

Abstract
Objective
SCN2A encodes the voltage‐gated sodium (Na+) channel α subunit NaV1.2, which is important for the generation and forward and back propagation of action potentials in neurons. Genetic variants in SCN2A are associated with a spectrum of neurodevelopmental disorders. However, the mechanisms whereby variation in SCN2A leads to disease remains incompletely understood, and the full spectrum of SCN2A‐related disorders may not be fully delineated.
Methods
Here, we identified seven de novo heterozygous variants in SCN2A in eight individuals with developmental and epileptic encephalopathy (DEE) accompanied by prominent malformation of cortical development (MCD). We characterized the electrophysiological properties of Na + currents in human embryonic kidney (HEK) cells transfected with the adult (A) or neonatal (N) isoform of wild‐type (WT) and variant NaV1.2 using manual and automated whole‐cell voltage clamp recording.
Results
The neonatal isoforms of all SCN2A variants studied exhibit gain of function (GoF) with a large depolarized shift in steady‐state inactivation, creating a markedly enhanced window current common across all four variants tested. Computational modeling demonstrated that expression of the NaV1.2‐p.Met1770Leu‐N variant in a developing neocortical pyramidal neuron results in hyperexcitability.
Significance
These results support expansion of the clinical spectrum of SCN2A‐related disorders and the association of genetic variation in SCN2A with MCD, which suggests previously undescribed roles for SCN2A in fetal brain development.
Keywords: developmental and epileptic encephalopathy, malformation of cortical development, Nav1.2, SCN2A, voltage‐gated sodium channel
Key points.
The SCN2A gene encodes the voltage‐gated sodium channel subunit NaV1.2.
Variants in SCN2A have been associated previously with a spectrum of neurological dysfunction including epilepsy, intellectual disability, and autism spectrum disorder.
Dysfunctional NaV1.2 variants were identified in a novel cohort of eight individuals with epilepsy and malformation of cortical development (MCD), with prominent effects on the voltage dependence of steady‐state inactivation.
This study supports an association between SCN2A and MCD.
1. INTRODUCTION
Voltage‐gated sodium (Na+) channels are responsible for action potential generation and propagation in excitable tissues. Of the nine Na + channel α subunit genes, at least four (SCN1A, 2A, 3A, and 8A) are expressed in brain, and pathogenic variation in each brain‐expressed Na + channel gene is a known cause of a spectrum of neurological dysfunction including epilepsy and autism spectrum disorder (ASD). 1 In general, loss of function (LoF) variants in SCN2A cause intellectual disability (ID) and ASD with or without epilepsy, whereas gain of function (GoF) variants are associated with early‐onset epilepsy which ranges in severity from benign familial neonatal and infantile seizures (BFNIS) to developmental and epileptic encephalopathy (DEE). 2 However, many variants exhibit complex effects that cannot be simply or easily classified as gain vs loss of function 3 , 4 in a straightforward manner. Consequently, the precise relationship between the effects of disease‐associated variants and clinical phenotype remains unclear.
Epilepsy‐related Na + channelopathies are thought to result in neurological dysfunction via alteration in neuronal excitability. However, we and others recently identified variants in the embryonic Na + channel gene SCN3A as associated with malformation of cortical development (MCD) including polymicrogyria (PMG). 5 , 6 , 7 Although the role of Na + channels in early brain development and the mechanism whereby dysfunction of Na + channels might lead to brain malformation remain unclear, the basis of this association is hypothesized to relate to the early expression of SCN3A in migrating neurons. 8 PMG is an MCD defined by abnormal lamination and excessive gyration of the neocortex and can be due to genetic and acquired causes. 9 The genetic basis of PMG is heterogenous and has been reported in association with variation in more than 40 genes, but most typically in genes in the mammalian target of rapamycin (mTOR) pathway (MTOR, AKT3, PIK3CA, and so on) and the tubulinopathies (TUBA1A, TUBB2B, TUBB3). 10 PMG is also seen in a subset of individuals with variants in the glutamate receptor genes GRIN1 11 and GRIN2B. 12 Variants in SCN2A have also been reported in association with MCD in individual case reports 13 , 14 , 15 including one individual in a large cohort with PMG undergoing exome sequencing, 16 although SCN2A has yet to be definitively established as a genetic cause of PMG because the underlying mechanism of this association is unclear. These reports remain descriptive and did not include functional analysis of the associated NaV1.2 variants or synthesize such findings into a unifying explanation for how Na + channel dysfunction might result in MCD.
Here, we report eight patients with a severe phenotype of DEE and diffuse MCD found to harbor de novo heterozygous variants in SCN2A. Electrophysiological studies demonstrate that such variants exert complex effects on Na + channel function yet share a common signature of a large depolarizing shift in the voltage dependence of steady‐state inactivation. These results support the conclusion that rare disease‐associated variants in SCN2A with a specific electrophysiological fingerprint can be associated with MCD, perhaps by a mechanism involving early expression in developing neurons.
2. MATERIALS AND METHODS
2.1. Patients, clinical assessment, and genetic analysis
Individuals were enrolled in a study approved by the institutional review board of the Children's Hospital of Philadelphia (CHOP; #15–12 226). Informed consent for participation was provided by parents, as all participants were minors with ID. Patients were ascertained through an international collaborative network. Four patients were identified during clinical care in the Epilepsy Neurogenetics Initiative (ENGIN) Clinic at CHOP, which includes a cohort of more than 5000 individuals seen for initial or secondary evaluation or management since 2019, and who underwent or had previously undergone genetic testing (epilepsy gene panel or exome sequencing). Of these, we identified 63 patients with neurodevelopmental disorder (epilepsy and/or developmental delay/ID or ASD) and de novo heterozygous variants in SCN2A (1.0% of all patients), a small but distinct subset (three; 4.7%) of whom were also noted to have MCD. Two patients were from Italy (Meyer Children's Hospital, Instituto di Ricovero e Cura a Carattere Scientifico (IRCSS); Children's Hospital, ASST Spedali Civli of Brescia) and one was from the United Kingdom (University College London Hospitals). Two additional patients with SCN2A variants and epilepsy accompanied by MCD were ascertained through the FamiliesSCN2A Foundation and enrolled in the research study. Since 2019, 65 patients with an International Classification of Diseases, Tenth Edition (ICD‐10) code of Q04.3 (PMG) were seen/evaluated at CHOP, of whom 3 (4.6%) were found to harbor de novo heterozygous variants in SCN2A.
Patient 1 was a 2‐month‐old, ex‐33‐week‐gestation male born to non‐consanguineous parents. The infant was apneic and bradycardic at birth and was intubated for respiratory support. The patient exhibited clinical seizure activity on the first day of life that consisted of clonic limb movements, repetitive blinking, and lip smacking, associated with oxygen desaturation. Initial electroencephalography (EEG) showed multifocal seizures with onset originating independently from the right centrotemporal and left occipital regions. Seizures responded to phenobarbital and levetiracetam and the patient was extubated on the second day of life. Brain magnetic resonance imaging (MRI) (Figure 1A) demonstrated a simplified gyral pattern most prominent in the frontal lobes, with abnormal gyrification and suggestive of PMG in the bilateral sylvian fissures, along with partial fusion of the thalami, a dysplastic corpus callosum, and bilateral optic nerve hypoplasia. Trio whole exome sequencing identified a de novo heterozygous variant in SCN2A c.5308A>T (p.Met1770Leu). This variant converts a larger sulfur‐containing methionine to the smaller leucine in segment 6 (S6) of domain IV (DIV) of NaV1.2, which contributes to the channel pore; the variant is not present in the Exome Aggregation Consortium (ExAC) or genome Aggregation Database (gnomAD; http://gnomad.broadinstitute.org) and has a Combined Annotation Dependent Depletion (CADD) score 17 of 23.6. This variant was reported previously in an individual with SCN2A‐related disorders, 15 but detailed clinical phenotyping was not available. SCN2A‐p.Met1770Leu is paralogous to SCN3A‐Met1765Ile identified in an individual with DEE and MCD,18 and SCN8A‐p.Met1760Ile identified as de novo in an individual with in utero onset seizures, Ohtahara syndrome, severe ID, and cortical blindness shown electrophysiologically to exhibit a GoF effect. 19 After initial positive response to anti‐seizure medications (ASMs), patient 1 continued to have epilepsy that was resistant to treatment with multiple ASMs in various combinations. Subsequent continuous video‐EEG showed a burst‐suppression pattern with multifocal‐onset seizures, which evolved to hypsarrhythmia with multifocal and generalized epileptiform discharges by age 2 months.
FIGURE 1.

Magnetic resonance imaging (MRI) of the brain of patients in the cohort with SCN2A encephalopathy and malformation of cortical development. (A) Patient 1. T2 axial and coronal MRI images at age 6 weeks show diminutive size of the intraorbital and prechiasmatic optic nerves (at left; right optic nerve indicated by arrowhead); marked cortical and subcortical atrophy with extensive abnormality of gyrification. (B) Patient 2. T2 axial MRI at age 3 weeks shows extensive malformation consistent with polymicrogyria (PMG) of the right frontal and parietal lobes. (C) Patient 3. T2 axial and coronal MRI at age 7 months shows extensive malformation consistent with PMG throughout the neocortex, predominantly affecting both parietal lobes. (D) Patient 4. Axial T2 fluid‐attenuated inversion recovery (FLAIR) and coronal T2 MRI images illustrate diffuse PMG. (E) Patient 5. Coronal and axial T2 and coronal T2 FLAIR MRI images at age 3 years showing marked cortical and subcortical atrophy with extensive dysgyria and areas of extensive malformation with presumptive PMG involving the bilateral frontal and parietal lobes. Note as well the appearance of cerebellar atrophy on the T2 coronal image. (F) Patient 6. Coronal, sagittal, and axial T2 MRI images at age 13 days showing simplified gyral pattern of the left hemisphere consistent with PMG, mostly involving the temporo‐parieto‐insular region (white arrows). (G) Patient 7. Coronal and axial T2 MRI images at age 1 day showing PMG of the left hemisphere, mostly affecting the frontal and temporal lobes (white arrows). (H) Schematic representation of Nav1.2 indicating the locations of the A263V variant in domain I (DI) S4 (red circle), Thr400Arg in the DI S5–6 extracellular linker (orange), I1488V in the DII–DII linker (green), L1614P in the extracellular DIII S3–4 linker (raspberry), Leu1639Pro (light blue) and Ile1640Asn (dark blue) in the DIV voltage‐sensing region, and Met1770Leu variants (purple) in DIV S6. (I) Topology diagram of Nav1.2 showing the predicted location of the affected residues.
Patient 2 was an ex‐full‐term male born to non‐consanguineous parents who presented with episodes of respiratory impairment, bradycardia, and tonic extension of the bilateral upper extremities on the first day of life, with events confirmed to be seizures. Subsequent continuous video‐EEG showed hypsarrhythmia with periods of excessive discontinuity and multifocal‐onset seizures accompanied by tonic–clonic movements of the left or right leg followed by unilateral or bilateral arm extension and head deviation. MRI of the brain showed extensive MCD suggestive of PMG involving the right frontoparietal lobes and bilateral peri‐rolandic regions (Figure 1B). Next‐generation sequencing of epilepsy‐associated genes identified a de novo variant in SCN2A c.1199C>G (p.Thr400Arg). This variant converts a small polar uncharged threonine residue in the intracellular S5‐S6 linker of domain I of NaV1.2 to a large, positively charged arginine residue. This variant is not present in gnomAD, not previously seen in other individuals with SCN2A‐related disorders, and is associated with a CADD score of 26.7.
Patient 3 was an ex‐35 and 5/7 week gestation male born to non‐consanguineous parents who presented with apneic episodes at birth that were accompanied by tonic posturing of the bilateral upper extremities and bicycling movements of the lower extremities, confirmed to be seizures. Continuous video‐EEG showed a slow and disorganized background with multifocal epileptiform discharges. MRI of the brain showed bilateral frontoparietal perisylvian PMG with reduced white matter volume and proportionate prominence of the lateral ventricles with an immature myelination pattern. Physical examination revealed central hypotonia and peripheral spasticity. Trio whole exome sequencing identified a de novo heterozygous SCN2A variant c.4919A>T (p.Ile1640Asn), which was reported previously as an isolated case of SCN2A encephalopathy accompanied by PMG. 16 This variant converts a small hydrophobic isoleucine residue in the S4 segment of domain IV to a polar but neutral and hydrophilic asparagine. This variant is not present in gnomAD and has a CADD score of 26.5. SCN2A‐p.Ile1640Asn is paralogous to SCN8A‐p.Ile1631Asn discovered in a patient with epilepsy and ID. 20 At last follow‐up at age 2 years, the patient continued to have treatment‐resistant epilepsy and severe global developmental delay, and was non‐verbal and non‐ambulatory, with a gastrostomy tube required for feeding.
Patient 4 was an ex‐full‐term gestation male who exhibited epilepsy onset at 12 days of life after an unremarkable pregnancy and perinatal course. EEG recording subsequently exhibited diffuse spike‐and‐wave discharges on day 6 of life and a burst‐suppression pattern at age 3 weeks, leading to a clinical diagnosis of Ohtahara syndrome. MRI of the brain revealed diffuse MCD suggestive of PMG. Genome sequencing identified a de novo heterozygous SCN2A variant c.4916T>C (p.Leu1639Pro), which converts the hydrophobic leucine in the S4 segment of domain IV to a proline. This variant is not present in gnomAD and is associated with a CADD score of 25.7. SCN2A‐p.Leu1639Pro is paralogous to SCN8A‐p.Leu1630Pro identified in a patient with epilepsy. 21
Patient 5 was an ex‐35‐week‐gestation male and exhibited seizures on the first day of life after an unremarkable pregnancy and perinatal course. EEG on the first day of life showed a suppression‐burst pattern. Seizures at onset were characterized by asymmetric tonic contraction of the bilateral upper extremities with bradycardia. Later, epileptic spasms, bilateral tonic seizures, and non‐motor seizures with oxygen desaturation and bradycardia appeared. Seizures were refractory to multiple ASMs including nitrazepam, pimozide, pregabalin, vigabatrin, levetiracetam, phenytoin, and topiramate, in various combinations. The greatest reduction in seizure frequency was observed with a combination of phenobarbital, carbamazepine, and cenobamate. MRI of the brain revealed abnormal thickening and irregularity of the bilateral fronto‐parietal neocortex suggestive of PMG, along with thinning of the corpus callosum and diffuse abnormal T1 signal with the subcortical white matter and posterior arm of the internal capsule bilaterally, as well as reduction of thalamic volume (Figure 1E). Whole exome sequencing revealed a de novo heterozygous SCN2A variant c.4916T>C (p.Leu1639Pro). At age 3 years, there was profound global developmental impairment with central visual impairment, non‐verbal, non‐ambulatory, and hypotonia with poor head control. In addition, the patient presented with a severe movement disorder, experiencing dystonic episodes accompanied by autonomic signs such as bradycardia and diaphoresis. The individual received a tracheostomy and required feeding via gastrostomy tube.
Patient 6 was a 6‐month‐old ex‐35‐week‐gestation female who exhibited clinical seizure activity on the first day of life, consisting of asymmetric tonic posturing, with eye and head deviation to the right, accompanied by oxygen desaturation. She continued to experience seizures almost daily, which were refractory to multiple antiseizure medications, including carbamazepine, levetiracetam, phenobarbital, and phenytoin. At age 6 months, there was evidence of mild global developmental delay. Prolonged EEG monitoring showed a slow and asymmetric background with bilateral asynchronous multifocal polyspikes and spike–wave abnormalities. Brain MRI (Figure 1F) demonstrated unilateral left PMG, mostly involving the temporo‐parieto‐insular region. Trio whole exome sequencing identified a de novo heterozygous variant in SCN2A c.788C > T (p.Ala263Val). This variant is not present in gnomAD and is associated with a CADD score of 28.0. This variant was functionally studied previously and classified as GoF using dynamic action potential clamp. 22
Patient 7 was a male born at 35 weeks and 6 days gestation who presented with startle episodes and asynchronous clonic limb movements on the first day of life. He continued to have multiple daily focal seizures with autonomic and motor components, with up to 50 events per day. His seizures did not respond to multiple treatments, including phenobarbitone, phenytoin, topiramate, midazolam, hydrocortisone, and ketogenic diet. At 1 month and 19 days of age, the patient exhibited severe developmental delay. Seizure frequency was lower (10–20 per day) on a combination of carbamazepine, lacosamide, clonazepam, and levetiracetam. A prolonged EEG study showed poor background activity during wakefulness and sleep. Seizures were recorded arising from the left temporal lobe. Brain MRI (Figure 1G) showed left‐sided PMG, along with thinning of the corpus callosum. Next‐generation sequencing of epilepsy‐associated genes identified a de novo variant in SCN2A c.4462A>G (p.Ile1488Val). This variant is not present in gnomAD and is associated with a CADD score of 24.3.
Patient 8 was an 18‐year‐old male born at 42 weeks of gestation who exhibited seizures starting in the neonatal period. His mother recalled unusually forceful fetal movements during pregnancy, followed by fetal “hiccups.” He exhibited episodes of body stiffening followed by hiccups on day one of life. Over time, he continued to experience focal‐onset seizures, often progressing to bilateral tonic–clonic seizures, including recurrent episodes of status epilepticus. His seizures were refractory to treatments, including carbamazepine, clobazam, levetiracetam, phenobarbital, phenytoin, and valproate. Brain MRI scan showed left posterior perisylvian and parietal PMG. Surgical treatment (left anterior temporal lobectomy with partial amygdalo‐hippocampectomy and functional disconnection of the parieto‐occipital and temporal lobes), and insertion of a vagus nerve stimulator (VNS) were not effective. EEG recordings continued to show slow asymmetric background (left more than right), left temporal and parietal epileptiform abnormalities, and ictal seizure onset from either hemisphere. On the post‐operative MRI, regions of PMG appeared to be disconnected, with suspicion of a residual connection on the medial parietal lobe. Histology confirmed PMG of the temporal lobe. The patient met criteria for developmental and epileptic encephalopathy, with no expressive language, accompanied by behavioral and sleep disturbances, and by dysphagia requiring placement of a gastrostomy tube at age 17 years. Next‐generation sequencing of epilepsy‐associated genes identified a de novo variant in SCN2A c.4841T>C (p.Leu1614Pro). This variant is not present in gnomAD and is associated with a CADD score of 24.9.
The locations of the identified variants are presented in a two‐dimensional schematic of the NaV1.2 protein (Figure 1I) and using the molecular visualization software PyMOL (Schrödinger, New York, NY) (Figure 1J). Additional clinical details for each patient are provided in Table S2.
2.2. Variant interpretation
SCN2A variants were interpreted according to the American College of Medical Genetics and Genomics standards and guidelines for the interpretation of sequence variants. 23 All SCN2A variants reported here were classified as likely pathogenic, as they were confirmed to have occurred de novo in the affected individual with established parental relationships and testing, were not observed in a control cohort of individuals in the Exome Aggregation Consortium (ExAC) or v2 release of the genome Aggregation Database (gnomAD; http://gnomad.broadinstitute.org), 24 and demonstrated by electrophysiology to exert an abnormal effect on the function of NaV1.2‐containing Na + channels.
2.3. cDNA cloning and preparation
The human coding sequence of SCN2A (NC_000002.12) WT was subcloned in a p.CMV plasmid resulting in a p.CMV‐SCN2A construct. Variants were then introduced by directed site mutagenesis as described previously. 3 , 6
2.4. Cell culture and transfection
Human embryonic kidney (HEK)‐293 cells were grown at 37°C with 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 1% penicillin–streptomycin. Transient transfections were performed with 1600 ng cDNA total by co‐transfecting pCMV‐SCN2A, pRFP‐IRES‐SCN1B, and pGFP‐IRES‐SCN2B using 1000 ng/300 ng/300 ng respectively using PolyFect transfection reagent (Invitrogen) according to the manufacturer's instruction.
2.5. Electrophysiology
Forty‐eight hours after transfection, human embryonic kidney (HEK) 293T cells were lifted from the culture dish with trypsin and re‐seeded to a density that enabled identification and isolation of single cells. Green and red double‐positive cells were chosen for patch‐clamp experiments. During patch‐clamp recordings, cells were bathed in an extracellular Tyrode's solution containing in mM: NaCl, 150; KCl, 2; MgCl2, 1; CaCl2, 1.5; NaH2PO4, 1; glucose, 10; HEPES, 10; adjusted to pH 7.4 with CsOH. Intracellular solution was (in mM): NaCl, 35; CsF, 105; MgCl2, 2; EGTA, 10; HEPES, 10; adjusted to pH 7.4 with CsOH and 285 mOsm/L with sucrose. Patch‐clamp recordings were carried out in the whole‐cell configuration at room temperature.
Ionic currents were recorded with an Axopatch 200B amplifier (Molecular Devices, San Jose, CA, USA). Patch pipettes (Corning Kovar Sealing code 7052, WPI, Sarasota, FL, USA) had resistances of 1.7–2.5 MΩ in the bath. Voltage errors were reduced via partial series resistance compensation. Currents were filtered at 5 kHz (−3 dB, 4‐pole low‐pass Bessel filter) and digitized at 33.3 kHz (Digidata 1550B, Molecular Devices); data were acquired with pClamp 11.1 and analyzed with Clampfit 11.1 (Molecular Devices), SigmaPlot 11 (Systat Software, Inc., San Jose, CA, USA), and MATLAB (MathWorks).
For current–voltage relationships and calculation of the voltage dependence of activation, currents were elicited by 300 ms test potentials of 0.2 Hz frequency to potentials ranging from −80 to 65 mV in 5 mV increments from a holding potential of −120 mV. Currents were converted to current density (pA/pF) by normalizing to cell capacitance. The steady‐state inactivation protocol was performed from a holding potential of −120 mV, and a 500 ms conditioning prepulse was applied in 5 mV increments between −140 and + 5 mV at 0.2 Hz, followed by a 400‐ms test pulse to 0 mV. Data for the activation curve and voltage dependence of steady‐state availability were fitted to the Boltzmann equation:
where V m is the membrane potential, V 1/2 is the half‐activation voltage, and k is the slope factor. For activation curves, Y represents the relative conductance; for steady‐state availability, Y represents the relative current (I Na/I Na(max)).
Automated patch‐clamp recording was performed as described previously. 3 Briefly, HEK‐β cells were maintained in DMEM(GIBCO/Invitrogen; San Diego, CA, USA) supplemented with 10% fetal bovine serum (Atlanta Biologicals; Norcross, GA, USA), 2 mM L‐glutamine, 50 units/mL penicillin, 50 mg/mL streptomycin, and 3 μg/mL puromycin, at 37°C in 5% CO2.
Full‐length WT or variant SCN2A (NaV1.2) cDNAs were electroporated into HEK‐β cells using the MaxCyte STX electroporation system (MaxCyte Inc., Gaithersburg, MD, USA) as described previously. 3
Automated patch‐clamp recording was performed using the Nanion Syncropatch 768PE platform (Nanion Technologies; Munich, Germany) using single‐hole thin‐wall low‐resistance (2–4 MΩ) recording chips. Pulse generation and data collection were performed using PatchControl384 v1.6.6 and DataControl384 v1.6.0 software (Nanion Technologies). Whole‐cell currents were acquired at 10 kHz, series resistance was compensated 80%, and leak currents were subtracted using P/4 subtraction. The external solution contained (in mM): NaCl, 140; KCl, 4; CaCl2, 2; MgCl2, 1; HEPES, 1; glucose, 5; with the final pH adjusted to 7.4 with NaOH, and osmolality adjusted to 300 mOsm/L with sucrose. The composition of the internal solution was (in mM): CsF, 100; CsCl, 10; NaCl, 10; EGTA, 20; HEPES, 10; with the final pH adjusted to 7.2 with CsOH, and osmolality adjusted to 300 mOsm/L with sucrose. High‐resistance seals were obtained by addition of seal enhancer solutions, which contained (in mM): NaCl, 80; KCl, 3; CaCl2, 35; MgCl2, 10; HEPES, 10; with the final pH adjusted to 7.4 with NaOH. Cells were washed twice with external solution prior to recording, and the final concentrations of CaCl2 and MgCl2 were 3 mM and 2 mM, respectively. Biophysical data were collected only from cells with inward currents larger than 200 pA. Stringent quality control criteria were set to select cells included in the final analysis as described previously. 3
Data were analyzed and plotted using a combination of DataControl384 v1.9.0 (Nanion Technologies), Clampfit 10.4 (Molecular Devices), Microsoft Excel (Microsoft Office 2019), and GraphPad Prism (GraphPad Software; San Diego, CA, USA). All data are reported as mean ± 95% confidence interval (CI). One‐way analysis of variance (ANOVA) with Dunn's post hoc test was used for statistical comparison, and the threshold for statistical significance was set at p ≤ .05.
2.6. Statistical analysis
Data were analyzed blind to experimental group using Clampfit 11.1 (Molecular Devices) and Sigma Plot 11. Data are presented as mean ± standard error of the mean (SEM) throughout. Statistical significance was estimated with SigmaPlot software by one‐way ANOVA, with post hoc correction for multiple comparisons as appropriate; significance was established as p < .05 with p values reported exactly.
2.7. Computational modeling
Hodgkin–Huxley (HH) models of NaV1.2 wild‐type (WT) and NaV1.2‐Met1770Leu variant‐containing Na + channels were developed by modifying the original NaV1.2 WT parameters from Ben‐Shalom et al. 25 The NaV1.2‐Met1770Leu variant model was optimized using a parallel genetic algorithm to match the differences in biophysical properties observed between the WT and the variant in whole‐cell voltage‐clamp recordings. 26 , 27 The fitted HH models of NaV1.2 channels were then incorporated into a well‐established computational model of a layer V cortical pyramidal cell. 25 , 28 , 29 , 30 , 31 The model was implemented using the NEURON simulation environment 32 and incorporated morphological parameters and channel distributions as described by Ramaswamy and Markram. 33
Electrophysiological properties were based on previously reported values. 30 To simulate an early developing neuronal model, we modified the model to include 50% neonatal variant and 50% adult variant wherever NaV1.2 was expressed in the model 34 ; for the NaV1.2‐Met1770Leu variant model, both 50% neonatal and 50% adult NaV1.2 were included to obtain an equal contribution of WT and NaV1.2‐Met1770Leu subunits representing the heterozygous condition observed in patients (with the total amount of sodium channels the same between the WT and NaV1.2‐Met1770Leu condition). Synaptic stimulation was modeled as a series of excitatory postsynaptic conductances (EPSCs) following the approach described by Clarkson et al. 35
3. RESULTS
3.1. Electrophysiological characterization of NaV1.2 variants associated with epilepsy and brain malformation using manual whole‐cell patch clamp
HEK‐293 T cells were transfected with cDNA constructs encoding WT or variant human adult and neonatal isoforms of SCN2A along with β1 and β2 subunits as described (see Material and Methods). The biophysical properties of Na+ channels containing WT vs variant subunits were then determined using whole‐cell voltage‐clamp electrophysiology.
Peak current density (normalized to cell capacitance, reported as pA/pF; I Na) was the same for SCN2A‐WT‐N (−140.3 ± 26.3 pA/pF; n = 14) and WT‐A (−187.5 ± 46.2 pA/pF; n = 13; p = .627 vs WT‐A via one‐way ANOVA) (Figure 2; Table 1). Peak current density for SCN2A‐p.Thr400Arg‐N and ‐A, SCN2A‐p.Leu1639Pro‐N and ‐A, and SCN2A‐p.Met1770Leu‐N and ‐A were not different from WT. However, SCN2A‐p.Ile1640Asn‐N (−39.4 ± 9.1; n = 7; p = .016 vs WT‐N) and SCN2A‐p.Ile1640Asn‐A (−46.1 ± 11.8; n = 13; p = .002 vs WT‐A) exhibited significantly smaller I Na relative to WT.
FIGURE 2.

Current–voltage relationship of sodium currents recorded from human embryonic kidney (HEK)‐293 cells expressing wild‐type (WT) SCN2A vs variants associated with early‐onset epilepsy and malformation of cortical development. (A) Representative families of currents recorded from WT and variants SCN2A‐p.Thr400Arg (T400R), Leu1639Pro (L1639P), Ile1640Asn (I1640N), and Met1770Leu (M1770L) adult (A; top) and neonatal (N; bottom) isoforms. (B) Current–voltage (I–V) relationships. Note that only SCN2A‐p.Ile1640Asn displayed significant loss in peak current density compared to WT.
TABLE 1.
Biophysical properties of Nav1.2 variants associated with epilepsy and brain malformation.
| Variant | Biophysical properties | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Condition | Isoform | Peak I Na (pA) | n | V 1/2 activation (mV) | V 1/2 Inctivation (mV) | Slope inactivation | Tau fast (ms) | Tau slow (ms) | Persistent current (%) |
| WT | N | 140.3 ± 26.3 | 14 | −10.8 ± 1.1**** | −57.0 ± .9 | 8 ± .2 | 1.0 ± .15***** | 4.9 ± 0.01 | 0.7 ± 0.2 |
| T400R | N | 13.7 ± 13.2 | 15 | −13.2 ± 1.4 | −5.8 ± 1.4** | 7 ± 0.3 | 1.3 ± 0.3 | 6.9 ± 1.7 | 0.3 ± 0.2 |
| L1639P | N | 118.4 ± 11.6 | 14 | −16.2 ± 1.4* | −41.2 ± 1.1*** | 4 ± 0.3*** | 2.7 ± 0.2*** | 12.7 ± 2.2* | 2.1 ± 0.1 |
| I1640N | N | 39.4 ± 3.5* | 7 | −3.5 ± 1.4* | −42.0 ± 1.9*** | 5 ± 0.5*** | 3.3 ± 0.3***, ***** | 14.5 ± 3.5** | 1.1 ± 0.2 |
| M1770L | N | 139.8 ± 11.5 | 11 | −11.5 ± 1.2 | −49.1 ± 0.9*** | 7 ± 0.3 | 0.9 ± 0.1 | 10.3 ± 2.5* | 4.6 ± 2.1* |
| WT | A | 187.5 ± 46.2 | 13 | −14.9 ± 1.3 | −57.2 ± 1.2 | 7 ± 0.3 | 0.7 ± 0.1 | 5.5 ± 1.4 | 1.2 ± 0.5 |
| T400R | A | 120.9 ± 20.3 | 14 | −16.8 ± 1.5 | −53.5 ± 0.5 | 7 ± 0.3 | 0.9 ± 0.1** | 8.9 ± 3.4 | 0.7 ± 0.3 |
| L1639P | N | 176.5 ± 15.1 | 12 | −15.05 ± 1.2 | −34.5 ± 1.1*** | 6 ± .2*** | 3.6 ± .4*** | 28.1 ± 9.5** | 1.6 ± .1 |
| I1640N | A | 46.1 ± 11.8* | 13 | −7.9 ± 1.5** | −46.2 ± 2.5** | 4 ± .2*** | 2.1 ± .2*** | 29.7 ± 4.5*** | 1.1 ± .7 |
| M1770L | A | 80.9 ± 17.7 | 16 | −6.9 ± 1.9** | −45.7 ± 1.2*** | 6 ± 0.3 | 1.1 ± 0.2*** | 14.1 ± 3.2*** | 4.5 ± 2.1* |
p < .05 vs WT of the same isoform.
p < .01 vs WT of the same isoform.
p < .001 vs WT of the same isoform.
p < .05 comparing neonatal vs adult isoform.
p < .01 comparing neonatal vs adult isoform.
p < .001 comparing neonatal vs adult isoform.
Abbreviations: A, adult isoform; N, neonatal isoform; WT, wild type.
SCN2A‐p.Thr400Arg‐N and ‐A had no effect on the voltage‐dependence of activation relative to WT (Figure 3 and Table 1). SCN2A‐p.Ile1640Asn‐A and ‐N and SCN2A‐p.Met1770Leu‐A yielded currents that displayed a right‐shift of the voltage‐dependence of activation of 7–8 mV, whereas SCN2A‐p.Leu1639Pro‐N (but not ‐A) exhibited a − 5 mV hyperpolarizing shift (Figure 3 and Table 1).
FIGURE 3.
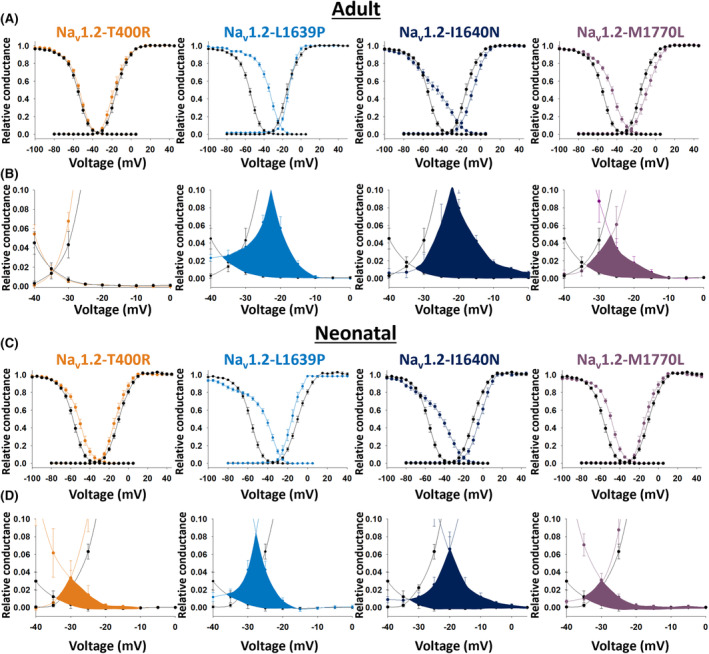
Voltage dependence of activation and steady‐state inactivation recorded from human embryonic kidney (HEK)‐293 cells expressing wild‐type (WT) SCN2A vs variants associated with early‐onset epilepsy and malformation of cortical development. (A) Shown are the voltage dependence of activation and steady‐state inactivation for the WT SCN2A adult isoform vs variants SCN2A‐p.Thr400Arg (T400R; orange), Ile1640Asn (I1640N; dark blue), and Met1770Leu (M1770L; purple). (B) Insets from (A) showing the window currents. (C) Shown are the voltage dependence of activation and steady‐state inactivation for the WT SCN2A neonatal isoform vs variants SCN2A‐p.Thr400Arg (T400R; orange), Leu1639Pro (L1639P; light blue), Ile1640Asn (I1640N; dark blue), and Met1770Leu (M1770L; purple). (D) Insets from (C) showing the window currents. Note that window currents are significantly augmented for the neonatal isoform of all four variants.
The most consistent finding across variants was a marked depolarized shift in the voltage dependence of steady‐state inactivation (Figure 3). SCN3A‐p.Thr400Arg‐A was associated with a 6.2 mV shift relative to WT, although this did not reach statistical significance. However, SCN2A‐Thr400Arg‐N, and both ‐N and ‐A isoforms of SCN2A‐p.Leu1639Pro, SCN2A‐p.Ile1640Asn, and p.Met1770Leu, exhibited significant depolarizing shifts of between 6 and 22 mV relative to WT (Figure 3 and Table 1).
We calculated the window current as the area under the voltage‐dependence of activation and steady‐state inactivation curves (highlighted in yellow in Figure 3). Again, the SCN2A‐p.Thr400Arg‐A window current was not significantly different from WT, consistent with the non‐significant difference in either voltage‐dependence of activation or steady‐state inactivation. However, window currents of p.Leu1639Pro‐A and ‐N, p.Ile1640Asn‐A and ‐N, and p.Met1770Leu‐A and ‐N were 2‐ to 6‐fold larger than WT (Figure 3).
To further complete our biophysical analysis, we investigated the gating kinetics of the different channels; that is, time to peak as well as fast‐ and slow‐inactivation components. The Thr400Arg variant was mildly abnormal relative to WT, and only the fast inactivation decay of the adult isoform reached significance (Table 1). The p.Leu1639Pro and p.Ile1640Asn variants were the most affected, and displayed marked slower gating kinetics, including delayed time to peak and deceleration of fast and slow inactivation decay for both neonatal and adult isoforms (Table 1).The Met1770Leu‐A variant displayed a delayed time to peak, as well as a slowing of fast‐ and slow‐inactivation decay (Table 1). However, differences in kinetics exhibited by the Met1770Leu‐N variant failed to reach significance.
3.2. Electrophysiological characterization of NaV1.2 variants associated with epilepsy and brain malformation using automated patch clamp
Recent work validated the use of automated patch‐clamp recording for classification of epilepsy‐associated ion channel variants. 3 We studied three SCN2A variants associated with epilepsy and MCD using this approach, which allows for standardization of recording conditions and is free of operator bias. We compared sodium currents mediated by SCN2A‐WT‐N and ‐A with SCN2A‐p.Thr400Arg, Ile1640Asn, and Met1770‐Leu‐N and ‐A. Similar to the results obtained with manual patch‐clamp recording, the automated patch‐clamp recording showed that Ile1640Asn, in both the adult and neonatal splice isoforms, showed smaller currents relative with WT, whereas current density for both Thr400Arg and Met1770Leu was similar to WT (Figure 4A,B and Table SS1). We observed no changes in voltage dependence of activation for any variant using automated patch clamp. However, we did observe depolarized shifts in voltage dependence of inactivation in both splice isoforms, manifested as larger window currents for all variants (Figure 4C,D; Table SS1). Detailed biophysical data are presented in Table SS1. These results suggest that differences in biophysical properties associated with these variants are consistent across experimental platforms.
FIGURE 4.

Automated patch‐clamp recordings from human embryonic kidney (HEK)‐293 cells expressing wild‐type (WT) SCN2A vs variants associated with early‐onset epilepsy and malformation of cortical development. (A) Average normalized whole‐cell current families of SCN2A‐WT (black), T400R (orange), I1640N (dark blue), and M1770L (purple) in the adult (top) and neonatal (bottom) splice isoforms. Summary data showing deviation from WT of each variant for (B) whole‐cell current density, (C) voltage dependence of inactivation, and (D) window current. Circles represent data recorded from the adult splice isoform, whereas squares represent data from the neonatal splice isoform. Open symbols represent data from each recorded cell, and solid symbols show mean ± 95% confidence interval. Asterisks (*) denote p < .05.
3.3. A computational model of a developing neocortical pyramidal neuron
To determine the physiological impact of NaV1.2 variants associated with epilepsy and MCD, we introduced the SCN2A‐p.Met1700Leu‐N variant into a computational model of a developing layer 5 neocortical pyramidal neuron. The model was based on a previously‐established framework and incorporated detailed morphological and electrophysiological properties (see Materials and Methods). To mimic the developmental changes in SCN2A expression, we modified the model to express 50% neonatal and 50% adult NaV1.2 isoforms wherever SCN2A was present. 34 To simulate the effects of the NaV1.2‐Met1770Leu variant on neuronal excitability, we fitted an HH ion channel model to mimic the biophysical properties of NaV1.2‐Met1770Leu from whole‐cell voltage‐clamp recordings obtained in HEK‐293 T cells. The model accurately recapitulated the depolarized voltage dependence for both activation and inactivation observed for NaV1.2‐Met1770Leu compared to WT for both the adult and neonatal isoforms (Figure 5A).
FIGURE 5.

Simulated neuronal excitability in a neocortical pyramidal cell model expressing wild‐type (WT; black) or M1770L variant (purple). (A) Voltage dependence of activation and inactivation for the adult (top) and neonatal (bottom) isoforms of Nav1.2‐WT and Nav1.2‐M1770L. (B) Firing of the model WT (black) and M1770L cell (purple) in response to stimulation using a 500 ms step stimulation (top) or a synaptic stimulation (bottom). F–I curves (left) and example traces (right) of modeled firing in response to 0.8 and 1.6 nA current injections (top) and different multiplications of the synaptic stimulation (bottom).
After incorporating the ion channel model into the neocortical pyramidal neuron model, we simulated the firing in response to somatic current injection. The model expressing NaV1.2‐Met1770Leu showed more firing compared to WT across a range of injected currents (Figure 5B; top). Example traces at 0.8 and 1.6 nA injected current highlight the greater firing in the NaV1.2‐Met1770Leu model. To test the effects under more physiological conditions, we simulated synaptic input to the model using stochastic excitatory conductances (see Materials and Methods). Consistent with the results from step current injection, the NaV1.2‐Met1770Leu model exhibited higher firing rates compared to WT across different synaptic stimulation intensities (Figure 5B; bottom). Representative voltage traces illustrate the increased firing in the NaV1.2‐Met1770Leu model.
In summary, our computational modeling—which considers the developmental expression of neonatal and adult isoforms—predicts that the complex biophysical changes caused by the NaV1.2‐p.Met1770Leu variant lead to an overall increase in neuronal excitability in a developing neocortical pyramidal neuron model.
4. DISCUSSION
Here we report seven de novo variants in SCN2A encoding the voltage‐gated sodium channel α subunit NaV1.2 identified in eight individuals with epilepsy, developmental disability, and MCD. Four of these variants are new, whereas three were reported previously in the literature. We provide detailed clinical, neuroimaging, and genetic data on the affected individuals, along with electrophysiological characterization of the associated variants using manual as well as automated whole‐cell voltage‐clamp recording in heterologous cells, and construct a biologically‐informed computational model of a developing neocortical pyramidal neuron with WT vs variant NaV1.2. We also examined all variants in the neonatal (N) and adult (A) isoforms based on known functional differences between the isoforms and evidence of isoform‐specific variants as a cause of early‐onset SCN2A‐related disorders. 28
Variants in SCN2A are associated with a range of neurological dysfunction, including developmental delay/ID, ASD, and childhood‐onset epilepsy of varying severity. Variants identified in patients with ASD are more likely to be truncating (leading to a frameshift or premature stop codon; i.e., LoF) whereas variants identified in patients with DEE are missense with predominantly GoF effects on channel function in heterologous cell systems. 36 We hypothesized that Na + channels containing SCN2A variants associated with epilepsy and MCD might exhibit a specific signature or set of biophysical properties in a heterologous system. However, recent work indicates a more complex relationship between clinical phenotype and any simple GoF/LoF framework. 3 Here, we found this to be the case for the SCN2A‐p.Ile1640Asn‐N variant, which was associated with lower current density and a depolarized shift in the voltage dependence of activation (consistent with LoF) accompanied by a hyperpolarized shift in the voltage dependence of steady‐state channel availability and slowed kinetics of inactivation (GoF).
A feature common among all four of the neonatal isoforms of the identified SCN2A variants was the large shift in the voltage dependence of steady‐state inactivation, which led to a large window current for each variant. How this feature might contribute to MCD currently remains unclear. However, a large (>5 mV) depolarized shift in the voltage dependence of inactivation is not unique to SCN2A variants associated with MCD, as this finding has been observed in at least one other SCN2A variant associated with early‐onset epilepsy but purportedly without MCD (SCN2A‐p.Met1879Thr). 3 , 37 It is further possible that a shift in the voltage dependence of inactivation is a feature of other SCN2A variants associated with epilepsy without MCD, but this particular electrophysiological parameter may not have been assessed in all studies. It is also possible for this finding to be missed if only present in the neonatal isoform—as with the SCN2A‐p.Thr400Asn variant—as some studies evaluate the effect of disease‐associated variants in the adult isoform only. This association could be further clarified in future work.
Collectively, our results identify de novo heterozygous pathogenic variants in SCN2A in eight individuals with early‐onset epilepsy accompanied by diffuse developmental brain malformation. Findings support the conclusion that SCN2A may be a genetic cause of MCD, joining other ion channels and ionotropic neurotransmitter receptor genes. 5 , 7 , 11 , 12 Yet, the mechanism whereby variants in SCN2A might lead to MCD, and why only a small subset of SCN2A variants are associated with MCD, is unclear. One possibility is that such variants share a combination of biophysical properties unique among disease‐associated variants in SCN2A, although this conclusion is not supported by our data and a thorough review of the literature. Instead, it may be the case that the relative proportions of SCN2A and SCN3A expressed by developing neurons differ from individual to individual, such that developing neurons in some individuals express relatively more SCN2A, 34 rendering them more vulnerable to the effects of abnormal NaV1.2 during fetal development. Future studies will be required to investigate the mechanism whereby variation in SCN2A leads to MCD.
AUTHOR CONTRIBUTIONS
Jérôme Clatot: data curation (co‐lead); formal analysis (co‐lead); investigation (co‐lead); writing—original draft preparation (supporting); writing—review & editing (supporting). Christopher H. Thompson: data curation (supporting); formal analysis (co‐lead); investigation (co‐lead); writing—review & editing (supporting); writing—original draft preparation (supporting); writing—review & editing (supporting). Susan Sotardi: data curation (supporting); investigation (supporting). Jinan Jiang: investigation (supporting); Angeliki Vakrinou: data curation (supporting). Marina Trivisano: data curation (supporting); investigation (supporting). Simona Balestrini: data curation (supporting); investigation (supporting). D. Isum Ward: data curation (supporting). Natalie Ginn: data curation (supporting). Brunetta Guaragni: data curation (supporting). Laura Malerba: data curation (supporting). Mia Sherer: formal analysis (co‐lead); Ingo Helbig: data curation (supporting); writing—review & editing (supporting). Ala Somarowthu: formal analysis (supporting); software (supporting). Sanjay M. Sisodiya: data curation (supporting). Roy Ben‐Shalom: data curation (supporting); formal analysis (co‐lead); investigation (co‐lead); software (supporting); supervision (supporting); writing—original draft preparation (supporting); writing—review & editing (supporting). Renzo Guerrini: data curation (supporting); supervision (supporting); writing‐ review & editing (supporting). Nicola Specchio: data curation (supporting); supervision (supporting); writing—review & editing (supporting). Alfred L. George, Jr.: data curation (supporting); funding acquisition (co‐lead); methodology (supporting); project administration (supporting); resources (supporting); supervision (supporting); writing—review & editing (supporting). Ethan M. Goldberg: conceptualization (lead); data curation (lead); formal analysis (co‐lead); funding acquisition (co‐lead); writing—original draft preparation (lead); investigation (supporting); project administration (lead); resources (lead); supervision (lead); writing—review & editing (lead).
CONFLICT OF INTEREST STATEMENT
None of the authors has any conflict of interest to disclose.
ETHICS STATEMENT
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Table S1.
Table S2.
ACKNOWLEDGMENTS
This work was supported by the National Institute of Neurological Disorders and Stroke (NINDS) R01 NS119977 to E.M.G. and U54 NS108874 to A.L.G.; Current Research 2023 of the Italian Ministry of Health (to R.G. and S.B.); Ministry of University and Research (MIUR), National Recovery and Resilience Plan (NRRP), project MNESYS (PE0000006) (to R.G. and S.B.); Brain Optical Mapping by Fondazione CARIFI (to R.G.); and DECODEE Call Health 2018 of the Tuscany Region (to R.G.). We would like to thank the patients enrolled in the study and their families for participation in research, referring providers and clinicians involved in the care of these patients, as well as Leah Schust for her leadership of the FamilieSCN2A Foundation and involvement in the project.
Clatot J, Thompson CH, Sotardi S, Jiang J, Trivisano M, Balestrini S, et al. Rare dysfunctional SCN2A variants are associated with malformation of cortical development. Epilepsia. 2025;66:914–928. 10.1111/epi.18234
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Meisler MH, Hill SF, Yu W. Sodium channelopathies in neurodevelopmental disorders. Nat Rev Neurosci. 2021;22(3):152–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wolff M, Brunklaus A, Zuberi SM. Phenotypic spectrum and genetics of SCN2A‐related disorders, treatment options, and outcomes in epilepsy and beyond. Epilepsia. 2019;60(Suppl 3):S59–S67. [DOI] [PubMed] [Google Scholar]
- 3. Thompson CH, Potet F, Abramova TV, DeKeyser J‐M, Ghabra NF, Vanoye CG, et al. Epilepsy‐associated SCN2A (NaV1.2) variants exhibit diverse and complex functional properties. J Gen Physiol. 2023;155(10):e202313375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berg AT, Thompson CH, Myers LS, Anderson E, Evans L, Kaiser AJE, et al. Expanded clinical phenotype spectrum correlates with variant function in SCN2A‐related disorders. Brain. 2024;awae125:2761–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Smith RS, Kenny CJ, Ganesh V, Jang A, Borges‐Monroy R, Partlow JN, et al. Sodium Channel SCN3A (NaV1.3) regulation of human cerebral cortical folding and Oral motor development. Neuron. 2018;99(5):905–913.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zaman T, Helbig KL, Clatot J, Thompson CH, Kang SK, Stouffs K, et al. SCN3A‐related neurodevelopmental disorder: a Spectrum of epilepsy and brain malformation. Ann Neurol. 2020;88(2):348–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zaman T, Helbig I, Božović IB, DeBrosse SD, Bergqvist ACC, Wallis K, et al. Mutations in SCN3A cause early infantile epileptic encephalopathy. Ann Neurol. 2018;83(4):703–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Smith RS, Walsh CA. Ion Channel functions in early brain development. Trends Neurosci. 2020;43(2):103–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barkovich AJ, Guerrini R, Kuzniecky RI, Jackson GD, Dobyns WB. A developmental and genetic classification for malformations of cortical development: update 2012. Brain. 2012;135(Pt 5):1348–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bahi‐Buisson N, Poirier K, Fourniol F, Saillour Y, Valence S, Lebrun N, et al. The wide spectrum of tubulinopathies: what are the key features for the diagnosis? Brain. 2014;137(Pt 6):1676–1700. [DOI] [PubMed] [Google Scholar]
- 11. Fry AE, Fawcett KA, Zelnik N, Yuan H, Thompson BAN, Shemer‐Meiri L, et al. De novo mutations in GRIN1 cause extensive bilateral polymicrogyria. Brain. 2018;141(3):698–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Platzer K, Yuan H, Schütz H, Winschel A, Chen W, Hu C, et al. GRIN2B encephalopathy: novel findings on phenotype, variant clustering, functional consequences and treatment aspects. J Med Genet. 2017;54(7):104509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bernardo S, Marchionni E, Prudente S, De Liso P, Spalice A, Giancotti A, et al. Unusual association of SCN2A epileptic encephalopathy with severe cortical dysplasia detected by prenatal MRI. Eur J Paediatr Neurol. 2017;21(3):587–590. [DOI] [PubMed] [Google Scholar]
- 14. Epilepsy Phenome/Genome Project, Epi4K Consortium . Diverse genetic causes of polymicrogyria with epilepsy. Epilepsia. 2021;62(4):973–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vlachou V, Larsen L, Pavlidou E, Ismayilova N, Mazarakis ND, Scala M, et al. SCN2A mutation in an infant with Ohtahara syndrome and neuroimaging findings: expanding the phenotype of neuronal migration disorders. J Genet. 2019;98(2):54. [PubMed] [Google Scholar]
- 16. Akula SK, Chen AY, Neil JE, Shao DD, Mo A, Hylton NK, et al. Exome sequencing and the identification of new genes and shared mechanisms in polymicrogyria. JAMA Neurol. 2023;80(9):980–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Inuzuka LM, Macedo‐Souza LI, Della‐Ripa B, Cabral KSS, Monteiro F, Kitajima JP, et al. Neurodevelopmental disorder associated with de novo SCN3A pathogenic variants: two new cases and review of the literature. Brain and Development. 2020;42(2):211–216. [DOI] [PubMed] [Google Scholar]
- 19. Liu Y, Schubert J, Sonnenberg L, Helbig KL, Hoei‐Hansen CE, Koko M, et al. Neuronal mechanisms of mutations in SCN8A causing epilepsy or intellectual disability. Brain. 2019;142(2):376–390. [DOI] [PubMed] [Google Scholar]
- 20. Lindy AS, Stosser MB, Butler E, Downtain‐Pickersgill C, Shanmugham A, Retterer K, et al. Diagnostic outcomes for genetic testing of 70 genes in 8565 patients with epilepsy and neurodevelopmental disorders. Epilepsia. 2018;59(5):1062–1071. [DOI] [PubMed] [Google Scholar]
- 21. Retterer K, Juusola J, Cho MT, Vitazka P, Millan F, Gibellini F, et al. Clinical application of whole‐exome sequencing across clinical indications. Genet Med. 2016;18(7):696–704. [DOI] [PubMed] [Google Scholar]
- 22. Berecki G, Howell KB, Heighway J, Olivier N, Rodda J, Overmars I, et al. Functional correlates of clinical phenotype and severity in recurrent SCN2A variants. Commun Biol. 2022;5(1):515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ben‐Shalom R, Keeshen CM, Berrios KN, An JY, Sanders SJ, Bender KJ. Opposing effects on NaV1.2 function underlie differences between SCN2A variants observed in individuals with autism Spectrum disorder or infantile seizures. Biol Psychiatry. 2017;82(3):224–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ben‐Shalom R, Aviv A, Razon B, Korngreen A. Optimizing ion channel models using a parallel genetic algorithm on graphical processors. J Neurosci Methods. 2012;206(2):183–194. [DOI] [PubMed] [Google Scholar]
- 27. Van Geit W, Gevaert M, Chindemi G, Rössert C, Courcol J‐D, Muller EB, et al. BluePyOpt: leveraging open source software and cloud infrastructure to optimise model parameters in neuroscience. Front Neuroinform. 2016;10:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thompson CH, Ben‐Shalom R, Bender KJ, George AL. Alternative splicing potentiates dysfunction of early‐onset epileptic encephalopathy SCN2A variants. J Gen Physiol. 2020;152(3):e201912442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Echevarria‐Cooper DM, Hawkins NA, Misra SN, Huffman AM, Thaxton T, Thompson CH, et al. Cellular and behavioral effects of altered NaV1.2 sodium channel ion permeability in Scn2aK1422E mice. Hum Mol Genet. 2022;31(17):2964–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Spratt PWE, Alexander RPD, Ben‐Shalom R, Sahagun A, Kyoung H, Keeshen CM, et al. Paradoxical hyperexcitability from NaV1.2 sodium channel loss in neocortical pyramidal cells. Cell Rep. 2021;36(5):109483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Spratt PWE, Ben‐Shalom R, Keeshen CM, Burke KJ, Clarkson RL, Sanders SJ, et al. The autism‐associated gene Scn2a contributes to dendritic excitability and synaptic function in the prefrontal cortex. Neuron. 2019;103(4):673–685.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Awile O, Kumbhar P, Cornu N, Dura‐Bernal S, King JG, Lupton O, et al. Modernizing the NEURON simulator for sustainability, portability, and performance. Front Neuroinform. 2022;16:884046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ramaswamy S, Courcol J‐D, Abdellah M, Adaszewski SR, Antille N, Arsever S, et al. The neocortical microcircuit collaboration portal: a resource for rat somatosensory cortex. Front Neural Circuits. 2015;9:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liang L, Fazel Darbandi S, Pochareddy S, Gulden FO, Gilson MC, Sheppard BK, et al. Developmental dynamics of voltage‐gated sodium channel isoform expression in the human and mouse brain. Genome Med. 2021;13(1):135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Clarkson RL, Liptak AT, Gee SM, Sohal VS, Bender KJ. D3 receptors regulate excitability in a unique class of prefrontal pyramidal cells. J Neurosci. 2017;37(24):5846–5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sanders SJ, Campbell AJ, Cottrell JR, Moller RS, Wagner FF, Auldridge AL, et al. Progress in understanding and treating SCN2A‐mediated disorders. Trends Neurosci. 2018;41(7):442–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Adney SK, Millichap JJ, DeKeyser J‐M, Abramova T, Thompson CH, George AL. Functional and pharmacological evaluation of a novel SCN2A variant linked to early‐onset epilepsy. Ann Clin Transl Neurol. 2020;7(9):1488–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1.
Table S2.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.