

Abstract
Sixteen thio/semicarbazide-based benzyloxy derivatives (BT1-BT16) were synthesized and evaluated for their inhibitory activities against monoamine oxidases (MAOs). Most compounds showed better inhibitory activity against MAO-B than against MAO-A. BT1, BT3, and BT5 showed the greatest inhibitory activity with an identical IC50 value of 0.11 µM against MAO-B, followed by BT6 and BT7 (IC50 = 0.12 µM) and BT2 (IC50 = 1.68 µM). The selectivity index of BT5 was the highest (363.64) for MAO-B, whereas that of BT1 was 88.73. BT1 and BT5 were reversible MAO-B inhibitors, based on the results of dialysis experiments. In inhibition kinetics, BT1 and BT5 were competitive MAO-B inhibitors with Ki values of 0.074 ± 0.0020 and 0.072 ± 0.0079 µM, respectively. Additionally, in the in-vitro parallel artificial membrane penetration assay, BT1 and BT5 crossed the blood–brain barrier. Cytotoxicity and possible neuroprotective effects of the lead compounds were assessed using IMR 32 cells. Levels of the antioxidant superoxide dismutase, catalase, and glutathione peroxidase in IMR 32 cells were increased by pretreatment with lead compounds. Five lead molecules (BT1, BT3, BT5, BT6, and BT7) were used for the docking studies. A significant pi–pi interaction with Tyr 326 was observed and molecular dynamics studies were performed for the most promising BT1-MAO-B complex. These results suggested that BT1 and BT5 could be used therapeutically for the treatment of various neurodegenerative diseases.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-82771-3.
Keywords: Benzyloxy benzene-based thio/Semicarbazide derivatives, Monoamine oxidase, Kinetics, Reversibility, Molecular dynamics
Subject terms: Biochemistry, Biological techniques, Drug discovery
Introduction
Neurodegenerative disorders (NDDs) are a broad category of neurological conditions that induce the progressive and selective loss of neurons, compromising the cognitive and physical capacities of the affected individuals1. As a component of age-related disorders, NDDs are becoming a major public health problem2. Monoamine oxidases (MAOs) are enzymes affixed to the outer mitochondrial membrane in the central and peripheral regions. Two expressed isoforms, MAO-A and MAO-B, are encoded by distinct genes3. MAO-B metabolizes dopamine, which is taken up by glial cells and released into the synaptic cleft. Glial cell numbers increase with age and the activity of MAO-B increases, as expected in NDDs. MAO-B drugs prevent dopamine catabolism, reduce brain MAO-B activity, increase dopamine levels in synaptic clefts, and enhance the dopamine signaling pathway4. Physicians primarily treat patients with early Parkinson’s disease (PD) and motor deficits using MAO-B inhibiting agents. MAO-B inhibitors, including safinamide, selegiline, and rasagiline, are used to treat PD5. There is much hope, particularly for safinamide, which is being evaluated in clinical trials. A dual strategy could pave the way for new treatments in cases of non-motor dysfunctions6. Numerous studies have documented that molecules with isatin, chromone, coumarin, pyrazoline, and piperidine nuclei may be reversible MAO-B inhibitors7–11.
Safinamide is an aminoamide derivative discovered by Neuron Pharmaceuticals (Bresso, Italy). The drug was approved to treat PD by the Food and Drug Administration in 2017; it is a potential inhibitor of MAO-B12,13. Following several preclinical and clinical studies, safinamide was identified as an intriguing therapy to control the progression of PD. Safinamide is currently used as the first-line treatment in patients with mid-to late-stage PD, together with levodopa and a peripheral decarboxylase inhibitor14. Safinamide, the only known reversible MAO-B inhibitor, acts through two different mechanisms to influence the dopaminergic and glutamatergic systems15. Within the active site of the enzyme, (S)-safinamide forms a non-covalent bond with the flavin adenine dinucleotide (FAD) cofactor.
In FAD, the fluorobenzene ring is positioned away from the site, and the amine group is oriented towards the flavin group of FAD. Furthermore, the fluorobenzene ring interacts with the hydrophobic residues Ile199, Phe168, and Leu167, whereas the carbonyl group of safinamide is oriented towards the hydrophobic residues Tyr60, Phe343, and Tyr 398. The interaction of safinamide with MAO-B at the hydrophobic active site has been confirmed, which is a structural prerequisite for enzyme inhibition16. The long-range binding of safinamide to human MAO-B allows it to attach to the active site cavities, which provides insight into the selectivity of the inhibitors. Moreover, unlike other cavity-spanning MAO-B-selective inhibitors, they have polar substituents that align their binding mode to the hydrophilic region in front of the flavin to generate H-bond interactions with both conserved water molecules and protein residues17. Human clinical trials in their late stages have indicated that safinamide has not yet reached its clinical endpoint. However, no unfavorable side effects have been observed. Owing to the overproduction of biogenic amines, the design and lead optimization of this scaffold is a successful drug design method for treating many neurodegenerative illnesses18.
Occasionally, the benzyloxy group is employed as a protective group in chemical synthesis19. Oxadiazolones, indolalkylamines, safinamide, caffeines, benzofurans, α-tetralones, β-nitrostyrene, benzoquinones, coumarins, indoles, chromones, and chromanone analogs are among the moieties that are linked to benzyloxy MAO-B inhibitors20. Structure-affinity relationship (SAR) data and docking simulations have demonstrated that the benzyloxy group at the meta position of the coumarin ring provides affirmative hydrophobic interactions, whereas substitution at the para position of the ring offers strong negative steric hindrance, lowering the inhibition potential as an outcome21. The importance of the conformationally flexible terminal phenyl group has been confirmed by the addition of the benzyloxy group to various frameworks, thus serving as a MAO-B inhibitor. This was further strengthened by the addition of a conformationally malleable -CH2O- bridge22,23. There is greater MAO-B inhibition when the benzyloxy pharmacophore serves as a substitute at the para position of the B ring of chalcones than when it is substituted at the ortho position24. Therefore, the ability of a compound to increase MAO-B inhibitory activity makes it a more promising molecule for additional inhibition investigations in the presence of a benzyloxy pharmacophore.
The semicarbazone and thiosemicarbazone imine units have been shown to have anti-protozoal, anti-convulsant, anti-cancer, urease, and monoamine oxidase inhibitory properties25,26. Thiosemicarbazides are molecules that can pass through the blood–brain barrier, making them promising candidates for use as medications to treat neurological disorders27. The distinct terminal hydrazine unit of the thiosemicarbazide/methylthiosemicarbazide groups allows them to react with carbonyl compounds. Data from recent pharmacophore-based studies have shown that MAO-A/B inhibitors contain one or more hydrophobic rings, hydrogen bond acceptor (HBA), and hydrogen bond donor (HBD). Moreover, substantial MAO inhibition occurs when HBA and HBD are present in thiosemicarbazone units with different aryl or heteroaryl units. A recent study suggested the presence of methylthiosemicarbazones as a new class of reversible MAO-B inhibitors for neurodegenerative diseases28. The discovery that a methyl group can be added to the terminal amino of 5-phenyl substituted thiosemicarbazones to enhance their lipophilicity led to the discovery that these novel adducts could be used as MAO-B inhibitors for the treat neurological disorders29. Studies on distinct aryl thiosemicarbazones under varying conditions for electron withdrawal and donation have reported the inhibition of multiple monoamine oxidase30.
In this study, benzyloxybenzene-derived thio/semicarbazones were synthesized and developed as a new class of MAO-B inhibitors. Our modified design strategy utilizes the benzyloxy benzene moiety from safinamide, which is a MAO-B inhibitor, by giving the four different benzyloxy substitutions including 4-(3-fluorobenzyloxy), 4-benzyloxy, 3-benzyloxy and 2-benzyloxy moieties, along with the utilization of thiosemicarbazone, methyl thiosemicarbazone, semicarbazone, and phenyl thiosemicarbazone. The corresponding benzyloxy benzene derived thio/semicarbazones represent a new class of MAO-B inhibitors (Fig. 1). We confirmed the formation of compounds by spectral characterization and performed inhibition, enzyme kinetics, reversibility, and parallel artificial membrane permeability assay (PAMPA). The lead molecule was further used in docking and molecular dynamics (MD) simulation studies.
Fig. 1.

Design strategy of benzyloxy benzene-based thio/semicarbazide derivatives.
Materials and methods
Chemicals
For the synthesis of 4-(3-fluorobenzyloxy)benzaldehyde, 4-benzyloxy benzaldehyde, 3-benzyloxy benzaldehyde, and 2-benzyloxy benzaldehyde were purchased from TCI Chemicals (Chennai, Tamil Nadu, India). Thiosemicarbazide, methyl thiosemicarbazide, semicarbazide hydrochloride, phenyl thiosemicarbazide, recombinant human MAO-A, MAO-B, benzylamine, kynuramine, safinamide mesylate salt, pargyline, clorgyline, and toloxatone were purchased from Sigma-Aldrich (St. Louis, MO, USA). Sodium phosphate (dibasic and monobasic anhydrous) was purchased from Daejung Chemicals and Metals Co. Ltd. (Siheung, Korea). The DiaEasy™ Dialysis kit (6–8 kDa) was purchased from BioVision (St. Louis, MA, USA)31. Melting point (Digital Melting Point Apparatus, Amtech India, Ambala Cantt Haryana, India, 133001), 1H nuclear magnetic resonance (NMR), 13C NMR (Bruker Advance Neo 500 MHz NMR spectrometer, Billerica, MA, USA), and mass spectra (Waters Xevo G2-XS QTOF, Milford, MA, USA) were recorded to characterize the synthesized compounds.
Chemical synthesis
Benzyloxybenzene-derived thio-/semicarbazide-based derivatives were synthesized in a microwave reactor Monowave 50 (Anton Paar, Graz, Austria) by reacting benzyloxybenzene derivatives (1 equivalent) with thio-/semicarbazide derivatives (1.3 equivalent) using ethanol as the solvent and glacial acetic acid as the catalyst. The preparation was maintained at 80 ֯C for 5 min for thio derivatives and between 80 and 120 ֯C for 6 min for semicarbazide derivatives (Scheme 1). The solid product obtained as a precipitate was washed with ethanol and the dried product was purified by flash chromatography. Thin-layer chromatography (TLC) was performed to confirm each reaction using precoated TLC plates (silica gel, 60–120 #). Hexane: ethyl acetate (2:1) was used as the solvent system for the thio derivatives, whereas hexane: ethyl acetate (1:2) was used for the semicarbazide derivatives.
Scheme 1.

Synthesis of the BT series of compounds. Reaction condition: For thio derivatives: MW at 80 ֯C (5 min), For semicarbazide derivatives: MW at 80 ֯C to 120 ֯C (5–6 min).
2-(4-((3-fluorobenzyl)oxy)benzylidene)hydrazine-1-carbothioamide (BT1)
Pale yellow; Percentage Yield: 86%; MP: 154–156 °C; Rf: 0.67. 1H NMR (500 MHz, CDCl3) δ: 10.03 (s, 1 H, -NH-N=), 7.88 (s, 1 H, -N = CH-), 7.60–7.57 (d, 2 H, J = 15 Hz, Ar-H), 7.37–7.32 (m, 1 H, J = 25 Hz, Ar-H), 7.20–7.17 (m, 2 H, J = 15 Hz, Ar-H), 7.15–7.13 (d, 1 H, J = 10 Hz, Ar-H), 7.04-7.00 (m, 1 H, J = 20 Hz, Ar-H), 6.98–6.95 (d, 2 H, J = 15 Hz, Ar-H), 6.50 (s, 1 H, H2N-C = S), 5.14 (s, 2 H, -O-CH2-Ph). 13C NMR (125 MHz, CDCl3) δ: 178.02, 163.98, 162.02, 160.59, 143.97, 138.98, 138.92, 130.28, 130.21, 129.20, 126.12, 122.73, 115.14, 69.26, 29.70. Molecular formula: C15H14FN3OS, (ESI) Calculated MW = 303.35, observed MW = 303.3498.
2-(4-((3-fluorobenzyl)oxy)benzylidene)-N-methylhydrazine-1-carbothioamide (BT2)
White; Percentage Yield: 82%; MP: 135–137 °C; Rf: 0.70. 1H NMR (500 MHz, CDCl3) δ: 9.23 (s, 1 H, -NH-N=), 7.73 (s, 1 H, -N = CH-), 7.60–7.57 (d, 2 H, J = 15 Hz, Ar-H), 7.43 (s, 1 H, CH3-NH-), 7.37–7.33 (m, 1 H, J = 20 Hz, Ar-H), 7.19–7.18 (d, 1 H, J = 5 Hz, Ar-H), 7.16–7.14 (d, 1 H, J = 10 Hz, Ar-H), 7.08-7.00 (m, 1 H, J = 40 Hz, Ar-H), 6.99–6.96 (d, 2 H, J = 15 Hz, Ar-H), 5.10 (s, 2 H, -O-CH2-Ph), 3.26–3.25 (d, 3 H, CH3). 13C NMR (125 MHz, CDCl3) δ: 178.26, 164.08, 162.12, 160.46, 142.17, 139.09, 132.11, 130.36, 129.03, 126.40, 122.80, 115.33, 114.23, 69.36, 32.01, 31.28. Molecular formula: C16H16FN3OS, (ESI) Calculated MW = 317.38, observed MW = 317.3799.
2-(4-((3-fluorobenzyl)oxy)benzylidene)hydrazine-1-carboxamide (BT3)
White; Percentage Yield: 81%; MP: 1110 –112 °C; Rf: 0.58. 1H NMR (500 MHz, DMSO) δ: 10.12 (s, 1 H, -N = CH-), 7.79 (s, 1 H, -NH-N=), 7.67–7.65 (d, 2 H, J = 10 Hz, Ar-H), 7.46–7.42 (m, 1 H, J = 10 Hz, Ar-H), 7.30–7.27 (m, 2 H, J = 15 Hz, Ar-H), 7.18–7.14 (m, 1 H, J = 10 Hz, Ar-H), 7.04–7.02 (d, 2 H, J = 10 Hz, Ar-H), 6.43 (s, 2 H, H2N-C = O), 5.16 (s, 2 H, -O-CH2-Ph). 13C NMR (125 MHz, DMSO) δ: 163.05, 161.12, 158.74, 156.74, 139.77, 138.93, 130.40, 127.93, 127.73, 123.42, 114.83, 114.41, 114.22, 114.05, 68.31. Molecular formula: C15H14FN3O2, (ESI) Calculated MW = 287.28, observed MW = 287.2798.
2-(4-((3-fluorobenzyl)oxy)benzylidene)-N-phenylhydrazine-1-carbothioamide (BT4)
Pale brown; Percentage Yield: 85%; MP: 1124 –126 °C; Rf: 0.63. 1H NMR (500 MHz, DMSO) δ: 11.72 (s, 1 H, -NH-N=), 10.04 (s, 1 H, Ph-NH-C = S), 8.12 (s, 1 H, -N = CH-), 7.87–7.85 (d, 2 H, J = 10 Hz, Ar-H), 7.59–7.57 (d, 2 H, J = 10 Hz, Ar-H), 7.46–7.42 (m, 1 H, J = 20 Hz, Ar-H), 7.38–7.35 (m, 2 H, J = 15 Hz, Ar-H), 7.31–7.28 (m, 2 H, J = 15 Hz, Ar-H), 7.21–7.18 (m, 1 H, J = 15 Hz, Ar-H), 7.16–7.14 (m, 1 H, J = 10 Hz, Ar-H), 7.08–7.06 (d, 2 H, J = 10 Hz, Ar-H), 5.19 (s, 2 H, -O-CH2-Ph). 13C NMR (125 MHz, DMSO) δ: 175.55, 163.07, 161.13, 159.58, 142.63, 139. 66, 139.02, 130.43, 130.36, 129.21, 127.91, 126.85, 125.66, 125.10, 123.48, 123.46, 114.92, 114.63, 114.47, 114.10, 68.39. Molecular formula: C21H18FN3OS, (ESI) Calculated MW = 379.45, observed MW = 379.4498.
2-(4-(benzyloxy)benzylidene)hydrazine-1-carbothioamide (BT5)
Pale yellow; Percentage Yield: 86%; MP: 132–134 °C; Rf: 0.51. 1H NMR (500 MHz, CDCl3) δ: 9.86 (s, 1 H, -NH-N=), 7.85 (s, 1 H, -N = CH-), 7.59–7.57 (d, 2 H, J = 10 Hz, Ar-H), 7.42–7.38 (m, 4 H, J = 20 Hz, Ar-H), 7.37–7.32 (m, 1 H, J = 25 Hz, Ar-H), 7.08–7.07 (d, 1 H, J = 5 Hz, Ar-H), 6.99–6.97 (d, 2 H, J = 10 Hz, Ar-H), 6.43 (s, 1 H, H2N-C = S), 5.09 (s, 2 H, -O-CH2-Ph). 13C NMR (125 MHz, CDCl3) δ: 178.14, 161.02, 143.98, 136.40, 132.09, 129.23, 128.81, 128.75, 128.41, 128.27, 127.54, 125.89, 115.34, 115.24, 70.20. Molecular formula: C15H15N3OS, (ESI) Calculated MW = 285.36, observed MW = 285.3599.
2-(4-(benzyloxy)benzylidene)-N-methylhydrazine-1-carbothioamide (BT6)
Pale yellow; Percentage Yield: 81%; MP: 139–141 °C; Rf: 0.55. 1H NMR (500 MHz, DMSO) δ: 11.36 (s, 1 H, -NH-N=), 8.43–8.42 (d, 1 H, J = 10 Hz, Ar-H), 8.00 (s, 1 H, -N = CH-), 7.76–7.73 (d, 2 H, J = 15 Hz, Ar-H), 7.47–7.45 (d, 2 H, J = 10 Hz, Ar-H), 7.41–7.38 (m, 2 H, J = 15 Hz, Ar-H), 7.35–7.32 (m, 1 H, -NH-), 7.07–7.04 (d, 2 H, J = 15 Hz, Ar-H), 5.15 (s, 2 H, -O-CH2-Ph), 3.02–3.01 (d, 3 H, CH3). 13C NMR (125 MHz, DMSO) δ: 177.42, 159.57, 141.44, 136.66, 128.66, 128.32, 127.79, 127.64, 126.96, 114.87, 69.24, 30.65. Molecular formula: C16H17N3OS, (ESI) Calculated MW = 299.39, observed MW = 299.3899.
2-(4-(benzyloxy)benzylidene)hydrazine-1-carboxamide (BT7)
White; Percentage Yield: 78%; MP: 117–119 °C; Rf: 0.56. 1H NMR (500 MHz, DMSO) δ: 10.11 (s, 1 H, -N = CH-), 7.79 (s, 1 H, -NH-N=), 7.66–7.64 (d, 2 H, J = 10 Hz, Ar-H), 7.46–7.44 (d, 2 H, J = 10 Hz, Ar-H), 7.40–7.38 (m, 2 H, J = 10 Hz, Ar-H), 7.35–7.31 (m, 1 H, J = 20 Hz, Ar-H), 7.03-7.00 (d, 2 H, J = 15 Hz, Ar-H), 6.43 ( s, 2 H, H2N-C = O), 5.13 (s, 2 H, -O-CH2-Ph). 13C NMR (125 MHz, DMSO) δ: 158.99, 156.75, 138.99, 136.76, 128.32, 127.91, 127.76, 127.62, 127.55, 114.81, 69.18. Molecular formula: C15H15N3O2, (ESI) Calculated MW = 269.29, observed MW = 269.2898.
2-(4-(benzyloxy)benzylidene)-N-phenylhydrazine-1-carbothioamide (BT8)
Pale brown; Percentage Yield: 79%; MP: 130–132 °C; Rf: 0.50. 1H NMR (500 MHz, CDCl3) δ: 10.28 (s, 1 H, -NH-N=), 9.17 (s, 1 H, Ph-NH-C = S), 7.91 (s, 1 H, -N = CH-), 7.66–7.64 (d, 2 H, J = 10 Hz, Ar-H), 7.61–7.59 (d, 2 H, J = 10 Hz, Ar-H), 7.42–7.37 (m, 6 H, J = 25 Hz, Ar-H), 7.34–7.31 (t, 1 H, J = 15 Hz, Ar-H), 7.25–7.22 (t, 1 H, J = 15 Hz, Ar-H), 7.00-6.98 (d, 2 H, J = 10 Hz, Ar-H), 5.09 (s, 2 H, -O-CH2-Ph). 13C NMR (125 MHz, CDCl3) δ: 175.41, 160.88, 143.11, 137.89, 136.35, 129.15, 128.78, 128.72, 128.67, 128.19, 127.48, 126.15, 125.97, 124.59, 115.27, 70.13. Molecular formula: C21H19N3OS, (ESI) Calculated MW = 361.46, observed MW = 361.4598.
2-(3-(benzyloxy)benzylidene)hydrazine-1-carbothioamide (BT9)
Pale yellow; Percentage Yield: 79%; MP: 127–129 °C; Rf: 0.63. 1H NMR (500 MHz, CDCl3) δ: 10.18 (s, 1 H, -NH-N=), 7.89 (s, 1 H, -N = CH-), 7.44–7.42 (d, 3 H, J = 10 Hz, Ar-H), 7.41–7.38 (m, 3 H, J = 15 Hz, Ar-H), 7.34–7.31 (m, 2 H, J = 15 Hz, Ar-H), 7.30–7.28 (d, 2 H, J = 10 Hz, Ar-H), 7.20–7.19 (d, 2 H, J = 5 Hz, Ar-H), 7.03–7.01 (d, 1 H, J = 10 Hz, Ar-H), 6.55 (s, 1 H, H2N-C = S), 5.08 (s, 2 H, -O-CH2-Ph). 13C NMR (125 MHz, CDCl3) δ: 159.18, 143.99, 136.65, 134.42, 130.02, 128.72, 128.21, 127.57, 120.98, 117.66, 113.01, 70.28. Molecular formula: C15H15N3OS, (ESI) Calculated MW = 285.36, observed MW = 285.3598.
2-(3-(benzyloxy)benzylidene)-N-methylhydrazine-1-carbothioamide (BT10)
Pale yellow; Percentage Yield: 80%; MP: 145–146 °C; Rf: 0.65. 1H NMR (500 MHz, CDCl3) δ: 9.79 (s, 1 H, -NH-N=), 7.80 (s, 1 H, -N = CH-), 7.47–7.45 (d, 3 H, J = 10 Hz, Ar-H), 7.44–7.41 (m, 2 H, J = 15 Hz, Ar-H), 7.40–7.38 (d, 1 H, J = 10 Hz, Ar-H), 7.35–7.33 (d, 1 H, J = 10 Hz, Ar-H), 7.32–7.28 (m, 1 H, -NH-), 7.20–7.18 (d, 1 H, J = 10 Hz, Ar-H), 7.02-7.00 (m, IH, J = 10 Hz, Ar-H), 5.09 (s, 2 H, -O-CH2-Ph), 3.26–3.25 (d, 3 H, CH3). 13C NMR (125 MHz, CDCl3) δ: 178.29, 159.14, 142.26, 136.63, 134.71, 129.93, 128.66, 128.15, 127.53, 120.70, 117.06, 112.93, 70.25, 31.19. Molecular formula: C16H17N3OS, (ESI) Calculated MW = 299.39, observed MW = 299.3899.
2-(3-(benzyloxy)benzylidene)hydrazine-1-carboxamide (BT11)
White; Percentage Yield: 80%; MP: 102–104 °C; Rf: 0.52. 1H NMR (500 MHz, DMSO) δ: 10.27 (s, 1 H, -N = CH-), 7.82 (s, 1 H, -NH-N=), 7.48–7.46 (m, 3 H, J = 10 Hz, Ar-H), 7.41–7.38 (m, 2 H, J = 15 Hz, Ar-H), 7.35–7.33 (d, 1 H, J = 10 Hz, Ar-H), 7.32–7.28 (m, 1 H, J = 20 Hz, Ar-H), 7.22–7.20 (d, 1 H, J = 10 Hz, Ar-H), 7.00-6.97 (m, 1 H, J = 15 Hz, Ar-H), 6.55 (s, 2 H, H2N-C = O), 5.14 (s, 2 H, -O-CH2-Ph). 13C NMR (125 MHz, DMSO) δ: 158.59, 156.74, 139.06, 136.96, 136.19, 129.60, 128.35, 127.79, 127.75, 119.85, 115.95, 111.44, 69.25. Molecular formula: C15H15N3O2, (ESI) Calculated MW = 269.29, observed MW = 269.2899.
2-(3-(benzyloxy)benzylidene)-N-phenylhydrazine-1-carbothioamide (BT-12)
Pale yellow; Percentage Yield: 77%; MP: 156–158 °C; Rf: 0.62. 1H NMR (500 MHz, CDCl3) δ: 10.40 (s, 1 H, -NH-N=), 9.15 (s, 1 H, Ph-NH-C = S), 7.93 (s, 1 H, -N = CH-), 7.64–7.63 (d, 2 H, J = 5 Hz, Ar-H), 7.44–7.40 (m, 3 H, J = 20 Hz, Ar-H), 7.39–7.35 (m, 3 H, J = 20 Hz, Ar-H), 7.33–7.29 (t, 3 H, J = 20 Hz, Ar-H), 7.27–7.21 (m, 2 H, J = 30 Hz, Ar-H), 7.04–7.02 (m, 1 H, J = 10 Hz, Ar-H), 5.09 (s, 2 H, -O-CH2-Ph). 13C NMR (125 MHz, CDCl3) δ: 175.83, 159.19, 143.10, 137.80, 136.66, 134.54, 130.03, 128.89, 128.71, 128.18, 127.53, 126.41, 124.82, 120.93, 117.54, 113.05, 70.28. Molecular formula: C21H19N3OS, (ESI) Calculated MW = 361.46, observed MW = 361.4599.
2-(2-(benzyloxy)benzylidene)hydrazine-1-carbothioamide (BT13)
Pale yellow; Percentage Yield: 86%; MP: 125–127 °C; Rf: 0.48. 1H NMR (500 MHz, CDCl3) δ: 9.84 (s, 1 H, -NH-N=), 8.34 (s, 1 H, -N = CH-), 7.80–7.79 (d, 1 H, J = 5 Hz, Ar-H), 7.41–7.39 (d, 4 H, J = 10 Hz, Ar-H), 7.36–7.32 (m, 2 H, J = 20 Hz, Ar-H), 7.14 (s, 1 H, Ar-H), 6.99–6.93 (m, 2 H, J = 30 Hz, Ar-H), 6.56 (s, 1 H, H2N-C = S), 5.06 (s, 2 H, -O-CH2-Ph). 13C NMR (125 MHz, CDCl3) δ: 178.23, 157.62, 140.32, 136.29, 131.99, 128.77, 128.23, 127.43, 126.59, 121.96, 121.13, 112.68, 70.45. Molecular formula: C15H15N3OS, (ESI) Calculated MW = 285.36, observed MW = 285.3598.
2-(2-(benzyloxy)benzylidene)-N-methylhydrazine-1-carbothioamide (BT14)
Pale yellow; Percentage Yield: 76%; MP: 142–144 °C; Rf: 0.66. 1H NMR (500 MHz, CDCl3) δ: 9.36 (s, 1 H, -NH-N=), 8.23 (s, 1 H, -N = CH-), 7.83–7.81 (d, 1 H, J = 10 Hz, Ar-H), 7.42–7.39 (d, 5 H, J = 15 Hz, Ar-H), 7.37–7.32 (m, 2 H, J = 25 Hz, Ar-H), 7.00-6.97 (t, 2 H, J = 15 Hz, Ar-H), 5.09 (s, 2 H, -O-CH2-Ph), 3.21–3.20 (d, 3 H, CH3). 13C NMR (125 MHz, CDCl3) δ: 178.34, 157.46, 138.70, 136.31, 131.67, 128.75, 128.24, 127.42, 126.44, 122.17, 121.12, 112.67, 70.49, 31.14. Molecular formula: C16H17N3OS, (ESI) Calculated MW = 299.39, observed MW = 299.3898.
2-(2-(benzyloxy)benzylidene)hydrazine-1-carboxamide (BT15)
White; Percentage Yield: 77%; MP: 119–121 °C; Rf: 0.56. 1H NMR (500 MHz, CDCl3) δ: 9.04 (s, 1 H, -N = CH-), 8.19 (s, 1 H, -NH-N=), 7.81–7.80 (d, 1 H, J = 5 Hz, Ar-H), 7.41–7.36 (m, 4 H, J = 25 Hz, Ar-H), 7.33–7.28 (m, 2 H, J = 25 Hz, Ar-H), 6.98–6.96 (d, 1 H, J = 10 Hz, Ar-H), 6.95–6.92 (t, 1 H, J = 15 Hz, Ar-H), 5.19 (s, 2 H, H2N-C = O), 5.09 (s, 2 H, -O-CH2-Ph). 13C NMR (125 MHz, CDCl3) δ: 157.55, 156.98, 137.88, 136.59, 130.94, 128.66, 128.12, 127.38, 126.21, 122.89, 121.10, 112.69, 70.46. Molecular formula: C15H15N3O2, (ESI) Calculated MW = 269.29, observed MW = 269.2897.
2-(2-(benzyloxy)benzylidene)-N-phenylhydrazine-1-carbothioamide (BT16)
Pale yellow; Percentage Yield: 80%; MP: 145–147 °C; Rf: 0.67. 1H NMR (500 MHz, CDCl3) δ: 9.39 (s, 1 H, -NH-N=), 9.18 (s, 1 H, Ph-NH-C = S), 8.31 (s, 1 H, -N = CH-), 7.88–7.87 (d, 1 H, J = 5 Hz, Ar-H), 7.66–7.64 (d, 2 H, J = 10 Hz, Ar-H), 7.42–7.32 (m, 8 H, Ar-H), 7.25–7.21 (m, 1 H, J = 20 Hz, Ar-H), 7.03–6.99 (m, 2 H, J = 20 Hz, Ar-H), 5.12 (s, 2 H, -O-CH2-Ph). 13C NMR (125 MHz, CDCl3) δ: 175.73, 157.67, 139.11, 137.92, 136.21, 132.07, 128.78, 128.76, 128.31, 127.40, 126.46, 126.05, 124.29, 121.90, 121.22, 112.74, 70.56. Molecular formula: C21H19N3OS, (ESI) Calculated MW = 361.46, observed MW = 361.4598.
Enzyme assays and kinetics
The activities of MAO-A and MAO-B were assayed using kynuramine (0.06 mM) and benzylamine (0.30 mM), respectively, by continuously measuring the absorbance changes at 316 and 250 nm, respectively32. Km values were determined by assaying five substrate concentrations and analyzing Lineweaver–Burk (LB) plots.
Inhibition studies of MAO-A and MAO-B
As an initial screening step for inhibition study and inhibition kinetics, residual activity was analyzed by measuring the absorbance change in the presence of 10 µM of the inhibitor. IC50 values were determined for potential compounds with residual activity < 80% using GraphPad Prism software 5 (San Diego, CA, USA) [27]. The limit of IC50 was set to 40 µM. The selectivity index (SI) for MAO-B was calculated by IC50 of MAO-A the IC50 of MAO-B33. The inhibition types of the leading compounds, BT1 and BT5, for MAO-B were determined at three inhibitor concentrations (~ 1/2×, 1×, and 2× IC50) as well as five different substrate concentrations ranging from 0.0375 to 0.6 µM34. Inhibition patterns and Ki values were determined by comparing the Lineweaver–Burk lots with their secondary plots, respectively35. Toloxatone, clorgyline, safinamide, and pargyline were used as the reference compounds.
Reversibility studies
The reversibilities of BT1 and BT5 for MAO-B were evaluated by comparing the undialyzed (AU) and dialyzed (AD) residual activities at a concentration of about 2-times the IC50 after pre-incubation for 30 min prior to measurement. Two reference inhibitors were used: a reversible MAO-B inhibitor, safinamide mesylate, and an irreversible MAO-B inhibitor pargyline36.
PAMPA for blood–brain barrier (BBB) permeation study
Early drug research employed a PAMPA to predict passive drug transcellular permeability across the BBB. A sandwich-like structure was established in the PAMPA using a 96-well microtiter plate and 96-well filter plate (IPVH, 125 μm thick filter, 0.45 μm pore; Millipore, Billerica, MA, USA). The plates were submerged in 0.1 mL of n-dodecane. Stock solutions of drug samples in Dimethyl sulfoxide (DMSO) were prepared at 10 mM dosages and stored at 0 °C before use. The stock solution was further diluted in a buffer (pH 7.4) before being added to a 96-well filter plate. This allowed for the attainment of a final sample concentrations of 0.01, 0.1, 0.5, and 1 mM and the restriction of the DMSO content to 1% (v/v). Two hundred microliters of a pH 7.4 buffer were added to the acceptor well and the final diluted solutions were transferred in 270 µL aliquots to the donor wells. In order for the plates to create a “sandwich,” the donor plate had to be precisely positioned over the acceptor filter plate. The donor plate contains an artificial lipid barrier in the middle, an aqueous recipient on top, and an aqueous donor carrying an analyte on bottom. Via the lipid membrane, the test material diffuses from the donor well and into the acceptor well. At the time of the breach, the “sandwich” was allegedly intact. Ultraviolet spectroscopy was used to determine drug concentrations in the donor, recipient, and reference wells. The degree of penetration was calculated as follows37:
Log Pe = -ln [1-CA /Equilibrium]/A× (1/VD + 1/VA) × t.
where Pe is permeability (cm. s− 1), CA is the receptor concentration, A is the area of effective filtering (4.8 cm2), VD is the donor volume (mL), VA is the acceptor volume (mL), t is the time of incubation (s), and Equilibrium= (CD×VD + CA×VA)/(VD+VA).
In-vitro cytotoxicity and antioxidant assays
The cytotoxicity of the lead molecules BT1, BT3, BT5, BT6, and BT7 were analyzed using the 3-(4, 5-Dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay. The antioxidant effects were evaluated by determining the activities of superoxide dismutase (SOD), glutathione peroxidase (GPX), and catalase (CAT) in IMR 32 cells38–40. The detailed procedure is described in the Supporting Information.
MAO-B prediction
The recently created web program MAO-B-pred (https://mao-b-pred.streamlit.app/) was used to evaluate the bioactivity of each compound against MAO-B. The expected values for the compounds were compared with those of standard MAO-B inhibitors (Safinamide and Pargyline)41.
Molecular docking
The promising compounds, BT1, BT3, BT5, BT6, and BT7 were subjected to molecular docking analysis using the Schrödinger Glide docking module within the hMAO-B enzyme (PDB ID: 2V5Z) -crystallized with safinamide42. The protein crystal structure was generated using a three-step Protein Preparation Wizard, which includes preprocessing, optimization, and protein energy minimization phases43. LigPrep was used to prepare the ligands for the docking studies. A receptor grid-generating module and grid file were generated44. The extra-precision (XP) mode was employed for ligand docking.
MD simulation
MD studies were performed for the top-docking postures (lowest negative scores) of the lead compound BT1 by applying the Desmond MD simulation program and NVIDIA Quadro 6000 graphics processing unit45. Using the default values for the systems under investigation, 1000 frame trajectories were created to investigate interactions between the protein and ligand. The MD generation was 100 ns, with coordinates captured at 100 ps. Prior research has indicated the value of additional information for MD investigations, such as box type, estimates of long- and short-range interactions, and thermostat and barometer settings46–49.
Results
Chemistry
Benzyloxybenzene-derived thio/semicarbazones were synthesized (Scheme 1) using different substituted benzyloxybenzene derivatives and thiosemicarbazide or semicarbazide derivatives via a Schiff base mechanism in the microwave reactor. Sixteen derivatives were synthesized and characterized using1H nuclear magnetic resonance (NMR)13, C NMR (Bruker Advance Neo 500 MHz NMR spectrometer, Billerica, MA, USA), and mass spectroscopy (Waters Xevo G2-XS QTOF, Milford, MA, USA). The imine hydrogen and the –NH–group are detected in the deshielded region between 7.64 and 11.36 ppm. The aromatic hydrogen is in the region from 6.96 to 7.87 ppm. The benzyloxy-CH2 proton shielded region was between 5.09 and 5.19 ppm. The extensively deshielded region of C = S is between 170 and 180 ppm. The deshielded region at 150 to 160 ppm includes the C = O group of semicarbazone. –CH = N is in the region from 130 to 150 ppm. The most shielded region of benzyloxy-CH2 is in the range of 60 to 80 ppm (Supplementary Materials, Figures S1-S16).
Inhibition studies of MAO-A and MAO-B
Among the 16 compounds tested at 10 µM, 13 compounds showed low residual activity of < 50% for MAO-B, while five compounds showed low residual activity of < 50% for MAO-A (Table 1). Compounds BT1, BT3, and BT5 showed the greatest inhibitory ability against MAO-B with an identical IC50 value of 0.11 µM, followed by BT6 and BT7 (IC50 = 0.12 µM) and BT2 (IC50 = 1.68 µM). Compound BT9 exhibited the highest MAO-A inhibition, with an IC50 of 2.32 µM. Fourteen compounds showed more effective inhibition of MAO-B than MAO-A, with high SI values for MAO-B: BT5 with 363.64, followed by BT6 and BT7 with 333.33. The potencies of BT1, BT3, and BT5 were comparable to those of the reference compound pargyline (IC50 = 0.11 µM).
Table 1.
Inhibitions of MAO-A and MAO-B by thio/semicarbazide-based benzyloxy derivatives.
| Compound | Residual activity (%) | IC50 (µM) | SI | ||
|---|---|---|---|---|---|
| MAO-A (10 µM) | MAO-B (10 µM) | MAO-A | MAO-B | ||
| BT1 | 46.06 ± 1.16 | -10.16 ± 1.66 | 9.76 ± 0.44 | 0.11 ± 0.003 | 88.73 |
| BT2 | 88.81 ± 4.16 | 16.63 ± 0.39 | > 40 | 1.68 ± 0.027 | 23.81 |
| BT3 | 45.79 ± 0.78 | 3.57 ± 2.53 | 8.44 ± 2.02 | 0.11 ± 0.018 | 76.73 |
| BT4 | 97.42 ± 3.64 | 54.41 ± 1.44 | > 40 | 12.25 ± 1.19 | 3.27 |
| BT5 | 124.33 ± 0.59 | 0.91 ± 0.64 | > 40 | 0.11 ± 0.023 | 363.64 |
| BT6 | 91.09 ± 7.61 | 3.81 ± 1.41 | > 40 | 0.12 ± 0.0043 | 333.33 |
| BT7 | 81.55 ± 2.53 | 13.43 ± 1.78 | > 40 | 0.12 ± 0.0035 | 333.33 |
| BT8 | 60.33 ± 0.18 | 38.06 ± 3.77 | 17.65 ± 1.91 | 8.22 ± 1.36 | 2.15 |
| BT9 | 10.00 ± 0.28 | 13.64 ± 2.75 | 2.32 ± 0.066 | 2.27 ± 0.27 | 1.02 |
| BT10 | 36.60 ± 7.34 | 39.58 ± 1.47 | 4.53 ± 0.67 | 4.75 ± 0.41 | 0.95 |
| BT11 | 66.61 ± 4.80 | 33.05 ± 0.20 | 32.87 ± 8.21 | 1.55 ± 0.092 | 21.21 |
| BT12 | 2.13 ± 0.23 | -0.86 ± 1.22 | 4.49 ± 0.42 | 0.46 ± 0.12 | 9.70 |
| BT13 | 65.98 ± 3.91 | 46.59 ± 8.04 | > 40 | 9.65 ± 0.95 | 4.15 |
| BT14 | 64.94 ± 8.93 | 24.68 ± 1.84 | > 40 | 5.77 ± 1.02 | 6.93 |
| BT15 | 169.11 ± 8.33 | 92.03 ± 5.64 | > 40 | > 40 | – |
| BT16 | 98.26 ± 0.82 | 64.29 ± 4.04 | > 40 | 15.39 ± 1.58 | 2.60 |
| Toloxatone | – | – | 1.65 ± 0.094 | > 40 | 0.041 |
| Safinamide | – | – | > 40 | 0.019 ± 0.0019 | 2105.26 |
| Clorgyline | – | – | 0.008 ± 0.001 | 2.43 ± 0.71 | 0.0033 |
| Pargyline | – | – | 2.15 ± 0.23 | 0.11 ± 0.01 | 19.55 |
Results are expressed as the mean ± SD of triplicate experiments. Selectivity index (SI) values are expressed for MAO-B as compared with MAO-A.
The compound series was classified into four subtypes, first (BT1-BT4), second (BT5-BT8), third (BT9-BT12), and fourth (BT13-BT16), consisting of 4(3-benzyloxy), 4-benzyloxy, 3-benzyloxy, and 2-benzyloxy, respectively, of the parent structure connected with thio/semicarbazide units connected to the imine linker.
For MAO-B inhibition, overall, the 4(3-benzyloxy) and 4-benzyloxy groups with thio/semicarbazide substituents showed high inhibitory activity, followed by the inhibitory activity in the order of the other series. The fourth subtype showed lower inhibitory activity than the other subtypes. When comparing the leading substances, BT1 and BT5, there was little difference in the IC50 levels depending on whether fluorine was bound. However, when BT2 and BT6 were compared, enzyme inhibition occurred when fluorine was not bound to the structure. When benzene was bound to the thiourea moiety, the overall inhibitory activity was low for all compounds, except BT12. Considering the IC50 of BT3, BT7, and BT11 confirmed that semicarbazide had inhibitory activity similar to that of thiosemicarbazide. Based on the inhibitory profiles of MAO-B by thio/semicarbazide-based benzyloxy imines, some SARs were observed (Fig. 2).
Fig. 2.

Structure–activity relationship of thio/semicarbazide-based benzyloxy derivatives.
Enzyme kinetics and inhibition studies
Enzyme kinetics and inhibition studies were performed using five substrate concentrations and three inhibitor concentrations of BT1 or BT5. In the Lineweaver–Burk plot, BT1 and BT5 appeared to be competitive MAO-B inhibitors (Fig. 3A and C, respectively). Through the secondary plots, Ki values were 0.074 ± 0.0020 and 0.072 ± 0.0079 μm, respectively (Fig. 3B and D).
Fig. 3.

Lineweaver–Burk plots for MAO-B inhibitions by BT1 and BT5 (A and C), and their secondary plots (B and D) of the slopes vs. inhibitor concentrations. Results are expressed as the mean ± SD of triplicate experiments.
Reversibility studies
The reversibility of MAO-B inhibition by BT1 and BT5 was analyzed by dialysis after 30 min of pre-incubation. The concentrations of BT1, BT5, safinamide, and pargyline used were 2-times their IC50 values (0. 22, 0. 22, 0.038, and 0.22 µM, respectively). Recovery patterns were compared using undialyzed (AU) and dialyzed (AD) relative activities. inhibition of MAO-B by BT1 and BT5 were recovered from 25.88 to 98.91% and from 27.83 to 64.52%, respectively. The recovery values of the compounds were similar those of safinamide (reversible type, from 23.06 to 52.85%) and could be distingushed from pargyline (irreversible type, 25.17–29.65%). These results suggest that BT1 and BT5 are reversible MAO-B inhibitors (Fig. 4).
Fig. 4.

Recovery of MAO-B inhibition by BT1 and BT5 in residual activities measeured before and after dialysis. Concentrations used were 2-times of their IC50 values: BT1, BT5, and pargyline, 0.22 µM; safinamide, 0.038 µM.
PAMPA for permeation of BBB
Compounds BT1 and BT5 showed a significant degree of permeation and central nervous system (CNS) bioavailability, according to the PAMPA data. BT1 and BT5 showed near Pe values that were greater than 4.0 × 10− 6 cm/s, indicating a higher BBB penetration rate of 4.08 × 10− 6 cm/s and 4.03 × 10− 6 cm/s, respectively (Table 2). Brain penetration is required for the delivery of medications that act on the CNS50. This experiment used the PAMPA-BBB assay to assess the brain permeability of all derivatives. Using the formula and the effective permeability (Log Pe) of the material, the penetration depth can be calculated. Pe values for compounds assessed as probably non-BBB penetrating (CNS-) were < 2.0 × 10− 6 cm/s, whereas compounds classified as conceivably permeating (CNS+) had Pe values > 4.0 × 10− 6 cm/s.
Table 2.
BBB assay of key compounds by PAMPA.
| Compound | Experimental, Pe (×10− 6 cm/s) | Prediction |
|---|---|---|
| BT1 | 4.08 ± 0.12 | CNS+ |
| BT5 | 4.03 ± 0.09 | CNS+ |
| Selegiline | 5.69 ± 0.04 | CNS+ |
Pe (10− 6 cm/s) > 4.00: CNS+ (high permeation); Pe (10− 6 cm/s) < 2.00: CNS− (low permeation); Pe (10− 6 cm/s) from 2.00 to 4.00: CNS± (permeation uncertain). Results are expressed as the mean ± SD of triplicate experiments.
In-vitro cytotoxicity and antioxidant assays
MTT cytotoxicity assay
The cytotoxicity of BT1, BT3, BT5, BT6, and BT7 for IMR 32 cells was evaluated using the MTT colorimetric assay that is based on cellular metabolic activity. It relies on the reduction of MTT by mitochondrial dehydrogenase in viable cells to form an insoluble formazan product, which can be quantified by measuring the absorbance. The percentage cell viability was calculated by comparing the optical density (OD) values of the treated samples with those of the control. The respective IC50 values of BT1, BT3, BT5, BT6, and BT7 of 47.21, 49.71, 42.13, 49.41, and 42.00 µM, equivalent to 14.32, 14.28, 12.02, 14.79, and 11.31 µg/mL, respectively. These results indicate that the compounds are non-cytotoxic and may promote the proliferation of IMR-32 cells at lower concentrations when used at a volume of 100 µL (Fig. 5).
Fig. 5.

MTT viability assay of the lead compounds BT1, BT3, BT5, BT6, and BT7 using IMR 32 cells.
SOD activity
The SOD activities of BT1, BT3, BT5, BT6, and BT7 at various concentrations were compared to standard values in IMR 32 cells (Fig. 6). These values were graphed. Each compound’s activity at different concentrations is represented by distinct bars or markers, with the x-axis indicating concentration (µg/ml) and the y-axis representing activity (measured values). Although it remained below the normal values, BT1 showed a progressive increase in activity with increasing concentrations, starting at 0.119 at 50 µg/ml and reaching 0.407 at 500 µg/ml. Of all the compounds, BT3 showed the lowest activity. The results were much lower than the standard, beginning at 0.018 at 50 µg, and reaching only 0.159 at 500 µg/ml. Low activity was observed for BT5, which increased slightly with concentration (from 0.027 at 50 µg/ml to 0.182 at 500 µg/ml), but remained far below the standard. BT6 showed moderate activity with values ranging from 0.025 at 50 µg to 0.177 at 500 µg/ml. Among all the compounds, BT7 showed the highest activity. The values ranged from 0.128 at 50 µg/ml to 0.398 at 500 µg/ml, almost matching and often surpassing the standard. The relative efficacy of various drugs in the SOD assay was better understood based on this thorough analysis, which also highlighted BT1 and BT7 as promising candidates for further investigation.
Fig. 6.

Superoxide dismutase (SOD) levels observed in different concentrations of BT1, BT3, BT5, BT6, and BT7 treatment of IMR 32 cells. Std denotes standard.
GPX activity
The OD at 450 nm for IMR 32 cells in the GPX assay are plotted in Fig. 7. OD values were obtained for BT1, BT3, BT5, BT6, and BT7, and the standard at various concentrations. The standard OD values increased with increasing concentration, suggesting a dose-dependent response. This was anticipated because increasing standard concentrations are expected to increase GPX activity, which in turn should increase OD readings. Plotting the data with the OD at 450 nm on the y-axis and the concentration on the x-axis revealed a smooth, upward-sloping curve of the standard that showed an increase in GPX activity in a dose-dependent manner. BT1 and BT7 displayed bars that were roughly identical to the standard, but consistently lower, indicating reduced GPX activity. BT3 and BT5 exhibited relatively low GPX activities. At 200 µg/ml, BT6 exhibited an increase in activity, further investigation is required to clarify whether the observation was an anomaly.
Fig. 7.

Glutathione peroxidase (GPX) levels observed in different concentrations of BT1, BT3, BT5, BT6, and BT7 treatment against the IMR 32 cells. Std denotes standard.
CAT activity
The amount of catalase activity (measured as OD at 450 nm) in IMR 32 cells at different concentrations of BT1, BT3, BT5, BT6, and BT7 relative to a standard was assessed. The raw data clearly demonstrated that CAT activity increased as the concentration increased from 50 to 500 µg/ml. The graph displays the CAT activity on the y-axis and drug concentration (µg/ml) on the x-axis, with different colored bars denoting various compounds and standard (Fig. 8). The standard (blue bar) showed the strongest catalytic response, with the largest and steepest increase in CAT activity with increasing concentration. The test compounds with the most encouraging results were BT1 and BT7, followed by BT3, BT5 and BT6, which showed the least activity, indicating that they were the least effective at inducing CAT.
Fig. 8.

Catalase (CAT) levels observed in different concentrations of BT1, BT3, BT5, BT6, and BT7 treatment against IMR 32 cells. Std denotes standard.
MAO-B prediction
The bioactivity of the compounds against MAO-B was examined using the recently created web application MAO-B-pred. For all compounds, we computed the observed and predicted pIC50 values, and compared them with those of the reference drugs (Safinamide and Pargyline). Using the formula pIC50 = –log (IC50 × 10− 6), the observed pIC50 was determined. The data from Table 3 indicate the comparable observed and predicted pIC50 values for all compounds (BT1-BT16).
Table 3.
Observed and predicted pIC50 of compounds.
| Compound | Observed pIC50 | Predicted pIC50 |
|---|---|---|
| BT1 | 6.958607 | 5.6771 |
| BT2 | 5.774691 | 5.7083 |
| BT3 | 6.958607 | 6.0073 |
| BT4 | 4.911864 | 5.7875 |
| BT5 | 6.958607 | 5.6053 |
| BT6 | 6.920819 | 5.5937 |
| BT7 | 6.920819 | 5.8976 |
| BT8 | 5.085128 | 5.736 |
| BT9 | 5.643974 | 5.5839 |
| BT10 | 5.323306 | 5.5678 |
| BT11 | 5.809668 | 5.806 |
| BT12 | 6.337242 | 5.719 |
| BT13 | 5.015473 | 5.5711 |
| BT14 | 5.238824 | 5.556 |
| BT15 | 4.39794 | 5.8575 |
| BT16 | 4.812761 | 5.7304 |
| Safinamide | 7.721246 | 6.9317 |
| Pargyline | 6.958607 | 5.4266 |
Molecular docking
Based on the results of the enzyme assay, BT1, BT3, BT5, BT6, and BT7 had the highest efficacy and selectivity for human MAO-B. The top five lead compounds were subjected to docking studies to investigate their binding patterns and assess the impact of structural alterations on their inhibitory activity against MAO-B using PDB ID: 2V5Z. All five compounds showed significant docking scores for MAO-B, suggesting their selectivity for MAO-B, corroborating the findings of the MAO-B activity (Table 4). Among the five compounds, the highest docking score was for BT1 (-11.105 kcal/mol). Figures 9 and 10 present the two- and three-dimensional interactions of the lead molecules with MAO-B, respectively. The binding interactions between BT1 and MAO-B are shown in Fig. 9. Each of the five molecules exhibited polar interactions with Gln 206 and hydrophobic interactions with Tyr435, Tyr60, Tyr326, Tyr188, Tyr398, Phe343, Phe99, Phe103, Phe168, Pro102, Pro104, Leu164, Leu167, Leu171, Cys172, Ile198, Ile199, Ile316, and Trp119 (Supplementary Information). The aromatic benzene ring of benzyloxy benzaldehyde displays π–π stacking with Tyr 326 for BT1, BT6 and BT7. For BT3, the human MAO-B docking postures showed the attachment of the NH2 group of the semicarbazide to Gly 434 via a water molecule and hydrogen bonding. The oxygen atom in the carbonyl group of BT3 and BT7 is linked to Tyr188. BT1, BT3, BT7, BT6 and BT5 display hydrophobic interaction in the binding site with surrounding 5 Å residues. This indicates the likelihood that the reversible MAO-B inhibitor, safinamide, also exhibits such types of interactions.
Table 4.
XP docking scores of the lead compounds to the MAO-B active site.
| Compound | Docking score (kcal/mol) |
|---|---|
| BT1 | − 11.105 |
| BT3 | − 10.861 |
| BT7 | − 10.583 |
| BT6 | − 10.143 |
| BT5 | − 9.646 |
| Safinamide | − 12.671 |
Fig. 9.
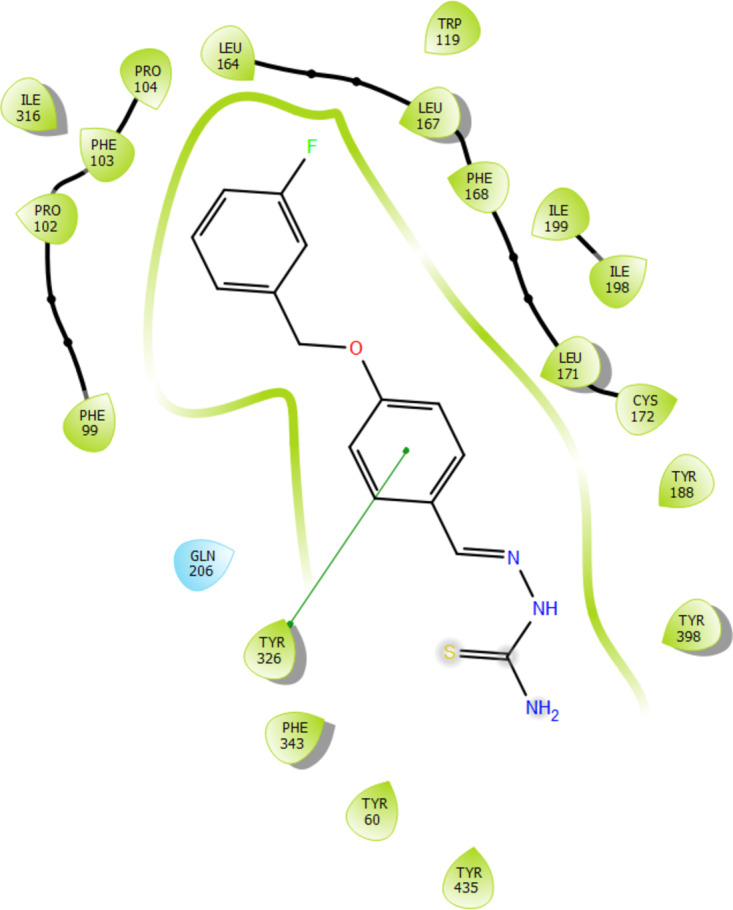
Two-dimensional interaction of compound BT1 to the active site of MAO-B (PDB ID: 2V5Z).
Fig. 10.

Three-dimensional interaction of compound BT1 to the active site of MAO-B (PDB ID: 2V5Z).
MD simulation
MD simulation data provide an overall picture of the interactions between a protein and its ligands. The approach can be used to represent all biological systems as dynamic networks of molecular interactions. An MD simulation analysis of the docked BT1-MAO-B complex was performed over the course of 100 ns. The generated MD trajectories were thoroughly analyzed using key metrics, including root mean square deviation (RMSD), root mean square fluctuation (RMSF), and radius of gyration (RGyr). These investigations are essential for understanding the intricate interactions between BT1 and the corresponding enzyme-binding sites and to provide insights into the underlying mechanisms influencing the observed selectivity.
RMSD
In MD simulations, RMSD is a key metric that shows changes in the structural conformation of the protein backbone over time during system equilibration. Low and steady RMSD values reflect the stability of the protein structure. Throughout the 100 ns MD simulation, BT1 bound to MAO-B. The RMSD of MAO-B with BT1 ranged from 2.00 to 3.50 Å. Over the course of 45 to 55 ns, the protein Cα atoms in this complex showed notable fluctuations, with the RMSD peaking at 4.50 Å (Fig. 11A).
Fig. 11.

MD simulation studies of BT1-MAO-B complex. The panels display root mean square deviation (RMSD) (A), root mean square fluctuation (RMSF) (B), two-dimensional interaction diagram of the complex (C), protein–ligand contact histogram of MD trajectory (D), and time-dependent RGyr of BT1-hMAO-B (E).
RMSF and protein–ligand contacts
In MD simulations, RMSF is frequently used to evaluate changes in the internal chains of proteins caused by ligands. It is conceivable that there is greater binding between the ligand and protein when the RMSF value is low at residues in the active site or where the ligand interacts with the receptor protein. On the other hand, lower values indicate the predominance of secondary structures, such as alpha-helices and beta-sheets, and evidences of structural stability, despite higher values (peaks) revealing the existence of twists, loosely bound loops, and terminal ends, indicative of structural flexibility51. The flexibility of the amino acid residues in the MAO-B active site when BT1 was bound was evaluated using RMSF analysis. The RMSF values of each interacting residue in the BT1-hMAO-B complex ranged from 0.3 Å to 2.0 Å. Greater fluctuations were observed between Gly425 and Ser465, which belong to the loop region (Fig. 11B). The data indicate that the essential amino acids for the binding site are Leu171 and Cys172, with hydrogen bond interactions of 68% and 48% respectively (Fig. 11C). Contact of BT1 with the Glu84, Leu88, Phe99, Pro104, Pro105, Leu164, Phe168, Leu171, Cys172, Tyr188, Ile198, Ile199, Gly205, Gln206, Ile316, Tyr326, Tyr398, Ser433, and Tyr435 residues was evident throughout the 100 ns simulation period (Fig. 11D). The amino acid Leu171 demonstrates 75% hydrophobic interactions, 20% hydrogen bond interactions, and almost 2% water bridges. Cys172 displays approximately 60% hydrogen bond interactions and 10% water bridges. Ile198, Ile199, Ile316, and Tyr326 have hydrophobic interactions. Overall, the MD simulation findings confirm that BT1 and human MAO-B have stable and suitable binding interactions.
RGyr
The compactness or globularity of a protein–ligand complex is determined by its RGyr. A longer or more open structural shape of a protein is often indicated by a higher RGyr value, whereas a more compact structure is indicated by a lower RGyr value52. A graph was created by plotting the RGyr values of BT1 to human MAO-B versus simulation time (Fig. 11E). The values ranging from 4.65 to 5.10 Å indicate the consistent compact behavior during the 100 ns timeframe.
Conclusion
Sixteen thio/semicarbazide-based benzyloxy compounds were synthesized and their inhibitory effectiveness against human MAOs was assessed. Each of the sixteen derivatives displayed higher MAO-B inhibition compared to safinamide standard. BT1 ((E)-2-(4-((3-fluorobenzyl)oxy)benzylidene)hydrazine-1-carbothioamide) and BT5 ((E)-2-(4-(benzyloxy)benzylidene)hydrazine-1-carbothioamide) most potently inhibit MAO-B, with an IC50 value of 0.11 ± 0.003 and 0.11 ± 0.023 µM, respectively. BT1 and BT5 are thus potentially highly competitive and reversible MAO-B inhibitors. The BBB penetration of BT1 and BT5 was similar to that of CNS drugs. In addition, BT1 and BT5 are neuroprotective and non-cytotoxic. The docking data revealed a pi–pi interaction in the aromatic benzene moiety of benzyloxy benzaldehyde of BT1 and the binding interaction of BT1 with MAO-B. MD simulation data provide novel insights into the binding mode, clearly indicating the stability of BT1-MAO-B complex. Our collective findings suggest that BT1 and BT5 could be used therapeutically to treat various neurodegenerative diseases.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Author contributions
Conceptualization, BM and HK; synthesis, NC, SK, and MP; biological tests, JL, PP, DGT, STS, TGA and MP; Computer simulation study, SK, and NC.; writing—original draft preparation, NC SK, and BM; writing—review and editing, BM and HK; supervision, BM and HK; funding acquisition, HK. All authors have read and agreed to the published version of the manuscript.
Data availability
Data is provided within the manuscript or supplementary information files.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Namitha Chandran and Jiseong Lee contributed equally to this work.
Contributor Information
Hoon Kim, Email: hoon@sunchon.ac.kr.
Bijo Mathew, Email: bijomathew@aims.amrita.edu, Email: bijovilaventgu@gmail.com.
References
- 1.Parambi, D. G. T. et al. Gene therapy approach with an emphasis on growth factors: theoretical and clinical outcomes in neurodegenerative diseases. In Molecular Neurobiologyvol. 59 (Springer US, 2022). [DOI] [PMC free article] [PubMed]
- 2.Rodríguez-Enríquez, F. et al. Novel coumarin-pyridazine hybrids as selective MAO-B inhibitors for the Parkinson’s disease therapy. Bioorg. Chem.104, 104203 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Abid, S. M. A. et al. Sulfonyl hydrazones derived from 3-formylchromone as non-selective inhibitors of MAO-A and MAO-B: synthesis, molecular modelling and in-silico ADME evaluation. Bioorg. Chem.75, 291–302 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Chen, J. J. & Swope, D. M. Clinical pharmacology of rasagiline: a novel, second-generation propargylamine for the treatment of Parkinson disease. J. Clin. Pharmacol.45, 878–894 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Jost, W. H. A critical appraisal of MAO-B inhibitors in the treatment of Parkinson’s disease. J. Neural Transm. 129, 723–736 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pagonabarraga, J., Tinazzi, M., Caccia, C. & Jost, W. H. The role of glutamatergic neurotransmission in the motor and non-motor symptoms in Parkinson’s disease: clinical cases and a review of the literature. J. Clin. Neurosci.90, 178–183 (2021). [DOI] [PubMed] [Google Scholar]
- 7.Kumar, S. et al. Development of Isopropyl-Tailed Chalcones as a new class of selective MAO-B inhibitors for the treatment of Parkinson’s disorder. ACS Omega. 8, 6908–6917 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ipe, R. S. et al. A Concise Review of the Recent Structural Explorations of Chromones as MAO-B Inhibitors: Update from 2017 to 2023 (2023). [DOI] [PMC free article] [PubMed]
- 9.Koyiparambath, V. P. et al. Deciphering the detailed structure–activity relationship of coumarins as Monoamine oxidase enzyme inhibitors—An updated review. Chem. Biol. Drug Des.98, 655–673 (2021). [DOI] [PubMed] [Google Scholar]
- 10.Jayan, J. et al. Piperidine: a versatile heterocyclic ring for developing monoamine oxidase inhibitors. ACS Omega8, 37731–37751 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rangarajan, T. M. & Mathew, B. Recent updates on Pyrazoline Derivatives as promising candidates for neuropsychiatric and neurodegenerative disorders. Curr. Top. Med. Chem.21, 2695–2714 (2021). [DOI] [PubMed] [Google Scholar]
- 12.Pevarello, P. et al. Synthesis and anticonvulsant activity of a new class of 2- [(arylalkyl)amino]alkanamide derivatives. J. Med. Chem.41, 579–590 (1998). [DOI] [PubMed] [Google Scholar]
- 13.Ahmad, A. M. Potential pharmacokinetic interactions between antiretrovirals and medicinal plants used as complementary and African traditional medicines. Biopharm. Drug Dispos.28, 135–143 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Wasan, H., Singh, D. & KH, R. Safinamide in neurological disorders and beyond: evidence from preclinical and clinical studies. Brain Res. Bull.168, 165–177 (2021). [DOI] [PubMed] [Google Scholar]
- 15.Abbruzzese, G., Barone, P., Lopiano, L. & Stocchi, F. The current evidence for the use of safinamide for the treatment of parkinson’s disease. Drug Des. Dev. Ther.15, 2507–2517 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ledesma, A. E., Catalán, C. A. N. & Brandán, S. A. A combined theoretical and experimental study on the structure, vibrational, and electronic properties of antiparkinsonian drug safinamide. SN Appl. Sci.2, 1–16 (2020). [Google Scholar]
- 17.Binda, C. et al. Structures of human monoamine oxidase B complexes with selective noncovalent inhibitors: Safinamide and coumarin analogs. J. Med. Chem.50, 5848–5852 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Joy, M., Mathew, B. & Sudarsanakumar, C. Structural features of Safinamide: a combined Hirshfeld surface analysis & quantum chemical treatment. Chem. Data Collect.17–18, 404–414 (2018). [Google Scholar]
- 19.Pisani, L. et al. A twenty-year journey exploring coumarin-based derivatives as bioactive molecules. Front. Chem.10, 1–8 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sudevan, S. T. et al. Revealing the role of the benzyloxy pharmacophore in the design of a new class of monoamine oxidase-B inhibitors. Arch. Pharm. (Weinheim)355, 2200084 (2022). [DOI] [PubMed] [Google Scholar]
- 21.Catto, M. et al. Structural insights into monoamine oxidase inhibitory potency and selectivity of 7-substituted coumarins from ligand- and target-based approaches. J. Med. Chem.49, 4912–4925 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Rao, Y. J., Abhijit, K., Mallikarjun, G. & Hemasri, Y. Design and synthesis of novel benzyloxy-tethered-chromone-carboxamide derivatives as potent and selective human monoamine oxidase-b inhibitors. Chem. Pap. 75, 703–716 (2021). [Google Scholar]
- 23.Knez, D. et al. Indoles and 1-(3-(benzyloxy)benzyl)piperazines: reversible and selective monoamine oxidase B inhibitors identified by screening an in-house compound library. Bioorg. Chem.119, 105581 (2022). [DOI] [PubMed] [Google Scholar]
- 24.Singh, A. K. et al. Exploration of a new class of monoamine oxidase B inhibitors by assembling benzyloxy pharmacophore on halogenated chalcones. Chem. Biol. Drug Des.102, 271–284 (2023). [DOI] [PubMed] [Google Scholar]
- 25.Qazi, S. U. et al. Semicarbazone derivatives as urease inhibitors: synthesis, biological evaluation, molecular docking studies and in-silico ADME evaluation. Bioorg. Chem.79, 19–26 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Qazi, S. U. et al. Semicarbazones, thiosemicarbazone, thiazole and oxazole analogues as monoamine oxidase inhibitors: synthesis, characterization, biological evaluation, molecular docking, and kinetic studies. Bioorg. Chem.115, 105209 (2021). [DOI] [PubMed] [Google Scholar]
- 27.Mitkov, J., Kondeva-Burdina, M., Peikova, L., Georgieva, M. & Zlatkov, A. Design, synthesis and evaluation of semi- and thiosemicarbazides containing a methylxanthine moiety with in vitro neuroprotective and MAO-B inhibitory activities. Biotechnol. Biotechnol. Equip.36, 486–499 (2022). [Google Scholar]
- 28.Mathew, G. E. et al. Development of methylthiosemicarbazones as new reversible monoamine oxidase-B inhibitors for the treatment of Parkinson’s disease. J. Biomol. Struct. Dyn.39, 4786–4794 (2021). [DOI] [PubMed] [Google Scholar]
- 29.Mathew, G. E. et al. Inhibitions of monoamine oxidases and acetylcholinesterase by 1-methyl, 5-phenyl substituted thiosemicarbazones: synthesis, biochemical, and computational investigations. Process. Biochem.99, 246–253 (2020). [Google Scholar]
- 30.Mathew, B. et al. Selected aryl thiosemicarbazones as a new class of multi-targeted monoamine oxidase inhibitors. Medchemcomm9, 1871–1881 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oh, J. M. et al. Novel class of chalcone oxime ethers as potent monoamine oxidase-B and acetylcholinesterase inhibitors. Molecules25 (2020). [DOI] [PMC free article] [PubMed]
- 32.Mathew, B. et al. Enzyme inhibition assays for monoamine oxidase. Methods Mol. Biol.2761, 329–336 (2024). [DOI] [PubMed] [Google Scholar]
- 33.Baek, S. C. et al. Rhamnocitrin isolated from Prunus padus var. Seoulensis: a potent and selective reversible inhibitor of human monoamine oxidase A. Bioorg. Chem.83, 317–325 (2019). [DOI] [PubMed] [Google Scholar]
- 34.Baek, S. C. et al. Selective inhibition of monoamine oxidase A by hispidol. Bioorg. Med. Chem. Lett.28, 584–588 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Oh, J. M. et al. Calycosin and 8-O-methylretusin isolated from Maackia amurensis as potent and selective reversible inhibitors of human monoamine oxidase-B. Int. J. Biol. Macromol.151, 441–448 (2020). [DOI] [PubMed] [Google Scholar]
- 36.Lee, H. W. et al. Potent selective monoamine oxidase B inhibition by maackiain, a pterocarpan from the roots of Sophora flavescens. Bioorg. Med. Chem. Lett.26, 4714–4719 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Di, L., Kerns, E. H., Fan, K., McConnell, O. J. & Carter, G. T. High throughput artificial membrane permeability assay for blood–brain barrier. Eur. J. Med. Chem.38, 223–232 (2003). [DOI] [PubMed] [Google Scholar]
- 38.Ameen, N. & Shafi, S. Biochemical and In-Vivo antioxidant parameters for evaluation of memory enchancing activity. Int. J. Pharm. Chem. Biol. Sci.6, 265–270 (2016). [Google Scholar]
- 39.Tiwari, P. C. et al. Biochemical and immunological studies on Protective Effect of Mangiferin in 6-Hydroxydopamine (6-OHDA)-Induced Parkinson’s disease in rats. Ann. Neurosci.28, 137–149 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alqurashi, M. M. et al. Protective effect of sterubin against neurochemical and behavioral impairments in rotenone-induced Parkinson’s disease. Braz. J. Med. Biol. Res57 (2024). [DOI] [PMC free article] [PubMed]
- 41.Kumar, S. et al. Machine learning driven web-based app platform for the discovery of monoamine oxidase B inhibitors. Sci. Rep.14, 1–20 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Son, S. Y. et al. Structure of human monoamine oxidase A at 2.2-Å resolution: The control of opening the entry for substrates/inhibitors. Proc. Natl. Acad. Sci. U. S. A. 105, 5739–5744 (2008). [DOI] [PMC free article] [PubMed]
- 43.Schrödinger Release 2024-2: BioLuminate, Schrödinger (LLC, 2024).
- 44.Girase, R., Ahmad, I., Pawara, R. & Patel, H. Optimizing cardio, hepato and phospholipidosis toxicity of the Bedaquiline by chemoinformatics and molecular modelling approach. SAR QSAR Environ. Res.33, 215–235 (2022). [DOI] [PubMed] [Google Scholar]
- 45.Schrödinger Release 2024-2: Desmond Molecular Dynamics System, Shaw, D. E. & Research New York, NY, Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY (2024).
- 46.Mathew, B. et al. Pharmacophore-based 3D-QSAR Analysis of thienyl chalcones as a new class of human MAO-B inhibitors: investigation of combined quantum chemical and molecular dynamics approach. J. Phys. Chem. B121, 1186–1203 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Kumar, S. et al. Isatin-tethered halogen-containing acylhydrazone derivatives as monoamine oxidase inhibitor with neuroprotective effect. Sci. Rep.141 (14), 1–20 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jayan, J. et al. Development of a new class of monoamine oxidase-B inhibitors by fine-tuning the halogens on the acylhydrazones. ACS Omega8, 47606–47615 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thomas Parambi, D. G. et al. Halogenated class of oximes as a new class of monoamine oxidase-B inhibitors for the treatment of Parkinson’s disease: synthesis, biochemistry, and molecular dynamics study. Comput. Biol. Chem.105 (2023). [DOI] [PubMed]
- 50.Mathew, B. et al. Perspective design of chalcones for the management of CNS disorders: a mini-review. CNS Neurol. Disord. Drug Targets18, 432–445 (2019). [DOI] [PubMed] [Google Scholar]
- 51.Krishnendu, P. R. et al. A structure-based approach to explore novel COX-2 inhibitors using pharmacophore modelling, 3D-QSAR analysis, virtual screening and dynamics simulation study. J. Mol. Struct.1295, 136634 (2024). [Google Scholar]
- 52.Zrieq, R. et al. Tomatidine and patchouli alcohol as inhibitors of SARS-CoV-2 enzymes (3CLpro, PLpro and NSP15) by molecular docking and molecular dynamics simulations. Int. J. Mol. Sci.22 (2021). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data is provided within the manuscript or supplementary information files.