

Abstract

The NRF2-KEAP1 interaction is central for cytoprotection against stresses, giving it high clinical significance. Covalent modification of KEAP1 is an efficient approach, but the covalent inhibitors used in the clinic carry undesired side effects originating in their moderate selectivity. Starting with a phenotypic screen, we identified a new covalent inhibitor chemotype that was optimized to deliver a series of potent and highly selective KEAP1 binders. While the developed compounds showed both cellular and in vivo activity, upregulating antioxidant response element-dependent target genes, they showed no genotoxicity in vitro. The lead compound exhibited broad selectivity in activity-based protein profiling and showed no significant interaction with a panel of commonly studied receptors nor with a broad panel of kinases. The nature of its interaction with KEAP1 and the origin of its selectivity were revealed by X-ray crystallography.
Introduction
Kelch-like ECH-associated protein 1 (KEAP1), an adaptor protein of the CUL3 E3 ubiquitin ligase complex, is a key regulator of the cellular response to oxidative and electrophilic stress.1 Its best known substrate is the key transcription factor NRF2 that regulates the expression of antioxidant proteins to protect cells from oxidative damage.2 Under basal conditions, KEAP1 homodimers bind to both the DLG and ETGE motifs within NRF2 and facilitate NRF2 ubiquitination and subsequent proteasomal degradation, thereby blocking the NRF2-mediated transcriptional activity.3 Upon accumulation of oxidative or electrophilic species, key cysteine residues in KEAP1 are modified, leading to NRF2 dissociation and nuclear translocation.4 Thus, upon oxidative or electrophilic stress, NRF2 levels increase, resulting in increased expression of antioxidant response element (ARE)-directed genes.5 Activation of NRF2 has been shown to protect against many diseases in preclinical models across multiple therapeutic areas.6 Given this disease relevance, targeting the KEAP1-NRF2 pathway by blocking the KEAP1-NRF2 interaction has been of high interest over the last two decades, resulting in numerous disclosed inhibitors.7
Some of the compounds that entered clinical trials or are in clinical practice today induce NRF2 by covalently modifying KEAP1 to block its E3 activity (Figure 1). Dimethyl fumarate (DMF), a very simple compound approved for the treatment of multiple sclerosis by the FDA in 2013, as well as some of its mixed ester analogues, binds covalently to multiple cysteines of the BTB and DC domains in KEAP1,8 including Cys151, and upregulates the activity of NRF2.9 The reversible covalent inhibitor of KEAP1 omaveloxolone was approved in 2023 for Friedreich’s ataxia.10 Both omaveloxolone and its predecessor bardoxolone methyl (also named as CDDO-Me), a compound that reached Phase III clinical trials, act through Cys151 in a mechanism similar to fumarate esters.11 There are other natural products, including curcumin, sulforaphane, chalcone, resveratrol, and caffeic acid derivatives,12 as well as synthetic analogues,13 which also exhibit chemo preventive effects through activating the NRF2 pathway. A major concern with respect to any of the covalent KEAP1 inhibitors is their selectivity against other intracellular proteins, which could compromise their therapeutic margin. Therefore, we initiated a program to identify covalent KEAP1 binders.
Figure 1.

Selected covalent inhibitors of the NRF2-KEAP1 interaction.
Results and Discussion
Our screening strategy for the identification of KEAP1-NRF2 interaction disruptors was based on the downstream elements of the KEAP1-NRF2 pathway. A library of 193k compounds was screened in HepG2 human liver carcinoma cells at a single dose of 10 μM in a FRET β-lactamase reporter assay, under the control of the interaction of NRF2 and the ARE promoter regions, the DNA sequence being specifically bound by activated NRF2.14 As a positive control, we also tested the clinical candidate bardoxolone in this assay and observed an EC50 of 9.2 nM. As a secondary screen, we used a complementation assay relying on the translocation of NRF2 from the cytoplasm into the nucleus also at the single dose of 10 μM. Active compounds were progressed in dose response studies in both assays, and the resulting 162 hits were further validated by SPR measurements. Additionally, DLS and cytotoxicity in HepG2 cells were assessed to provide around 50 validated hits. Some of the hits were found to be covalent modifiers of KEAP1, of which bis(arylsulfonyl)thiazoles 1 and 2 were selected for further optimization (Figure 2). The general electrophilic reactivity of 2 was moderate, as in a pH 7.4 phosphate buffer containing 1 mM 2, 10 mM glutathione, and 10% DMSO as cosolvent, only a moderate 30% conversion was achieved in 54 h at 37 °C. It is interesting to note that recently 2 representatives of this compound class were identified as bioactive compounds, although no mention was made of their potential covalent character. One of them was reported to have antifungal effects through targeting thioredoxin reductase of the Paracoccidioides genus,15 while the other was found to stimulate STING-mediated innate immune activity in an allele-specific manner.16
Figure 2.

Validated hits selected for further optimization and sites targeted for modification.
Since we expected these compounds to react with Keap1 through the 2-position, we outlined a design strategy that included the optimization of the aryl group in the 4-position, the fine-tuning of the reactivity in the 2-position through broader modifications, and the adjusting of the compound properties through modulation of the substitution pattern in the 5-position (Figure 2). To enable the exploration of this compound class, we developed an efficient and modular synthesis route using easily accessible building blocks (Figure 3). The key transformations included the sequential activation of the 4, 2, and 5-positions of the thiazole ring through halogenation, the establishment of the C–S bonds in a palladium catalyzed coupling for position 4 or nucleophilic substitution for position 2 with the appropriate thiol, and the introduction of the substituent in the 5-position in a nucleophilic substitution.
Figure 3.

General synthesis of 2,4-bis(aryl/alkyl)sulphonyl-thiazoles. Reagents and conditions: (i) Pd(dppf)Cl2, K3PO4, DMA, reflux; (ii) TMPMgCl·LiCl, THF, −40 °C; (iii) CBr4, −78 to −10 °C; (iv) R2-SNa, DMF, rt or R2-SH, K2CO3, MeCN, 45 °C; (v) NCS, MeCN, reflux; (vi) H2O2, AcOH, reflux; (vii) MeCN or MeCN/EtOH, reflux; (viii) formamide, toluene, rt; (ix) PCl5, DCM, 10 °C; (x) R1SO2Na, MeCN, rt; (xi) PCl5, 120 °C; (xii) thiourea, MeCN, reflux; and (xiii) pyridine, toluene, rt.
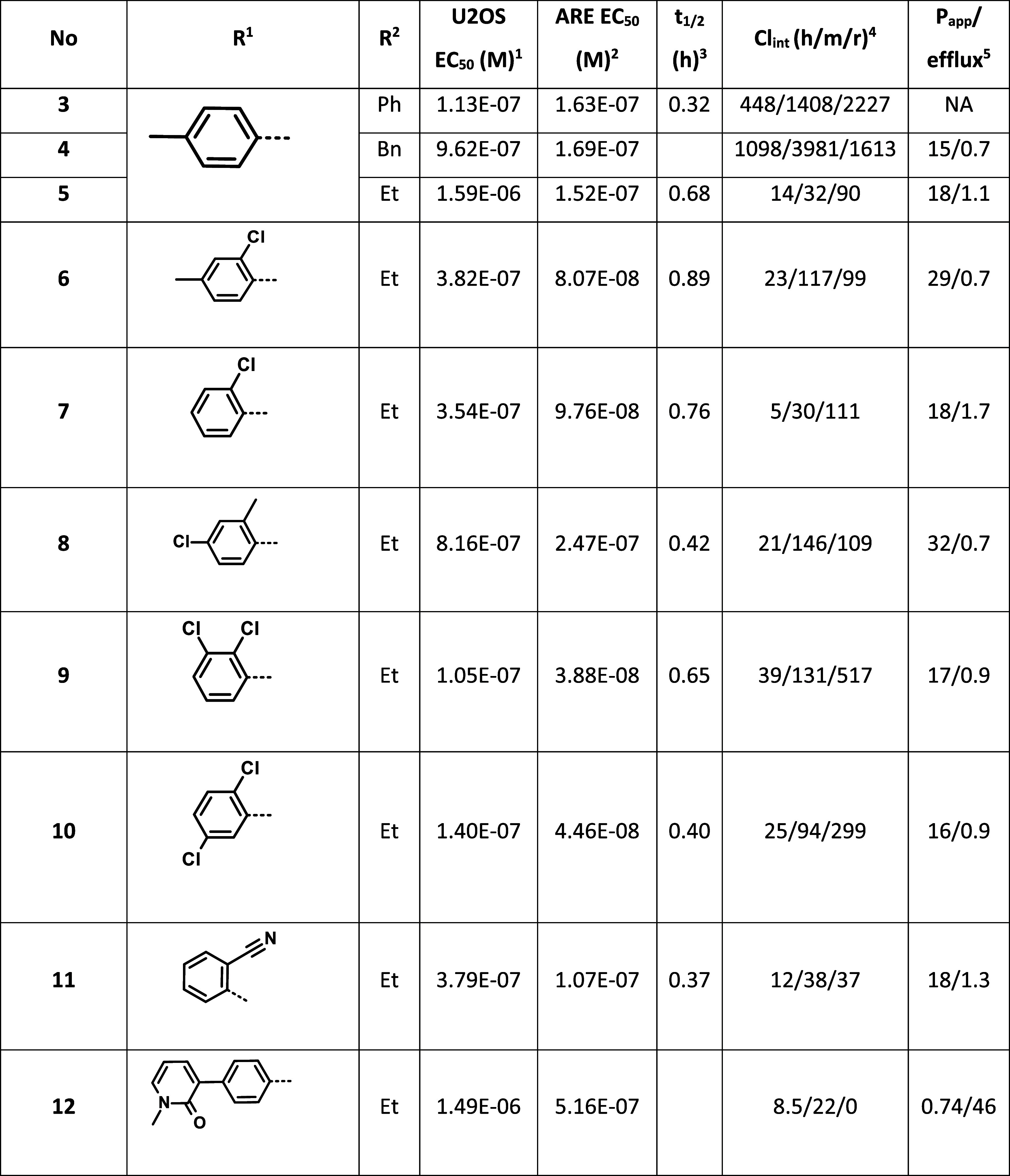
We found early on that modification of the piperazine ring to the lactam analogue 3 improved activity (Table 1). Besides the cellular experiments, we also wanted to assess the electrophilic reactivity of the selected compounds. To adjust the time window of this transformation to 2–4 h, we had to use a large excess of glutathione and increase its nucleophilicity by adding some base, so we incubated the compounds with 10 equiv of glutathione in the presence of 20 equiv of triethylamine and determined kinetics of the compound’s disappearance. For compound 3, this came to about 0.32 h. Unfortunately, the in vitro metabolic stability of 3 was poor, which we tried to address by modifying the R2 group to benzyl (4) and ethyl (5), respectively. In spite of some loss of activity in the complementation assay, coupled with decreased chemical reactivity, the other properties of 5 were clearly superior; therefore, we decided to fix the substituents in the 2-, and 5-positions and embark on a systematic variation of R1. Addition of a chlorine to the ortho-position (6) boosted potency but not through increased chemical reactivity and slightly deteriorated stability, which could be recovered by omission of the para-methyl substituent (7). Compound 7 possesses a balanced profile of activity and stability. We also tested its mode of action and found that on incubation with the BTB domain of KEAP1, a single adduct was formed, presumably through Cys151. Incubation of 7 with the C151A mutant of the same protein gave no covalent adduct.
Table 1. Hit Exploration and Variation of the Ra Substituent.

Measured activity in the Nrf2 translocation assay in the engineered U2OS cell line.
Measured activity in the ARE reporter assay in the HepG2 cell line.
Reactivity against 10 equiv of glutathione in the presence of 20 equiv of TEA expressed as the time needed to reach 50% conversion.
Predicted Clblood in microsomes in mL min–1 kg–1.
Apparent permeability (10–6 cm/s) and efflux ratio measured in Caco-2 cells using 10 μM compound concentration.
Swapping the methyl and chloro substituents in compound 6 to yield (8) had a detrimental effect on activity, coupled with the undesired increase of chemical reactivity. The presence of two chlorine atoms on the aromatic ring either in the 2,3 (9) or 2,5-positions (10) resulted in the most active compounds so far, with a favorable intermediate chemical reactivity, but unfortunately their microsomal clearance was inferior to 7. Swapping the chlorine of 7 with a cyano group (11) resulted in a compound with acceptable activity and good in vitro ADME profile, although with a moderately increased chemical reactivity. Finally, we wanted to probe the flexibility of the binding region by introducing a bulkier substituent (12), which led to about an order of magnitude loss in activity. To assess if the general reactivity of our compounds as electrophiles could interfere with the biological experiments, we incubated compounds 7 and 11 in mouse, rat, or human plasma for 5 h. Under these conditions, none of the compounds showed any decomposition, increasing our confidence in the studied chemotype. We further characterized 7 in vitro. In Nrf2 KO mouse embryonic fibroblast cells following treatment with 7, we did not observe any of the ARE signal, suggesting that 7 acted selectively through the Keap1-Nrf2 axis. Running a screen on the 36 receptor Cerep panel, we did not observe any significant off-target activity at a dose of 10 μM. We also checked the in vivo PK of 7. Administering the compound at 100 mg/kg to db/db mice, we observed a cmax of 2.7 μM after 30 min. The half-life of 1.4 h and AUC of 4.45 μM × h suggested that at this dose, we should also be able to observe its pharmacological effect. Indeed, after 6 and 24 h, we observed 199% and 379% overexpression of the mRNA of NQO1 (NAD(P)H quinone dehydrogenase 1), a gene of the ARE,17 respectively. We also repeated this study, along with bardoxolone as a positive control, in WT and Nrf2 −/– C57BI6 mice. In the WT mice, both compounds induced substantial Noq1 induction, which was completely absent in the KO mice in spite of normal exposure (for details see Supporting Information).
In the next wave of optimization, we fixed the ortho-chlorophenylsulfonyl substituent in the 4-position and modified the presumed leaving group in the 2-position (13–16). Introducing the smaller methylsulfonyl leaving group (13) increased activity in the cellular assays, probably through a considerable increase of chemical reactivity, as apparent by the shortest half-life observed so far. With respect to the balance of cellular activity and general chemical reactivity, the cyclopropyl (14) and cyclobutyl (15) analogues appeared to be the best so far. They showed similar activity to our lead 7 but with a significant drop of general reactivity with half-lives of 1.43 and 1.52 h, respectively (Table 3). Unfortunately, their in vitro ADME parameters deteriorated significantly compared to those of 7. We also tested the hydroxyethyl analogue 16 to observe that it possessed an unfavorable shift of the activity/reactivity ratio.
Table 3. Variation of the R2 and R3 Substituents.


Measured activity in the Nrf2 translocation assay in the engineered U2OS cell line.
Measured activity in the ARE reporter assay in the HepG2 cell line.
Reactivity against 10 equiv of glutathione in the presence of 20 equiv of TEA expressed as the time needed to reach 50% conversion.
Predicted Clblood in microsomes in mL min–1 kg–1.
Apparent permeability (10–6 cm/s) and efflux ratio measured in Caco-2 cells using 10 μM compound concentration.
Encouraged by the in vitro results, we progressed 11 and 14 into in vivo PK/PD studies. As expected on the basis of the in vitro ADME properties, both compounds showed a PK profile similar to that of 7 with slightly elevated cmax and AUC values (Table 2). Looking at the mRNA levels of NOQ1, we observed a strong response after 6 h (299%) for 11, while 14 was less active (149%). The activity of 11 decreased moderately (209%) after 24 h, while for 14, it remained practically unchanged (164%).
Table 2. In Vivo PK–PD and In Vitro Toxicity Data of Selected Compounds.
| Cmax (μM)a | AUC (μM h)a | NQO1 (6 h)b (%) | NQO1 (24 h)b (%) | DDR (TK6)/S9c | |
|---|---|---|---|---|---|
| 7 | 2.68 | 4.45 | +199 | +379 | clastogen/NA |
| 11 | 4.86 | 5.53 | +299 | +209 | clastogen/NA |
| 14 | 5.43 | 10.2 | +149 | +164 | negative/negative |
| 23 | 4.12 | 3.75 | +344 | +171 | negative/negative |
| 25 | 1.51 | 1.36 | +88 | +214 | negative/negative |
| 27 | 6.39 | 8.5 | +125 | +116 | negative/aneugen |
In vivo PK parameters measured in db/db mice following oral administration of 100 mg/kg of the compound in 1% HEC.
Changes in the Nqo1 mRNA levels in the kidney of db/db mice following compound treatment (100 mg/kg, 1% HEC, p.o.) compared to vehicle treatment.
DNA damage response assay results measured in TK6 cells.
In the last round of optimization, we varied the electron donating group in position 5 while fixing the ethylsulfonyl and ortho-chlorophenylsulfonyl moieties in positions 2 and 4, respectively (17–24). We found that the nature of this substituent had a profound effect on the activity. The 3-pyridyl analogue (17) was, for example, completely inactive. The formal opening of the piperazine ring at the distant nitrogen (18) was well tolerated, although it resulted in some loss of activity (Table 3). Introducing a condensed [1,2,4]triazole analogue (19) led to a compound whose activity was in the 100 nm range in both assays, while it possessed an increased chemical reactivity with a half-life of 0.2 h. Modifications of the piperazine ring through converting it to a sultam (20) or replacing the distant nitrogen by a methylphosphinyl group (21) had a similar detrimental effect. The activity dropped in both assays to the high nanomolar–low micromolar range. Introduction of a spirobicyclic piperidine analogue (22) decreased the activity even further, and we obtained only partial inhibition at the highest tested doses of 32 and 12 μM, respectively. We obtained the best results by adding a methyl group between the carbonyl group and the proximal ring nitrogen of the piperidone moiety. This modification generates chirality, and we isolated and tested both enantiomers (23, 24). We found that 23 was around 4 times more active than 24 (136 and 62 vs nM and 201 nM), while their general chemical reactivities were very similar. Unfortunately, this chemical modification also led to increased microsomal clearance. Before embarking on in vivo experiments, we tested the stability of 23 in whole blood for multiple species. The measured half-life for mouse was 298 min, while in rat, it decreased to 23 min. For nonrodent species, we observed half-lives of 127 min for dog, 147 min for monkey, and over 300 min for human. Testing 23 in our in vivo PK/PD model, we observed a good exposure with a very strong PD response after 6 h (NOQ1 mRNA at 344%), which was halved after 24 h (171%).
Having identified the methyl piperidone motif, we combined it with the 2-cyanophenyl moiety to get the pair of enantiomeric compounds 25 and 26 (Table 4). Similarly to the 23–24 pair, the R-enantiomer (25) was around 4 times more active than the S-enantiomer (26), and they both showed a decreased microsomal stability compared to the parent compound 11. We also prepared another combination compound carrying the 2,3-dichlorophenyl moiety in the 4-position and the 4-methyl-1,4-azaphosphinane-4-oxide in the 5-position (27). We were curious how the outstanding activity of the former (cf. 9) and the increased stability of the latter moiety (cf. 21) combine. The results were quite satisfactory since the cellular activity of 27 was around 8-fold better than that of 21, and its in vitro ADME properties were between those of 9 and 21. Although the enantiomer pairs showed some difference in activity, for the pharmacological studies, we needed a negative control of similar structure but significantly decreased activity. For this purpose, we prepared 28, a compound in which the ethylsulfonyl leaving group was replaced by an ethylene moiety. The utility of this compound was confirmed by the lack of any activity in the cellular assays.
Table 4. Combination Compounds 25–27 and Negative Control 28.


Measured activity in the Nrf2 translocation assay in the engineered U2OS cell line.
Measured activity in the ARE reporter assay in the HepG2 cell line.
Predicted Clblood in microsomes in mL min–1 kg–1.
Apparent permeability (10–6 cm/s) and efflux ratio measured in Caco-2 cells using 10 μM compound concentration.
We tested 25 and 27 in our in vivo PK/PD model too. While the high clearance of 25 translated into decreased exposure, the cmax and AUC values of 27 were quite favorable (Table 2). Looking at the NOQ1 mRNA levels, we found only a moderate increase for both compounds at the studied time points, underlining that for increased activity, one needs a combination of good exposure and high cellular activity.
Having some potent covalent KEAP1 inhibitors in hand, we set out to characterize their binding interactions by X-ray crystallography. Co-crystals with compounds 23 (2.33 Å, PDB code: 9ETX) and 25 (1.87 Å, PDB code: 9ETY) were generated after incubating the KEAP1 BTB+3box region for several hours with 2-fold excess compound. Clear adduct formation to Cys151 Sγ was observed for both compounds, with a consistent thiazole position that occupied the adjoining shallow groove (Figure 4 and Table S1). Additionally, both compounds interacted with a histidine pair His129 and His154, forming an extended π-stack. This may effectively restrict compound flexibility once bound and stabilize the adduct. A weak potential hydrogen bond was present between the sulphonyl oxygen and Arg135 (3.6 and 3.4 Å for 23 and 25, respectively). In the case of 25, the nitrile group participated in an additional interaction with Arg135 that may explain the increase in potency. The piperidone in both compounds had fewer interactions with the protein, with the ketone oxygen having the potential to form a weak electrostatic interaction with Lys150 (>3.6 Å).
Figure 4.

X-ray structure of the KEAP1 BTB+3 box dimer with bound inhibitors 23 and 25. Electron density 2Fo–Fc map contoured at 1.5 σ, colored orange showing covalent linkage between Cys151 and ligands. Models and electron densities shown are from chain A in both structures. KEAP1 protein and compounds are colored blue and orange, respectively.
Sequence alignments within the BTB-Kelch family revealed that Cys151 of KEAP1 was conserved only in KLHL26. To understand the basis for specificity and determine if KLHL26 could be targeted through the same mechanism, the BTB domain from KLHL26 was purified and crystallized (1.51 Å, PDB code: 9ETW). Determination of the structure showed good conservation of the BTB fold compared to that of KEAP1 (Figure S1 and Table S1). However, the structure revealed that the conserved KLHL26 Cys132 located on α5 was shielded from solvent by a salt bridge formed between KLHL26 Arg112 and Asp131. With this packing, the KLHL26 Cys132 side chain was rotated 156° away from the surface compared to KEAP1 Cys151. As such, the cysteine was not available for adduct formation and no binding of the tested compounds 23, 25, or 27 to the KLHL26 BTB domain was observed by LC–MS, in contrast to KEAP1 (Figures S2–S3).
Next, we sought to evaluate the selectivity of 23 on a proteome-wide scale using the isoTOP ABPP approach. We treated live MDA-MB-231 cells with 23, its companion negative control 28, or DMSO vehicle, respectively, for 3 h at 1 μM concentration, followed by addition of 100 μM iodoacetamide-alkyne (IAA) as a clickable and promiscuous, cysteine-reactive probe. For both 23 and the negative control 28, we found that, overall, less than 1% of the detected proteins were competed with a ratio of R > 4.5 (Figure 5). For 23, only two targets (RBM12B_C204, PLIN3_C39, and PLIN3_C60) were above the threshold out of a total of more than 3000 identified sites, suggesting good selectivity. In case of the KEAP1 negative control 28, only RBM12B_C204 was competed with a ratio >4.5.
Figure 5.

Waterfall plots of the isoTOP ABPP results for 23 and the negative control 28 in MDA-MB-231 cells. Competed peptides with a ligandability cutoff R > 4.5 are labeled.
To further study the selectivity and safety of 23, it was screened by DSF at 10 μM against a panel of 96 kinases,18 and the ΔTm values ranged between 0.9 and −1.9 °C (see Table S3 for details), suggesting no significant interaction. 23 was also tested at 10 μM against a panel of 45 receptors in a radioligand binding assay format,19 and following a 90 min incubation at room temperature, the maximal inhibition observed was 31% (see Table S2 for details), underlining the exquisite selectivity of this compound. Another way to assess the safety of our covalent KEAP1 inhibitors was to establish their in vitro genotoxicity in the DNA damage response (DDR) assays using the well-established human TK6 and insect SF9 cell lines.20 Of the tested compounds, 7 and 11 showed clastogenicity at 10 μM in the TK6 cell line, while the later compounds (14, 23, 25, and 27) were void of any genotoxic behavior in either cell lines (Table 2), further highlighting their safety.
Conclusions
The inhibition of the NRF2-KEAP1 interaction has high clinical significance and therefore represents an active area of intense research. Covalent modification of the BTB domain in KEAP1, which harbors a significant number of Cys residues, is a proven strategy to disrupt KEAP1 activity toward NRF2, but most of the known covalent inhibitors carry undesired side effects originating in their covalent nature. Running a phenotypic HTS campaign, we identified a new covalent inhibitor chemotype, and with systematic optimization, we delivered a series of potent KEAP1 binders that facilitated the dissociation and nuclear translocation of NRF2. The potent cellular activity of our inhibitors was also recapitulated in in vivo PK–PD experiments through the marked upregulation of ARE-dependent target genes. The molecular mechanism of KEAP1 inhibition was demonstrated through X-ray crystallography and mass spectrometry measurements. The new covalent inhibitor chemotype also conferred unprecedented selectivity to our inhibitors. The lead compound showed no significant interaction with a panel of commonly studied receptors or with a broad panel of kinases. Furthermore, it was remarkably selective in an activity-based protein profiling experiment on a cohort of over 6000 proteins and showed no genotoxicity in in vitro DDR studies on cell lines of different origin. Together, these results highlight the exquisite selectivity of our new KEAP1 covalent inhibitor chemotype and suggest that this chemical motif might have great potential in the future for the design of selective covalent inhibitors for other targets.
Experimental Section
Chemical Reactivity Measurement of Selected Compounds toward GSH
To the freshly prepared solution of the test compound (1 micromol) and 6.2 mg glutathione (20 micromol) in 1 mL of DMSO, 5.6 μL of triethylamine (40 micromol) was added, and the reaction was followed by measuring the decrease of the concentration of the test compound by injecting the same aliquots into LC-UV/MS. First time point was acquired immediately after the addition of the base. Under the described conditions or solubility issues, no precipitation was observed during the experiments.
Metabolic Stability Measurement
Compounds were tested at a final concentration of 0.1 μM in duplicate. Compounds were preincubated with pooled liver microsomes (0.1 mg/mL) for 10 min in a 37 °C shaking water bath.
The reaction was initiated by adding an NADPH-generating system and incubating at 37 °C in a shaking water bath. Several aliquots (0, 5, 15, 30, and 45 min) of the incubation were collected and transferred to a quench solution. Then, samples were centrifuged, and the supernatants were analyzed by HPLC-MS/MS for the determination of the in vitro half-life (t1/2). The intrinsic clearance (Clint) was calculated as follows
Caco2 Permeability Measurement
Compound was incubated at 10 μM in the acceptor chamber in HBSS containing 0.1% BSA. Samples (in duplicate) were collected at 120 min in the receiver chamber and analyzed by LC–MS/MS. The apparent permeability in apical to basolateral direction (Papp) and in basolateral to apical direction (PappBA) was calculated as follows
where Vr: volume of the receiver chamber; A: surface area of the membrane.
The efflux ratio was calculated as follows: Efflux ratio = Papp BA/Papp.
Cellular Experiments
HepG2—Gene Reporter Assay
Nrf2 transcription factor will activate protective genes from oxidative damage through binding on the ARE. We used the transfection factor property of Nrf2 to develop a gene reporter assay based on the activation of the beta-lactamase reporter gene under the control of the ARE and Nrf2 activation.
The CellSensor ARE-bla HepG2 cell line (ref.K1208, Invitrogen) containing a beta-lactamase reporter gene under control of the ARE was stably integrated into HepG2 cells.
CellSensor ARE-bla HepG2 cells were grown to confluence in DMEM GlutaMAXTM (ref.61965–026, Thermo Fisher), 10% dialyzed FBS (ref.P30–193306, PanBiotech), 12 mM HEPES (ref.15630–056, Gibco), 0.1 mM non-essential amino acid (NEAA) cell culture supplement (ref.11140–35, Gibco), 1% Na-pyruvate (ref.S8636, Sigma), 2.5 μg/mL blasticidin (ref.210–01, Invitrogen), and 1% penicillin/streptomycin (15070–063, Gibco) in collagen I (50 μg/mL, ref.A10483–1, Life Technologies) coated flasks, 37 °C, 5% CO2.
Eighteen hours before the experiment, cells were harvested using TrypLE Xpress (ref.126905, Bibco) for 10 min at 37 °C, resuspended in DMEM GlutaMAXTM (ref.61965–026, Thermo Fisher), 1% dialyzed FBS (ref.P30–193306, PanBiotech), 25 mM HEPES (ref.15630–056, Gibco), 0.1 mM NEAA (ref.11140–35, Gibco), 1% Na-pyruvate (ref.S8636, Sigma), 2.5 μg/mL blasticidin (ref.210–01, Invitrogen), and 1% penicillin/streptomycin (15070–063, Gibco) and then plated into 384 wells cell culture microclear plates (ref.781091, Greiner) at the density of 30 000 cells/well in 32 μL. Cell were stored at 37 °C with 5% CO2 until used.
Compounds in 100% DMSO (0.315 μL/well) were resuspended in 20 μL of DMEM GlutaMAXTM, 1% dialyzed FBS, 25 mM HEPES, 0.1 mM NEAA, 1% Na-pyruvate (ref.S8636, Sigma), 2.5 μg/mL blasticidin, 3.4% DMSO, and 1% penicillin/streptomycin (15070–063, Gibco). Compounds and andrographolide (10 μM final concentration; ref.365645–500MG, Aldrich) as positive control were dispensed on cell, 8 μL/well, and then incubated for 16 h at 37 °C and 5% CO2. The day after, Live Blazer reagent (Live Blazer FRET B/G (CCF4-AM), ref.K1089, Invitrogen) was dispensed on cells (8 μL/well) and incubated for 2 h at room temperature in the dark.
Then, fluorescence resonance energy transfer (FRET) signal was measured using multimodal reader (Ex 409 nM/Em 460 nM and 530 nM; Envision, PerkinElmer). Data were normalized between 1% DMSO (basal signal) and 10 μM andrographolide (positive signal) and analyzed using Activity Base software.
U2OS—Translocation Assay
U2OS cells have been stably transduced using MMLV-derived retroviral vector to overexpress Nrf2-enzyme donor fusion protein and acceptor enzyme in the nucleus (PathHunter U2OS Keap1-NRF2 Nuclear Translocation Cell Line, ref.93–0821C3, DiscoverX).
PathHunter U2OS cells were grown in minimum essential medium (MEM) (ref.30–2003, ATCC), 10% FBS (ref.P30–193306, PanBiotech), 500 μg/mL geneticin, 250 μg/mL hygromycin, and 0.25 μg/mL puromycin (10131–027, 10687–010, and ref.A1113802, respectively, Life Technologies).
The night before experiment, cells were harvested using TrypLE Xpress (ref.126905, Bibco) for 5 min at 37 °C, resuspended in Opti-MEMTM (ref.31985–047, Gibco) and 1% FBS (ref.P30–193306, PanBiotech) and then plated into 384 well plates (ref.6007680, PerkinElmer) at the density of 7500 cells/well in 20 μL. Cell were stored overnight at 37 °C and 5% CO2 until used.
Compounds in 100% DMSO (0.315 μL/well) were resuspended in 20 μL of MEM, 1% FBS, and 3.4% DMSO. Compounds and andrographolide (10 μM final concentration; ref.365645–500MG, Aldrich) as positive control were dispensed on cell, 5 μL/well, and then incubated for 3h at 37 °C and 5% CO2. After incubation, PathHunter reagents were dispensed on cells (12 μL/well, ref.93–0001, DiscoverX) and incubated for 60 min at room temperature in the dark.
Then, the luminescence signal was measured using a multimodal reader (PHERAstar, BMG Labtech). Data were normalized between 1% DMSO (basal signal) and 10 μM andrographolide (positive signal) and analyzed using Activity Base software.
DDR Assay
The aim of the study is to assess DDR in TK6 cells treated with the compounds with and without metabolic activation using a Multiflow DNA Damage Kit. TK6 cells from ATCC (American Type Culture Collection) were cultured in RPMI 10 (RPMI supplemented with 10% inactivated horse serum, pluronic acid, l-glutamine, sodium pyruvate, and amphotericin B) in a 75 mL flask at 37 °C with 5% CO2 and 95% humidity.
S9 mix preparation: a mixture of 50 mM of KCl, 1 mL of 5 mg/mL NADP, and 1 mL of 60 mg/mL glucose-6-phosphate is prepared and filtered. For every 3 mL of the mix, 2 mL of S9 is added and directly diluted in RPMI 10 (1/10 v/v) prior to cell treatment.
Stock solution of the tested compound was prepared in DMSO at a concentration of 100 mg/mL. This solution was diluted in RPMI10 ∓ S9 at 1/50 (v/v) to obtain the desired test concentration of up to 1000 μg/mL in the main assay.
In a 96-well plate, 6 × 104 cells were incubated with the solution of the tested compound at different concentrations ranging from 4.23 to 1000 μg/mL (DMSO concentration not higher than 1%) in the following 4 conditions: 4 h with compound ± S9 followed by 20h recovery, 4 h with compound and S9, and 24 h with compound without S9, all in a final volume of 200 μL. Methyl methanesulfonate (200 μM), vinblastine (24 nM), and griseofulvin (60 μg/mL) were used as positive controls in duplicate cell cultures in the 4 h and 24 treatment schedules without metabolic activation. Cyclophosphamide (40 μg/mL) was used as positive control in duplicate cell cultures in 4 h treatment with metabolic activation.
Following the treatment, a Multiflow kit was used to label the nucleus (DNA stain), p53, yH2AX, and phosphor-histone H3. Briefly, cells in the 96 plates were resuspended, and 50 μL aliquots of the suspension were transferred into a 96 V bottom well plate and centrifuged for 6 min at 1000 rpm. Supernatants were discarded, and the cells were resuspended in 25 μL of RPMI 10 and transferred into a 96 U-bottom plate containing 50 μL of the complete labeling solution in every well and incubated for 30 min at room temperature protected from light. For the cell incubated with S9, an additional washing step with 150 μL of RPMI10 was performed before the incubation.
Data acquisition was performed by flow cytometry to assess cytotoxicity (at 24 h), fold induction of polyploidy (at 24 h), phospho-histone H3 (at 4 and 24 h), nuclear p53 (at 4 and 24 h), and yH2AX (at 4 and 24 h). Classification of the test item as clastogen and aneugen was based on a comparison of the fold induction to the global evaluation factor (GEF) reported in the table below. The tested compound was considered clastogen or aneugen if 2 successive concentrations met or exceed the GEF for at least 2 out of 4 clastogen or aneugen markers listed below.
Clastogenicity −4 h-γH2AX: 1.51, 4 h-p53:1.4, 24 h-γH2AX: 2.11, 24 h-p53:1.45.
Aneugenicity −4 h-p-H3:1.71, 24 h-p-H3:2.52, 24 h-polyploidy: 5.86, 24 h-p53:1.45.
In Vivo Studies
Male db/db mice (8–10 weeks old, Janvier Laboratories) were maintained on a 12:12 h light/dark cycle at 21 ± 2 °C and had ad libitum access to tap water and 5K52 diet. All procedures were performed according to the ethical protocol that has been approved by the Servier Institutional Animal Care and Use Committee in accordance with the French regulations (Decree no 2013–118 from 01 February 2013 relative to the protection of animals used for scientific purposes and 4 orders of 01 February 2013).
PK/PD Study
Fed mice received either the treatment with compounds (100 mg/kg) or vehicle (hydroxyethyl cellulose (HEC) 1% for all compounds except for 7, for which the vehicle was Phosal 50PG/Ethanol/PEG400 (m/m/m)) by oral gavage. Blood sampling was performed at different time points after treatment (0.5, 1, 2, 4, 6, and 24 h, with 2 blood sampling per mouse) for PK measurement. Mice were then euthanized at different time points (4, 6, and 24 h), and the liver was sampled and snap-frozen for PD measurement (liver NQO1 mRNA).
Synthesis
The general synthetic remarks as well as the synthesis of the intermediates are described in the Supporting Information. All obtained products had an LC purity above 96% that was corroborated by their 1H/13C NMR spectra unless specifically mentioned otherwise.
General Procedure I–SNAr Reaction on Chlorothiazoles
1 equiv of the appropriate 5-chloro-sulfonyl-thiazole derivative, 1.5–2 equiv of the appropriate amine or its HCl salt, and 1.5–3 equiv of DIPEA if the amine is HCl salt were refluxed in acetonitrile or in a 1:1 mixture of acetonitrile and ethanol until no further conversion was observed. Celite was added to the reaction mixture, and the volatiles were evaporated under reduced pressure. The solid residue was purified by flash chromatography on silica gel using DCM and methanol as eluents.
4-[2-(Benzenesulfonyl)-4-(p-tolylsulfonyl)thiazol-5-yl]morpholine (1)
Starting from 0.300 g of 1f (0.72 mmol) and 0.126 mL of morpholine (1.44 mmol) in 4 mL of acetonitrile–ethanol following General Procedure I, 0.231 g (69%) of 1 was obtained.
1H NMR (400 MHz, DMSO-d6): δ ppm 7.82–7.72 (m, 3H), 7.67 (d, J = 8.3 Hz, 2H), 7.64–7.54 (m, 2H), 7.38 (d, J = 8.2 Hz, 2H), 3.75 (m, 4H), 3.4 (m, 4H), 2.42 (s, 3H); HRMS (TOF, ESI) m/z: calcd for C20H21N2O5S3 [M + H]+ 465.0607. Found: 465.0606.
2-(Benzenesulfonyl)-5-(4-methylpiperazin-1-yl)-4-(p-tolylsulfonyl)thiazole (2)
Starting from 0.190 g of 1f (0.72 mmol) and 0.10 mL of 1-methylpiperazine (0.92 mmol) in 2 mL of acetonitrile–ethanol following General Procedure I, 0.164 g (75%) of 2 was obtained.
1H NMR (500 MHz, DMSO-d6): δ ppm 7.76 (m, 1H), 7.73 (m, 2H), 7.65 (m, 2H), 7.58 (m, 2H), 7.37 (d, J = 8.5 Hz, 2H), 3.4 (m, 4H), 2.47 (m, 4H), 2.42 (s, 3H), 2.22 (s, 3H); 13C NMR (125 MHz, DMSO-d6): δ ppm 164, 149.5, 145.1, 138.6, 137.9, 135.15, 135.1, 130.3, 130.2, 128.2, 128.1, 55.3, 53.9, 45.9, 21.6; HRMS (TOF, ESI) m/z: calcd for C21H24N3O4S3 [M + H]+ 478.0923. Found: 478.0925.
4-[2-(Benzenesulfonyl)-4-(p-tolylsulfonyl)thiazol-5-yl]-1-methyl-piperazin-2-one (3)
Starting from 0.190 g of 1f (0.72 mmol) and 0.105 mg of 1-methylpiperazin-2-one (0.92 mmol) in 2 mL of acetonitrile–ethanol following General Procedure I, 0.165 g (73%) of 3 was obtained.
1H NMR (500 MHz, DMSO-d6): δ ppm 7.8–7.54 (m, 5H), 7.67 (m, 2H), 7.37 (d, J = 8.5 Hz, 2H), 4.03 (s, 2H), 3.74 (m, 2H), 3.51 (m, 2H), 2.9 (s, 3H), 2.42 (s, 3H); 13C NMR (125 MHz, DMSO-d6): δ ppm 163.7, 162.0, 149.7, 145.2, 138.5, 137.9, 135.2, 134.9, 130.3, 130.2, 128.2, 128.1, 57, 52.7, 46.9, 34, 21.6; HRMS (TOF, ESI) m/z: calcd for C21H22N3O5S3 [M + H]+ 492.0716. Found: 492.0715.
4-[2-Benzylsulfonyl-4-(p-tolylsulfonyl)thiazol-5-yl]-1-methyl-piperazin-2-one (4)
Starting from 0.200 g of 2-benzylsulfonyl-5-chloro-4-(p-tolylsulfonyl)thiazole (0.47 mmol) and 0.081 g of 1-methylpiperazin-2-one (0.71 mmol) in 3 mL of acetonitrile following General Procedure I, 0.186 g (78%) of 4 was obtained.
1H NMR (400 MHz, DMSO-d6): δ ppm 7.87 (d, J = 8.3 Hz, 2H), 7.52 (d, J = 8.1 Hz, 2H), 7.31 (m, 1H), 7.20 (t, J = 7.6 Hz, 2H), 6.89 (d, J = 7.4 Hz, 2H), 4.78 (s, 2H), 3.89 (s, 2H), 3.65 (m, 2H), 3.48 (m, 2H), 2.89 (s, 3H), 2.41 (s, 3H); 13C NMR (400 MHz, DMSO-d6): δ ppm 163.2, 161.8, 149.3, 145.0, 137.6, 135.2, 131.0, 130.0, 128.8, 128.5, 127.8, 127.2, 59.8, 56.7, 52.2, 46.4, 33.5, 21.1; HRMS (TOF, ESI) m/z: calcd for C22H24N3O5S3 [M + H]+ 506.0873. Found: 506.0848.
4-[2-Ethylsulfonyl-4-(p-tolylsulfonyl)thiazol-5-yl]-1-methyl-piperazin-2-one (5)
Starting from 0.175 g of 5-chloro-2-ethylsulfonyl-4-(p-tolylsulfonyl)thiazole (5b, 0.48 mmol) and 0.082 g of 1-methylpiperazin-2-one (0.72 mmol) in 2 mL of acetonitrile following General Procedure I, 0.167 g (78%) of 5 was obtained.
1H NMR (500 MHz, DMSO-d6): δ ppm 7.8 (m, 2H), 7.45 (m, 2H), 4.03 (s, 2H), 3.74 (m, 2H), 3.52 (m, 2H), 3.37 (q, J = 7.3 Hz, 2H), 2.91 (s, 3H), 2.4 (s, 3H), 1.01 (t, J = 7.3 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ ppm 163.8, 162.1, 149.2, 145.3, 138.0, 135.0, 130.3, 128.1, 57.1, 52.6, 49.3, 47, 34, 21.6, 7.4; HRMS (TOF, ESI) m/z: calcd for C17H22N3O5S3 [M + H]+ 444.0716. Found: 444.0696.
4-[4-(2-Chloro-4-methyl-phenyl)sulfonyl-2-ethylsulfonyl-thiazol-5-yl]-1-methyl-piperazin-2-one (6)
Starting from 0.143 g of 6e (0.36 mmol) and 0.082 g of 1-methylpiperazin-2-one (0.72 mmol) in 3 mL of acetonitrile following General Procedure I, 0.151 g (88%) of 6 was obtained.
1H NMR (500 MHz, DMSO-d6): δ ppm 8.07 (d, J = 8.1 Hz, 1H), 7.52 (m, 1H), 7.46 (m, 1H), 4.09 (s, 2H), 3.79 (m, 2H), 3.49 (m, 2H), 3.28 (q, J = 7.3 Hz, 2H), 2.9 (s, 3H), 2.41 (s, 3H), 0.99 (t, J = 7.3 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ ppm 163.7, 162.7, 147.8, 147.4, 135.2, 133.0, 132.5, 132.1, 131.0, 128.8, 57.2, 52.6, 49.4, 46.8, 34.0, 21.1, 7.4; HRMS (TOF, ESI) m/z: calcd for C17H21ClN3O5S3 [M + H]+ 478.0326. Found: 478.0338.
4-[4-(2-Chlorophenyl)sulfonyl-2-ethylsulfonyl-thiazol-5-yl]-1-methyl-piperazin-2-one (7)
Starting from 0.279 g of 7e (0.72 mmol) and 0.223 g of 1-methylpiperazin-2-one (1.08 mmol) in 10 mL of acetonitrile following General Procedure I, 0.256 g (77%) of 7 was obtained.
1H NMR (500 MHz, DMSO-d6): δ ppm 8.20 (dd, J = 8.0, 1.4 Hz, 1H), 7.76 (m, 1H), 7.67 (d, J = 7.5 Hz, 1H), 7.66 (m, 1H), 4.11 (s, 2H), 3.8 (m, 2H), 3.51 (m, 2H), 3.26 (q, J = 7.3 Hz, 2H), 2.9 (s, 3H), 0.96 (t, J = 7.3 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ ppm 163.7, 162.9, 147.8, 138.1, 136.2, 132.5, 132.33, 132.27, 131.1, 128.4, 57.3, 52.7, 49.4, 46.8, 34.0, 7.4; HRMS (TOF, ESI) m/z: calcd for C16H19ClN3O5S3 [M + H]+ 464.017. Found: 464.0169.
4-[4-(4-Chloro-2-methyl-phenyl)sulfonyl-2-ethylsulfonyl-thiazol-5-yl]-1-methyl-piperazin-2-one (8)
Starting from 0.150 g of 8c (0.37 mmol) and 0.084 g of 1-methylpiperazin-2-one (0.74 mmol) in 5 mL of acetonitrile following General Procedure I, 0.101 g (57%) of 8 was obtained.
1H NMR (400 MHz, DMSO-d6): δ ppm 8.00 (d, J = 8.3 Hz, 1H), 7.58 (brs., 1H), 7.57 (dd, J = 8.3, 2.1 Hz, 1H), 4.06 (s, 2H), 3.76 (m, 2H), 3.33 (q, J = 7.3 Hz, 2H), 2.9 (s, 3H), 2.31 (s, 3H), 0.97 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ ppm 163.7, 162.3, 148.5, 140.8, 139.3, 137.9, 133.7, 132.6, 131.4, 127.1, 57.1, 52.6, 49.4, 46.8, 34.0, 19.5, 7.3; HRMS (TOF, ESI) m/z: calcd for C17H21ClN3O5S3 [M + H]+ 478.0326. Found: 478.0326.
4-[4-(2,3-Dichlorophenyl)sulfonyl-2-ethylsulfonyl-thiazol-5-yl]-1-methyl-piperazin-2-one (9)
Starting from 0.200 g of 9c (0.48 mmol) and 0.110 g of 1-methylpiperazin-2-one (0.96 mmol) in 3 mL of acetonitrile following General Procedure I, 0.191 g (80%) of 9 was obtained.
1H NMR (500 MHz, DMSO-d6): δ ppm 8.21 (dd, J = 8.0, 1.5 Hz, 1H), 8.06 (dd, J = 8.0, 1.5 Hz, 1H), 7.69 (t, J = 8.0 Hz, 1H), 4.13 (s, 2H), 3.82 (m, 2H), 3.53 (m, 2H), 3.27 (q, J = 7.3 Hz, 2H), 2.91 (s, 3H), 0.94 (t, J = 7.3 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ ppm 163.6, 163.3, 147.5, 140.3, 136.4, 134.7, 131.3, 130.4, 130.0, 129.4, 57.3, 52.8, 49.4, 46.7, 34.0, 7.4; HRMS (TOF, ESI) m/z: calcd for C16H18Cl2N3O5S3 [M + H]+ 497.978. Found: 497.9774.
4-[4-(2,5-Dichlorophenyl)sulfonyl-2-ethylsulfonyl-thiazol-5-yl]-1-methyl-piperazin-2-one (10)
Starting from 0.250 g of 10e (0.59 mmol) and 0.136 g of 1-methylpiperazin-2-one (1.19 mmol) in 5 mL of acetonitrile following General Procedure I, 0.21 g (71%) of 10 was obtained.
1H NMR (500 MHz, DMSO-d6): δ ppm 8.14 (d, J = 2.6 Hz, 1H), 8.87 (dd, J = 8.6, 2.6 Hz, 1H), 7.73 (d, J = 8.6 Hz, 1H), 4.10 (s, 2H), 3.79 (m, 2H), 3.51 (m, 2H), 3.31 (q, J = 7.3 Hz, 2H), 2.91 (s, 3H), 0.99 (t, J = 7.3 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ ppm 163.6, 163.4, 148.1, 139.6, 135.9, 134.1, 133.0, 131.5, 131.2, 130.2, 57.3, 52.7, 49.4, 46.7, 34.0, 7.5; HRMS (TOF, ESI) m/z: calcd for C16H18Cl2N3O5S3 [M + H]+ 497.978. Found: 497.9765.
2-[2-Ethylsulfonyl-5-(4-methyl-3-oxo-piperazin-1-yl)thiazol-4-yl]sulfonylbenzonitrile (11)
Starting from 0.150 g of 11e (0.4 mmol) and 0.91 g of 1-methylpiperazin-2-one (0.8 mmol) in 3 mL of acetonitrile following General Procedure I, 0.147 g (81%) of 11 was obtained.
1H NMR (500 MHz, DMSO-d6): δ ppm 8.24 (dd, J = 7.9, 1.2 Hz, 1H), 8.16 (dd, J = 7.6, 1.3 Hz, 1H), 8 (td, J = 7.8, 1.4 Hz, 1H), 7.94 (td, J = 7.6, 1.3 Hz, 1H), 4.1 (s, 2H), 3.83 (m, 2H), 3.54 (m, 2H), 3.29 (q, J = 7.3 Hz, 2H), 2.91 (s, 3H), 0.95 (t, J = 7.3 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ ppm 163.6, 163.4, 148, 142.3, 136.3, 135.1, 134.5, 131.4, 130.1, 115.5, 110.6, 57.3, 52.7, 49.3, 46.7, 34.1, 7.4; HRMS (TOF, ESI) m/z: calcd for C17H19N4O5S3 [M + H]+ 455.0512. Found: 455.0520.
4-[2-Ethylsulfonyl-4-[4-(1-methyl-2-oxo-3-pyridyl)phenyl]sulfonyl-thiazol-5-yl]-1-methyl-piperazin-2-one (12)
Starting from 0.140 g of 12e (0.31 mmol) and 0.07 g of 1-methylpiperazin-2-one (0.61 mmol) in 1.2 mL of acetonitrile following General Procedure I, 0.075 g (46%) of 12 was obtained.
1H NMR (400 MHz, DMSO-d6): δ ppm 7.98 (dm, 2H), 7.91 (dm, 2H), 7.84 (dd, J = 6.7, 2.0 Hz, 1H), 7.75 (dd, J = 7.1, 2.0 Hz, 1H), 6.38 (t, J = 6.9 Hz, 1H), 4.06 (s, 2H), 3.77 (m, 2H), 3.54 (m, 2H), 3.52 (s, 3H), 3.37 (q, J = 7.3 Hz, 2H), 2.92 (s, 3H), 1.00 (t, J = 7.3 Hz, 3H); HRMS (TOF, ESI) m/z: calcd for C22H25N4O6S3 [M + H]+ 537.0931. Found: 537.0932.
4-[4-(2-Chlorophenyl)sulfonyl-2-methylsulfonyl-thiazol-5-yl]-1-methyl-piperazin-2-one (13)
Starting from 0.219 g of 13a (0.59 mmol) and 0.101 g of 1-methylpiperazin-2-one (0.89 mmol) in 3 mL of acetonitrile following General Procedure I, 0.186 g (70%) of 13 was obtained.
1H NMR (500 MHz, DMSO-d6): δ ppm 8.2 (dm, 1H), 7.77 (m, 1H), 7.68 (dm, 1H), 7.66 (m, 1H), 4.07 (s, 2H), 3.77 (m, 2H), 3.48 (m, 2H), 3.22 (s, 3H), 2.9 (s, 3H); 13C NMR (125 MHz, DMSO-d6): δ ppm 163.6, 162.5, 150.1, 138.0, 136.2, 132.8, 132.4, 132.3, 131.0, 128.4, 57.2, 52.5, 46.8, 42.8, 34.0; HRMS (TOF, ESI) m/z: calcd for C15H17ClN3O5S3 [M + H]+ 450.0013. Found: 450.0011.
4-[4-(2-Chlorophenyl)sulfonyl-2-cyclopropylsulfonyl-thiazol-5-yl]-1-methyl-piperazin-2-one (14)
Starting from 0.180 g of 14c (0.45 mmol) and 0.103 g of 1-methylpiperazin-2-one (0.90 mmol) in 3 mL of acetonitrile following General Procedure I, 0.179 g (84%) of 14 was obtained.
1H NMR (500 MHz, DMSO-d6): δ ppm 8.2 (dm, 1H), 7.8–7.62 (m, 3H), 4.11 (s, 2H), 3.81 (m, 2H), 3.51 (m, 2H), 2.9 (s, 3H), 2.77 (m, 1H), 1.03–0.89 (m, 4H); 13C NMR (125 MHz, DMSO-d6): δ ppm 163.7, 162.4, 148.7, 138.1, 136.1, 132.5, 132.4, 132.3, 131.0, 128.3, 57.2, 52.6, 46.8, 34.0, 31.9, 6.4; HRMS (TOF, ESI) m/z: calcd for C17H19ClN3O5S3 [M + H]+ 476.017. Found: 476.0171.
4-[4-(2-Chlorophenyl)sulfonyl-2-cyclobutylsulfonyl-thiazol-5-yl]-1-methyl-piperazin-2-one (15)
Starting from 0.880 g of 15c (2.13 mmol) and 0.486 g of 1-methylpiperazin-2-one (4.26 mmol) in 30 mL of acetonitrile following General Procedure I, 0.810 g (78%) of 15 was obtained.
1H NMR (500 MHz, DMSO-d6): δ ppm 8.21 (dm, 1H), 7.76 (m, 1H), 7.69 (dm, 1H), 7.66 (m, 1H), 4.12 (s, 2H), 4.04 (qui, J = 8.2 Hz, 1H), 3.82 (m, 2H), 3.52 (m, 2H), 2.91 (s, 3H), 2.09 (m, 2H), 2.01 (m, 2H), 1.85 (m, 1H), 1.72 (m, 1H); 13C NMR (125 MHz, DMSO-d6): δ ppm 163.7, 162.4, 148.7, 138.1, 136.1, 132.5, 132.4, 132.3, 131.0, 128.3, 57.2, 52.6, 46.8, 34.0, 31.9, 6.4; HRMS (TOF, ESI) m/z: calcd for C18H21ClN3O5S3 [M + H]+ 490.0326. Found: 490.0329.
4-[4-(2-Chlorophenyl)sulfonyl-2-(2-hydroxyethylsulfonyl)thiazol-5-yl]-1-methyl-piperazin-2-one (16)
The mixture of 0.17 g of 16c (0.42 mmol) and 0.12 g of 1-methylpiperazin-2-one (1.05 mmol) in 10 mL of acetonitrile was refluxed for 4 h. The reaction mixture was concentrated under reduced pressure. The crude product was purified by preparative HPLC (C18) using a 5 mM aqueous NH4HCO3 solution and acetonitrile as eluents to give 0.095 g (47%) of 16.
1H NMR (500 MHz, DMSO-d6): δ ppm 8.18 (dm, 1H), 7.76 (m, 1H), 7.66 (m, 2H), 4.97 (s, J = 5.3 Hz, 1H), 4.05 (s, 2H), 3.75 (m, 2H), 3.6 (q, J = 5.9 Hz, 2H), 3.46 (m, 2H), 3.44 (t, J = 6.2 Hz, 2H), 2.89 (s, 3H); 13C NMR (125 MHz, DMSO-d6): δ ppm 163.6, 162.7, 149.8, 138.0, 136.2, 132.9, 132.4, 132.3, 131.0, 128.4, 57.4, 57.2, 55.0, 52.4, 46.8, 34.0; HRMS (TOF, ESI) m/z: calcd for C16H19ClN3O6S3 [M + H]+ 480.0119. Found: 480.0126.
4-(2-Chlorophenyl)sulfonyl-2-ethylsulfonyl-5-(3-pyridyl)thiazole Hydrochloride (17)
To 0.386 g of 7e (1.0 mmol), 0.226 g of 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine (1.1 mmol), and 0.106 g of Na2CO3 (1.0 mmol) in 5 mL of acetonitrile and 2.5 mL of water, 0.058 g of Pd(PPh3)4 (0.05 mmol) was added, and the mixture was heated at 120 °C under nitrogen in a microwave reactor for 20 min. The reaction mixture was filtered through a Celite pad. The filtrate was concentrated under reduced pressure. The residue was extracted with DCM, and the organic phase was dried over Na2SO4 and then filtered. The filtrate was concentrated under reduced pressure, and the residue was purified by column chromatography on silica gel (0% → 3% methanol/DCM). The product was dissolved in 0.05 M aqueous HCl and then lyophilized to afford 0.046 g (10%) of 17.
1H NMR (500 MHz, DMSO-d6): δ ppm 8.78 (dd, J = 2.3, 0.7 Hz, 1H), 8.73 (dd, J = 4.9, 1.6 Hz, 1H), 8.08 (dm, 1H), 8.05 (dd, J = 7.9, 1.6 Hz, 1H), 7.75 (dt, J = 7.7, 1.7 Hz, 1H), 7.67 (dd, J = 8.0, 1.1 Hz, 1H), 7.59 (m, 2H), 3.58 (q, J = 7.4 Hz, 2H), 1.15 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ ppm 164.5, 151.5, 150.0, 149.0, 147.4, 139.0, 136.8, 136.6, 132.4, 132.2, 131.3, 128.6, 123.9, 123.7, 49.5, 7.4; HRMS (TOF, ESI) m/z: calcd for C16H14ClN2O4S3 [M + H]+ 428.9799. Found: 428.9802.
2-[[4-(2-Chlorophenyl)sulfonyl-2-ethylsulfonyl-thiazol-5-yl]-ethyl-amino]-N-methyl-acetamide (18)
Starting from 0.200 g of 7e (0.52 mmol) and 0.151 g of 2-(ethylamino)-N-methyl-acetamide (1.3 mmol) in 3 mL of acetonitrile following General Procedure I, 0.202 g (83%) of 18 was obtained.
1H NMR (500 MHz, DMSO-d6): δ ppm 8.18 (dm, 1H), 8.02 (brq., J = 4.6 Hz, 1H), 7.75 (m, 1H), 7.66 (dm, 1H), 7.65 (m, 1H), 4.23 (s, 2H), 3.5 (q, J = 7.2 Hz, 2H), 3.25 (q, J = 7.3 Hz, 2H), 2.6 (d, J = 4.6 Hz, 3H), 1.12 (t, J = 7.1 Hz, 3H), 0.95 (t, J = 7.3 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ ppm 167.9, 164.8, 147.1, 138.4, 136, 132.5, 132.3, 132.2, 131, 128.4, 58.5, 53.8, 49.3, 25.9, 12.7, 7.4; HRMS (TOF, ESI) m/z: calcd for C16H21ClN3O5S3 [M + H]+ 466.0326. Found: 466.0325.
4-(2-Chlorophenyl)sulfonyl-2-ethylsulfonyl-5-(2-methyl-6,8-dihydro-5H-[1,2,4]triazolo[1,5-a]pyrazin-7-yl)thiazole (19)
Starting from 0.150 g of 7e (0.39 mmol), 0.102 g of 2-methyl-5,6,7,8-tetrahydro-[1,2,4]triazolo[1,5-a]pyrazine hydrochloride (0.59 mmol), and 0.13 mL of DIPEA (0.78 mmol) in 3 mL of acetonitrile following General Procedure I, 0.128 g (67%) of 19 was obtained.
1H NMR (500 MHz, DMSO-d6): δ ppm 8.20 (dd, J = 7.9, 1.6 Hz, 1H), 7.75 (dt, J = 7.7, 1.7 Hz, 1H), 7.67 (dd, J = 8.0, 1.1 Hz, 1H), 7.62 (dt, J = 7.7, 1.1 Hz, 1H), 4.74 (s, 2H), 4.24 (m, 2H), 4.02 (m, 2H), 3.32 (q, J = 7.3 Hz, 2H), 2.26 (s, 3H), 0.99 (t, J = 7.3 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ ppm 163.6, 159.9, 150.1, 148.4, 137.7, 136.3, 134.8, 132.3, 132.25, 131.2, 128.4, 53.5, 51.9, 49.4, 45.3, 14.1, 7.4; HRMS (TOF, ESI) m/z: calcd for C17H19ClN5O4S3 [M + H]+ 488.02822. Found: 488.02815.
5-[4-(2-Chlorophenyl)sulfonyl-2-ethylsulfonyl-thiazol-5-yl]-2-methyl-1,2,5-thiadiazinane 1,1-Dioxide (20)
A mixture of 0.0262 g of 20c (0.049 mmol), 0.047 g of Cs2CO3 (0.14 mmol), and 0.036 g of KI (0.22 mmol) in 2 mL of acetonitrile was heated at 130 °C under nitrogen in a microwave reactor for 1h. This procedure was repeated an additional five times. The combined reaction mixture was filtered, the filtrate was evaporated directly onto Celite, and then, it was purified by flash chromatography on silica gel (0% → 1.5% methanol/DCM) to give 0.040 g (25%) of 20.
1H NMR (500 MHz, DMSO-d6): δ ppm 8.18 (dd, J = 8.0, 1.2 Hz, 1H), 7.85 (td, J = 8.0, 1.2 Hz, 1H), 7.71 (d, J = 8.8 Hz, 1H), 7.53 (t, J = 7.6 Hz, 1H), 5.11 (s, 2H), 4.52 (br t, 2H), 3.7 (t, J = 5.9 Hz, 2H), 3.67 (q, J = 7.3 Hz, 2H), 3 (s, 3H), 1.27 (t, J = 7.3 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ ppm 153.2, 152.9, 136.3, 134.3, 129.9, 126.7, 124.8, 124.7, 116.7, 53.9, 53.6, 49.7, 47.5, 36.8, 7.6; HRMS (TOF, ESI) m/z: calcd for C15H22ClN4O6S4 [M + NH4]+ 517.0105; found: 517.0109.
1-[4-(2-Chlorophenyl)sulfonyl-2-ethylsulfonyl-thiazol-5-yl]-4-methyl-1,4λ5-azaphosphinane 4-Oxide (21)
Starting from 0.200 g of 7e (0.52 mmol), 0.176 g of 4-methyl-1,4λ5-azaphosphinane 4-oxide hydrochloride (1.04 mmol), and 0.18 mL of DIPEA (1.04 mmol) in 3 mL of acetonitrile following General Procedure I, 0.187 g (74%) of 21 was obtained.
1H NMR (500 MHz, DMSO-d6): δ ppm 8.23 (dd, J = 8.1, 1.5 Hz, 1H), 7.79–7.65 (m, 3H), 3.82 (m, 2H), 3.72 (m, 2H), 3.27 (q, J = 7.3 Hz, 2H), 2.08 (m, 2H), 1.99 (m, 2H), 1.56 (d, J = 13.4 Hz, 3H), 0.96 (t, J = 7.3 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ ppm 164.6, 148.1, 138.2, 136.1, 133.0, 132.3, 132.2, 131.1, 128.5, 53.6, 49.3, 28.5, 13.9, 7.4; HRMS (TOF, ESI) m/z: calcd for C16H21ClN2O5PS3 [M + H]+ 483.0033. Found: 483.0034.
5-(5-Azaspiro[2.3]hexan-5-yl)-4-(2-chlorophenyl)sulfonyl-2-ethylsulfonyl-thiazole (22)
Starting from 0.150 g of 7e (0.388 mmol), 0.069 g of 5-azaspiro[2.3]hexane hydrochloride (0.577 mmol), and 0.096 mL of DIPEA (0.744 mmol) in 3 mL of acetonitrile following General Procedure I, 0.078 g (46%) of 22 was obtained.
1H NMR (500 MHz, DMSO-d6): δ ppm 8.16 (dd, J = 7.9, 1.6 Hz, 1H), 7.76–7.6 (m, 3H), 4.39 (s, 4H), 3.31 (q, J = 7.3 Hz, 2H), 1.02 (t, J = 7.3 Hz, 3H), 0.72 (s, 4H); 13C NMR (125 MHz, DMSO-d6): δ ppm 163.4, 143.3, 138.9, 135.8, 132.3, 132.0, 130.8, 128.5, 126.5, 67.0, 49.6, 16.4, 9.4, 7.5; HRMS (TOF, ESI) m/z: calcd for C16H18ClN2O4S3 [M + H]+ 433.0112. Found: 433.0104.
(3R)-4-[4-(2-Chlorophenyl)sulfonyl-2-ethylsulfonyl-thiazol-5-yl]-1,3-dimethyl-piperazin-2-one (23) and (3S)-4-[4-(2-Chlorophenyl)sulfonyl-2-ethylsulfonyl-thiazol-5-yl]-1,3-dimethyl-piperazin-2-one (24)
Starting from 4.60 g of 7e (12 mmol) and 2.30 g of 1,3-dimethylpiperazin-2-one (18 mmol) in 50 mL of acetonitrile following General Procedure I, a mixture of 23 and 24 was obtained. The enantiomers were separated on a CHIRALPAK IC Preparative HPLC Chiral Column (50 × 500 mm, 20 μm) using EtOH as an eluent to obtain 2.30 g (40%) of 23 as the first eluting enantiomer and 2.30 g of (40%) 24 as the second eluting enantiomer.
23: 1H NMR (500 MHz, DMSO-d6): δ ppm 8.22 (dm, 1H), 7.8–7.64 (m, 3H), 4.25 (q, J = 6.9 Hz, 1H), 3.79 (m, 1H), 3.62 (m, 1H), 3.49 (tm, 1H), 3.35 (m, 1H), 3.35 (q, J = 7.3 Hz, 2H), 2.87 (s, 3H), 1.22 (d, J = 6.9 Hz, 3H), 1.02 (t, J = 7.3 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ ppm 166.8, 162.0, 150.4, 137.8, 136.3, 135.7, 132.3, 132.2, 131.3, 128.4, 62.5, 49.4, 49.3, 47, 34.4, 16.1, 7.4; HRMS (TOF, ESI) m/z: calcd for C17H21ClN3O5S3 [M + H]+ 478.0326. Found: 478.0326; ee >99.8%.
24: 1H NMR (500 MHz, DMSO-d6): δ ppm 8.22 (dm, 1H), 7.8–7.64 (m, 3H), 4.25 (q, J = 6.9 Hz, 1H), 3.79 (m, 1H), 3.62 (m, 1H), 3.49 (tm, 1H), 3.35 (m, 1H), 3.35 (q, J = 7.3 Hz, 2H), 2.87 (s, 3H), 1.22 (d, J = 6.9 Hz, 3H), 1.02 (t, J = 7.3 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ ppm 166.8, 162.0, 150.4, 137.8, 136.3, 135.7, 132.3, 132.2, 131.3, 128.4, 62.5, 49.4, 49.3, 47, 34.4, 16.1, 7.4; HRMS (TOF, ESI) m/z: calcd for C17H21ClN3O5S3 [M + H]+ 478.0326. Found: 478.0331; ee >99.8%.
2-[5-[(2R)-2,4-Dimethyl-3-oxo-piperazin-1-yl]-2-ethylsulfonyl-thiazol-4-yl]sulfonylbenzonitrile (25) and 2-[5-[(2S)-2,4-Dimethyl-3-oxo-piperazin-1-yl]-2-ethylsulfonyl-thiazol-4-yl]sulfonylbenzonitrile (26)
Starting from 0.50 g of 11e (1.33 mmol), 0.328 g of 1,3-dimethylpiperazin-2-one (1.99 mmol), and 0.68 mL of DIPEA (3.9 mmol) in 8 mL of acetonitrile following General Procedure I, a mixture of 25 and 26 was obtained. The enantiomers were separated on a CHIRALPAK AD Preparative HPLC Chiral Column (50 × 500 mm, 20 μm) using 50:50 MeOH/EtOH as an eluent to obtain 0.129 g (21%) of 25 as the first eluting enantiomer and 0.127 g of (20%) 26 as the second eluting enantiomer.
25: 1H NMR (500 MHz, DMSO-d6): δ ppm 8.26 (dm, 1H), 8.16 (dm, 1H), 8.01 (td, J = 7.7, 1.4 Hz, 1H), 7.95 (td, J = 7.7, 1.4 Hz, 1H), 4.31 (q, J = 6.9 Hz, 1H), 3.89 (m, 1H), 3.72 (m, 1H), 3.59 (m, 1H), 3.35 (m, 1H), 3.32 (m, 2H), 2.88 (s, 3H), 1.36 (d, J = 6.9 Hz, 3H), 0.98 (t, J = 7.3 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ ppm 166.7, 162.5, 149.4, 142.1, 136.4, 135.1, 134.5, 133.5, 130.2, 115.4, 110.5, 62.9, 49.3, 49.2, 46.9, 34.5, 16.1, 7.4; HRMS (TOF, ESI) m/z: calcd for C18H21N4O5S3 [M + H]+ 469.0669. Found: 469.0666; ee >99.8%.
26: 1H NMR (500 MHz, DMSO-d6): δ ppm 8.26 (dm, 1H), 8.16 (dm, 1H), 8.01 (td, J = 7.7, 1.4 Hz, 1H), 7.95 (td, J = 7.7, 1.4 Hz, 1H), 4.31 (q, J = 6.9 Hz, 1H), 3.89 (m, 1H), 3.72 (m, 1H), 3.59 (m, 1H), 3.35 (m, 1H), 3.32 (m, 2H), 2.88 (s, 3H), 1.36 (d, J = 6.9 Hz, 3H), 0.98 (t, J = 7.3 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ ppm 166.7, 162.5, 149.4, 142.1, 136.4, 135.1, 134.5, 133.5, 130.2, 115.4, 110.5, 62.9, 49.3, 49.2, 46.9, 34.5, 16.1, 7.4; HRMS (TOF, ESI) m/z: calcd for C18H21N4O5S3 [M + H]+ 469.0669. Found: 469.0664; ee >99%.
1-[4-(2,3-Dichlorophenyl)sulfonyl-2-ethylsulfonyl-thiazol-5-yl]-4-ethyl-1,4λ5-azaphosphinane 4-Oxide (27)
Starting from 0.150 g of 9c (0.36 mmol), 0.099 g of 4-ethyl-1,4λ5-azaphosphinane 4-oxide hydrochloride (0.54 mmol), and 0.18 mL of DIPEA (1.0 mmol) in 5 mL of acetonitrile following General Procedure I, 0.122 g (64%) of 27 was obtained.
1H NMR (500 MHz, DMSO-d6): δ ppm 8.23 (dd, J = 8.0, 1.4 Hz, 1H), 8.06 (dd, J = 8.0, 1.4 Hz, 1H), 7.7 (t, J = 8.0 Hz, 1H), 3.8 (m, 4H), 3.26 (q, J = 7.3 Hz, 2H), 2.1 (m, 2H), 1.99 (m, 2H), 1.84 (dq, J = 11.5, 7.7 Hz, 2H), 1.09 (dt, J = 17.1, 7.7 Hz, 3H), 0.93 (t, J = 7.3 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ ppm 165.0, 147.7, 140.5, 136.3, 134.7, 131.8, 130.3, 129.9, 129.4, 53.5, 49.3, 26.3, 20.1, 7.4, 5.3; HRMS (TOF, ESI) m/z: calcd for C17H22Cl2N2O5PS3 [M + H]+ 530.98. Found: 530.9797.
4-[4-(2-Chlorophenyl)sulfonyl-2-vinyl-thiazol-5-yl]-1-methyl-piperazin-2-one (28)
The mixture of 0.250 g of 7 (0.539 mmol), 0.145 g of potassium trifluoro(vinyl)borate (1.53 mmol), 0.15 mL of N,N-diethylethanamine (1.0 mmol), and 20 mg of Pd(dppf)Cl2 (0.027 mmol) in 1-propanol (7.5 mL) was refluxed under nitrogen for 18 h. The solvent was evaporated under reduced pressure, and the crude product was purified by preparative HPLC (C18) using 5 mM aqueous NH4HCO3 solution and acetonitrile as eluents to give 0.02 g (16%) of 28.
1H NMR (500 MHz, DMSO-d6): δ ppm 8.16 (dd, J = 7.9, 1.6 Hz, 1H), 7.75–7.6 (m, 3H), 6.67 (dd, J = 17.4, 10.9 Hz, 1H), 5.85 (d, J = 17.4 Hz, 1H), 5.56 (d, J = 10.9 Hz, 1H), 3.82 (s, 2H), 3.55 (m, 2H), 3.37 (m, 2H), 2.87 (s, 3H); 13C NMR (125 MHz, DMSO-d6): δ ppm 164.1, 157.4, 155.8, 138.4, 135.8, 134.3, 132.25, 132.0, 131.0, 130.5, 128.2, 121.8, 57.3, 52.3, 47.4, 33.9; HRMS (TOF, ESI) m/z: calcd for C16H17ClN3O3S2 [M + H]+ 398.0394. Found: 398.0390.
Acknowledgments
The authors would like to thank Diamond Light Source for beamtime (proposal mx28172), as well as the staff of beamline I03 and i04 for assistance with crystal testing and data collection. This project has received funding from the Innovative Medicines Initiative 2 Joint Undertaking (JU) under grant agreement No 875510. The JU receives support from the European Union’s Horizon 2020 research and innovation programme, EFPIA, Ontario Institute for Cancer Research, Royal Institution for the Advancement of Learning McGill University, Kungliga Tekniska Hoegskolan, and Diamond Light Source Limited. Stefan Knapp’s help is gratefully acknowledged for providing the kinase selectivity data. The authors thank co-workers at the Analytical Division of the Servier Research Institute of Medicinal Chemistry for providing the detailed chemical analysis of the compounds.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.4c02019.
Author Contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- a Baird L.; Dinkova-Kostova A. T. The cytoprotective role of the Keap1–Nrf2 pathway. Arch. Toxicol. 2011, 85, 241–272. 10.1007/s00204-011-0674-5. [DOI] [PubMed] [Google Scholar]; b Itoh K.; Chiba T.; Takahashi S.; Ishii T.; Igarashi K.; Katoh Y.; Oyake T.; Hayashi N.; Satoh K.; Hatayama I. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]; c Kensler T. W.; Wakabayashi N.; Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- Hayes J. D.; Dinkova-Kostova A. T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. 10.1016/j.tibs.2014.02.002. [DOI] [PubMed] [Google Scholar]
- Lo S. C.; Li X.; Henzl M. T.; Beamer L. J.; Hannink M. Structure of the Keap1: Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J. 2006, 25, 3605–3617. 10.1038/sj.emboj.7601243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie Y.; Suzuki T.; Inoue J.; Iso T.; Wells G.; Moore T. W.; Mizushima T.; Dinkova-Kostova A. T.; Kasai T.; Kamei T.; Koshiba S.; Yamamoto M. Molecular basis for the disruption of Keap1-Nrf2 interaction via Hinge & Latch mechanism. Commun. Biol. 2021, 4, 576. 10.1038/s42003-021-02100-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaya K.; Suzuki T.; Motohashi H.; Onodera K.; Satomi S.; Kensler T. W.; Yamamoto M. Validation of the multiple sensor mechanism of the Keap1-Nrf2 system. Free Radic. Biol. Med. 2012, 53, 817–827. 10.1016/j.freeradbiomed.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a O’Connell M. A.; Hayes J. D. The Keap1/Nrf2 pathway in health and disease: from the bench to the clinic. Biochem. Soc. Trans. 2015, 43, 687–689. 10.1042/BST20150069. [DOI] [PubMed] [Google Scholar]; b Wu W. L.; Papagiannakopoulos T. The pleiotropic role of the KEAP1/NRF2 pathway in cancer. Free Radic. Biol. Med. 2020, 4, 413–435. 10.1146/annurev-cancerbio-030518-055627. [DOI] [Google Scholar]
- a Abed D. A.; Goldstein M.; Albanyan H.; Jin H.; Hu L. Discovery of direct inhibitors of Keap1-Nrf2 protein-protein interaction as potential therapeutic and preventive agents. Acta Pharm. Sin. B 2015, 5, 285–299. 10.1016/j.apsb.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hur W.; Gray N. S. Small molecule modulators of antioxidant response pathway. Curr. Opin. Chem. Biol. 2011, 15, 162–173. 10.1016/j.cbpa.2010.12.009. [DOI] [PubMed] [Google Scholar]; c Jang J.; To C.; De Clercq D. J.; Park E.; Ponthier C. M.; Shin B. H.; Mushajiang M.; Nowak R. P.; Fischer E. S.; Eck M. J.; et al. Mutant-selective allosteric EGFR degraders are effective against a broad range of drug-resistant mutations. Angew. Chem., Int. Ed. Engl. 2020, 59, 14481–14489. 10.1002/anie.202003500. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Magesh S.; Chen Y.; Hu L. Small molecule modulators of Keap1-Nrf2-ARE pathway as potential preventive and therapeutic agents. Med. Res. Rev. 2012, 32, 687–726. 10.1002/med.21257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Structural insights into the multiple binding modes of Dimethyl Fumarate (DMF) and its analogs to the Kelch domain of Keap1Unni S.; Deshmukh P.; Krishnappa G.; Kommu P.; Padmanabhan B. Structural insights into the multiple binding modes of Dimethyl Fumarate (DMF) and its analogs to the Kelch domain of Keap1. FEBS J. 2021, 288, 1599–1613. 10.1111/febs.15485. [DOI] [PubMed] [Google Scholar]
- Brennan M. S.; Matos M. F.; Li B.; Hronowski X.; Gao B.; Juhasz P.; Rhodes K. J.; Scannevin R. H. Dimethyl Fumarate and Monoethyl Fumarate Exhibit Differential Effects on KEAP1, NRF2 Activation, and Glutathione Depletion In Vitro. PLoS One 2015, 10, e0120254 10.1371/journal.pone.0120254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch D. R.; Perlman S.; Schadt K. Omaveloxolone for the treatment of Friedreich ataxia: clinical trial results and practical considerations. Expert Rev. Neurother. 2024, 24, 251–258. 10.1080/14737175.2024.2310617. [DOI] [PubMed] [Google Scholar]
- Dinkova-Kostova A. T.; Liby K. T.; Stephenson K. K.; Holtzclaw W. D.; Gao X.; Suh N.; Williams C.; Risingsong R.; Honda T.; Gribble G. W.; Sporn M. B.; Talalay P. Extremely potent triterpenoid inducers of the phase 2 response: Correlations of protection against oxidant and inflammatory stress. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 4584–4589. 10.1073/pnas.0500815102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuadrado A.; Rojo A. I.; Wells G.; Hayes J. D.; Cousin S. P.; Rumsey W. L.; Attucks O. C.; Franklin S.; Levonen A. L.; Kensler T. W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discovery 2019, 18, 295–317. 10.1038/s41573-018-0008-x. [DOI] [PubMed] [Google Scholar]
- Markovtsov V.; Duncton M. A. J.; Bagos A.; Yi S.; Braselmann S.; Bhamidipati S.; Darwish I. S.; Yu J.; Owyang A. M.; Fernandez B.; Samant B.; Park G.; Masuda E. S.; Shaw S. J. Tuning the Reactivity of Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2) Activators for Optimal in Vivo Efficacy. ACS Med. Chem. Lett. 2023, 14, 1700–1706. 10.1021/acsmedchemlett.3c00336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallesen J. S.; Tran K. T.; Bach A. Non-Covalent Small-Molecule Kelch-like ECH-Associated Protein 1–Nuclear Factor Erythroid 2-Related Factor 2 (Keap1–Nrf2) Inhibitors and Their Potential for Targeting Central Nervous System Diseases. J. Med. Chem. 2018, 61, 8088–8103. 10.1021/acs.jmedchem.8b00358. [DOI] [PubMed] [Google Scholar]
- Abadio A. K. R.; Kioshima E. S.; Leroux V.; Martins N. F.; Maigret B.; Felipe M. S. S. Identification of New Antifungal Compounds Targeting Thioredoxin Reductase of Paracoccidioides Genus. PLoS One 2015, 10, e0142926 10.1371/journal.pone.0142926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham J.; Botto S.; Mizuno N.; Pryke K.; Gall B.; Boehm D.; Sali T. M.; Jin H.; Nilsen A.; Gough M.; Baird J.; Chakhtoura M.; Subra C.; Trautmann L.; Haddad E. K.; DeFilippis V. R. Characterization of a Novel Compound That Stimulates STING-Mediated Innate Immune Activity in an Allele-Specific Manner. Front. Immunol. 2020, 11, 1430. 10.3389/fimmu.2020.01430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross D.; Siegel D. The diverse functionality of NQO1 and its roles in redox control. Redox Biol. 2021, 41, 101950. 10.1016/j.redox.2021.101950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrhein J. A.; Berger L. M.; Balourdas D. I.; Joerger A. C.; Menge A.; Krämer A.; Frischkorn J. M.; Berger B. T.; Elson L.; Kaiser A.; Schubert-Zsilavecz M.; Müller S.; Knapp S.; Hanke T. Synthesis of Pyrazole-Based Macrocycles Leads to a Highly Selective Inhibitor for MST3. J. Med. Chem. 2024, 67, 674–690. 10.1021/acs.jmedchem.3c01980. [DOI] [PubMed] [Google Scholar]
- Besnard J.; Ruda G. F.; Setola V.; Abecassis K.; Rodriguiz R. M.; Huang X. P.; Norval S.; Sassano M. F.; Shin A. I.; Webster L. A.; Simeons F. R.; Stojanovski L.; Prat A.; Seidah N. G.; Constam D. B.; Bickerton G. R.; Read K. D.; Wetsel W. C.; Gilbert I. H.; Roth B. L.; Hopkins A. L. Automated design of ligands to polypharmacological profiles. Nature 2012, 492, 215–220. 10.1038/nature11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart D. J.; Helbling F. R.; Verardo M.; Huber A.; McHugh D.; Vanscheeuwijck P. Development of an integrated assay in human TK6 cells to permit comprehensive genotoxicity analysis in vitro. Mutat. Res., Genet. Toxicol. Environ. 2020, 849, 503129. 10.1016/j.mrgentox.2019.503129. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.