed] [Google Scholar]
- 5.Royster, R. L., Groban, L., Locke, A. Q., Morris, B. N. & Slaughter, T. F. Cardiovascular Pharmacology. in Kaplan’s Essentials of Cardiac Anesthesia 132–166, Elsevier (2018). doi: 10.1016/B978-0-323-49798-5.00008-5
- 6.Fuchs, S. et al. Angiotensin-converting enzyme C-terminal catalytic domain is the main site of angiotensin I cleavage in vivo. Hypertension. 51, 267–274 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Olalere, O. A., Yap, P. G. & Gan, C. Y. Comprehensive review on some food-derived bioactive peptides with anti-hypertension therapeutic potential for angiotensin-converting enzyme (ACE) inhibition. J. Proteins Proteom.14, 129–161 (2023). [Google Scholar]
- 8.Khan, M. A. H. & Imig, J. D. Antihypertensive Drugs. in Reference Module in Biomedical Sciences. Elsevier (2018), doi: 10.1016/B978-0-12-801238-3.96704-7
- 9.Yilmaz, I. Angiotensin-converting enzyme inhibitors induce cough. Turk. Thorac. J.20, 36–42 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Na Takuathung, M. et al. Adverse effects of angiotensin-converting enzyme inhibitors in humans: a systematic review and meta-analysis of 378 randomized controlled trials. IJERPH 19, 8373 (2022). [DOI] [PMC free article] [PubMed]
- 11.Wei, C. C. et al. Mast cell chymase limits the cardiac efficacy of Ang I-converting enzyme inhibitor therapy in rodents. J. Clin. Invest.120, 1229–1239 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elijovich, F. & Laffer, C. A role for single-pill triple therapy in hypertension. Ther. Adv. Cardiovasc. Dis.3, 231–240 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Yang, D. et al. Formation and inhibition mechanism of novel angiotensin I converting enzyme inhibitory peptides from Chouguiyu. Front. Nutr.9, 920945 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miralles, B., Amigo, L. & Recio, I. Critical review and perspectives on food-derived antihypertensive peptides. J. Agric. Food Chem.66, 9384–9390 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Kaur, A., Kehinde, B. A., Sharma, P., Sharma, D. & Kaur, S. Recently isolated food-derived antihypertensive hydrolysates and peptides: a review. Food Chem.346, 128719 (2021). [DOI] [PubMed] [Google Scholar]
- 16.Daskaya-Dikmen, C., Yucetepe, A., Karbancioglu-Guler, F., Daskaya, H. & Ozcelik, B. Angiotensin-I-converting enzyme (ACE)-inhibitory peptides from plants. Nutrients. 9, 316 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang, Q. et al. The Antihypertensive effects and potential molecular mechanism of microalgal angiotensin I-converting enzyme inhibitor-like peptides: a mini review. Int. J. Mol. Sci.22, 4068 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manoharan, S., Shuib, A. S., Abdullah, N. Structural characteristics and antihypertensive effects of angiotensin-I-converting enzyme inhibitory peptides in the renin-angiotensin and kallikrein kinin systems. Afr. J. Tradit Complement. Altern. Med.14, 383–406 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sornwatana, T. et al. Terminalia chebula Fruit fruit-derived peptide with angiotensin‐I–converting enzyme inhibitory activity. Biotech. App Biochem.62, 746–753 (2015). Chebulin. [DOI] [PubMed] [Google Scholar]
- 20.Chasanah, E., Martosuyono, P., Budiari, S. & Kurnianto, M. A. Detection of native peptides from Channa striata extract using de novo sequencing. IOP Conf. Ser. : Earth Environ. Sci.967, 012041 (2022). [Google Scholar]
- 21.Heo, S. Y. et al. A heptameric peptide purified from Spirulina sp. gastrointestinal hydrolysate inhibits angiotensin I-converting enzyme- and angiotensin II-induced vascular dysfunction in human endothelial cells. Int. J. Mol. Med.39, 1072–1082 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lau, C. C., Abdullah, N. & Shuib, A. S. Novel angiotensin I-converting enzyme inhibitory peptides derived from an edible mushroom, Pleurotus cystidiosus O.K. Miller identified by LC-MS/MS. BMC Complement. Altern. Med.13, 313 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang, R., Yun, J., Wu, S., Bi, Y. & Zhao, F. Optimisation and characterisation of novel angiotensin-converting enzyme inhibitory peptides prepared by double enzymatic hydrolysis from Agaricus bisporus scraps. Foods. 11, 394 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lafarga, T., Rai, D., O’Connor, K. & Hayes, M. Generation of bioactive hydrolysates and peptides from bovine hemoglobin with in vitro renin, angiotensin-I-converting enzyme and dipeptidyl peptidase-IV inhibitory activities J. Food Biochem.40, 673–685 (2016). [Google Scholar]
- 25.Mielczarek, P. et al. Hemorphins—from discovery to functions and pharmacology. Molecules. 26, 3879 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ali, A., Baby, B., Soman, S. S. & Vijayan, R. Molecular insights into the interaction of hemorphin and its targets. Sci. Rep.9, 14747 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cohen, M., Fruitier-Arnaudin, I. & Piot, J. M. Hemorphins: substrates and/or inhibitors of dipeptidyl peptidase IV. Biochimie. 86, 31–37 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Ayoub, M. A. & Vijayan, R. Hemorphins targeting G protein-coupled receptors. Pharmaceuticals. 14, 225 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heimann, A. S. et al. Hemopressin as a breakthrough for the cannabinoid field. Neuropharmacology. 183, 108406 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heimann, A. S. et al. Hemopressin is an inverse agonist of CB 1 cannabinoid receptors. Proc. Natl. Acad. Sci. U S A. 104, 20588–20593 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gomes, I. et al. Hemoglobin-derived peptides as novel type of bioactive signaling molecules. AAPS J.12, 658–669 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petrucci, V. et al. Pepcan-12 (RVD-hemopressin) is a CB2 receptor positive allosteric modulator constitutively secreted by adrenals and in liver upon tissue damage. Sci. Rep.7, 9560 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bauer, M. et al. Identification and quantification of a new family of peptide endocannabinoids (pepcans) showing negative allosteric modulation at CB1 receptors. J. Biol. Chem.287, 36944–36967 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rioli, V. et al. Novel natural peptide substrates for endopeptidase 24.15, neurolysin, and angiotensin-converting enzyme. J. Biol. Chem.278, 8547–8555 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Acharya, K. R., Sturrock, E. D., Riordan, J. F. & Ehlers, M. R. W. ACE revisited: a new target for structure-based drug design. Nat. Rev. Drug Discov. 2, 891–902 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Herman, L. L. et al. Angiotensin-Converting Enzyme Inhibitors (ACEI). in: StatPearls (StatPearls Publishing, 2023). Available from: https://www.ncbi.nlm.nih.gov/books/NBK431051/ [PubMed] [Google Scholar]
- 37.Alvíz-Amador, A., Contreras‐Puentes, N. & Márquez‐Lázaro, J. Bioactive peptides against angiotensin‐converting enzyme I: an in silico study. Pept. Sci.116, e24332 (2024). [Google Scholar]
- 38.Ningrum, S., Sutrisno, A. & Hsu, J. L. An exploration of angiotensin-converting enzyme (ACE) inhibitory peptides derived from gastrointestinal protease hydrolysate of milk using a modified bioassay-guided fractionation approach coupled with in silico analysis. J. Dairy Sci.105, 1913–1928 (2022). [DOI] [PubMed] [Google Scholar]
- 39.Jao, C. L., Huang, S. L. & Hsu, K. C. Angiotensin I-converting enzyme inhibitory peptides: inhibition mode, bioavailability, and antihypertensive effects. BioMedicine. 2, 130–136 (2012). [Google Scholar]
- 40.Natesh, R., Schwager, S. L. U., Sturrock, E. D. & Acharya, K. R. Crystal structure of the human angiotensin-converting enzyme–lisinopril complex. Nature. 421, 551–554 (2003). [DOI] [PubMed] [Google Scholar]
- 41.Yue, S. et al. Highly selective and pH responsive adsorption of ZIF-8 for angiotensin-converting enzyme (ACE) inhibitory active peptides and its mechanism. Sep. Purif. Technol.324, 124620 (2023). [Google Scholar]
- 42.Sanjukta, S. et al. Production, characterization and molecular docking of antioxidant peptides from peptidome of kinema fermented with proteolytic Bacillus spp. Food Res. Int.141, 110161 (2021). [DOI] [PubMed] [Google Scholar]
- 43.Iwaniak, A., Minkiewicz, P., Darewicz, M., Food-originating ACE inhibitors, including antihypertensive peptides, as preventive food components in blood pressure reduction. Comp. Rev. Food Sci. Food Safe. 13, 114–134 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Wu, J., Aluko, R. E. & Nakai, S. Structural requirements of angiotensin I-converting enzyme inhibitory peptides: quantitative structure – activity relationship study of di- and tripeptides. J. Agric. Food Chem.54, 732–738 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Bünning, P. The functional role of zinc in angiotensin converting enzyme: implications for the enzyme mechanism. J. Inorg. Biochem.24, 183–198 (1985). [DOI] [PubMed] [Google Scholar]
- 46.Zheng, W. et al. Small molecule angiotensin converting enzyme inhibitors: a medicinal chemistry perspective. Front. Pharmacol.13, 968104 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li, J. et al. Novel angiotensin-converting enzyme-inhibitory peptides from fermented bovine milk started by Lactobacillus helveticus KLDS.31 and Lactobacillus casei KLDS.105: purification, identification, and Interaction mechanisms. Front. Microbiol.10, 2643 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wei, G. et al. Identification, structural characterization, and molecular dynamic simulation of ACE inhibitory peptides in whey hydrolysates from Chinese Rushan cheese by-product. Food Chemistry: X. 21, 101211 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li, J. et al. Novel natural angiotensin converting enzyme (ACE)-Inhibitory peptides derived from sea cucumber-modified hydrolysates by adding exogenous proline and a study of their structure–activity relationship. Mar. Drugs. 16, 271 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schrödinger, L. L. C. New York, NY, USA.
- 51.Madhavi Sastry, G., Adzhigirey, M., Day, T., Annabhimoju, R. & Sherman, W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des.27, 221–234 (2013). [DOI] [PubMed] [Google Scholar]
- 52.Schrödinger Release 2022-4. Protein Preparation Wizard; Epik, Schrödinger, LLC: New York, NY, USA, (2022).
- 53.Riordan, J. F. Angiotensin-I-converting enzyme and its relatives. Genome Biol.4, 225 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Friesner, R. A. et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem.47, 1739–1749 (2004). [DOI] [PubMed] [Google Scholar]
- 55.Halgren, T. A. et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem.47, 1750–1759 (2004). [DOI] [PubMed] [Google Scholar]
- 56.Friesner, R. A. et al. Extra precision Glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein – ligand complexes. J. Med. Chem.49, 6177–6196 (2006). [DOI] [PubMed] [Google Scholar]
- 57.Schrödinger Release 2020-4: Desmond Molecular Dynamics System & Shaw Research, D. E. New York, NY, ; Maestro-Desmond Interoperability Tools, Schrödinger: New York, NY, USA, 2020. (2021).
- 58.Bowers, K. J. et al. IEEE, Tampa, FL,. Scalable algorithms for molecular dynamics simulations on commodity clusters. in ACM/IEEE SC 2006 Conference (SC’06) 43–43 doi: (2006). 10.1109/SC.2006.54
- 59.Essmann, U. et al. A smooth particle mesh Ewald method. J. Chem. Phys.103, 8577–8593 (1995). [Google Scholar]
- 60.Martyna, G. J., Tobias, D. J. & Klein, M. L. Constant pressure molecular dynamics algorithms. J. Chem. Phys.101, 4177–4189 (1994). [Google Scholar]
- 61.Martyna, G. J., Klein, M. L. & Tuckerman, M. Nosé–Hoover chains: the canonical ensemble via continuous dynamics. J. Chem. Phys.97, 2635–2643 (1992). [Google Scholar]
- 62.Tuckerman, M., Berne, B. J. & Martyna, G. J. Reversible multiple time scale molecular dynamics. J. Chem. Phys.97, 1990–2001 (1992). [Google Scholar]
- 63.Kräutler, V., Van Gunsteren, W.F., Hünenberger, P.H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem., 22, 501–508 (2001).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated during and /or analyzed during the current study are available from the corresponding author on reasonable request.