

Abstract
目的
探讨E3 泛素连接酶Parkin缺失造成的线粒体自噬障碍在帕金森病(PD)神经炎症中的作用。
方法
建立MPTP-PD模型,野生型(WT)小鼠和Parkin-/-小鼠连续腹腔注射MPTP 5 d建立PD小鼠模型,分别设置对照组:WT-PBS组、Parkin-/--PBS组;PD建模组:WT-MPTP组、Parkin-/--MPTP组,8只/组,对照组连续5 d注射等量的PBS。建模1周后,通过旷场实验评估WT小鼠与Parkin-/-小鼠的运动行为;通过脑切片免疫荧光和Western blotting检测 Parkin缺失对PD发展以及神经炎症的影响;通过PINK 1/Parkin信号通路变化,探讨Parkin调控线粒体自噬对PD神经炎症发生的分子机制。
结果
与WT-MPTP组相比,Parkin-/--MPTP组小鼠运动功能下降(P<0.001),脑内TH+神经元减少并且α-突触核蛋白(α-syn)积累增加;神经炎症相关GFAP和I-ba1阳性细胞数量增加(P<0.001)。Parkin缺失影响PINK1/Parkin介导的线粒体自噬,导致mtDNA增多以及炎症相关蛋白STING和NLRP3炎症小体的表达上调(P<0.01)。
结论
Parkin通过调节PINK1/Parkin信号通路加速了小鼠帕金森病发展及神经炎症发生,为后期研究Parkin基因隐性遗传早发性帕金森病发病机制及其治疗奠定了实验基础。
Keywords: 帕金森病, Parkin, 神经炎症, 线粒体自噬
Abstract
Objective
To investigate the role of mitochondrial autophagy disorder caused by deletion of E3 ubiquitin ligase Parkin in neuroinflammation in a mouse model of MPTP-induced Parkinson's disease (PD).
Methods
Wild-type (WT) male C57BL/6 mice and Parkin-/- mice were given intraperitoneal injections with MPTP or PBS for 5 consecutive days, and the changes in motor behaviors of the mice were observed using open field test. The effects of Parkin deletion on PD development and neuroinflammation were evaluated using immunofluorescence and Western blotting. The changes of the PINK 1/Parkin signaling pathway in the midbrain substantia nigra of the mice were examined to explore the molecular mechanism of Parkin-mediated regulation of mitochondrial autophagy and its effect on neuroinflammation in PD mice.
Results
Compared with their WT counterparts, the Parkin-/- mice with MPTP injections exhibited significant impairment of motor function with decreased TH+ neurons, increased α-synuclein (α-syn) accumulation, and increased numbers of GFAP+ and I-ba1+ cells in the midbrain substantia nigra. Parkin deletion obviously affected PINK1/Parkin-mediated mitochondrial autophagy to result in significantly increased mtDNA and upregulated expressions of STING and NLRP3 inflammatosomes in the midbrain substantia nigra of MPTP-treated transgenic mice.
Conclusion
Parkin deletion causes mitochondrial autophagy disorder to accelerate PD progression and exacerbates neuroinflammation in mice by affecting the PINK1/Parkin signaling pathway, suggesting the important role of Parkin in early pathogenesis of PD.
Keywords: Parkinson's disease, Parkin, neuroinflammation, mitochondrial autophagy
帕金森病(PD)是老年人中第2常见的神经退行性疾病,对老年人身体健康造成重大影响[1]。PD核的心病理特征包括黑质致密部多巴胺能神经元过量死亡以及运动功能障碍,并伴有α-突触核蛋白的异常积累[2, 3]。PD现在被认为是一种多系统疾病,具有显著的神经炎症和免疫功能障碍[4, 5]。
线粒体功能障碍在散发型和家族性帕金森病中发挥核心作用。有研究表明,线粒体自噬可以清除功能失调的线粒体来维持细胞稳态,线粒体自噬失调会造成线粒体ROS上升、氧化应激损伤以及细胞死亡[6-8]。黑质致密部的多巴胺能神经元由于基础能量代谢高等特征,更易受氧化应激损伤的影响,常用于帕金森小鼠建模的药物如1-甲基-4-苯基-1,2,3,6-四氢吡啶(MPTP)、鱼藤酮素等可以引发线粒体氧化应激导致多巴胺神经元死亡,而线粒体自噬提供一种保护机制,可防止多巴胺神经元的过度死亡[9, 10]。因此,探究线粒体自噬的调控机制对研究PD的发生和治疗有重要意义。
Parkin(PARK2)是一种 E3 泛素连接酶,与常染色体隐性遗传家族型帕金森病的发生有关,受 PINK1的调节,在维持线粒体功能和完整性中起重要作用[11, 12]。PINK1/Parkin信号通路是线粒体自噬的主要通路,在线粒体自噬发生时,PINK1将Parkin募集到线粒体膜上,然后Parkin泛素化线粒体外膜上的蛋白质并募集p62(一种衔接蛋白),靶向功能失调的线粒体及线粒体自噬清除[13, 14]。有研究表明在Parkin 突变的 PD 患者中生成了诱导多能干细胞衍生的中脑神经元,Parkin 被证明可以防止线粒体膜通透性,阻碍线粒体 DNA (mtDNA) 逃逸并检测到小胶质细胞过度表达促炎细胞因子的上调[15]。
Parkin介导的线粒体自噬已被广泛研究,但Parkin突变是否通过调控线粒体自噬途径促进帕金森病发病和神经炎症发生的机制仍未完全明确[16, 17]。实际使用Parkin敲除鼠模型较少,这是由于Parkin缺陷小鼠表现出与人类不同的表型,大多数PD小鼠模型不表现出在这些基因突变的患者中观察到的早发性PD症状[18, 19]。因此本研究尝试在Parkin敲除的基础上通过腹腔注射神经毒素MPTP,寻求建立一种稳健的Parkin敲除鼠早发性PD模型,并且将神经炎症发生和线粒体自噬的关键基因Parkin联系起来,探究了Parkin缺失引发的线粒体自噬障碍在帕金森病及其神经炎症的发展中的作用。以期为研究Parkin缺失的早发性帕金森病提供潜在的治疗靶点。
1. 材料和方法
1.1. 材料
1.1.1. 抗体及试剂
MPTP(Sigma-Aldrich,兔抗GFAP、兔抗Iba-1(BIOSS);鼠抗NLRP3(Affinity Biosciences);兔抗TH、兔抗LC3B(abcam)、鼠抗β-actin、鼠抗p62、鼠抗Parkin、鼠抗PINK1、鼠抗STING、鼠抗dsDNA(Santa Cruz Biotechnology);驴抗兔Cy3、驴抗兔Alexa Fluor 488、驴抗鼠Alexa Fluor 488(Jackson Lab);DAPI染液(碧云天);抗荧光淬灭封片剂(Invitrogen)。
1.1.2. 实验动物
成年雄性C57BL/6小鼠(8~10周)购自合肥青源动物科技有限公司,Parkin+/-杂合小鼠购自赛业(苏州)生物科技有限公司,Parkin-/-纯和基因敲除小鼠由Parkin+/-杂合小鼠交配繁殖而来。所有动物护理和程序均按照国家和国际指导方针进行,实验中动物的使用和实验程序均经蚌埠医科大学实验动物管理和伦理委员会批准(伦理批号:2023-178)。
1.2. 方法
1.2.1. MPTP-PD小鼠模型构建与行为学检测
将野生型(WT)小鼠和Parkin-/-小鼠分为4组,WT-PBS组、WT-MPTP组、Parkin-/--PBS组、Parkin-/--MPTP组,8只/组。
采用亚急性中毒模型研究Parkin缺失对小鼠PD发展以及神经炎症的作用。向野生型(WT)和 Parkin-/-小鼠腹腔分别注射MPTP 30 mg/kg,1次/d,连续5 d。对照组采用连续5 d注射相同体积的PBS。采用旷场实验测试小鼠的运动行为。测试前,先让小鼠在试验室不受干扰地适应30 min,然后置于旷场箱中央,让其自由探索10 min,通过Any-maze视频跟踪软件(V7.33,Stoelting)测量行动距离和中心区停留时间。
1.2.2. 小鼠脑冷冻切片及免疫荧光
小鼠腹腔注射1%戊巴比妥麻醉后进行心脏灌注取脑,大脑在4%多聚甲醛中固定24 h,然后采用梯度脱水的方法,将鼠脑依次放在15%、20%、25%的蔗糖溶液中脱水,-80 ℃冷冻放置。冷冻脑包埋在OCT化合物(Sakura, 4583)中,采用冷冻切片机(CM1950,Leica)将灌注的大脑切成13 µm厚的冠状面切片,并收集在涂有明胶的载玻片上。
经 2% BSA、10%驴血清和0.2%Triton X-100封闭1 h后,将以下一抗与样本置于4 ℃孵育过夜,检测小鼠脑内炎症水平:兔抗IBA1(1∶200),鼠抗NRPL3(1∶200)和兔抗GFAP(1∶300);黑质TH+神经元损失和α-syn积累:兔抗TH(1∶500)和兔抗α-syn(1∶500);线粒体自噬功能:鼠抗dsDNA(1∶500)。次日,室温与二抗Alexa Fluor 488(1∶500)或Cy3(1∶800)避光孵育1 h后,DAPI孵育15 min抗荧光淬灭封片剂封片,双光子激光共聚焦显微镜Olympus(FV-1200MPE SHARE)检测荧光表达[20]。
1.2.3. Western blotting检测
提取WT-PBS组、WT-MPTP组、Parkin-/--PBS组、Parkin-/--MPTP组小鼠中脑组织,用含有蛋白酶抑制剂和磷酸酶抑制剂的RIPA缓冲液制备小鼠中脑脑蛋白提取物,经超声破碎后,4 ℃,12 000 r/min离心30 min,收集上清。加入5×SDS PAGE蛋白上样缓冲液稀并煮沸。用BCA法测定蛋白质浓度,各组样品均取20 μg总蛋白进行SDS-PAGE(恒压120 V),电转至PVDF膜上(恒流340 mA),加入封闭液(3% BSA)封闭1 h,一抗分别使用鼠抗β-actin(1∶1000)、 鼠抗p62(1∶1000)、鼠抗Parkin(1∶1000)、鼠抗PINK1(1∶1000)、兔抗LC3B(1∶5000),鼠抗STING(1∶1000),鼠抗NRPL3(1∶1000)孵育, 4 ℃过夜。次日,洗去一抗,加入二抗羊抗小鼠IgG-HRP和羊抗兔IgG-HRP抗体,室温孵育1 h,ECL化学显影液显影条带,经凝胶成像仪(BIO×RAD)进行图像采集,用ImageJ软件对蛋白表达情况进行分析,检测多巴胺神经元标志物TH蛋白表达量以及PINK1/Parkin信号通路以及炎症相关蛋白表达量的变化。
1.3. 统计学分析
采用GraphPad Prism8.01软件进行统计学分析,实验数据均用均数±标准差表示,组间差异采用单因素方差分析检验,以P<0.05为差异具有统计学意义。
2. 结果
2.1. Parkin缺失对MPTP-PD小鼠运动功能的影响
WT和Parkin -/-小鼠建立PD模型及旷场行为学测试时间轴 (图1A) ,与WT-PBS组和Parkin -/--PBS组相比,WT-MPTP组和Parkin-/--MPTP组小鼠在旷场中的自主行为,探究行为均减少并且运动距离降低(P<0.001,图1B、C)。与WT-MPTP组相比,Parkin-/--MPTP组小鼠在整个场地移动距更低(图1C、D),在旷场中心区域的停留时间减少(P<0.01,图1E)。
图1.
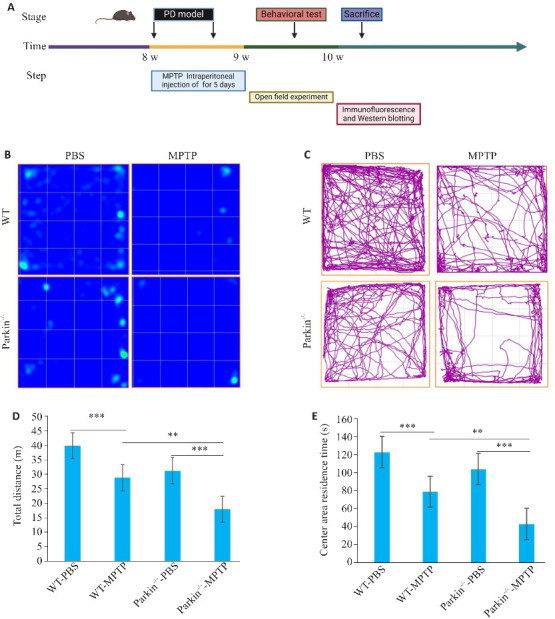
WT及Parkin-/-小鼠注射MPTP后运动行为测试
Fig.1 Behavioral test of wild-type (WT) and Parkin-/- mice after MPTP injection. A: Flow chart of MPTP-PD modeling in the mice. B: Heat map of movement of the mice in open field tests. C: Motion trajectory graph of the mice. D, E: Statistics of total movement distance and the distance in the central area of the mice in open field test. **P<0.01, ***P<0.001.
2.2. Parkin缺失对MPTP小鼠帕金森病变及α-syn变化的影响
Western blotting结果显示WT-MPTP组和Parkin -/--MPTP组小鼠中脑黑质部位TH蛋白表达量均减少,并且Parkin -/--MPTP的小鼠中TH蛋白表达量下降更多(P<0.001, 图2C)。与此同时,对各组小鼠黑质部位TH+神经元免疫荧光染色代表图也显示Parkin敲除后TH+神经元的减少,而WT-PBS组和Parkin-/--PBS组TH+神经元没有明显差异(图2 A、B)。
图2.

WT和Parkin-/-小鼠MPTP诱导的帕金森病变
Fig.2 MPTP-induced Parkinson's disease in WT and Parkin-/- mice. A: Immunofluorescence staining of dopaminergic neuron markers TH (red) and DAPI (blue). B: Protein expression level of TH in the midbrain substantia nigra. C: Analysis of TH protein expression. D: Immunofluorescence staining of α-syn (red) and DAPI (blue) in the mouse midbrain. **P<0.01, ***P<0.001.
免疫荧光检测结果显示MPTP处理导致WT-MPTP组和Parkin-/--MPTP组小鼠α‑syn的积累,并且 Parkin -/--MPTP小鼠显示出α-syn积累增多(图2C)。
2.3. Parkin缺失对脑内胶质细胞激活和神经炎症发展的影响
免疫荧光检测结果显示,与对照组相比,WT-MPTP组和Parkin-/--MPTP组小鼠中脑部位GFAP+和Iba-1+细胞数量增多(P<0.001,图3C、D)。与WT-MPTP小鼠相比,Parkin-/-小鼠给药MPTP后脑内GFAP和Iba-1数量增加(P<0.001,图3C、D)。同时,通过NLRP3炎症小体和Iba-1的三重免疫荧光检测,Parkin-/--MPTP组NLRP3的平均荧光强度比WT-MPTP组水平升高(图3B)。Parkin-/--PBS组与WT-PBS组相比,在没有MPTP刺激的情况下小胶质细胞的数量也有所上升 (P<0.05),并伴随NLRP3炎症小体的增加。
图3.

Parkin-/-小鼠MPTP诱导神经炎症加剧
Fig.3 Parkin deletion increased neuroinflammation induced by MPTP in mice. A: Immunofluorescence staining of astrocyte markers GFAP (red) and DAPI (blue). B: Co-staining of microglial marker I-ba1 (red) and NLRP3 (green) inflammasomes. C: Analysis of GFAP+ cell count (%). D: Analysis of I-ba1+ cell count (%).**P<0.01, ***P<0.001, ****P<0.0001.
2.4. PINK1/Parkin信号通路影响帕金森病发展的分子机制
对线粒体自噬相关蛋白的检测结果显示,与WT-PBS小鼠相比,WT-MPTP组小鼠中脑黑质部位与线粒体自噬激活有关的PINK1和Parkin表达水平均升高(P<0.01,图4B、C)。与WT-MPTP小鼠相比,Parkin-/--MPTP小鼠脑内线粒体自噬调节蛋白p62增多(P<0.01,图4B、C),与线粒体自噬程度呈负相关;但与自噬体清除有关的LC3B表达量下降(P<0.01,图4B、C)。
图4.

Parkin调控线粒体自噬的分子机制
Fig.4 Molecular mechanism of Parkin-mediated regulation of mitochondrial autophagy in midbrain substantia nigra. A: Immunofluorescence assay showing abnormal mitochondrial autophagy (white arrows) in Parkin-/- mice with MPTP injection. B: Western blotting for detecting protein changes in Parkin-mediated regulation of mitochondrial autophagy and neuroinflammation. C: Scanning densitometry for semi-quantitative analysis of the protein expression levels. *P<0.05, **P<0.01 vs WT-MPTP.
免疫荧光检测结果显示,在WT-PBS中dsDNA主要呈现圆形核轮廓,而注射MPTP 2周后,WT-MPTP组和Parkin-/--MPTP组中dsDNA的强度均升高,在细胞质和细胞核中的分布均增加,在 Parkin-/--MPTP组dsDNA的强度增加更明显,范围更大(图4A)。而与炎症相关的基因STING和NLRP3表达均上调(P<0.05),与dsDNA的强度呈正相关(图4B、C)。
3. 讨论
编码蛋白Parkin 的 PARK2基因突变常导致常染色体隐性遗传青少年帕金森病(ARJP)[21, 22]。但Parkin缺陷的小鼠和人类的表型不同,本研究发现Parkin小鼠早期(8~10周)并不表现出PD的典型特征,如明显的运动障碍以及黑质TH+神经元的减少等,并且脑内炎症反应也较低,不能直接作为PD的模型来使用。而利用MPTP注射Parkin基因敲除小鼠建立的早发性Parkin-/--PD小鼠,为阐明PD发展过程所涉及的机制提供了一个很好的模型。
炎症是一种针对致病理性刺激或组织损伤的高度调节机制,旨在保护宿主免受致损伤因子的侵害并促进组织修复[23, 24]。在生理条件下,中枢神经系统(CNS)小胶质细胞和星形胶质细胞通过释放神经营养因子、突触重塑等方式不断监视脑实质以维持神经系统稳态。而毒素或者环境因素影响导致小胶质细胞慢性和过度激活增加了炎症风险,被认为是PD的驱动因素之一[25, 26]。本研究显示Parkin敲除和MPTP处理均可导致慢性星形胶质细胞和小胶质细胞活化,而Parkin敲除能够加速小鼠星型胶质细胞和小胶质细胞激活,显著促进小鼠运动功能障碍和黑质多巴胺能神经元的丢失以及α-syn积累,表明了Parkin基因的缺陷增加了神经元的脆弱性,对神经炎症的发展也起到重要的促进作用,成为促PD发展的重要因素[27]。
目前许多研究都集中在Parkin泛素化与小胶质细胞激活及NLRP3炎症小体之间的关系。如有报道称NLRP3炎症小体是神经元中Parkin的底物,而炎症小体的激活与包括PD在内的几种神经退行性疾病的进展有关[28, 29]。在LPS处理的小鼠中,Parkin缺乏加剧了小胶质NLRP3炎症小体的过度活化,促进了运动功能障碍和多巴胺能神经元的丢失[30]。在此基础上,本研究发现在MPTP-PD小鼠模型中通过Parkin缺失导致的线粒自噬体功能障碍,可以引发NLRP3炎症小体过度激活并参与了PD发展。另外,有研究结果显示小鼠受到急性(竭力运动诱导)或慢性(mtDNA 突变诱导)线粒体氧化应激,在没有 Parkin 或 PINK1 的情况下,STING的遗传性失活这种能够拯救了黑质中的多巴胺能神经元变性,并降低中的促炎细胞因子[31]。本研究同时也验证了Parkin缺失后,PINK1/Parkin信号通路受阻可导致未被降解的dsDNA(mtDNA)增加触发cGAS-STING信号途径的激活,表明Parkin靶向STING通路可能是减轻PD神经炎症病理的潜在治疗策略。
Parkin和神经炎症的发生密切相关,但Parkin依赖的线粒体自噬和PD神经炎症发生之间的关系未完全探索[32]。 因此,本研究使用Parkin敲除小鼠并通过腹腔注射神经毒素MPTP,将Parkin敲除引发的PD病理表型和炎症表型表现出来,还将Parkin缺失带来的PD病理和炎症反应与Parkin依赖的线粒体自噬联系起来。更重要的是我们通过对PINK1/Parkin线粒体自噬信号通路的研究以及胞质mtDNA的变化,很好地解释了Parkin依赖的线粒体自噬在PD中的重要作用,建立的Parkin基因敲除鼠早发性PD模型也反向验证了Parkin介导线粒体自噬障碍时能够加剧PD神经炎症发展。
总之,本研究表明Parkin通过介导PINK1/Parkin线粒体自噬信号通路调控帕金森病以及神经炎症发展的机制,为Parkin缺陷遗传性帕金森病的治疗奠定基础。
基金资助
国家自然科学基金(82371382,81771381);安徽省重点研究与开发计划项目(2022e07020030,2022e07020032);安徽省自然科学基金(2308085MH256);蚌埠医科大学重大科技项目孵育计划(2021byfy002);国家级大学生创新创业训练项目资助 (202310367005,202310367043)
Supported by National Natural Science Foundation of China (82371382,81771381).
参考文献
- 1. Tolosa E, Garrido A, Scholz SW, et al. Challenges in the diagnosis of Parkinson's disease[J]. Lancet Neurol, 2021, 20(5): 385-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jankovic J, Tan EK. Parkinson's disease: etiopathogenesis and treatment[J]. J Neurol Neurosurg Psychiatry, 2020, 91(8): 795-808. [DOI] [PubMed] [Google Scholar]
- 3. Kwon EH, Steininger J, Scherbaum R, et al. Large-fiber neuropathy in Parkinson's disease: a narrative review[J]. Neurol Res Pract, 2024, 6(1): 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Araújo B, Caridade-Silva R, Soares-Guedes C, et al. Neuro-inflammation and Parkinson's disease-from neurodegeneration to therapeutic opportunities[J]. Cells, 2022, 11(18): 2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pajares M, Rojo AI, Manda G, et al. Inflammation in Parkinson's disease: mechanisms and therapeutic implications[J]. Cells, 2020, 9(7): 1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moradi Vastegani S, Nasrolahi A, Ghaderi S, et al. Mitochondrial dysfunction and Parkinson's disease: pathogenesis and therapeutic strategies[J]. Neurochem Res, 2023, 48(8): 2285-308. [DOI] [PubMed] [Google Scholar]
- 7. Ryan BJ, Hoek S, Fon EA, et al. Mitochondrial dysfunction and mitophagy in Parkinson's: from familial to sporadic disease[J]. Trends Biochem Sci, 2015, 40(4): 200-10. [DOI] [PubMed] [Google Scholar]
- 8. Chen CY, Yang C, Wang J, et al. Melatonin ameliorates cognitive deficits through improving mitophagy in a mouse model of Alzheimer's disease[J]. J Pineal Res, 2021, 71(4): e12774. [DOI] [PubMed] [Google Scholar]
- 9. Zuo L, Motherwell MS. The impact of reactive oxygen species and genetic mitochondrial mutations in Parkinson's disease[J]. Gene, 2013, 532(1): 18-23. [DOI] [PubMed] [Google Scholar]
- 10. O’Hanlon ME, Tweedy C, Scialo F, et al. Mitochondrial electron transport chain defects modify Parkinson's disease phenotypes in a Drosophila model[J]. Neurobiol Dis, 2022, 171: 105803. [DOI] [PubMed] [Google Scholar]
- 11. Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease[J]. Neuron, 2015, 85(2): 257-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kamienieva I, Duszyński J, Szczepanowska J. Multitasking guardian of mitochondrial quality: Parkin function and Parkinson's disease[J]. Transl Neurodegener, 2021, 10(1): 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu J, Liu WJ, Lu YQ, et al. Piperlongumine restores the balance of autophagy and apoptosis by increasing BCL2 phosphorylation in rotenone-induced Parkinson disease models[J]. Autophagy, 2018, 14(5): 845-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Barazzuol L, Giamogante F, Brini M, et al. PINK1/parkin mediated mitophagy, Ca2+ signalling, and ER-mitochondria contacts in Parkinson's disease[J]. Int J Mol Sci, 2020, 21(5): 1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eiyama A, Okamoto K. PINK1/Parkin-mediated mitophagy in mammalian cells[J]. Curr Opin Cell Biol, 2015, 33: 95-101. [DOI] [PubMed] [Google Scholar]
- 16. Wasner K, Smajic S, Ghelfi J, et al. Parkin deficiency impairs mitochondrial DNA dynamics and propagates inflammation[J]. Mov Disord, 2022, 37(7): 1405-15. [DOI] [PubMed] [Google Scholar]
- 17. Geisler S, Holmström KM, Skujat D, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1[J]. Nat Cell Biol, 2010, 12(2): 119-31. [DOI] [PubMed] [Google Scholar]
- 18. Ren ZL, Wang CD, Wang T, et al. Ganoderma lucidum extract ameliorates MPTP-induced Parkinsonism and protects dopaminergic neurons from oxidative stress via regulating mitochondrial function, autophagy, and apoptosis[J]. Acta Pharmacol Sin, 2019, 40(4): 441-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chia SJ, Tan EK, Chao YX. Historical perspective: models of Parkinson's disease[J]. Int J Mol Sci, 2020, 21(7): E2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu JJ, Li YY, Zhu H, et al. Therapeutic function of a novel rat induced pluripotent stem cell line in a 6-OHDA-induced rat model of Parkinson's disease[J]. Int J Mol Med, 2022, 50(6): 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Noda S, Sato S, Fukuda T, et al. Loss of Parkin contributes to mitochondrial turnover and dopaminergic neuronal loss in aged mice[J]. Neurobiol Dis, 2020, 136: 104717. [DOI] [PubMed] [Google Scholar]
- 22. Li J, Yang DM, Li ZP, et al. PINK1/Parkin-mediated mitophagy in neurodegenerative diseases[J]. Ageing Res Rev, 2023, 84: 101817. [DOI] [PubMed] [Google Scholar]
- 23. Zhang XW, Feng N, Liu YC, et al. Neuroinflammation inhibition by small-molecule targeting USP7 noncatalytic domain for neurodegenerative disease therapy[J]. Sci Adv, 2022, 8(32): eabo0789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jurcau A, Andronie-Cioara FL, Nistor-Cseppento DC, et al. The involvement of neuroinflammation in the onset and progression of Parkinson's disease[J]. Int J Mol Sci, 2023, 24(19): 14582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Quinn PMJ, Moreira PI, Ambrósio AF, et al. PINK1/PARKIN signalling in neurodegeneration and neuroinflammation[J]. Acta Neuropathol Commun, 2020, 8(1): 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Iorio R, Celenza G, Petricca S. Multi-target effects of β‑caryo-phyllene and carnosic acid at the crossroads of mitochondrial dysfunction and neurodegeneration: from oxidative stress to microglia-mediated neuroinflammation[J]. Antioxidants, 2022, 11(6): 1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sun K, Jing XZ, Guo JC, et al. Mitophagy in degenerative joint diseases[J]. Autophagy, 2021, 17(9): 2082-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Panicker N, Kam TI, Wang H, et al. Neuronal NLRP3 is a parkin substrate that drives neurodegeneration in Parkinson's disease[J]. Neuron, 2022, 110(15): 2422-37.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee E, Hwang I, Park S, et al. MPTP-driven NLRP3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration[J]. Cell Death Differ, 2019, 26(2): 213-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zengeler KE, Lukens JR. Taking the parkin brakes off of neuronal NLRP3 drives inflammasome activation and neurodegeneration in Parkinson's disease[J]. Neuron, 2022, 110(15): 2356-8. [DOI] [PubMed] [Google Scholar]
- 31. Sliter DA, Martinez J, Hao L, et al. Parkin and PINK1 mitigate STING-induced inflammation[J]. Nature, 2018, 561(7722): 258-62. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32. Eldeeb MA, Thomas RA, Ragheb MA, et al. Mitochondrial quality control in health and in Parkinson's disease[J]. Physiol Rev, 2022, 102(4): 1721-55. [DOI] [PubMed] [Google Scholar]