

Abstract
Purpose:
Appendiceal adenocarcinoma (AA) is a rare malignancy with distinct histopathologic subtypes and a natural history with metastasis primarily limited to the peritoneum. Little is known about the molecular pathogenesis of AA relative to common tumors.
Experimental Design:
We analyzed molecular data for patients within the Guardant Health database with appendix cancer (n = 718). We then identified patients with AA at our institution (from 10/2004–9/2022) for whom circulating tumor DNA (ctDNA) mutation profiling (liquid biopsy) was performed (n=168) and extracted clinicopathologic and outcomes data. Of these 168 patients 57 also had tissue-based tumor mutational profiling allowing for evaluation of concordance between liquid and tissue assays.
Results:
The mutational landscape of ctDNA in AA is distinct from tissue-based sequencing, with TP53 being the most frequently mutated (46%). Relative to other tumors, AA appears less likely to shed ctDNA, with only 38% of metastatic AA patients having detectable ctDNA (OR 0.26, p < 0.0001 relative to CRC). When detectable the median VAF was significantly lower in AA (0.4% vs. 1.3% for CRC, p≤0.001). High grade, signet-ring or colonic-type histology, metastatic spread beyond the peritoneum, and TP53 mutation were associated with detectable ctDNA. With respect to clinical translation, patients with detectable ctDNA had worse overall survival (HR = 2.32, p = 0.048). In the Guardant Health cohort actionable mutations were found in 93 patients (13.0%).
Conclusions:
Although metastatic AA tumors are less likely to shed tumor DNA into the blood relative to CRC, ctDNA profiling in AA has clinical utility.
One Sentence Summary:
The landscape of ctDNA in appendiceal adenocarcinoma shows a low rate of ctDNA shedding and a mutational profile distinct from colorectal cancer.
INTRODUCTION
Appendiceal adenocarcinoma (AA) is a rare cancer with a unique clinical course stratified by high- and low-grade tumors.(1–4) Unlike colorectal cancer (CRC) and other gastrointestinal malignancies, AA rarely metastasizes hematogenously, but rather, has a strong tendency to metastasize to the peritoneum.(5–7) Patients with AA are therefore uniquely suited to benefit from cytoreductive surgery (CRS) and heated intraperitoneal chemotherapy (HIPEC), by which peritoneal metastatic disease can be resected with curative intent. Our incomplete understanding of the tumor-peritoneal interface and a tumor’s potential for distant spread, however, is a major limitation for clinicians in selecting appropriate operative candidates, clinical decision making around systemic therapies, and increasing our understanding of this rare disease.
Interrogating the landscape of somatic mutations in the peripheral blood of patients with AA provides an avenue to understand the molecular basis of tumor formation and progression. Given technical limitations and the rarity of these primary AA tumors, however, mutational characterization to date has been limited. Although AA was historically thought to be similar to CRC due to anatomic vicinity and a shared embryogenic origin (8, 9), recent studies have shown that AA is quite different from CRC at a molecular level.(10, 11) Appendiceal tumors appear to have distinct driver mutations as TP53, KRAS, and GNAS mutations are common while APC mutation, pathognomonic for CRC, are surprisingly rare in AA.(12–15) Moreover, in AA, the mutation status of key driver genes including KRAS, GNAS, and TP53 have been shown to be both prognostic and predictive biomarkers.(10, 16)
Beyond tumor mutational profiles, recent advances allowing for reliable sequencing of circulating tumor DNA (ctDNA) have the potential to inform tumoral mutational status as well as provide insight into the biologic interface of the tumor, peritoneum, and systemic circulation.(17) Circulating tumor DNA has demonstrated its utility as a biomarker in other cancer types for applications including genotyping patients with advanced cancer, prognostic stratification, disease detection, and serial monitoring (18), and is currently included in the treatment-guidelines of many tumor types.(19) The mechanisms and limitations of AA ctDNA shedding (i.e. the degree to which tumor DNA enters systemic circulation), however, are poorly understood. While ctDNA shedding is influenced by the anatomic location of metastatic disease as well as intrinsic properties of the tumor cells including rate of cell division and treatment pressure on ctDNA shed, further understanding of peritoneal metastases and AA specifically is needed.(20–22) Despite a great interest in the potential to measure circulating tumor DNA as a means for identifying driver somatic mutations (liquid biopsy), track treatment response, and identify minimal residual disease (MRD) in AA, prior reports regarding the sensitivity of detection of ctDNA in AA have been conflicting.(23–25) Few prior reports of ctDNA in AA exist, and those that do have contained small numbers of patients and lacked comparison of biomarkers detected from tumor tissue with those detected via ctDNA.(15, 23, 25–28) Through our analysis of tumoral and circulating mutations in AA, we aim to shed light on the tumor biology and mechanisms of metastatic spread of these rare tumors.
Materials and Methods
Patient Identification
Initial analysis was undertaken via a de-identified database of all patients who received ctDNA testing via Guardant Health as part of usual clinical care. Patients with a diagnosis of appendix carcinoma, appendix adenocarcinoma, appendix mucinous tumor, goblet cell carcinoid, or signet ring appendix carcinoma, as listed by the ordering provider on the test request form (TRF) were queried. Patient cancer type was extracted from the TRF, and the ordering provider had to confirm the patient had advanced disease (stage III or higher). No further clinical information was required. Retrospective, de-identified analysis of the Guardant Health database is IRB approved by Advarra Pro00034566.
This study was approved by the UTMDACC Institutional Review Board, a waiver of informed consent was granted per USA federal regulation 45 CFR 46.116(f) (Common Rule) given minimal risk to patients. Initial analysis was undertaken querying all patients with appendiceal adenocarcinoma included within the Guardant Health database. Given the limited associated clinical data held therein, we then retrospectively reviewed the demographic (including age, sex, and BMI), clinicopathologic, and outcomes data of 168 patients with appendiceal adenocarcinoma followed at The University of Texas MD Anderson Cancer Center, for whom molecular testing (liquid biopsy) had been performed on their blood (from October 2004 until September 2022). These analyses of the MDACC cohort were performed as secondary subset analyses distinct from the overall group analysis to avoid repeat measurements affecting our data. These patients had been identified from the electronic health record (EHR) at the University of Texas M.D. Anderson Cancer Center (UTMDACC) using the Foundry software system (Palantir Technologies, Denver, CO). 57 out of the 168 patients had PCR-based next generation sequencing (NGS) done on their tumor which allowed the evaluation of concordance with the assay. Time-difference between tissue sequence and liquid biopsy ranged from −21 months to 116 months (Supplementary Fig.1).
Guardant Health Database and Assay
Patients within the Guardant Health database had ctDNA testing via the Guardant360 assay. Guardant360 is a CLIA-certified, College of American Pathologists-accredited, New York State Department of Health-approved, targeted next-generation sequencing ctDNA assay, with analytical and clinical validation previously described. The assay included analysis of 73–83 genes, depending on the version of the assay used, with complete or critical exon coverage of single nucleotide variants and select insertion/deletions, amplifications, and fusions. The reportable range for SNVs, indels, fusions, and copy number amplifications is ≥0.04–0.06%, ≥0.02–0.04%, ≥0.04–0.06%, and ≥2.12 copies, respectively.(29, 30) In total, molecular data from 718 patients with AA diagnoses was included in the landscape of AA.
Statistical Analysis
All analyses focused on the 153 patients with metastatic AA. Patients’ disease status was determined from reviewing CT scans and was divided into No Evidence of Disease (NED) or Evidence of Disease (ED). Differences in Variant Allele Frequency (VAF) between AA and CRC were assessed with Wilcoxon-Mann-Whitney tests. A p<0.05 was considered significant. Treating tumor biopsy as the gold standard, the positive concordance of the assay was calculated using the following formula = (Mutation found in both blood and tumor)/Mutation found only in Tumor. When restricting to patients who had evidence of disease when liquid biopsy was done, calculating the positive concordance was applicable on 47 out of the 57 patients with tumor biopsy.
Chi-Square testing was done on 153 patients with metastatic AA to determine what factors predict a detectable ctDNA result. Variables included were: Age, Chemotherapy never administered or given before or during ctDNA drawn date, Completeness of Cytoreduction (CCR), Disease status, Gender, Grade, Histology, Lymph node involvement, Microsatellite instability status, Peritoneal Carcinomatosis Index (PCI), Presence of perineural invasion, Presence of lympho-vascular invasion, Extent of metastatic spread (limited to peritoneum vs hematogenous spread), and Tumor Markers (CEA, CA 19–9, CA 125). Overall Survival (OS) was determined using Kaplan-Meier analysis from the day ctDNA was drawn till death with censoring at the last follow up date. Comparison was performed using the log-rank test; p-value< 0.05 was considered significant. A p<0.05 was considered significant.
All analyses were performed using GraphPad Prism version 8.0 (GraphPad; La Jolla, CA, RRID:SCR_002798), and R version 4.0.1.
Clinical and Molecular data/ Sequencing
Molecular testing, both liquid biopsy and tumor biopsy, was performed at MD Anderson’s College of American Pathologists (CAP) accredited and Clinical Laboratory Improvement Amendments (CLIA) certified molecular diagnostics laboratory. For the tumor biopsy, PCR-based next generation sequencing (NGS) was used to test for mutations in the coding sequence of 134 genes and copy number variations (CNV) in 47 genes as previously described (31) using GRCh37/hg19 as reference sequence. For the liquid biopsy, all patients were tested utilizing 73–83 genes, depending on the version of the assay used (Guardant360, Guardant Health, Inc., Palo Alto, California), including coverage of single nucleotide variants and select insertion/deletions, amplifications, and fusions (see Supplemental Table S3).(32) Microsatellite status was demined by immunohistochemistry evaluation for mismatch repair proteins MLH1, MSH2, MSH6, and PMS2 per standard criteria.(33) Mutations that were determined by treating physicians to represent clonal hematopoiesis (CH) were excluded. For germline mutations, this was decided based on the report from the assay and for clonal hematopoiesis, this was decided based on either physician clinical notes, published literature,(34) or using ClinVar.(35)
DATA AND MATERIALS AVAILABILITY:
All data are available in the main text or the supplementary materials. Deidentified clinical level data may be made available by reasonable request to the corresponding author under a data use agreement.
RESULTS
Landscape of alterations detected in patients with AA
To assess the mutational landscape of ctDNA shed in patients with AA, we analyzed a cohort of 718 patients with confirmed diagnosis of AA within the Guardant Health genomic database. The overall oncogenic mutation detection rate of ctDNA in circulation within this Guardant Health cohort, was 68% (491/718 patients with mutations detected). Similar to previous descriptions of somatic mutation frequency TP53 (46%) and KRAS (29%) were the most commonly mutated genes (Figure 1A). Mutations in ATM (17%) were more common than expected based on previous tumoral mutational studies, while frequency of mutations in GNAS (11%) were much lower (36). Mutations in APC, canonical in colorectal cancers (CRC), were only noted in 11% of samples. Microsatellite instability (MSI-H) was found in 7 (1.0%) of 718 patients, consistent with prior report that found frequency of MSI-H in AA was 0.9% (12). From 732 blood samples a total of 1411 variants were detected with missense mutations being most common (54.2%) followed by synonymous mutations (18%) and indels (11.7%), with only one detected fusions event (0.07%). Copy number variation (CNV) was also quite rare with 65 amplifications (4.6%) and no deletions detected (Figure 1B). Although deletions were not commonly detected on the panel utilized. The median variant allele frequency (VAF) across the entire cohort was 0.5% (Range 0.01–82.8%). The circulating tumor mutational burden (TMB) score was low, median 6.3 mutations per megabase (mut/Mb), with only nine (1.2%) samples harboring a TMB greater than 20mut/Mb (Figure 1C). Similarly, the overall number of variants detected per tumor was low (median 4, IQR [2–7], with one outlier MSI-H sample with 95 variants (Figure 1D). As expected, the number of variants detected in the seven MSI-H patients was significantly higher than in the population in which MSI was not detected (22.9 vs. 2.41, p<0.0001). Using the OncoKB database to annotate actionable mutations (37), a total of 94 currently actionable mutations were identified in 87 unique patients, including six patients with BRAFV600E, 11 patients with KRASG12C, and 62 patients with KRASG12D (Figure 1E, Supplementary Table S1). Several gene pairs were significantly enriched for co-occurrence within the same sample: TP53 and KRAS; KRAS and APC; KRAS and PIK3CA; TP53 and APC; KRAS and GNAS; among others (p<0.001; q<0.001) suggesting cooperative oncogenesis and consistent with known mutational subtyping of AA (Figure 1F, Supplementary Table S2).(10) Given the limited power of the cohort, no gene pairs were statistically mutually exclusive. However, there was only one sample with both KRAS and NRAS mutations, known to be mutually exclusive activators of the Ras/MAPK pathway (38), and no samples with both SMAD4 and NF1 mutation. Interestingly, although not previously reported in AA, SMAD4 and NF1 are known to cooperate to form a repressor complex in response to TGF-β.(39) Overall, the circulating genomic profile of AA was a stark contrast to that of CRC whose mutational profile has been previously described (Supplementary Figure 1 A,B).(40)
Figure 1. The landscape of circulating tumor DNA in appendiceal adenocarcinoma.

A) Oncoprint of the top 11 genes noted to be mutated in the peripheral blood of patients with appendiceal adenocarcinoma in the Guardant Health cohort of 718 patients, along with microsatellite instability status and TMB. These were the only mutations noted in greater than 1% of samples. B) Pie chart of the types of circulating mutations noted in appendiceal adenocarcinoma patients. C) Histogram of the number of alterations noted within this tumor agnostic ctDNA assay run on circulating plasma of patients with AA. D) Histogram of the tumor mutational burden (TMB) score noted within the peripheral blood of appendiceal adenocarcinoma patients. E) Histogram of the clinical actionable mutations identified in circulating plasma. F) Co-mutational and mutual exclusivity plot of mutations noted in the peripheral blood of AA patients. Statistically significant co-occurrences are denoted with an * while there were no statistically significant mutually exclusive associations.
Clinical and pathological associations with ctDNA detection in AA
To further evaluate clinical correlations with ctDNA and mutational status, we queried a subset of AA samples that were sequenced by liquid biopsy as part of routine clinical care at the MD Anderson Cancer Center (MDACC) in whom more comprehensive clinical data and paired tumor mutational data were available (Table 1). Out of 153 patients from whom liquid biopsies were obtained, 58 (38%) had detectable ctDNA with the majority having a single somatic alteration detected. The majority of variants were missense substitutions (76.9%) similar to the Guardant cohort (Supplementary Figure 2A). This rate of detection was significantly lower relative to a similar cohort of patients with CRC (70% with detectable ctDNA; OR 0.26, p<0.0001, Figure 2A). We next chose to compare the landscape of somatic mutations seen in blood versus that from tumor tissue. In examining the ratio of tumor mutations to peripherally detected mutations in blood, we see that mutations noted in AA were distinct from those noted in a cohort of CRC patients at MDACC (n=4,941 tumoral, n=1,282 ctDNA), although this was dependent on the mutation itself, suggesting an association between somatic mutational profiles and DNA shedding dynamics. KRAS was the most frequently mutated gene in AA tumors (47.1%) but was mutated in only 8.5% of blood specimens, a ratio of 0.18 (Figure 2B, Supplementary Figure 2B). SMAD4 had the second lowest ratio of mutation frequency in tumor (14.1%) relative to blood (2.6%) of 0.18. Similarly, this ratio was 0.30 for GNAS (25.7% in tumor vs. 7.8% in blood). In contrast, TP53 was the most frequently mutated gene in ctDNA (18.3%) and the second most frequently mutated in tumor tissue sequencing (27.4%), a ratio of 0.67. In comparison, the most frequently mutated genes in CRC, TP53, APC, and KRAS, had relatively equal frequency of mutation in ctDNA and tumor tissue, with only ATM, FBXW7 and CTNNB1 being significantly depleted in terms of ctDNA mutation frequency (Supplementary Figure 1B, 2B). All observed mutations can be found in Supplemental Data.
Table 1:
MDACC Cohort Demographics
| Feature | n=153 |
|---|---|
| ctDNA (%) | |
| Positive (%) | 58 (37.9) |
| Negative (%) | 95 (62.1) |
| Age at Diagnosis (mean (SD)) | 55.7 (11.6) |
| Grade (%) | |
| Well differentiated | 49 (32.0) |
| Moderately differentiated | 44 (28.8) |
| Poorly differentiated | 60 (39.2) |
| Histology (%) | |
| Mucinous | 106 (69.3) |
| Non-mucinous | 21 (13.7) |
| Goblet cell | 11 (7.2) |
| Signet ring cell | 15 (9.8) |
| Hematogenous Spread (%) | |
| Yes | 10 (6.5) |
| No | 143 (93.5) |
| Lymph Node Involvement (%) | |
| Yes | 32 (20.9) |
| No | 121 (79.1) |
| Gender (%) | |
| Male | 63 (41.2) |
| Female | 90 (58.8) |
| Chemotherapy given relative to ctDNA draw (%) | |
| Never | 90 (58.8) |
| Before | 33 (21.6) |
| During | 30 (19.6) |
| CA19–9 Level (%) | |
| < 37 ng/mL | 81 (52.9) |
| ≥ 37 ng/mL | 42 (27.5) |
| NA | 30 (19.6) |
| CA125 Level (%) | |
| < 35 ng/mL | 93 (60.8) |
| ≥ 35 ng/mL | 31 (20.3) |
| NA | 29 (19.0) |
| CEA Level | |
| < 5 ng/mL | 78 (51.0) |
| ≥ 5 ng/mL | 71 (46.4) |
| NA | 4 (2.6) |
Figure 2: Clinical and mutational predictors of detectable circulating tumoral DNA in appendiceal adenocarcinoma.

A) Percent of total samples with positive ctDNA comparing AA and CRC. Rates of ctDNA positivity were significantly higher in CRC (OR=0.26, p<0.0001). B) Circulating mutational profiles when for AA (n=153) along with tumor mutational status in AA (n=569) from the MD Anderson cohort. C) Percent of total samples with positive ctDNA in AA by tumor grade comparing well, moderate, and poorly differentiated tumors. A trend exists for increasing ctDNA positivity by grade and there was a significantly higher rate comparing well and poorly differentiated tumors (p=0.01). D) Percent of total samples with positive ctDNA in AA by tumor histology comparing mucinous, goblet, non-mucinous, and signet ring histologies. Rates of ctDNA positivity was lower in mucinous tumors as compared to goblet (p=0.02) and signet ring tumors (p=0.03), all other associations were non-significant (p>0.05). E) Percent of total samples with positive ctDNA in AA by site of metastatic disease shows distant metastases had a higher rate of ctDNA positivity as compared to local disease (p=0.005). F) Circulating tumoral variant allele frequency comparing AA (n=58) to CRC (n=241) demonstrating a significantly higher VAF in CRC (p<0.001).
Focusing solely on AA, we next evaluated what clinical features were associated with detectable ctDNA. Grade is known to be the one of the most important predictors of survival in AA (12), and was also found to associated with detectable ctDNA with poorly differentiated tumors having the highest detectable rate of somatic alterations by ctDNA (48%), followed by moderately differentiated (39%) and well differentiated (24%) (p=0.01 between well and poorly differentiated, Figure 2C). Similarly, considering tumor histology, signet ring cell tumors had the highest detectable rate of ctDNA (60%) comparable to non-mucinous (57%) and goblet cell tumors (45%) while the detection rate was significantly lower in mucinous tumors (30%, vs. signet p=0.03, vs. goblet cell p = 0.02, Figure 2D). Patients with metastatic spread limited to the peritoneum had a lower detection rate than patients with hematogenous spread (35% vs. 80%, p=0.005, Figure 2E). These factors, all previously described proxies for aggressive disease biology (12), along with perineural invasion were confirmed as independent predictors of detectable ctDNA in a multivariate analysis (Supplemental Figure 2C, p<0.05 bold) suggesting ctDNA may provide a surrogate marker of blood-peritoneal barrier disruption and more aggressive local biology.
In terms of quantity of ctDNA, the median circulating variant allele frequency (VAF) was significantly lower for patients with AA compared to CRC (median VAF 0.4% vs. 1.3%, p≤0.001, Mann-Whitney test) with no variant detected with a VAF above 0.1% in AA (Figure 2F). The lower VAF relative to CRC was seen across all genes (Supplementary Figure 3A). Similarly there was no significant difference in VAF between well, moderately and poorly-differentiated tumors (Supplementary Figure 3B), and no correlation was found between peritoneal disease burden and VAF in those with pre-op ctDNA (R2 = 0.018, p = 0.46) (Supplementary Figure 3C), suggesting ctDNA status to be function of microscopic disease biology rather than gross disease burden. All VAFs observed can be found in Data Supplement.
Plasma vs tissue concordance analysis
Given the differences seen in the mutational landscape of tissue and ctDNA, we analyzed a cohort of 58 AA and 241 CRC patients with both tissue and ctDNA testing to directly measure concordance. The overall blood-tissue mutation detection concordance for all genes was 96.8%, however this value is driven by the large number of instances where a mutation was not detected in either blood or tissue (negative concordance). Ten mutations were found in the blood but not in tissue sequencing, suggesting that spatial tumor heterogeneity may result in false negatives if only one of many metastatic sites is biopsied.(41) Of 104 mutations detected in genes included in the ctDNA assay only 15 were detected for a positive concordance rate of 20% in AA. This was significantly lower compared to CRC (20% vs 50%, p < 0.001, Figure 3A). Poorly differentiated tumors had significantly higher positive concordance (27%) relative to moderately differentiated (11%) and well differentiated (7%) tumors (Figure 3B). Similarly, TP53 mutations, known to be associated with high-grade tumors (36), had higher positive concordance relative to KRAS or GNAS (Figure 3C). These data further suggest that blood-peritoneal barrier disruption and grade of disease influences the ability to detect ctDNA.
Figure 3. Concordance between tissue and circulating mutational status within the MDACC cohort.

A) Mutational concordance between circulating and tumoral mutations comparing AA and CRC demonstrates significantly lower concordance in AA (p<0.0001). B) Mutational concordance between circulating and tumoral mutations comparing AA by grade demonstrates significantly lower concordance in well or moderately differentiated cancers as compared to poor (p=0.0001 for both associations). C) Mutational concordance between circulating and tumoral mutations comparing in AA by mutation status demonstrates higher concordance for TP53 mutations as compared to GNAS or KRAS mutational status. All comparisons of sensitivities described above calculated using McNemar’s Chi-squared.
Survival analysis
The overall survival (OS) of patients with AA who had detectable ctDNA was significantly lower than those with undetectable ctDNA (median OS not-yet-reached for both groups, HR = 2.32, p = 0.048) (Figure 4A). The worse survival associated detectable ctDNA was not just a surrogate for grade; when restricting to poorly differentiated tumors, median OS was 21 months for patients with detectable ctDNA vs. 31 months for undetectable ctDNA (HR = 2.7, p = 0.035, Figure 4B). The median follow-up for our cohort of 37.4 months was not long enough to evaluate OS in patients with well or moderately differentiated tumors. Interestingly, patients with presence of a KRAS mutation in blood had particularly poor survival with all 13 patients expiring within 2.5 years (median OS 21.2 months vs. not-yet-reached for KRAS mutation not detected, HR = 6.38, p < 0.0001, (Fig 4C). This was not observed for other genes such as GNAS and TP53 and contrasts the survival association seen in tissue NGS (Supplementary Figure 4A, B).(10) These data highlight the prognostic utility of ctDNA detection in AA.
Figure 4: Associations between circulating mutational status and survival.
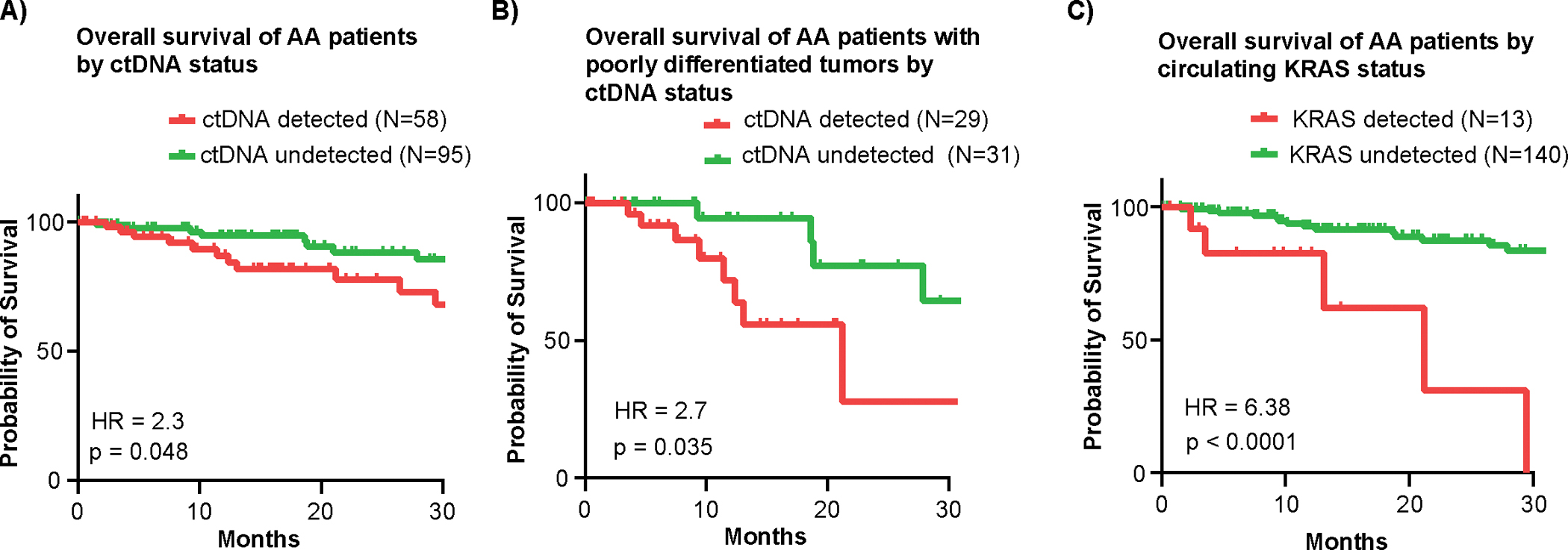
A) Overall survival of AA patients by ctDNA status demonstrates significantly impaired overall survival in the group of patients with detectable ctDNA (HR=2.324, p=0.0482). B) Overall survival of poorly differentiated AA patients by ctDNA status demonstrates significantly impaired overall survival in the group of patients with detectable ctDNA (HR=2.7, p=0.0351). C) Overall survival of AA patients by circulating KRAS status demonstrates significantly impaired overall survival in the group of patients with detectable ctDNA (HR=6.38, p<0.0001). All Hazard ratios calculated using Kaplan-Meier statistics.
Network Analysis
We next sought to study the role pathway alterations play in mutational subtyping of AA as well as associations with patient outcomes. Network analysis was conducted on co-mutated genes (co-occurring and mutually exclusive), calculated using Chi-square tests, followed by KEGG pathway enrichment analysis. The enrichment analysis grouped the selected gene interaction modules into pathways. (Supplementary Figure 5A). Notable overlap was observed across these groups in the patients' cohort of the Guardant Health genomic database (Supplementary Figure 6). We next correlated pathway alterations with patient outcomes in patients within our MDACC cohort and showed alterations in multiple pathways had worse overall survival compared to those with mutations restricted to the TP53/DNA Damage Response/Cell Cycle pathways (Supplementary Figure 5B).
DISCUSSION
This work is the largest description to date of tumor agnostic circulating tumoral DNA in appendiceal adenocarcinoma. We find that, compared to other malignancies the positive detection rate of ctDNA in the blood of patients with appendiceal adenocarcinoma is low. In the setting of metastatic disease, only 38% of patients in the MDACC cohort had a detected mutation.
Although rates of detection in the Guardant Health cohort were significantly higher (68%), this is likely due to resectable disease burden in the MDACC cohort often being considered for surgical resection with disease limited to the peritoneum.
Nevertheless, detectable ctDNA had a significant prognostic value with patients with a positive result having worse overall survival. We hypothesize this is related to microscopic changes within the tumor itself, with previously identified markers of invasion correlating with ctDNA positivity.(42, 43) Moreover, the lack of association with the peritoneal cancer index score underscores the barrier role of the poorly vascularized peritoneum.(44, 45) ctDNA status in AA therefore acts as a marker of biology rather than bulky disease burden and is reflective of more aggressive biology seen in a number of histopathologic factors.
Associating ctDNA positivity with mutational status, we found TP53 mut-predominant tumors, classically more aggressive and with more stromal invasion compared to RAS mut-predominant tumors (10), had higher rates of ctDNA positivity. This agrees with our reported higher concordance in TP53-mut tumors and our reported association of poor differentiation, signet ring cell histology, perineural invasion and hematogenous spread, were noted to be aggressive histopathological factors with high rates of ctDNA positivity. Across different cancer types, the detection of ctDNA is variable, with CRC having more than 75% detection rate versus other tumors such as primary CNS malignancies having less than 50%.(18) Even within the same cancer type, AA has had variable detection rates based on the assay used and cohort studied from 11% (46) of 24% (18, 27), while others reported a detection rate of 35.6% (47) comparable to our results here showing a detection rate of 38%. Here we highlight the potential of ctDNA to identify patients with poorly differentiated tumors who may have a comparatively favorable disease biology and prognosis (i.e. patients with poorly differentiated histology who are ctDNA negative). These patients may benefit from cytoreductive surgery after a period of disease response or stability with systemic therapy. Indeed, ctDNA may also help guide decisions regarding nature and duration of perioperative therapy in these patients. Trials are ongoing studying the role of ctDNA in aiding in therapeutic decision making in AA and CRC (NCT05947838) and alternative biomarkers utilizing peritoneal fluid or other ctDNA detection assays are being actively explored.(15, 48, 49)
The importance of circulating mutational status is here best exemplified by KRAS mutations that in themselves do not correlate with survival but when detected in blood was associated with a worse survival. This was observed in CRC as well, where pre-operative detection of KRAS mutation was associated with an earlier risk of relapse (50) and in pancreatic cancer, where it is noted as a negative predictive indicator (51). While RAS predominant mutated tumors were seen to have a better survival when compared to tumors with no RAS, TP53 or GNAS mutations (10), having a KRAS mutation in blood was associated with poor survival. Due to the low rate of detecting KRAS mutation in blood, a definite conclusion cannot be made; however, a trend, seen in other cancer types as well, is observed for patients with detectable ctDNA, whether pre- or post-operatively, tend to relapse earlier than those with undetectable ctDNA.
Several limitations of this study should be noted, including its retrospective nature, lack of clinical correlative data from the Guardant Health cohort and the single institution nature of the MDACC cohort. Samples within the Guardant Health cohort were tested across several years and thus ran on various version of the assay (spanning 73- to 83-genes), which means that certain genes and/or alterations may have only been assessed in a portion of patients. Additionally, given the nature of liquid biopsy, we cannot rule-out the possibility that additional alterations were occurring in samples below the assay’s limit of detection. (10, 30, 52, 53) Importantly as well, mOS was not reached in our survival analyses as the clinical follow-up time was relatively limited given the indolent nature of many appendiceal tumors and favorable outcomes after curative intent surgery; mOS may not be reached for some years in this cohort (Figure 4A). In the future ctDNA should be studied in other high-volume centers to build a cross-institutional database to better understand the natural history of ctDNA positivity in patients with AA across the clinical course of diagnosis and treatment, including surgical and chemotherapeutic interventions.
In summary, this work represents a critical piece of information of AA biology as patients undergoing cytoreduction and HIPEC are thought to benefit from this procedure primarily in the setting of disease confined to the peritoneal cavity. This is the first known study of the relationship between blood-based NGS concordance and tumor sequencing in AA. Moving forward, it will be important for additional and pooled data from multiple centers nationally and internationally to be analyzed as a whole for validation. Additionally, future work including increased sequencing coverage may help to better understand the relationship of these common mutations described here and potential novel biomarkers to outcomes. Ultimately, we hope to begin to apply these observations to clinical trials to understand their ability to personalize care for both patients with unresectable AA and those undergoing curative intent resection based on tumoral and/or circulating mutational status. These data suggest an important role for ctDNA status in defining disease biology in AA and highlight the potential for ctDNA to aid in decision-making around perioperative systemic therapy utilization and timing of definitive surgical resection.
Supplementary Material
STATEMENT OF TRANSLATIONAL RELEVANCE:
Here we describe for the first time the landscape of circulating tumoral DNA (ctDNA) in Appendiceal Adenocarcinoma (AA). For patients with AA, particularly low-grade disease confined to the peritoneum, cytoreductive surgery (CRS) with heated intraperitoneal chemotherapy (HIPEC) may be curative. Data, however, are lacking on how to optimize patient selection and ongoing algorithm of surveillance and future care. Here, we describe appendix cancer with a distinct mutational profile and ctDNA detection rate.
We propose the use of circulating tumoral DNA (ctDNA) as a means to inform the status of integrity or breakdown of the blood-peritoneal barrier and identify a population of patient with appendiceal adenocarcinomas with worse overall survival. This work has the potential to be in the very near-future clinically applicable to the practice of cancer medicine for patients suffering from appendix cancer.
FUNDING SOURCES AND ACKNOWLEDGEMENTS:
This work was supported by the Col. Daniel Connelly Memorial Fund, the National Cancer Institute (K22 CA234406 to J.P.S., T32 CA009599 to E.A.F., P50 CA221707 Career Enhancement Program to M.G.W., and the Cancer Center Support Grant (P30 CA016672), the Cancer Prevention & Research Institute of Texas (RR180035 & RP240392 to J.P.S., J.P.S. is a CPRIT Scholar in Cancer Research), the Appendiceal Cancer Pseudomyxoma Peritonei Research Foundation (Young Investigator Award to M.G.W., Catalyst Research Grant to J.P.S.), and a Conquer Cancer Career Development Award (2022CDA-7604125121). Any opinions, findings, and conclusions expressed in this material are those of the author(s) and do not necessarily reflect those of the American Society of Clinical Oncology® or Conquer Cancer. J.P.S. is an Andrew Sabin Family Foundation Fellow at The University of Texas MD Anderson Cancer Center.
Footnotes
CONFLICT OF INTEREST STATEMENT:
A.D. reports research support from Novartis, Eisai, Ipsen, Hutchison Pharma, Guardant Health, and Natera; and personal/consulting fees from AbbVie, Crinetics, Hutchison Pharma, Ipsen, Novartis, Personalis, and Voluntis.
B.K. reports Medtronic stock outside the submitted work.
M.O. reports personal fees from Jansen, Bristol-Myers Squibb, Merck, Abbvie, Medimmune, and Takeda and research support from Merck, Bristol-Myers Squibb, Lilly, Nouscom, Medimmune, and Genentech/Roche.
K.R. reports consulting or advisory roles: Abbvie, AstraZeneca, Bayer, Eisai, Daiichi Sankyo, Sanofi, Seagen. Research funding (institutional): Abbvie, Bayer, Roche/Genentech, Guardant Health, Daiichi Sankyo/Astra Zeneca, Janssen, HiberCell, Innovent, Merck Serono, Seagen, Xencor.
A.U. reports consulting with Bayer pharmaceutical.
L.M.D. is an employee of Guardant Health and holds stock options as such.
J.P.S. reports consulting with Engine Biosciences and NaDeNo Nanoscience.
References
- 1.Nitecki SS, Wolff BG, Schlinkert R, Sarr MG, The natural history of surgically treated primary adenocarcinoma of the appendix. Ann Surg 219, 51–57 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ang CS-P et al. , Genomic Landscape of Appendiceal Neoplasms. JCO Precision Oncology, 1–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grotz TE et al. , Stratification of outcomes for mucinous appendiceal adenocarcinoma with peritoneal metastasis by histological grade. World J Gastrointest Oncol 9, 354–362 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levine EA et al. , Prognostic Molecular Subtypes of Low-Grade Cancer of the Appendix. J Am Coll Surg 222, 493–503 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fernandez RN, Daly JM, Pseudomyxoma peritonei. Archives of surgery 115, 409–414 (1980). [DOI] [PubMed] [Google Scholar]
- 6.Sugarbaker PH et al. , Pseudomyxoma peritonei syndrome. Adv Surg 30, 233–280 (1996). [PubMed] [Google Scholar]
- 7.Smith JW et al. , Pseudomyxoma peritonei of appendiceal origin. The Memorial Sloan-Kettering Cancer Center experience. Cancer 70, 396–401 (1992). [DOI] [PubMed] [Google Scholar]
- 8.Tejani MA et al. , Systemic therapy for advanced appendiceal adenocarcinoma: an analysis from the NCCN Oncology Outcomes Database for colorectal cancer. J Natl Compr Canc Netw 12, 1123–1130 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Malas MA, Sulak O, Gökçimen A, Sari A, Development of the vermiform appendix during the fetal period. Surg Radiol Anat 26, 202–207 (2004). [DOI] [PubMed] [Google Scholar]
- 10.Foote MB et al. , Molecular Classification of Appendiceal Adenocarcinoma. J Clin Oncol 41, 1553–1564 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.White M et al. , O-11 The genomic landscape of appendiceal adenocarcinoma revealed by 855 whole exome sequences. Annals of Oncology 34, S185 (2023). [Google Scholar]
- 12.Raghav K et al. , Integrated clinico-molecular profiling of appendiceal adenocarcinoma reveals a unique grade-driven entity distinct from colorectal cancer. Br J Cancer 123, 1262–1270 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tokunaga R et al. , Molecular Profiling of Appendiceal Adenocarcinoma and Comparison with Right-sided and Left-sided Colorectal Cancer. Clin Cancer Res 25, 3096–3103 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Y.-q. Lan et al. , Combination chemotherapy with paclitaxel and oxaliplatin as first-line treatment in patients with advanced gastric cancer. Cancer Chemotherapy and Pharmacology 81, 1007–1015 (2018). [DOI] [PubMed] [Google Scholar]
- 15.White BHM, Rivero-Hinojosa S, Yousef A, Bhutiani N, Aushev V, Chowdhury S, Jurdi A, Liu M, Fournier K, Shen J, The genomic landscape of appendiceal adenocarcinoma revealed by 855 whole exome sequences. Annals of Oncology 31, S185 (2023). [Google Scholar]
- 16.Liao X et al. , Mutation profile of high-grade appendiceal mucinous neoplasm. Histopathology 76, 461–469 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Parsons HA, Beaver JA, Park BH, Circulating Plasma Tumor DNA. Adv Exp Med Biol 882, 259–276 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Bettegowda C et al. , Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med 6, 224ra224 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Mattos-Arruda L et al. , Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: a proof-of-principle. Ann Oncol 25, 1729–1735 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beagan JJ et al. , Circulating Tumor DNA as a Preoperative Marker of Recurrence in Patients with Peritoneal Metastases of Colorectal Cancer: A Clinical Feasibility Study. J Clin Med 9, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loupakis F et al. , Detection of Molecular Residual Disease Using Personalized Circulating Tumor DNA Assay in Patients With Colorectal Cancer Undergoing Resection of Metastases. JCO Precis Oncol 5, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tie JCJ, Lahouel K, Lo SN, Wang Y, Wong R, Shapiro J, Harris S, Khattak A, Burge M, Horvath LG, Karapetis CS, Shannon J, Singh M, Yip D, Papadopoulos N, Tomasetti C, Kinzler K, Vogelstein B, Circulating tumour DNA (ctDNA) dynamics, CEA and sites of recurrence for the randomised DYNAMIC study: Adjuvant chemotherapy (ACT) guided by ctDNA analysis in stage II colon cancer (CC). Annals of Oncology 33, S683 (2022). [Google Scholar]
- 23.Shen JP, Regarding Blood-Based Next-Generation Sequencing Analysis of Appendiceal Cancers. The oncologist 25, e2019–e2020 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dhiman A et al. , Role of Tumor-informed Personalized Circulating Tumor DNA Assay in Informing Recurrence in Patients With Peritoneal Metastases From Colorectal and High-grade Appendix Cancer Undergoing Curative-intent Surgery. Annals of surgery 278, 925–931 (2023). [DOI] [PubMed] [Google Scholar]
- 25.Shaib WL et al. , Blood-Based Next-Generation Sequencing Analysis of Appendiceal Cancers. Oncologist 25, 414–421 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lopez-Rojo I et al. , Liquid biopsy in peritoneal fluid and plasma as a prognostic factor in advanced colorectal and appendiceal tumors after complete cytoreduction and hyperthermic intraperitoneal chemotherapy. Ther Adv Med Oncol 12, 1758835920981351 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Riviere P et al. , The Mutational Landscape of Gastrointestinal Malignancies as Reflected by Circulating Tumor DNA. Mol Cancer Ther 17, 297–305 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh H et al. , Highly Sensitive Circulating Tumor DNA Assay Aids Clinical Management of Radiographically Occult Isolated Peritoneal Metastases in Patients With GI Cancer. JCO Precis Oncol 7, e2200572 (2023). [DOI] [PubMed] [Google Scholar]
- 29.Odegaard JI et al. , Validation of a Plasma-Based Comprehensive Cancer Genotyping Assay Utilizing Orthogonal Tissue- and Plasma-Based Methodologies. Clin Cancer Res 24, 3539–3549 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Zill OA et al. , The Landscape of Actionable Genomic Alterations in Cell-Free Circulating Tumor DNA from 21,807 Advanced Cancer Patients. Clin Cancer Res 24, 3528–3538 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Luthra R et al. , A Targeted High-Throughput Next-Generation Sequencing Panel for Clinical Screening of Mutations, Gene Amplifications, and Fusions in Solid Tumors. J Mol Diagn 19, 255–264 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Lanman RB et al. , Analytical and Clinical Validation of a Digital Sequencing Panel for Quantitative, Highly Accurate Evaluation of Cell-Free Circulating Tumor DNA. PLoS One 10, e0140712 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Umar A et al. , Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 96, 261–268 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jaiswal S et al. , Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 371, 2488–2498 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Landrum MJ et al. , ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res 46, D1062–D1067 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ang CS et al. , Genomic Landscape of Appendiceal Neoplasms. JCO Precis Oncol 2, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suehnholz SP et al. , Quantifying the Expanding Landscape of Clinical Actionability for Patients with Cancer. Cancer discovery 14, 49–65 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Loree JM et al. , Clinical and Functional Characterization of Atypical KRAS/NRAS Mutations in Metastatic Colorectal Cancer. Clin Cancer Res 27, 4587–4598 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luciakova K, Kollarovic G, Kretova M, Sabova L, Nelson BD, TGF-β signals the formation of a unique NF1/Smad4-dependent transcription repressor-complex in human diploid fibroblasts. Biochemical and biophysical research communications 411, 648–653 (2011). [DOI] [PubMed] [Google Scholar]
- 40.Kato S et al. , Genomic Assessment of Blood-Derived Circulating Tumor DNA in Patients With Colorectal Cancers: Correlation With Tissue Sequencing, Therapeutic Response, and Survival. JCO Precis Oncol 3, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Russo M et al. , Tumor Heterogeneity and Lesion-Specific Response to Targeted Therapy in Colorectal Cancer. Cancer Discov 6, 147–153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bredno J et al. , Tumor area and microscopic extent of invasion to determine circulating tumor DNA fraction in plasma and detectability of colorectal cancer (CRC). Journal of Clinical Oncology 38, 243–243 (2020). [Google Scholar]
- 43.Wang J et al. , Circulating tumor DNA correlates with microvascular invasion and predicts tumor recurrence of hepatocellular carcinoma. Ann Transl Med 8, 237 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Flessner MF, The transport barrier in intraperitoneal therapy. Am J Physiol Renal Physiol 288, F433–442 (2005). [DOI] [PubMed] [Google Scholar]
- 45.Kastelein AW et al. , Poor perfusion of the microvasculature in peritoneal metastases of ovarian cancer. Clin Exp Metastasis 37, 293–304 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bhutiani N et al. , Utility of Circulating Tumor DNA in Appendiceal Tumors. J Gastrointest Surg, (2023). [DOI] [PubMed] [Google Scholar]
- 47.Baumgartner JM et al. , Preoperative Circulating Tumor DNA in Patients with Peritoneal Carcinomatosis is an Independent Predictor of Progression-Free Survival. Ann Surg Oncol 25, 2400–2408 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leick KM et al. , Peritoneal Cell-Free Tumor DNA as Biomarker for Peritoneal Surface Malignancies. Ann Surg Oncol 27, 5065–5071 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vanť Erve I et al. , Detection of tumor-derived cell-free DNA from colorectal cancer peritoneal metastases in plasma and peritoneal fluid. J Pathol Clin Res 7, 203–208 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nakamura Y et al. , Preoperative detection of KRAS mutated circulating tumor DNA is an independent risk factor for recurrence in colorectal cancer. Sci Rep 11, 441 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kruger S et al. , Repeated mutKRAS ctDNA measurements represent a novel and promising tool for early response prediction and therapy monitoring in advanced pancreatic cancer. Ann Oncol 29, 2348–2355 (2018). [DOI] [PubMed] [Google Scholar]
- 52.Siravegna G et al. , Plasma HER2 (ERBB2) Copy Number Predicts Response to HER2-targeted Therapy in Metastatic Colorectal Cancer. Clin Cancer Res 25, 3046–3053 (2019). [DOI] [PubMed] [Google Scholar]
- 53.Davies KD, Aisner DL, Wake Up and Smell the Fusions: Single-Modality Molecular Testing Misses Drivers. Clin Cancer Res 25, 4586–4588 (2019). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.