

ABSTRACT
Background
Esophageal squamous cell carcinoma (ESCC) is a lethal malignancy, and the molecular underpinnings of its aggressive behavior are not fully understood. FYN proto‐oncogene, Src family tyrosine kinase (FYN) has been linked to cancer progression, yet its role in ESCC remains elusive. This study investigated the influence of FYN on ESCC malignancy.
Methods
Quantitative real‐time polymerase chain reaction was used to assess the mRNA expression of FYN, while western blotting and immunohistochemistry (IHC) assays were performed to detect the protein expression of FYN, ubiquitin specific peptidase 8 (USP8) and protein tyrosine kinase 2 (PTK2). Cell viability was measured with a cell counting kit‐8 assay, and cell apoptosis was evaluated using flow cytometry.
Results
FYN expression was increased in ESCC tissues and cells when compared with normal esophageal tissues and normal esophageal epithelial cells. Knockdown of FYN inhibited cell invasion, migration, stem‐like traits, and glycolysis, while promoting apoptosis. USP8 was shown to stabilize FYN protein expression through its deubiquitinating activity in ESCC cells. Overexpression of FYN reversed the effects of USP8 silencing on the malignant phenotypes of ESCC cells in vitro and in vivo. FYN upregulated PTK2 expression in both TE1 and KYSE150 cell lines. Furthermore, PTK2 overexpression reversed the effects of FYN silencing on the malignant phenotypes of ESCC cells. Further, USP8 silencing‐induced inhibitory effect on PTK2 protein expression was counteracted after FYN overexpression.
Conclusion
USP8‐dependent FYN contributed to the malignant progression of ESCC by interacting with PTK2. Targeting this pathway may offer a novel therapeutic strategy for ESCC treatment.
Keywords: ESCC, FYN, PTK2, USP8
USP8‐dependent deubiquitination of FYN promoted the malignant progression of ESCC by upregulating PTK2 expression.

1. Introduction
Esophageal cancer is a malignant tumor that originates in the esophagus [1]. Esophageal squamous cell carcinoma (ESCC) is more common in developing countries, particularly in regions where dietary habits and environmental factors contribute to a higher risk of this disease. According to the latest global cancer statistics, esophageal cancer ranks eleventh in the world for incidence, and it is the seventh leading cause of cancer‐related deaths [2]. Despite significant advancements in perioperative management and techniques, the 5‐year survival rate for patients with esophageal cancer is less than 20% [3, 4]. This low survival rate is attributed to the late stage at which many patients are diagnosed and the aggressive nature of the disease [5]. Understanding the molecular mechanisms can identify novel therapeutic targets and improve survival rates for patients.
Src family tyrosine kinase (FYN) is one of the key members of the tyrosine kinase family. It consists of approximately 59 kDa and its genetic information locates on chromosome 6q21 in humans [6]. FYN is primarily located in the cytoplasmic leaflets and is involved in the intracellular trafficking of various cell surface receptors within the lipid rafts of the plasma membrane [7]. This trafficking is crucial for the activation of signaling pathways that regulate a wide range of cellular biological behaviors, such as cell growth, survival, synaptic function, platelet activation, cytoskeletal remodeling, cell motility, and the formation of the central nervous system's myelin sheath [8]. In the context of cancer development, FYN has been found to play a significant role. It can contribute to cancer development by activating signaling pathways that promote cell growth, survival, and migration [8]. Overexpression or mutation of FYN has occurred in gastric cancer [9] and hematological malignancies [10]. However, its function in ESCC and the underlying mechanism remain unclear.
Ubiquitination is a reversible post‐translational modification process that involves the proteasome‐induced protein degradation, a cellular machinery responsible for breaking down proteins [11]. Ubiquitination can also affect protein stability, localization, and activity. However, deubiquitination is the process by which ubiquitin is removed from proteins, thereby rescuing them from degradation and restoring their function. Deubiquitinating enzymes (DUBs) are responsible for this process. Ubiquitin specific peptidase 8 (USP8) is a DUB that can regulate K6‐, K48‐, and K63‐linked polyubiquitin chains [12]. USP8 is involved in the stability of a variety of substrates, some of which include TGF‐β receptor TβRII [13] and Nrf2 [14]. USP8 may play a role in cancer development. Its dysregulation has been linked to tumor growth, metastasis, and resistance to chemotherapy [15]. In particular, previous evidence has revealed its promoting effect on ESCC progression [16].
Based on the above evidence, the study explored FYN's function in the malignant progression of ESCC. In addition, we investigated the association of FYN and USP8 with ESCC cell malignancy to reveal the mechanism deriving FYN to regulate the malignant behaviors of ESCC cells, aiming to providing potential targets to hinder ESCC progression.
2. Materials and Methods
2.1. Clinical Samples
The study enrolled ESCC patients (N = 37) who had undergone esophageal cancer resection at the First Affiliated Hospital of Hainan Medical College. All patients had undergone resection for esophageal cancer without a history of other malignancies. They had not received any anti‐tumor treatment prior to surgery, and complete tumor removal (R0 resection) was achieved during the operation. The tumor tissues and paracancerous healthy esophageal tissues were collected from these patients and stored at −80°C. The study was approved by the Ethics Committee of the First Affiliated Hospital of Hainan Medical College and obtained informed consent from the patients or their relatives.
2.2. Cell Lines
ESCC cell lines including KYSE30, KYSE‐180, TE‐1, and KYSE150 and normal esophageal epithelial cells (HEEC) were purchased from Procell (Wuhan, China). All cells were maintained in RPMI‐1640 (Biosun, Shanghai, China) supplemented with 10% fetal bovine serum (Biosun) and 1% penicillin/streptomycin (Millipore, Bradford, MA, USA) at 37°C with 5% CO2.
2.3. Cell Transfection
Small interfering RNAs (siRNAs) were synthesized by Tsingke Biotech (Shanghai, China) and used to downregulate gene expression in ESCC cells. The siRNAs included those targeting FYN (si‐FYN) and USP8 (si‐USP8) and a negative control (sh‐NC). Overexpression plasmids were used to upregulate USP8 or FYN expression in ESCC cells, including USP8 overexpression plasmid and FYN overexpression plasmid. One day before transfection, cells were seeded into six‐well plates. Lipofectamine 2000 (Thermo Fisher, Waltham, MA, USA) was diluted in serum‐free Opti‐MEM (Solarbio, Beijing, China) and used for cell transfection. The diluted mixtures were added to the six‐well plates, followed by light shaking. The cells were cultured for 6 h, and the culture medium was replaced with serum‐containing RPMI‐1640 (Biosun).
2.4. Quantitative Real‐Time Polymerase Chain Reaction
Approximately 40–50 mg of tissue samples were taken and minced, followed by treatment with TransZol (TransGen, Beijing, China) to extract RNA. For cell samples, TransZol reagent was directly added to the culture plates for RNA extraction. The RNA was solubilized in RNase‐free ddH2O. Genomic DNA was removed using the gDNA wiper Mix (Vazyme, Nanjing, China), and reverse transcription was performed with the HiScript III qRT SuperMix (Vazyme). The synthesized cDNA and SYBR qPCR Master Mix (Vazyme) were mixed and used for gene expression detection. Finally, gene expression was analyzed using the 2−ΔΔCt method with the normalization to β‐actin. Primer sequences are listed in Table 1.
TABLE 1.
Primer sequences used in qRT‐PCR.
| Name | Primers for qRT‐PCR (5′–3′) | |
|---|---|---|
| FYN | Forward | TCCTTCTCCTTTCAGTCAGGAAT |
| Reverse | CACACCTCCAAAGACGGTGA | |
| β‐actin | Forward | TGGATCAGCAAGCAGGAGTA |
| Reverse | TCGGCCACATTGTGAACTTT |
2.5. Western Blotting Assay
Tissue blocks were taken and lysed with RIPA buffer (Beyotime, Shanghai, China) at a ratio of 10 μL/1 mg. The supernatant was separated by centrifugation and subjected to a 10‐min reaction at 100°C in a metallic bath to denature the proteins. The protein concentration was calculated, and the protein samples were loaded at 30 μg per well, and electrophoresis was performed at 120 V, with the running time adjusted according to the molecular weight of the target protein. Transblotting was carried out, and the PVDF membranes (Millipore) were removed and placed in a blocking solution. The membranes were then incubated with primary and secondary antibodies including anti‐FYN (1:1000, Affinity), anti‐USP8 (1:2000, Abcam), anti‐PTK2 (1:1000, Affinity), anti‐β‐actin (1:5000, Affinity), and Goat Anti‐Rabbit IgG (1:5000, Abcam). ECL reagent (Beyotime) was dripped onto the PVDF membrane surface and evenly spread, and an image analysis system (Thermo Fisher) was used for detection of protein expression.
2.6. Cell Counting Kit‐8
Healthy ESCC cells were selected and digested with trypsin, which was then diluted with RPMI‐1640 (Biosun). In 96‐well plates, 100 μL of cell suspension was added to each well, and the plates were placed in an incubator for an additional 48 h. Subsequently, cell counting kit‐8 (CCK‐8) reagent (Beyotime) was added to the plates. After the medium turned orange‐yellow, samples were analyzed using a microplate reader.
2.7. Flow Cytometry Analysis
After cells in six‐well plates had been intervened, they were digested with trypsin at 0.25% concentration without EDTA in an appropriate volume. The digestion was stopped with complete RPMI‐1640 medium (Biosun) equal to three times the volume of trypsin. The cells were collected into a centrifuge tube and centrifuged. They were resuspended in PBS and centrifuged again using a centrifuge machine. The cells were then resuspended in binding buffer, and Annexin V‐FITC (Solarbio) and propidium iodide staining reagents (Solarbio) were added to the buffer. The staining process was carried out in the dark. The stained cells were analyzed using a flow cytometer.
2.8. Transwell Invasion Assay
Matrigel gels (Abwbio, Shanghai, China) was added to the upper chambers of the Transwell, which was then allowed to solidify for 3–4 h. The cell pellets were resuspended in serum‐free RPMI‐1640 medium (Biosun), and the upper chambers were filled with the fully resuspended cell suspension. The lower chambers were then filled with complete medium and placed in the incubator for culture. After 24 h, the Transwell chamber was removed, fixed with 4% paraformaldehyde, and stained with 0.1% crystal violet solution. The upper chambers were wiped clean of non‐passing cells with a moist cotton swab. The chambers were allowed to air dry, inverted, and examined under a microscope for observation and photography.
2.9. Wound‐Healing Assay
The cell density was adjusted to 5 × 105 cells/mL using RPMI‐1640 medium (Biosun), and the cell suspension was added to each well of scratch inserts (ibidi, Martinsried, Germany), which was then placed in the cell culture incubator for incubation. Once the cell density reached 95%–100%, the inserts were carefully removed using tweezers and washed three times with PBS to remove non‐adherent cells. The inserts were then placed in serum‐free RPMI‐1640 medium and returned to the incubator for further culture. Twelve hours later, the cells were observed and photographed under a microscope.
2.10. Sphere Formation Assay
Under aseptic conditions, 50 μL of FGF (10 μg/mL, PeproTech, Rocky Hill, NJ, USA) and 10 μL of EGF (100 μg/mL, PeproTech) were added to 50 mL of RPMI‐1640 culture medium, along with 1 mL of B27 (Thermo Fisher) and 1% fetal bovine serum (Biosun). After 24 h of transfection, the cells from each intervention group were digested and resuspended in serum‐free RPMI‐1640 for cell counting. About 50 μL of a 1 × 104 cells/mL cell suspension was added to each well of a low‐adherence six‐well plate. The cells were cultured at 37°C, with culture medium added to the edge of six‐well plates every 2–3 days, and the culture was terminated upon the appearance of distinct cell spheroids.
2.11. Glycolysis Analysis
Glucose consumption, lactate production, and ATP/ADP ratio were determined to evaluate glycolysis using colorimetric methods according to the guidebooks provided by respective commercial kits including Glucose Assay Kit (#ab65333, Abcam), Lactate Assay Kit (#ab65331; Abcam), and ADP/ATP Ratio Assay Kit (#ab65313, Abcam).
2.12. Cycloheximide Assay
ESCC cells were seeded in 24‐well plates and transfected with USP8 overexpression plasmid or vector for 48 h. Then, the cells were exposed to cycloheximide (CHX) reagent (MedChemExpress, Princeton, NJ, USA) for 5, 10, 20 h. The cells were harvested for the protein expression analysis of FYN and β‐actin according to the above methods.
2.13. Co‐Immunoprecipitation
NP‐40 lysis buffer (Beyotime) was added to cell culture wells, and the cells were scraped from the culture dish and collected by centrifugation. To each 1 mL of total protein, 20 μL of Protein A/G Magnetic Beads (Millipore) were added. A 10 μL of cell supernatant was taken as the input group. To the remaining supernatant, 1 μg of the antibodies against FYN (1:40, Abcam), USP8 (1:200, Abcam), and PTK2 (1:50, Thermo Fisher) for immunoprecipitation was added and incubated overnight. The next day, the antigen–antibody complexes were captured using magnetic beads. The EP tubes were placed on a magnetic stand, the supernatant was removed, and 60 μL of loading buffer was added to suspend the magnetic beads with the antigen–antibody complexes. The samples were taken for western blotting analysis using anti‐USP8 (1:2000, Abcam), anti‐FYN (1:1000, Affinity), and anti‐PTK2 (1:1000, Affinity).
2.14. Immunofluorescence Assay
ESCC cells were cultured in 15 mm confocal dishes until they reached a fusion rate of 60%–70%. The cells were treated with 4% paraformaldehyde and 0.5% Triton X‐100 for 20 min. Primary antibodies against FYN (1:500, Abcam) and PTK2 (1:250, Abcam) were added and the slides were incubated overnight in a humid box. Fluorescent secondary antibodies including Dylight 488/Dylight 533‐conjugated antibodies (Thermo Fisher) were added. DAPI (Beyotime) was dropped onto the samples to stain the nuclei in the dark. The slides were coverslipped and observed under a fluorescence microscope for photography.
2.15. Ubiquitination Assay
To directly detect the ubiquitination of FYN in cell extracts, si‐USP8 and sh‐NC were transfected into ESCC cells. About 48 h later, total protein was collected. The protein collection steps were the same as above, and the immunoprecipitation experiment used an anti‐FYN antibody (1:40, Abcam). Subsequently, western blotting analysis was performed using the antibodies against Ub (1:1000, Abcam), USP8 (1:2000, Abcam), and FYN (1:1000, Affinity).
2.16. Xenograft Mouse Model Assay
Ethical approval for the animal experiments in this study was obtained from the Animal Care Committee of the First Affiliated Hospital of Hainan Medical College. The nude mice used in this study were female BALB/c nude mice (4–5 weeks old, N = 15, five mice/group) purchased from Hunan Slyke Jingda Experimental Animal Co. Ltd. (Changsha, China). KYSE150 cells with a cell confluence of 80%–90% were resuspended in PBS and the cell density was adjusted to 4 × 107 cells/mL. The cells were injected into the right axillary of the nude mice. Tumor size measurements were performed after 8 days later, with recordings every 4 days. Four weeks post‐injection, the nude mice were intravenously anesthetized with pentobarbital (100 mg/kg, Lianshuo Biotech, Shanghai, China), followed by cervical dislocation to euthanize the mice. The tumor tissues were then resected for further analysis.
2.17. Immunohistochemistry (IHC) Assay
Wax‐embedded slices were dehydrated and hydrated. Sodium citrate buffer (Qcheng Biotech, Shanghai, China) was added to the slices and covered the tissues. Then, 0.3% H2O2 (Sigma, St. Louis, MO, USA) was dropped onto the slices to cover the tissues. A working solution of primary antibodies against Ki67 (1:200, Affinity), FYN (1:200, Affinity), and PTK2 (1:200, Affinity) were added to the slices and covered the entire tissues. Subsequently, enzyme‐labeled IgG antibody (Phygene, Fuzhou, China) was added to the slices and covered the tissues. 3,3‐diaminobenzidine (DAB, Phygene) was then dropped onto the slices and incubated for 1–2 min. The dehydration steps were conducted, and the slides were coverslipped with neutral resin. The slides were observed under a microscope.
2.18. Statistical Analysis
Statistical analysis was conducted using GraphPad Prism 8.0. Comparisons between two groups were made using Student's t‐test, while comparisons among three or more groups were performed using one‐way analysis of variance (ANOVA). Measurement data were presented as the mean ± standard deviation (SD). p < 0.05 indicated statistically significant. *p < 0.05, **p < 0.01, and ***p < 0.001.
3. Results
3.1. FYN Expression Was Upregulated in ESCC Tissues and Cells
The expression property of FYN in ESCC tissues and normal esophageal tissues was analyzed by quantitative real‐time polymerase chain reaction (qRT‐PCR). As shown in Figure 1A, FYN mRNA expression was upregulated in ESCC tissues when compared with normal tissues. In addition, the results revealed that ESCC patients with high FYN expression predicted a poor prognosis in comparison with those with low FYN expression (Figure 1B). The results also showed that FYN expression at mRNA and protein levels was higher in ESCC tissues and cells (KYSE30, KYSE180, TE1, and KYSE150) than in normal esophageal tissues and esophageal epithelial cells (HEEC) (Figure 1C,D). Both TE1 and KYSE150 cells were employed for the following study due to their higher expression of FYN. These data demonstrate that FYN is highly expressed in ESCC tissues and cells.
FIGURE 1.
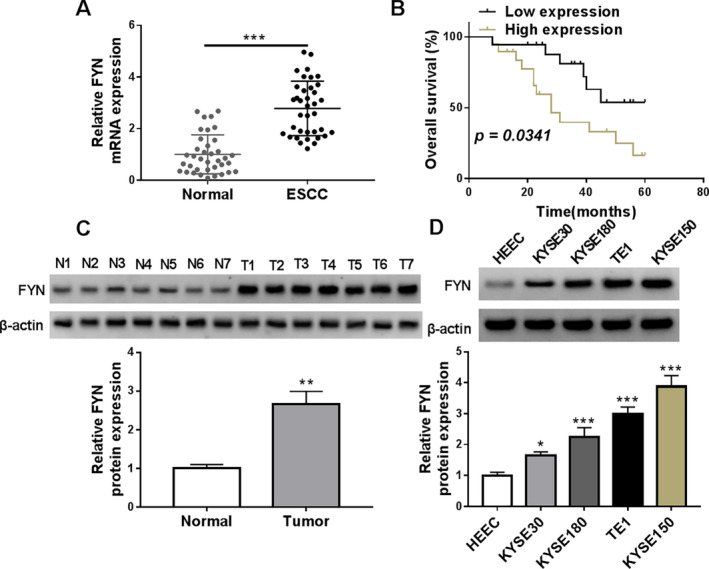
FYN expression was upregulated in ESCC tissues and cells. (A) The expression property of FYN in ESCC tissues and normal esophageal tissues was analyzed by qRT‐PCR. (B) The Kaplan–Meier method was performed to analyze the prognosis of ESCC patients with high or low FYN expression. The median expression of FYN in these ESCC patients was used as the cutoff value for high or low expression of FYN. (C and D) FYN protein expression was detected by western blotting assay in ESCC tissues (N = 7), normal esophageal tissues (N = 7), ESCC cells (KYSE30, KYSE180, TE1, and KYSE150), and normal esophageal epithelial cells (HEEC). *p < 0.05, **p < 0.01, and ***p < 0.001.
3.2. FYN Knockdown Inhibited ESCC Cell Invasion, Migration, Stem‐Like Trait, and Glycolysis and Induced Cell Apoptosis
The study then analyzed the effects of FYN silencing on the malignant property of ESCC cells by transfecting FYN siRNA into both TE1 and KYSE150 cells. The high efficiency of FYN knockdown in both cells is shown in Figure 2A. Subsequently, the results showed that FYN silencing inhibited cell viability but induced cell apoptosis (Figure 2B,C). In addition, the treatment inhibited cell invasion, cell migration, and the formation of microspheres (Figure 2D–G). Moreover, FYN silencing decreased glucose consumption, lactate production, and the value of ATP/ADP in both TE1 and KYSE150 cells (Figure 2H–J). Thus, FYN may contribute to the malignant growth of ESCC cells in vitro.
FIGURE 2.
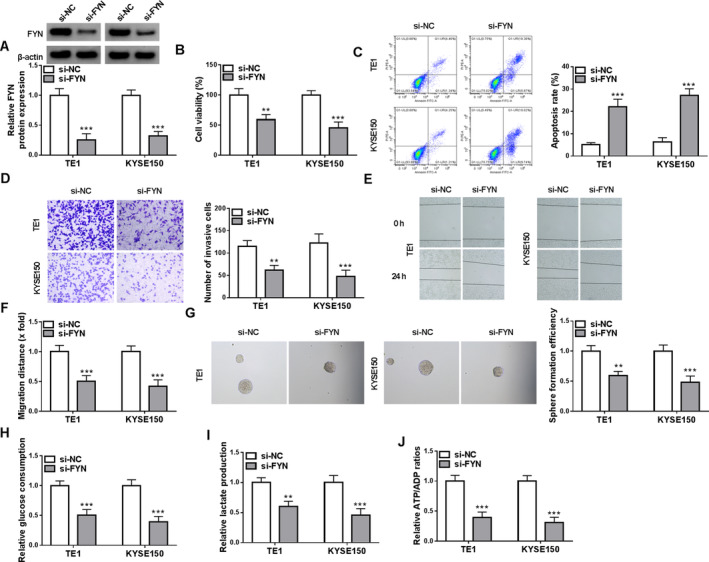
FYN knockdown inhibited ESCC cell invasion, migration, stem‐like trait, and glycolysis and induced cell apoptosis. Both TE1 and KYSE150 cells were divided into two groups, including si‐NC group and si‐FYN group. (A) FYN protein expression was analyzed by western blotting assay. (B) Cell viability was analyzed by CCK‐8 assay. (C) Cell apoptosis was analyzed by flow cytometry. (D) Cell invasion was assessed by transwell assays. (E and F) Cell migration was detected by wound‐healing assay. (G) Cancer stem‐like cell property was analyzed by sphere formation assay. (H–J) Colorimetric methods were used to analyze glucose consumption, lactate production, and the value of ATP/ADP. **p < 0.01 and ***p < 0.001.
3.3. USP8 Stabilized FYN Protein Expression Through Its Deubiquitinating Activity in ESCC Cells
The study then analyzed the upstream regulator of FYN through the UbiBrowser database. As shown in Figure 3A, USP8 might have a potential effect on the ubiquitination level of FYN. The study subsequently transfected USP8 siRNA and its control (si‐NC) into both TE1 and KYSE150 cells to explore the consequential effects on FYN expression. The high efficiency of USP8 knockdown in these cells is shown in Figure 3B. We discovered that USP8 silencing not affected FYN mRNA expression but significantly downregulated its protein expression (Figure 3C,D). Moreover, the inhibitory effect on FYN protein expression induced by USP8 silencing could be relieved after treatment with MG132, a proteasome inhibitor (Figure 3D). In addition, the study used CHX to analyze the effect of USP8 overexpression on the stability of FYN protein expression. The efficiency of USP8 overexpression was high in both TE1 and KYSE150 cells, and the result is shown in Figure 3E. As shown in Figure 3F, the half‐life of FYN protein was lengthened after USP8 overexpression. The co‐immunoprecipitation (Co‐IP) assay demonstrated that the USP8 protein could be immunoprecipitated using the FYN antibody, and conversely, the FYN protein could also be immunoprecipitated using the USP8 antibody (Figure 3G). Further, USP8 silencing significantly increased the levels of endogenous ubiquitinated FYN (Figure 3H). Thus, USP8 stabilized FYN protein expression through the deubiquitination process.
FIGURE 3.
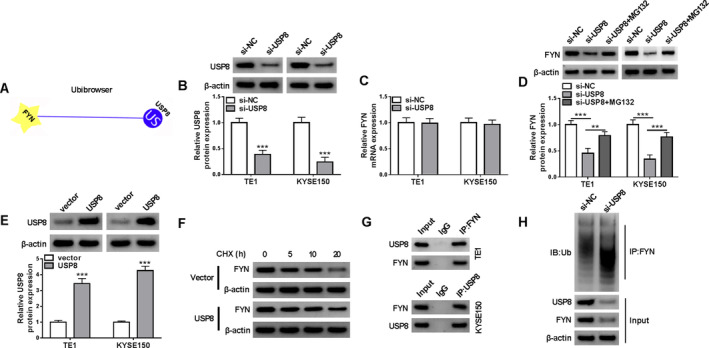
USP8 stabilized FYN protein expression through its deubiquitinating activity in ESCC cells. (A) The UbiBrowser database was used to analyze the association between FYN and USP8. (B) The efficiency of USP8 knockdown was analyzed through the western blotting assay. (C) The effect of USP8 knockdown on FYN mRNA expression was detected by qRT‐PCR. (D) Western blotting assay was used to analyze FYN protein levels in both TE1 and KYSE150 cells transfected with si‐USP8 or si‐NC, with or without MG132 treatment. (E) The efficiency of USP8 overexpression in both TE1 and KYSE150 cells was analyzed by western blotting assay. (F) CHX assay for the protein stability of FYN in both TE1 and KYSE150 cells transfected with USP8 overexpression plasmid or vector. (G and H) The association between FYN and USP8 was analyzed by the Co‐IP assays. **p < 0.01 and ***p < 0.001.
3.4. FYN Overexpression Attenuated USP8 Silencing‐Induced Effects on the Malignant Phenotypes of ESCC Cells In Vitro
The study then analyzed whether USP8 regulated the malignant behaviors of ESCC cells by increasing FYN expression. The result first showed the high efficiency of FYN overexpression plasmid in upregulating FYN protein expression in both TE1 and KYSE150 cells (Figure 4A). Subsequently, USP8 silencing inhibited cell viability and induced cell apoptosis, these effects were counteracted when FYN expression was upregulated (Figure 4B–D). The treatment inhibited cell invasion, cell migration, and the formation of microspheres, whereas these effects were relieved after FYN overexpression (Figure 4E–G). USP8 silencing decreased glucose consumption, lactate production and the value of ATP/ADP in both TE1 and KYSE150 cells, but these effects were restored after FYN expression was increased (Figure 4H–J). Thus, the USP8/FYN axis mediated the malignant growth of ESCC cells in vitro.
FIGURE 4.
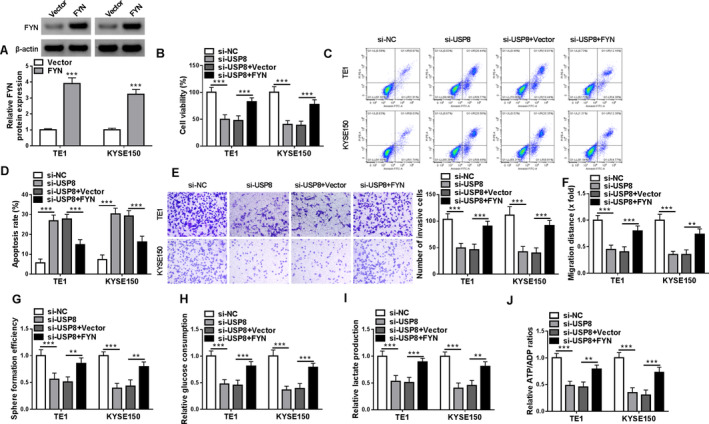
FYN overexpression attenuated USP8 silencing‐induced effects on the malignant phenotypes of ESCC cells in vitro. Both TE1 and KYSE150 cells were transfected with si‐NC, si‐USP8, si‐USP8 + vector, or si‐USP8 + FYN. (A) FYN protein expression was analyzed by western blotting assay. (B) Cell viability was analyzed by CCK‐8 assay. (C and D) Cell apoptosis was analyzed by flow cytometry. (E) Cell invasion was assessed by transwell assays. (F) Cell migration was detected by wound‐healing assay. (G) Cancer stem‐like cell property was analyzed by sphere formation assay. (H–J) Colorimetric methods were used to analyze glucose consumption, lactate production and the value of ATP/ADP. **p < 0.01 and ***p < 0.001.
3.5. FYN Upregulated PTK2 Expression in Both TE1 and KYSE150 Cells
Through the prediction of the STRING database, the study discovered that FYN potentially bound to PTK2 (Figure 5A). In addition, PTK2 expression was higher in ESCA tissues than in normal esophageal tissues, as analyzed using the online databases including the GEPIA and TCGA (Figure 5B,C). We also discovered that FYN silencing downregulated PTK2 protein expression in both TE1 and KYSE150 cells (Figure 5D). Moreover, the Co‐IP assay revealed that FYN and PTK2 interacted with each other directly or indirectly (Figure 5E). Further, the results revealed that FYN and PTK2 exhibited co‐localization in both TE1 and KYSE150 cells, but the phenomenon was weakened after FYN silencing (Figure 5F). Thus, FYN interacted with PTK2 in both TE1 and KYSE150 cells.
FIGURE 5.
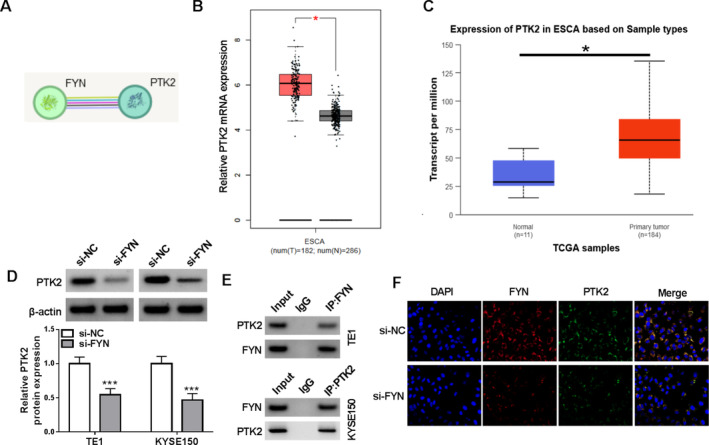
FYN upregulated PTK2 expression in both TE1 and KYSE150 cells. (A) The association between FYN and PTK2 was predicted through the STRING database. (B and C) PTK2 expression was analyzed through the online databases including the GEPIA and TCGA. (D) The effect of FYN silencing on PTK2 protein expression was detected by western blotting assay. (E and F) The Co‐IP assay and immunofluorescence assay were used to analyze the association between FYN and PTK2 in both TE1 and KYSE150 cells after FYN silencing. *p < 0.05 and ***p < 0.001.
3.6. PTK2 Overexpression Attenuated FYN Silencing‐Induced Effects on the Malignant Phenotypes of ESCC Cells
The study continued to analyze whether FYN regulated the malignant behaviors of ESCC cells by increasing PTK2 expression. The result first showed the high efficiency of PTK2 overexpression plasmid in upregulating PTK2 protein expression in both TE1 and KYSE150 cells (Figure 6A). Subsequently, FYN silencing inhibited cell viability and induced cell apoptosis, these effects were counteracted when PTK2 expression was upregulated (Figure 6B–D). The treatment inhibited cell invasion, cell migration, and the formation of microspheres, whereas these effects were relieved after PTK2 overexpression (Figure 6E–G). FYN silencing decreased glucose consumption, lactate production and the value of ATP/ADP in both TE1 and KYSE150 cells, but these effects were restored after PTK2 expression was increased (Figure 6H–J). Thus, the FYN/PTK2 axis mediated the malignant growth of ESCC cells in vitro.
FIGURE 6.
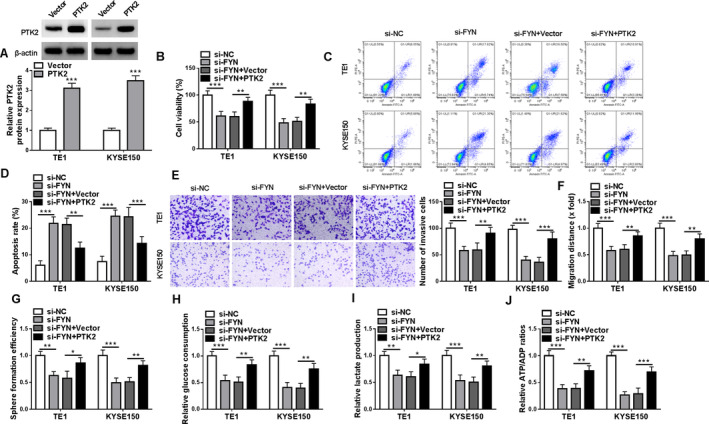
PTK2 overexpression attenuated FYN silencing‐induced effects on the malignant phenotypes of ESCC cells. Both TE1 and KYSE150 cells were transfected with si‐NC, si‐FYN, si‐FYN + vector, or si‐FYN + PTK2. (A) PTK2 protein expression was analyzed by western blotting assay. (B) Cell viability was analyzed by CCK‐8 assay. (C and D) Cell apoptosis was analyzed by flow cytometry. (E) Cell invasion was assessed by transwell assays. (F) Cell migration was detected by wound‐healing assay. (G) Cancer stem‐like cell property was analyzed by sphere formation assay. (H–J) Colorimetric methods were used to analyze glucose consumption, lactate production, and the value of ATP/ADP. *p < 0.05, **p < 0.01 and ***p < 0.001.
3.7. FYN Overexpression Attenuated USP8 Knockdown‐Induced Effects on the Malignant Growth of ESCC Cells In Vivo
A xenograft mouse model was further established using KYSE150 cells to validate the in vitro data regarding the effects of USP8 silencing and FYN overexpression on the malignant behaviors of ESCC cells. As shown in Figure 7A,B, the USP8 silencing group showed slower tumor growth rates and smaller sizes than the sh‐NC group, whereas these effects were relieved after FYN overexpression. In addition, the protein expression of FYN and PTK2 was downregulated in the resulting tumors from the KYSE150 cells transfected with USP8 shRNA when compared with those from the KYSE150 cells transfected with sh‐NC, however, the combination treatment of USP8 silencing and FYN overexpression relieved the effects induced by USP8 silencing alone (Figure 7C,D). Moreover, the result showed that the positive expression rates of Ki67, FYN, and PTK2 was lower in tumors from the sh‐USP8 group than in those from the sh‐NC group, but these values were higher in the tumors from the sh‐USP8 + FYN group than in those from the sh‐USP8 group (Figure 7E). Thus, the USP8/FYN axis mediated the malignant growth of ESCC cells in vivo.
FIGURE 7.
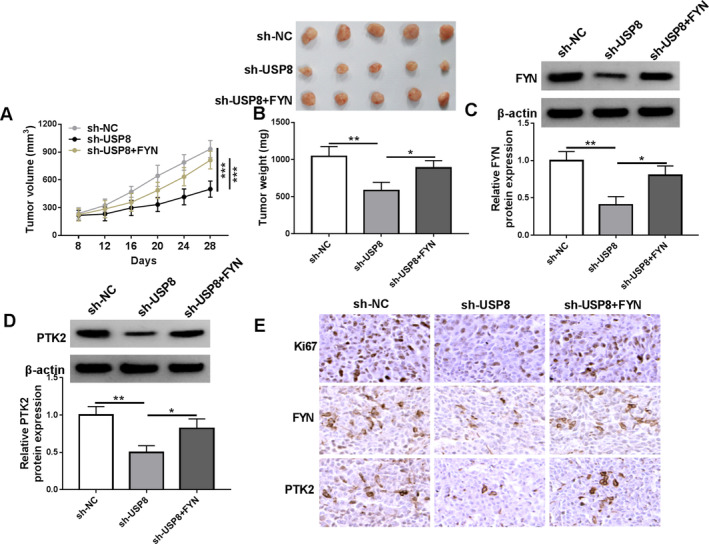
FYN overexpression attenuated USP8 knockdown‐induced effects on the malignant growth of ESCC cells in vivo. KYSE150 cells stably transfected with sh‐NC, sh‐USP8, or sh‐USP8 + FYN were injected into nude mice, and tumor volume was measured every 4 days after 8 days of injection (A). After 28 days, the forming tumors from KYSE150 cells were harvested for tumor weight (B) and FYN and PTK2 protein expression analysis (C and D). (E) The positive expression rates of Ki67, FYN, and PTK2 were detected by IHC assay in the forming tumors from the sh‐NC group, sh‐USP8 group, and sh‐USP8 + FYN group. *p < 0.05, **p < 0.01, and ***p < 0.001.
4. Discussion
Esophageal cancer is a significant public health concern worldwide, with a considerable annual burden of new cases and deaths [2]. The development of ESCC typically progresses through a multistage process, beginning with DNA damage and mutations that lead to the transformation of normal epithelial cells into precancerous lesions. Research in this area has revealed various epigenetic alterations that are related to ESCC, including mutations in genes such as P53 [17], HRAS [18], and KEAP1 [19], as well as alterations in ubiquitination patterns [20]. Despite significant advancements in the molecular biology of ESCC, the precise mechanisms underlying its development and progression are still not fully understood. The current study has uncovered a novel mechanism underlying the progression of ESCC where USP8‐dependent deubiquitination of FYN promoted ESCC progression by enhancing PTK2 expression.
It has been reported that FYN contributed to esophageal cancer cell proliferation and repressed cell apoptosis [21]. Yuan et al. indicated that interferon‐stimulated gene 15 (ISG15) promoted ESCC cell proliferation, invasion, and migration through the phosphorylation of FYN [22], indicating FYN's promoting effects on ESCC cell proliferation and motility. Another paper revealed that FYN inhibited cell apoptosis in an LINC00152 and miR‐153‐3p‐dependent manner [23]. Our data showed that FYN had a high expression in ESCC tissues and cells. Moreover, its high expression predicted poor prognosis of ESCC patients. We also found its promoting effects on ESCC cell migration and invasion and its inhibitory effect on cell apoptosis. In addition, the study observed that FYN could maintain cancer stem‐like cell properties and promote glucose metabolism. As reported by Peng et al., main factors regulating FYN expression include miRNAs, ubiquitination processes, and transcription factors [8]. In particular, Cbl and SNF2 histone linker PHD RING helicase (SHPRH) promoted degradation of FYN protein by their ubiquitinating activities [24, 25]. Our work identified that USP8 stabilized FYN protein expression through deubiquitination.
USP8 is a DUB that can protect the substrate from degradation and has an oncogenic role in cancer progression [26]. Recently, some studies pointed out its involvement in esophageal cancer progression. For example, USP8 expression was upregulated in ESCC tissue samples when compared with those tissues adjacent to carcinoma, and its inhibition induced by DUB‐IN‐1 led to cell cycle arrest and cell apoptosis promotion [27]. Li et al. revealed that USP8 stabilized ID1 protein expression to promote ESCC tumorigenesis [16]. Our work showed that USP8 silencing promoted ESCC cell apoptosis and inhibited cell invasion, migration, stem‐like cell properties, and glucose metabolism. The in vivo data also demonstrated its promoting effect on tumorigenesis. Moreover, the inhibitory effects of USP8 depletion on the malignant growth of ESCC cells were attenuated after FYN overexpression in vivo and in vitro, which indicated that USP8 regulated the malignant phenotypes of ESCC cells by inhibiting FYN degradation.
PTAK, also known as focal adhesion kinase (PTK2), serves as an integral component of the integrin‐mediated signaling axis [28]. PTAK is frequently overexpressed in various cancers and is associated with poor prognosis in these diseases [29]. FTAK induced ESCC proliferation and M2 macrophage infiltration through the PI3K/AKT pathway [30, 31]. Qiu et al. indicated that PTAK promoted esophageal cancer cell proliferation and migration by interacting with JNK signaling [32]. In addition, PTAK activated Rac1 to promote ESCC cell invasion [33]. We identified that FYN upregulated PTK2 expression in ESCC cells. Moreover, FYN regulated ESCC cell malignancy by increasing PTK2 expression. These data demonstrate that the FYN/PTK2 axis is responsible for the malignant growth of ESCC cells.
Taken together, USP8‐dependent deubiquitination of FYN promoted the malignant progression of ESCC by upregulating PTK2 expression. However, the study is limited by its cellular and in vitro focus, which may not fully reflect the complexity of ESCC progression in vivo. In addition, the mechanisms by which FYN and USP8 regulate PTK2 expression and function in ESCC cells are not fully elucidated and require further investigation. Despite these limitations, this study also suggests that targeting this axis could potentially lead to the development of new therapeutic strategies for ESCC treatment.
Author Contributions
Yuechang Wu and Dubiao Xian conceived and designed the study, and drafted the first draft of the manuscript. All experiments were completed by all authors. Yunzhong Liu, Ding Huang, Qingfeng Liu, Shubo Yang analyzed and collated the results. All authors reviewed and critiqued the manuscript, and agreed to the final submission of the manuscript. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgments
The authors have nothing to report.
Funding: The authors received no specific funding for this work.
Yuechang Wu and Dubiao Xian contributed equally to this work.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
References
- 1. Rogers J. E., Sewastjanow‐Silva M., Waters R. E., and Ajani J. A., “Esophageal cancer: Emerging Therapeutics,” Expert Opinion on Therapeutic Targets 26 (2022): 107–117. [DOI] [PubMed] [Google Scholar]
- 2. Bray F., Laversanne M., Sung H., et al., “Global cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries,” CA: A Cancer Journal for Clinicians 74 (2024): 229–263. [DOI] [PubMed] [Google Scholar]
- 3. Sheikh M., Roshandel G., McCormack V., and Malekzadeh R., “Current Status and Future Prospects for Esophageal Cancer,” Cancers (Basel) 15 (2023): 765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gronnier C. and Collet D., “New Trends in Esophageal Cancer Management,” Cancers (Basel) 13 (2021): 3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hagymási K. and Tulassay Z., “Risk Factors for Esophageal Cancer, and Possible Genetic Background,” Orvosi Hetilap 150 (2009): 407–413. [DOI] [PubMed] [Google Scholar]
- 6. Semba K., Nishizawa M., Miyajima N., et al., “Yes‐Related Protooncogene, syn, Belongs to the Protein‐Tyrosine Kinase Family,” Proceedings of the National Academy of Sciences of the United States of America 83 (1986): 5459–5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Goldsmith J. F., Hall C. G., and Atkinson T. P., “Identification of an Alternatively Spliced Isoform of the fyn Tyrosine Kinase,” Biochemical and Biophysical Research Communications 298 (2002): 501–504. [DOI] [PubMed] [Google Scholar]
- 8. Peng S. and Fu Y., “FYN: Emerging Biological Roles and Potential Therapeutic Targets in cancer,” Journal of Translational Medicine 21 (2023): 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Peng S., Yin Y., Zhang Y., Zhu F., Yang G., and Fu Y., “FYN/TOPK/HSPB1 axis Facilitates the Proliferation and Metastasis of Gastric cancer,” Journal of Experimental & Clinical Cancer Research 42 (2023): 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li S., Liu C., and Tang Y., “Role of Fyn in Hematological Malignancies,” Journal of Cancer Research and Clinical Oncology 149 (2023): 6759–6767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Han S., Wang R., Zhang Y., et al., “The Role of Ubiquitination and Deubiquitination in Tumor Invasion and Metastasis,” International Journal of Biological Sciences 18 (2022): 2292–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Durcan T. M., Tang M. Y., Pérusse J. R., et al., “USP8 Regulates Mitophagy by Removing K6‐Linked Ubiquitin Conjugates From Parkin,” EMBO Journal 33 (2014): 2473–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xie F., Zhou X., Li H., et al., “USP8 Promotes cancer Progression and Extracellular Vesicle‐Mediated CD8+ T Cell Exhaustion by Deubiquitinating the TGF‐β Receptor TβRII,” EMBO Journal 41 (2022): e108791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cui J., Guo Y., Yin T., et al., “USP8 Promotes Gemcitabine Resistance of Pancreatic cancer via Deubiquitinating and Stabilizing Nrf2,” Biomedicine & Pharmacotherapy 166 (2023): 115359. [DOI] [PubMed] [Google Scholar]
- 15. Xiong W., Gao X., Zhang T., et al., “USP8 Inhibition Reshapes an Inflamed Tumor Microenvironment That Potentiates the Immunotherapy,” Nature Communications 13 (2022): 1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li L., Liu Y., Zhao Y., et al., “Deubiquitinase USP8 Increases ID1 Stability and Promotes Esophageal Squamous Cell Carcinoma Tumorigenesis,” Cancer Letters 542 (2022): 215760. [DOI] [PubMed] [Google Scholar]
- 17. Efe G., Dunbar K. J., Sugiura K., et al., “p53 Gain‐of‐Function Mutation Induces Metastasis via BRD4‐Dependent CSF‐1 Expression,” Cancer Discovery 13 (2023): 2632–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang Y. X., Zhang X., Ma Q. Y., et al., “Adaptive Resistance to PI3Kα‐Selective Inhibitor CYH33 Is Mediated by Genomic and Transcriptomic Alterations in ESCC Cells,” Cell Death & Disease 12 (2021): 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Takahashi J., Suzuki T., Sato M., et al., “Differential Squamous Cell Fates Elicited by NRF2 Gain of Function Versus KEAP1 Loss of Function,” Cell Reports 43 (2024): 114104. [DOI] [PubMed] [Google Scholar]
- 20. Zhou X., Li Y., Wang W., et al., “Regulation of Hippo/YAP Signaling and Esophageal Squamous Carcinoma Progression by an E3 Ubiquitin Ligase PARK2,” Theranostics 10 (2020): 9443–9457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chu A., Sun C., Liu Z., et al., “Circ‐POSTN Promotes the Progression and Reduces Radiosensitivity in Esophageal cancer by Regulating the miR‐876‐5p/FYN axis,” Thorac Cancer 15 (2024): 1082–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yuan H., Zhou W., Yang Y., Xue L., Liu L., and Song Y., “ISG15 Promotes Esophageal Squamous Cell Carcinoma Tumorigenesis via c‐MET/Fyn/β‐Catenin Signaling Pathway,” Experimental Cell Research 367 (2018): 47–55. [DOI] [PubMed] [Google Scholar]
- 23. Liu D., Gao M., Wu K., Zhu D., Yang Y., and Zhao S., “LINC00152 Facilitates Tumorigenesis in Esophageal Squamous Cell Carcinoma via miR‐153‐3p/FYN axis,” Biomedicine & Pharmacotherapy 112 (2019): 108654. [DOI] [PubMed] [Google Scholar]
- 24. Rao N., Ghosh A. K., Douillard P., et al., “An Essential Role of Ubiquitination in Cbl‐Mediated Negative Regulation of the Src‐Family Kinase Fyn,” Signal Transduction 2 (2002): 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen W., Zhong M., Yu J., et al., “KMT2B Promotes SHPRH Expression to Regulate (131)I Sensitivity in Thyroid Carcinoma Cells by Affecting FYN Protein Stability,” Cellular Signalling 88 (2021): 110165. [DOI] [PubMed] [Google Scholar]
- 26. Islam M. T., Chen F., and Chen H., “The Oncogenic Role of Ubiquitin Specific Peptidase (USP8) and Its Signaling Pathways Targeting for cancer Therapeutics,” Archives of Biochemistry and Biophysics 701 (2021): 108811. [DOI] [PubMed] [Google Scholar]
- 27. Sha B., Sun Y., Zhao S., et al., “USP8 Inhibitor‐Induced DNA Damage Activates Cell Cycle Arrest, Apoptosis, and Autophagy in Esophageal Squamous Cell Carcinoma,” Cell Biology and Toxicology 39 (2023): 2011–2032. [DOI] [PubMed] [Google Scholar]
- 28. Guan J. L., Trevithick J. E., and Hynes R. O., “Fibronectin/Integrin Interaction Induces Tyrosine Phosphorylation of a 120‐kDa Protein,” Cell Regulation 2 (1991): 951–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chuang H. H., Zhen Y. Y., Tsai Y. C., et al., “FAK in Cancer: From Mechanisms to Therapeutic Strategies,” International Journal of Molecular Sciences 23 (2022): 1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang Y., Lyu Z., Qin Y., et al., “FOXO1 Promotes Tumor Progression by Increased M2 Macrophage Infiltration in Esophageal Squamous Cell Carcinoma,” Theranostics 10 (2020): 11535–11548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shi H., Luo J., Ye L., et al., “SH2D4A Inhibits Esophageal Squamous Cell Carcinoma Progression Through FAK/PI3K/AKT Signaling Pathway,” Cellular Signalling 114 (2024): 110997. [DOI] [PubMed] [Google Scholar]
- 32. Qiu Y., Fang B., Thuy N. T. T., et al., “Narciclasine Suppresses Esophageal cancer Cell Proliferation and Migration by Inhibiting the FAK Signaling Pathway,” European Journal of Pharmacology 921 (2022): 174669. [DOI] [PubMed] [Google Scholar]
- 33. Ma G., Jing C., Li L., et al., “MicroRNA‐92b Represses Invasion‐Metastasis Cascade of Esophageal Squamous Cell Carcinoma,” Oncotarget 7 (2016): 20209–20222. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.