

Abstract
Inflammatory myofibroblastic tumors (IMTs) are rare mesenchymal neoplasms characterized by spindle-cell morphology with accompanying inflammatory infiltrates. Originally described in 1939, these tumors can arise in various anatomic locations, with the urinary bladder being a rare site of occurrence but the most common within the genitourinary tract. IMTs typically present as polypoid masses or firm submucosal nodules, often with painless hematuria in bladder cases. Histopathologically, IMTs are composed of myofibroblasts with myxoid stroma and mixed inflammatory cells, predominantly lymphocytes and plasma cells. Immunohistochemically, these tumors commonly express anaplastic lymphoma kinase1 (ALK1), vimentin, smooth muscle actin (SMA), and cytokeratin, with ALK1 serving as a crucial marker for diagnosis. This report details the case of a 31-year-old female presenting with hematuria, found to have a soft tissue mass in the urinary bladder (5.0 × 3.0 cm). Imaging revealed a well-defined lesion with vascularity. Histopathological examination confirmed an IMT, with immunohistochemistry showing diffuse ALK1 positivity, patchy SMA staining, and variable desmin expression, consistent with the diagnosis. IMTs are generally considered neoplasms of intermediate malignant potential. While metastasis is exceedingly rare in bladder IMTs, local recurrence has been reported, particularly in cases of incomplete surgical resection. Recent advances highlight the role of ALK inhibitors in managing unresectable cases, enabling partial cystectomy in select patients. This article underscores the importance of achieving complete surgical excision and highlights the role of ALK expression in diagnosis and differentiation from other spindle-cell neoplasms. Further studies are needed to elucidate the molecular and clinical factors influencing prognosis and to refine treatment strategies for IMTs.
Keywords: Inflammatory myofibroblastic tumor (IMT), urinary bladder, ALK1, ALK
Introduction
In 1939, Bunn identified and described inflammatory myofibroblastic tumors (IMTs), a term later coined by Umiker and collaborators. 1 IMTs can arise in various regions of the body, with a predilection for the lungs, liver, and gastrointestinal tract, accounting for only 0.7% of reported tumors in the lung parenchyma and bronchus. 2 IMTs are mesenchymal tumors characterized by the proliferation of myofibroblasts accompanied by a diverse inflammatory infiltrate.3–4
Recognized by the World Health Organization in 2013, IMTs are diagnosed in approximately 150–200 new cases annually in the United States, although precise epidemiological data remain limited.5–7
IMTs primarily occur in middle-aged adults; however, development in children accounts for approximately 25% of reported cases. Anaplastic lymphoma kinase (ALK)-positive tumors exhibit a slight female predilection and tend to occur at a younger age compared to ALK-negative tumors. The exact etiology of IMTs remains unclear, but a significant proportion of patients report a history of prior procedures, raising the possibility that trauma may contribute to tumor development.
Despite being considered benign, IMTs tend to recur and are associated with specific genetic alterations. When IMTs involve the urinary tract, 25% are diagnosed at an average age of 13 years. 8 While IMTs of the urinary bladder are exceptionally rare, the bladder represents the most common site of IMT within the genitourinary tract.
Case presentation
A 31-year-old female presented to the emergency room with hematuria. The patient had no significant past medical history. Ultrasound imaging of the bladder revealed a soft tissue mass measuring 5.0 × 3.0 cm at the base of the bladder with abnormal vascularity (Figure 1). The lumen of the urinary bladder showed turbid urine with multiple blood clots. Multi-slice computed tomography revealed a round, well-defined, enhanced soft tissue lesion at the anterosuperior aspect of the urinary bladder, possibly arising from the bladder wall, yet inseparable from it. Few air loculi were identified in the bladder cavity. No signs of leakage from the urinary bladder were observed. The patient underwent transurethral resection of a bladder (TURB) followed by a partial cystectomy. Histopathological examination of the mass revealed a subepithelial tumor composed of spindle cells with a myofibroblastic appearance (Figure 2(a)). Tumor cells exhibited thin, fusiform morphology with elongated nuclei and eosinophilic cytoplasm. A background of myxoid stroma was noted. Inflammatory components were predominantly composed of lymphocytes. Mitotic figures were infrequent (Figure 2(b) and (d)). Immunohistochemical staining revealed positive staining for ALK1, displaying a diffuse, primarily cytoplasmic staining. Desmin staining was positive and diffuse. Smooth muscle actin (SMA) staining was positive and patchy (Figure 3(a) and (c)). A final diagnosis of an IMT of the urinary bladder was rendered.
Figure 1.
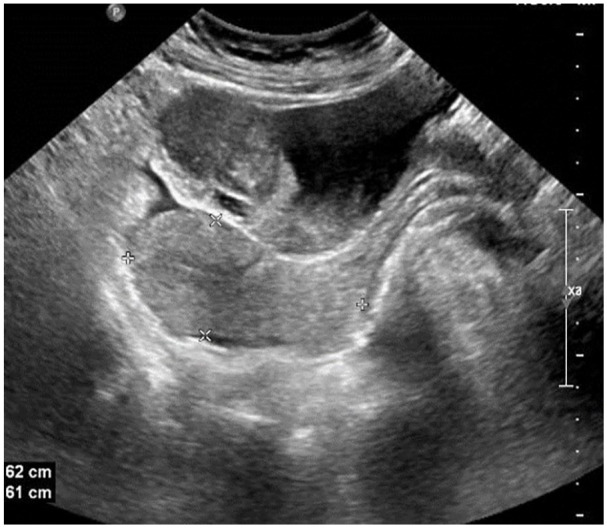
Inflammatory myofibroblastic tumor of the urinary bladder. Ultrasound imaging of the bladder reveals a urinary bladder soft tissue mass measuring 5.0 × 3.0 cm.
Figure 2.

(a–d) Inflammatory myofibroblastic tumor of the urinary bladder. Hematoxylin and Eosin staining reveal loose stellate cells with myxoid background and scattered inflammatory cells. Scanning magnification ((a) ×100, (b) ×200, (c) ×200, (d) ×200).
Figure 3.

(a–d) Inflammatory myofibroblastic tumor of the urinary bladder. Immunohistochemical staining shows positive staining for (a) Desmin: diffuse staining; (b) SMA: patchy staining; and (c) ALK: diffuse staining. (d) ALK Control. Scanning magnification ((a) ×100, (b) ×200, (c) ×200, (d) ×200).
ALK: anaplastic lymphoma kinase; SMA: smooth muscle actin.
Discussions
IMTs are rare mesenchymal spindle-cell neoplasms characterized by stellate myofibroblasts mixed with inflammatory infiltrates, predominantly composed of lymphocytes.
When occurring outside the bladder, IMTs are often associated with generalized symptoms such as pain, fever, weight loss, anemia, thrombocytosis, elevated erythrocyte sedimentation rate, and increased gamma globulins.9–11 In contrast, bladder IMTs typically present as painless hematuria.
The exact etiology of IMTs remains unclear. Proposed explanations include an infectious origin, chronic cystitis, or trauma, particularly surgical trauma. In adults, trauma related to TURB cancer is more frequently observed. In children, however, the cause is often less evident and typically lacks a history of preceding infectious or iatrogenic trauma. The infectious hypothesis is supported by the detection of Epstein-Barr Virus and Human Herpesvirus 8 in some cases, as demonstrated by immunohistochemical analyses.12,13–14
Clinically, IMTs typically present as a polypoid mass or a pale, firm submucosal nodule, occasionally with surface ulceration. Microscopically, IMTs are composed of spindle-shaped cells with a myofibroblastic appearance characterized by thin, fusiform cells with elongated cytoplasmic processes with no cytological abnormalities. A predominant admixture of inflammatory cells is often present, particularly lymphocytes. Many tumors extend to the urinary bladder muscularis propria. Rare cases with overtly sarcomatous morphology and necrosis have been reported, and such cases are associated with aggressive behavior. 15 Immunohistochemically, these tumors are positive for SMA, supporting their myofibroblastic phenotype, and they show variable positivity for cytokeratin, desmin, and h-caldesmon. Negative expression for other markers such as p63, GATA3, and high-molecular-weight cytokeratin, S100, CD34, KIT, CD21, and CD23 aids in excluding other diagnostic possibilities.
IMTs of the urinary bladder commonly express markers such as ALK1, vimentin, SMA, and cytokeratin. 14 Among these, ALK1 positivity is particularly significant. Previous studies have shown variable expression of desmin (0%–69%) in these tumors, indicating that while ALK1 positivity is a valuable diagnostic marker, desmin expression is not consistently observed in IMTs. 16 Approximately 35%–89% of inflammatory myofibroblastic bladder tumors express ALK1 protein. This marker is particularly useful in distinguishing IMTs from other neoplasms. For instance, ALK1 is also detected in approximately 20% of rhabdomyosarcomas, but not in leiomyosarcomas or sarcomatoid carcinomas, making it an important diagnostic tool. 17
IMTs are typically characterized by spindle-cell morphology accompanied by an inflammatory infiltrate, which was consistent with the findings in our case. In contrast, rhabdomyosarcomas often exhibit a more primitive cellular appearance with round cells and the presence of rhabdomyoblasts. 18 The spindle-cell morphology and associated inflammatory infiltrate observed in our case strongly supported a diagnosis of IMT. MyoD and Myogenin support the diagnosis of rhabdomyosarcoma in suspicious cases. ALK1 protein expression is present in a significant proportion of cases, which often correlates with genetically confirmed ALK rearrangement, commonly discovered by Fluorescence in situ hybridization (FISH). 12
The differential diagnosis includes nodular fasciitis, which typically presents in the extremities and is characterized by the MYH9::USP6 fusion, with a lack of ALK expression. Desmoid-type fibromatosis, which consists of spindle cells interspersed with collagen and minimal to no inflammation, is ALK-negative and associated with mutations in CTNNB1 (beta-catenin). Inflammatory leiomyosarcoma, distinguished by cigar-shaped nuclei and more organized fascicular growth, must also be considered. In addition, IgG4-related sclerosing disease should be included, as it exhibits a significantly higher IgG4+/IgG ratio compared to IMTs and is negative for ALK by immunohistochemistry. Spindle-cell melanoma, which can exhibit spindle-shaped tumor cells with pigment, should also be differentiated, as it may share a similar morphology with IMT. Immunohistochemical staining of S100 and SOX10 is essential in such cases. Rhabdomyosarcoma, often presenting with more primitive round cells and rhabdomyoblasts should also be considered, particularly in cases of spindle-cell morphology, as these tumors may overlap in appearance but differ in immunohistochemical markers and clinical behavior.
IMT is generally considered a neoplasm with intermediate malignant potential, characterized by rare occurrences of metastasis. However, in the urinary tract, it is regarded as having an even more favorable prognosis, with almost no documented cases of metastasis. Rare reports of potential metastasis originating in the urinary bladder have been described, though some historical accounts of malignant behavior may represent misdiagnosed cases of sarcomatoid urothelial carcinoma. Local recurrence has been observed in a minority of cases, primarily in adults, and is often associated with incomplete resection. Notably, local recurrence in IMTs has shown correlations with abdominopelvic sites, larger sizes, and advanced age. In contrast, metastases were associated with younger age, larger tumor size, and involvement of both abdominopelvic and pulmonary sites. Notably, according to Coffin et al., six instances of metastatic IMTs were found to be nonreactive for ALK1. Within the subset of ALK-positive IMTs, 54% exhibited recurrence, yet no instances of metastasis were reported in this specific subgroup. 9
Hussong et al. 17 investigated the histological features, DNA content, and protein expression in IMTs to identify potential indicators of aggressive behavior or malignant transformation. Among the 24 IMTs analyzed, six cases demonstrated recurrence, and two underwent malignant transformation. Cellular atypia and the presence of ganglion-like cells were more frequently observed in IMTs associated with recurrence or malignant transformation. However, no significant differences were noted in terms of cellularity, mitotic activity, or the extent of the inflammatory infiltrate.
All tumors expressed bax protein, while none exhibited c-myc expression. Aneuploidy was identified in two IMTs, one of which underwent malignant transformation. Bcl-2 expression was observed in nine of the 24 IMTs, without significant differences across outcome groups. The study concluded that a combination of cellular atypia, ganglion-like cells, p53 expression, and DNA ploidy analysis might help identify IMTs at higher risk for malignant transformation or a more aggressive clinical course with a tendency for recurrence.13,18
Complete surgical excision is the preferred treatment approach. Notably, in pediatric patients, no recurrence after successful resection was reported. 19 In contrast, local recurrence has been reported in adults when complete surgical excision has not been accomplished. 20 In unresectable cases, responses to ALK inhibitors have been reported, enabling subsequent resection via partial cystectomy. In some instances, treatment with anti-inflammatory agents may be employed.
Conclusion
In summary, IMTs are rare neoplasms with generally favorable outcomes, particularly in the urinary bladder, where metastasis is exceedingly uncommon. However, the potential for local recurrence underscores the importance of achieving complete surgical excision. Factors such as cellular atypia, ganglion-like cells, and DNA ploidy analysis may serve as useful markers to identify IMTs with a higher risk of recurrence or malignant transformation.
This report comprehensively outlines the clinical, radiological, and histopathological features of an IMT in the urinary bladder. The crucial role of positive immunohistochemical staining for ALK, alongside other markers, in confirming the diagnosis is highlighted. 21
Further research into the molecular and clinical characteristics of IMTs is essential to refine diagnostic criteria and guide treatment strategies, ensuring optimal management and patient outcomes. This contribution enhances our understanding of the atypical presentations of IMTs in the urinary bladder, underscoring the importance of a multidisciplinary approach for accurate diagnosis and effective management.
Acknowledgments
None.
Footnotes
Author contributions: All authors have reviewed the final version to be published and agreed to be accountable for all aspects of the work. Concept and design: F.N.U.P., I.M., R.M.A., N.S. Acquisition, analysis, or interpretation of data: F.N.U.P., I.M., R.M.A., N.S. Drafting of the manuscript: F.N.U.P., N.S. Critical review of the manuscript for important intellectual content: F.N.U.P., I.M., R.M.A., N.S. Supervision: N.S.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics approval: Our institution does not require ethical approval for reporting individual cases or case series.
Informed consent: Written informed consent was obtained from the patient(s) for their anonymized information to be published in this article.
ORCID iDs: FNU Poombal  https://orcid.org/0009-0005-8569-6805
https://orcid.org/0009-0005-8569-6805
Nada Shaker
https://orcid.org/0000-0001-6145-256X
References
- 1. Abu Asbeh Y, Naroditsky I, Katz A, et al. Unusual presentation of an inflammatory myofibroblastic tumor. CTSNet, Inc. 10.25373/ctsnet.5233969 (2017, accessed 21 July 2017). [DOI] [Google Scholar]
- 2. Golbert ZV, Pletnev SD. On pulmonary “pseudotumours”. Neoplasma 1967; 14(2): 189–198. [PubMed] [Google Scholar]
- 3. Palaskar S, Koshti S, Maralingannavar M, et al. Inflammatory myofibroblastic tumor. Contemp Clin Dent 2011; 2(4): 274–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Patel A. Pathology of inflammatory myofibroblastic tumour. Glob J Med Microbiol Sci 2022; 10(1): 166. [Google Scholar]
- 5. Fletcher CDM, Bridge JA, Hogendoorn PCW, et al. , eds. WHO classification of tumours of soft tissue and bone. pathology and genetics of tumours of soft tissue and bone. 4th ed. Lyon: IARC Press, 2013. [Google Scholar]
- 6. Casanova M, Brennan B, Alaggio R, et al. Inflammatory myofibroblastic tumor: the experience of the European pediatric Soft Tissue Sarcoma Study Group (EpSSG). Eur J Cancer 2020; 127: 123–129. [DOI] [PubMed] [Google Scholar]
- 7. Ferrari A, Alaggio R, Meazza C, et al. Fibroblastic tumors of intermediate malignancy in childhood. Expert Rev Anticancer Ther 2013; 13(2): 225–236. [DOI] [PubMed] [Google Scholar]
- 8. Williamson SR, Lopez-Beltran A, MacLennan GT, et al. (2013). Unique clinicopathologic and molecular characteristics of urinary bladder tumors in children and young adults. Urol Oncol Semin Orig Investig 31(4): 414–426. [DOI] [PubMed] [Google Scholar]
- 9. Coffin CM, Hornick JL, Fletcher CD. Inflammatory myofibroblastic tumor: comparison of clinicopathologic, histologic, and immunohistochemical features including ALK expression in atypical and aggressive cases. Am J Surg Pathol 2007;31(4): 509–520. [DOI] [PubMed] [Google Scholar]
- 10. Coffin CM, Watterson J, Priest JR, et al. Extrapulmonary inflammatory myofibroblastic tumor (inflammatory pseudotumor). A clinicopathologic and immunohistochemical study of 84 cases. Am J Surg Pathol 1995; 19(8): 859–872. [DOI] [PubMed] [Google Scholar]
- 11. Karnak I, Senocak ME, Ciftci AO, et al. Inflammatory myofibroblastic tumor in children: diagnosis and treatment. J Pediatr Surg 2001; 36(6): 908–912. [DOI] [PubMed] [Google Scholar]
- 12. Collin M, Charles A, Barker A, et al. Inflammatory myofibroblastic tumour of the bladder in children: a review. J Pediatr Urol 2015; 11(5): 239–245. [DOI] [PubMed] [Google Scholar]
- 13. Biselli R, Ferlini C, Fattorossi A, et al. Inflammatory myofibroblastic tumor (inflammatory pseudotumor): DNA flow cytometric analysis of nine pediatric cases. Cancer 1996; 77(4): 778-784. [DOI] [PubMed] [Google Scholar]
- 14. Mergan F, Jaubert F, Sauvat F, et al. Inflammatory myofibroblastic tumor in children: Clinical review with anaplastic lymphoma kinase, Epstein-Barr virus, and human herpesvirus 8 detection analysis. J Pediatr Surg 2005; 40(10): 1581–1586. [DOI] [PubMed] [Google Scholar]
- 15. Rao RN, Ranjan P, Singla N, et al. Inflammatory myofibroblastic tumor of the urinary bladder diagnosed by anaplastic lymphoma kinase immunostaining. Urol Ann 2012; 4(2): 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Freeman A, Geddes N, Munson P, et al. Anaplastic lymphoma kinase (ALK 1) staining and molecular analysis in inflammatory myofibroblastic tumours of the bladder: a preliminary clinicopathological study of nine cases and review of the literature. Mod Pathol 2004; 17(7): 765–771. [DOI] [PubMed] [Google Scholar]
- 17. Hussong JW, Brown M, Perkins SL, et al. Comparison of DNA ploidy, histologic, and immunohistochemical findings with clinical outcome in inflammatory myofibroblastic tumors. Mod Pathol 1999; 12(3): 279–286. [PubMed] [Google Scholar]
- 18. Corao DA, Biegel JA, Coffin CM, et al. ALK expression in rhabdomyosarcomas: correlation with histologic subtype and fusion status. Pediatr Dev Pathol 2009; 12(4): 275–283. [DOI] [PubMed] [Google Scholar]
- 19. Iczkowski KA, Shanks JH, Gadaleanu V, et al. Inflammatory pseudotumor and sarcoma of urinary bladder: differential diagnosis and outcome in thirty-eight spindle cell neoplasms. Mod Pathol 2001; 14(10): 1043–1051. [DOI] [PubMed] [Google Scholar]
- 20. Montgomery EA, Shuster DD, Burkart AL, et al. Inflammatory myofibroblastic tumors of the urinary tract: a clinicopathologic study of 46 cases, including a malignant example inflammatory fibrosarcoma and a subset associated with high-grade urothelial carcinoma. Am J Surg Pathol 2006; 30(12): 1502–1512. [DOI] [PubMed] [Google Scholar]
- 21. Li X-Q, Hisaoka M, Shi D-R, et al. Expression of anaplastic lymphoma kinase in soft tissue tumors: an immunohistochemical and molecular study of 249 cases. Hum Pathol 2004; 35(6): 711–721. [DOI] [PubMed] [Google Scholar]