

Abstract
The oxygen-sensitive molybdenum-dependent nitrogenase of Azotobacter vinelandii is protected from oxidative damage by a reversible ‘switch-off’ mechanism1. It forms a complex with a small ferredoxin, FeSII (ref. 2) or the ‘Shethna protein II’3, which acts as an O2 sensor and associates with the two component proteins of nitrogenase when its [2Fe:2S] cluster becomes oxidized4,5. Here we report the three-dimensional structure of the protective ternary complex of the catalytic subunit of Mo-nitrogenase, its cognate reductase and the FeSII protein, determined by single-particle cryo-electron microscopy. The dimeric FeSII protein associates with two copies of each component to assemble a 620 kDa core complex that then polymerizes into large, filamentous structures. This complex is catalytically inactive, but the enzyme components are quickly released and reactivated upon oxygen depletion. The first step in complex formation is the association of FeSII with the more O2-sensitive Fe protein component of nitrogenase during sudden oxidative stress. The action of this small ferredoxin represents a straightforward means of protection from O2 that may be crucial for the maintenance of recombinant nitrogenase in food crops.
Subject terms: Cryoelectron microscopy, Oxidoreductases, X-ray crystallography, Soil microbiology
A small ferredoxin (Shethna protein II) of Azotobacter vinelandii can provide protection from O2 stress that may be crucial for the maintenance of recombinant nitrogenase in food crops.
Main
The element nitrogen is a frequent limiting factor for organismic growth in habitats where organic detritus is not available; for instance, when pioneer organisms settle new habitats such as volcanic rocks or if biomass is systematically removed in an agricultural setting6. Diazotrophic bacteria and archaea then tap into the reservoir of atmospheric N2 and reduce the inert diatomic molecule to bioavailable ammonium, , in the process of biological nitrogen fixation7. With a bond dissociation energy of −946 kJ mol−1, the N2 triple bond is the most stable chemical bond to be broken in any enzymatic reaction, and only a single family of enzymes, the nitrogenases, has evolved to perform this task. Nitrogenases exist in three structurally similar isoforms containing molybdenum, vanadium or only iron at their active site8,9 and catalyse the same reaction with a minimal stoichiometry of
Nitrogenases are two-component metalloenzymes, consisting of a reductase component, or Fe protein that provides electrons through a [4Fe:4S] cluster and is the site of ATP hydrolysis, and the actual dinitrogenase, MFe protein (M = Mo, V, Fe), where catalysis occurs8. Electrons are delivered by Fe protein one at a time, and the two component proteins dynamically form a transient complex for every single transfer, that is, at least eight times for a single turnover10. Complex formation initially triggers the transfer of an electron from P-cluster in its all-ferrous PN state to the active site FeMo cofactor, and only subsequently is the P-cluster again reduced by Fe protein11. The catalysis of N2 reduction by nitrogenase is the focus of active research and has been reviewed extensively elsewhere8,12–16. The complex formed by the MoFe protein and two copies of the dimeric Fe protein has a total molecular mass of 360 kDa (ref. 17), and its synthesis under N-limited conditions is a considerable investment for the organism, justified primarily by the opportunity to outcompete non-diazotrophs in the same habitat. If bioavailable nitrogen in the environment is depleted, diazotrophs derepress their nitrogenase machinery and produce the enzyme system in large quantities that can amount to up to 20% of total cellular protein in efficient N2-fixers such as the free-living gammaproteobacterium Azotobacter vinelandii18,19. Production of the enzyme is downregulated swiftly once sufficient fixed N becomes available18. The ability to fix atmospheric N2 possibly predates the separation of prokaryotes and eukaryotes, whose last common ancestor was suggested to already possess a nitrogenase enzyme20. This was before the oxygenation of Earth’s atmosphere, in the absence of an evolutionary pressure to discriminate N2 from the chemically similar O2. This helps to rationalize why all extant nitrogenases are highly sensitive to dioxygen, and are rapidly and irreversibly inactivated in the presence of small amounts of O2 (ref. 21). Damage occurs at the sensitive metal clusters of the enzyme, most severely at the bridging [4Fe:4S] cluster of the Fe protein. Although not a high-priority issue for obligate anaerobic diazotrophs, it requires others, such as diazotrophic cyanobacteria, which even produce O2 during photosynthesis, to take extensive countermeasures. Among these are the formation of specialized, thick-walled heterocysts that abandon photosynthesis and differentiate towards nitrogen fixation, or the diversion of a fraction of photosynthetic electron flux towards O2 detoxification22. An even more striking case is found in free-living soil bacteria that have perfected a diazotrophic lifestyle while relying on aerobic respiration to satisfy the substantial energy requirement of the nitrogenase reaction23. A. vinelandii belongs to this group and has evolved two distinct strategies to protect the nitrogenase system from oxidative damage. As a ‘respiratory protection’, it produces a variety of highly active oxidases that react with the gas and keep the cytoplasm in an anoxic state24. In addition, the organism can rapidly and reversibly inactivate its nitrogenases (switch-off), forming a transient, protected state with substantially increased oxygen tolerance. This second process is dependent on the presence of a small ferredoxin, FeSII (ref. 1), that was discovered in 1968 as the ‘Shethna protein II’ by Beinert and co-workers3. FeSII reversibly forms a complex with both nitrogenase components25 that has been isolated and characterized2 and, even when recombinant FeSII was produced and isolated from Escherichia coli26 it had a strong protective effect on the isolated components of Mo-nitrogenase4,27. We observed that this protective effect was realized only when both components of nitrogenase were present, while FeSII did not stabilize either component alone5. The crystal structure of A. vinelandii FeSII showed that the protein was a member of the adrenodoxin type of [2Fe:2S] ferredoxins that are common in plants and mammals, but contained an extended, flexible ‘N-loop’ that constituted almost one-third of the protein chain and contained two short α-helices, hN1 and hN2. FeSII formed stable dimers. In the crystals, two out of three dimers had the N-loops in an extended conformation, packing against other monomers in the crystal lattice, while the remaining copy had them folded back to the protein core5. We further showed that the redox state of the protein altered its behaviour in size-exclusion chromatography (SEC), causing the oxidized protein to elute earlier, and indicate that this apparently larger form had its N-loops extended or flexible and should be the one to interact with the nitrogenase components.
A FeSII–nitrogenase complex eluted from SEC with a main peak corresponding to 320 kDa, but its architecture and the mode of its formation remained elusive5. We therefore proceeded to optimize the preparation of this O2-protected complex in vitro using recombinant FeSII and natively isolated nitrogenase components. Structural analysis by X-ray crystallography and cryo-electron microscopy (cryo-EM) single-particle analysis yielded high-resolution models for the reduced form of FeSII and for the complex of FeSII with both component proteins of the nitrogenase system and clarified its architecture.
The FeSII–nitrogenase complex
To investigate the interaction of FeSII with the nitrogenase components in vitro, we added 1 mM Na2S2O4 as a reductant and O2 scavenger to recombinant FeSII, isolated native MoFe and Fe proteins, and joined the components in molar ratios of 1:3:5 to produce MoFe:Fe:FeSII proteins under strict exclusion of dioxygen (Fig. 1a). ATP was added to allow for turnover conditions and O2 was supplied subsequently to the gas space to trigger oxic stress. After a short incubation, the sample was loaded onto a SEC column, where the formation of a large complex was evident. O2 was removed from a part of the sample and, after addition of dithionite, we observed separation into the component proteins, showing that complex formation is reversible (Fig. 1a). Using different amounts of O2, we found that reversible complex formation occurred from O2 concentrations between 0.1% and 20%, in samples exposed to the gas for 1 min or 5 min, and without visible damage or precipitation (Extended Data Fig. 1a). Complexes were identified on SDS–PAGE and could be dissociated by addition of dithionite (Extended Data Fig. 1b). In these samples, the protective effect of FeSII was clearly shown by activity assays (Extended Data Fig. 1c). The complexed sample was subsequently analysed by cryo-EM, where extended filaments were immediately apparent in negative staining (Fig. 1d and Extended Data Fig. 2a) and cryo-transmission electron microscopy (TEM) (Extended Data Fig. 2b) after SEC. Centring on the FeSII dimer, a core particle was built that included two copies each of Fe protein and MoFe protein (Fig. 1b) and refined to yield a 2.9 Å resolution structure of a ternary complex with two copies each of the Fe protein NifH2 and the MoFe protein NifD2K2 arranged around a central FeSII dimer (Fig. 1b,e, Extended Data Fig. 3 and Extended Data Table 1). All interactions were mediated exclusively by the small FeSII, which locked the components of the enzyme in place but kept them strictly separate, and the entire arrangement followed the C2 symmetry of the FeSII dimer (Fig. 1c). Alternatively, we could choose a larger core particle consisting of five copies of MoFe protein, four FeSII dimers and eight Fe protein dimers, with a total mass of 1.8 MDa that yielded a reconstruction to 5.4 Å resolution (Extended Data Fig. 2d). This structure followed the same architectural principle as the core complex, but reflected the fact that FeSII can bind to both sides of the C2-symmetric MoFe protein. We regularly observed filaments with a length of 150–180 nm (5–6 MDa) that assembled into a right-handed helix with a diameter of 24 nm (Fig. 1f). This assembly includes two Fe protein dimers per single MoFe protein heterotetramer, reflecting the minimal stoichiometric ratio that may exist in the cell, although for in vitro activity assays the more O2-sensitive Fe protein component is typically used in high molar excess. The FeSII–nitrogenase filament had a pitch of 30 nm with almost three basic units—or 1 MDa mass—per turn (Fig. 1g). In cells of A. vinelandii with a length of 2–5 µm, these are structures of considerable size28. Note also that, although A. vinelandii can produce all three isoforms of nitrogenase, this efficient protection by FeSII applies only to the Mo-dependent enzyme, which also has the highest catalytic activity and is produced preferentially by the organism4,9. Knowing the three-dimensional (3D) structures of the alternative nitrogenases, structural modelling shows that, in both VFe protein29 and FeFe protein15, the extra G-subunits would interfere directly with FeSII binding (Extended Data Fig. 6d–f). Accordingly, no complex formation was observed in the presence of dioxygen (Extended Data Fig. 6g). However, while Shethna protein II was known as an O2 protection system for Mo-nitrogenase1, it remained unclear whether the trigger for complex formation is the actual O2 concentration in the cytoplasm, other reactive oxygen species such as peroxide or superoxide radicals, or the overall redox state of the cell.
Fig. 1. Structure of the O2-protected FeSII–nitrogenase complex.

a, Complex formation monitored by SEC detected by absorption at 280 nm. In a dithionite (DT)-reduced sample (top), the nitrogenase component proteins and FeSII elute as separate peaks without detectable complex formation. Upon removal of reductant and addition of 0.1 vol% O2, complex formation is observed. Reduction and O2 removal lead to complex dissociation (bottom). b, Cryo-EM map of a particle consisting of two copies each of the MoFe protein NifDK and the Fe protein NifH held together by a dimer of the ferredoxin FeSII. c, Top view of the complex shown in a. FeSII is fully buried, and the entire particle follows the C2 symmetry of the ferredoxin. d, Negative-stain TEM image of a representative complex preparation. The assembly forms filaments of variable length and the refined particle structures are sub-averages of such filaments. e, Architecture of the FeSII-protected complex with labelled components. Each protomer of the small FeSII dimer directly contacts one NifH dimer and one MoFe protein. f, Filament formation occurs following the C2 symmetry of MoFe protein, leading to an extended structure with a diameter of 24 nm. g, The filament forms a right-handed helix with a pitch of approximately 30 nm, corresponding to about 1 MDa per helical turn. Scale bars, 50 nm (d), 24 nm (f), 10 nm (g).
Extended Data Fig. 1. Analysis of complex formation.

a, Variation of O2 concentration. Reversible complex formation was observed from 0.1% O2 (Fig. 1a) up to 20% O2 for exposure times of 1 and 5 min. b, SDS-PAGE for (a), showing stable, stoichiometric complexes under O2 and dissociation into the components upon O2 removal and reduction. c, Acetylene reduction assays for the samples in (a) in the absence (black) and presence (hatched) of FeSII. At incubation times of 1 and 5 min, the stabilizing effect of FeSII is substantial even at 20% of pO2. Error bars represent the standard deviation of three independent measurements. d, Detail of Fig. 1a, top trace (uncomplexed), with the peak positions for FeSII, the Fe protein NifH2 and the substantially larger peak for the MoFe protein NifD2K2. e, Detail of Fig. 1a, middle trace, after O2-induced complex formation. Due to its molar excess, residual free FeSII is present, but additional peaks for Fe protein are observed that are consistent with a FeSII2:NifH2 and a FeSII2:2NifH2 complex (Fig. 2h). f, Variation of molar ratios for complex formation. In each trace, one component is added in substoichiometric amounts (brown, MoFe; green, Fe; blue, FeSII). Lacking FeSII (blue trace), a shoulder appears (blue arrow) that represents a smaller complex. No residual Fe protein or FeSII are observed, and an aggregation peak in the void volume represents mostly Fe protein. With substoichiometric amounts of Fe protein (red trace), most FeSII cannot form a complex with MoFe protein alone. When substoichiomentric in MoFe protein, all available MoFe protein appears in a regular, broad complex peak, no aggregates are observed in the void volume. Some free FeSII and Fe protein remain, and a new peak emerges (black arrow), consistent with a FeSII2:NifH2 complex. g, Peak analysis of the bottom trace of panel (e) with a MoFe:Fe:FeSII ratio of 1:5:5. For the multi-Gaussian fit, only two complex peaks were modeled. A new peak representing a FeSII2:NifH2 complex can readily be identified.
Extended Data Fig. 2. The FeSII-nitrogenase complex in micrographs.

a, Negative-stain micrograph of the complex fraction of (a), showing filaments of variable length. b, Representative cryo-TEM micrograph from the data set used for structure solution, with most proteins present in the FeSII-complexed state.
Extended Data Fig. 3. Workflow for cryo-EM data collection and processing.

a, Representative micrograph out of 2,097 recorded movies. b, 2D class averages for the C2-symmetric core particle. c, 2D class averages for the filament fragment. d, A total of 1.9 million particles were picked and used for 2D classification, resulting in a 3D reconstruction for the C1 filament fragment at 5.36 Å resolution, and for the C2 core particle at 2.98 Å resolution. e, The core particle was most highly resolved in the central part surrounding the FeSII dimer. f, Angular distribution of particles used for the 3D reconstruction of the C2-symmetric core. g, Fourier shell correlation curves, using a 0.143 gold standard cutoff for overall resolution.
Extended Data Table 1.
Cryo-EM data collection, refinement, and validation statistics

Extended Data Fig. 6. The NifDK:NifH interface in the AMPPCP-stabilized complex of Mo-nitrogenase and the action of FeSII on alternative nitrogenases.

a, In the well-characterized complexes stabilized by non-hydrolyzable ATP analogs, two copies of Fe protein bind on either side of the C2-symmetric NifD2K2 heterotetramer such that the twofold symmetry axis of Fe protein coincides with the pseudo-twofold axis relating NifD and NifK. b, In its reduced state, the central S1 sulfide of P-cluster is located precisely on this axis. c, The binding interface of Fe protein and MoFe protein involves the conserved F125 in both NifD and NifK, as well as several direct or water-mediated hydrogen bonds. The strongest direct interaction, however, is mediated by R101H that forms salt bridges to NifD in one monomer of NifH and to NifK in the other. Note that these interactions show a slight asymmetry. While in both MoFe subunits residue E120 interacts with R101H, a second acceptor carboxylate is E184D in NifD, which is replaced by E156K in NifK. The binding of FeSII to Fe protein shields R101H in both protomers (Fig. 2f). Figure generated from the AMPPCP-stabilized complex of A. vinelandii NifHDK (PDB 4WZB)31. d, Binding of a FeSII monomer to NifD2K2. The top view shows the unobstructed binding of FeSII to the protein surface. e, Hypothetical model for the interaction of FeSII with VndD2K2G2 of V-nitrogenase. The additional subunit VnfG prevents FeSII at the same position as in MoFe protein (d). f, For Fe-nitrogenase, subunit AnfG causes analogous clashes to (e). g, While Mo-nitrogenase readily forms a protective complex with FeSII in the presence of oxygen, no such complex formation is observed with either V- or Fe-nitrogenase under the same conditions, highlighting that FeSII acts exclusively in conjunction with Mo-nitrogenase.
Conformational states of FeSII
FeSII follows the canonical topology of adrenodoxin-type ferredoxin (Extended Data Fig. 4a), and its flexible N-loop is formed primarily by insertion of helix hN2 (Extended Data Fig. 4b), which is absent in other members of the family. In our earlier structure for FeSII, we observed different conformations of the N-loop that we designated as ‘open’ and ‘closed’, which led us to suggest that its flexibility is a prerequisite for triggering complex formation5. As the crystals were grown in the absence of reductant, both forms should contain an oxidized [2Fe:2S] cluster. We therefore crystallized FeSII under reducing conditions and obtained a new crystal form with a single FeSII dimer per asymmetric unit that yielded a structural model at 1.45 Å resolution (Fig. 2a, Extended Data Fig. 5 and Extended Data Table 2), with a fully defined N-loop that differed from the previously observed states. Together with the conformation observed in the complex with the nitrogenase components, which differed from all others, four distinct structural snapshots now provide a sound basis for understanding FeSII activation.
Extended Data Fig. 4. Structural properties of FeSII.

a, Topology diagram of FeSII. b, Wall-eyed stereo image of the FeSII monomer in the locked conformation, highlighting the positioning of helix hN1 above the [2Fe:2S] cluster. c, In the reduced state the N-loop is fixed by two salt bridges to the body of FeSII (D44-K70 and E71-R99) and interacts with the other protomer via R61 and D93 and a salt bridge (R92-A122) that fixes the C-terminal carboxylate of the other chain. Helices hN1 and hN2 are separated. d, Upon oxidation, the interactions with D44, R99, and the C-terminal A122 are released, the N-loop folds into the paddle conformation and gains flexibility. e, In the locked state, the D44-K70 and E71-R99 are re-formed, but helix hN2 remains in the paddle conformation and does not interact with the other monomer of FeSII.
Fig. 2. Conformational states of FeSII.

a, Crystal structure of the reduced FeSII dimer viewed along the C2 axis, with the N-loops shown in grey and light blue. b, Closed dimer in the oxidized structure (PDB 5FFI). c, In the oxidized open state (PDB 5FFI), helices hN1 and hN2 of the N-loop extend outward, forming a paddle-like structure. d, As FeSII forms a complex with the nitrogenase component proteins, the N-loops in this locked state are retracted but the paddle arrangement is retained. hN1 interacts predominantly with NifH, while hN2 interacts with MoFe protein. e, In the reduced state, the cluster-binding loop P39–C50 faces inward, with K70 forming a hydrogen bond to D44. f, In all other structures, the cluster-binding loop faces outward, breaking the D44–K70 interaction and making E41 available to interact with Fe protein (Fig. 3f). g, Monomer structures of the four observed states. Helix hN1 remains close to the [2Fe:2S] cluster in the reduced and locked states, and also in the closed oxidized states, although the H-bond to K70 is broken (brown). Helix hN2 is disordered in the closed oxidized state, but moves only slightly between the reduced and open oxidized state (green). The cluster loop differs only in the reduced state (red in e). h, Upon activation, FeSII does not form a complex with MoFe protein as analysed by analytical SEC (upper panel) (masses: (1) MoFe, 227 kDa (calculated (calc.), 230 kDa); FeSII, 31 kDa (calc., 26 kDa)). By contrast, oxidized FeSII readily engages with Fe protein (lower panel), binding one or two copies per dimer (masses: (1) (NifH2)2:FeSII2, 152 kDa (calc., 152 kDa); (2) NifH2:FeSII2, 91 kDa (calc., 90 kDa); (3) NifH2, 61 kDa (calc., 64 kDa); FeSII, 26 kDa (calc., 26 kDa)).
Extended Data Fig. 5. Structural properties of FeSII.

a, FeSII monomer in cartoon representation, highlighting the residues mentioned throughout the manuscript. b, Top and side view of the [2Fe:2S] cluster of FeSII in the reduced state. Coordinative bond lengths around the iron ions are shown in Å. c, Wall-eyed stereo representation of the 2Fo–Fc difference electron density map around the [2Fe:2S] cluster, contoured at the 1σ level (grey) and the 5σ level (blue). d, Representation of the FeSII dimer with color and ribbon width representing local B-factors. Due to local crystal packing, chain B was less well defined than chain A. All distances and interactions discussed in the text refer to chain A.
Extended Data Table 2.
X-ray crystallography data collection and refinement statistics

a
b
Rfree is the cross-validation R value for a test set of 5 % of unique reflections.
Consistent in all four models are conformational changes in the N-loop, but also in the loop coordinating the iron–sulfur cluster containing the ligands C42, C47 and C50, and in the C terminus at residue A122. In the reduced-state structure that represents the inactive state of FeSII (Fig. 2a), the N-loops were folded back onto the core of the dimeric protein, fully ordered and resolved, including the α-helices hN1 (residues 69–78) and hN2 (84–91). The lid helix hN1 resides above the [2Fe:2S] cluster (Extended Data Fig. 5a) and hN2 points towards the dimer interface, allowing residues R92 and D93 to interact with the other FeSII protomer. D93 formed a short hydrogen bond to K53′ (that is, in the other monomer), while R92 contacted the C-terminal carboxylate of the second chain (A122′) that will play a vital role in complex formation with NifH (Extended Data Fig. 4c). In the oxidized closed state (Fig. 2b), helix hN1 remained close to the cluster, but residues K83–R92 that include helix hN2 were disordered, which also released the C terminus of the other protomer. The predominant conformation of FeSII in the oxidized state, however, had helices hN1 and hN2 in the parallel, paddle-like arrangement described earlier (Fig. 2c). Here, the N-loop was released as the interactions E71:R99 and D44:K70 were severed, and R92 moved away from the carboxylate of the other protomer, releasing its C terminus. The D93:K53′ interaction was retained, so that the N-loop gained flexibility and was released from the core of FeSII (ref. 5), and the formerly separate helices hN1 and hN2 rearranged into the parallel paddle arrangement (Extended Data Fig. 4d). In the cryo-EM structure of the complex with the nitrogenase components, the N-loops of FeSII showed yet another conformation that we designate ‘locked’, as it kept the complex stably in place (Fig. 2d). The N-loop was retracted to the FeSII core, re-establishing the E71:R99 salt bridge, but not the interaction of D44 and K70, as these residues were involved in protein–protein interactions with the enzyme. R92 and D93 rotated away from the other FeSII protomer, so that the C termini at A122 became accessible (Extended Data Fig. 4e).
Most recently, Murray and co-workers proposed that the arrangement observed in the original crystals, in particular the open conformation of the N-loops, might be due to domain swapping of the N-loop caused by crystal packing interactions30. This suggestion is consistent with the structural data, but nevertheless requires that, in the open state, the N-loop is able to leave its position at the same protomer to interact with another one. Comparing these earlier structures with the reduced state reported here, an important difference in the cluster environment becomes apparent in the cluster loop. The reduced state prominently features a salt bridge between residue D44 in the cluster loop and K70 in the N-loop (Fig. 2e). In all other structures, this salt bridge is broken due to a shift of the entire cluster loop that makes residues E41 and D44 in this loop available for interactions with the nitrogenase component proteins (Fig. 2f). This release of one of only three interaction of the N-loop with the core domain of FeSII (Extended Data Fig. 5a) is probably key to the increased flexibility observed upon oxidation.
Taken together, the different conformational states of FeSII thus outline the activation process of the ferredoxin (Fig. 2g). In the absence of O2, FeSII resides in the reduced state, where the separated N-loop helices hN1 and hN2 allow residue R92 to bind and block the C-terminus of the second protomer (Extended Data Fig. 4c). Oxidation of the cluster then changes the conformation of the cluster loop. However, while a transition to the oxidized closed state leaves hN1 in place but leads to flexible disorder of the C termini, the oxidized open state largely leaves hN2 in place, including the R92–A122′ interaction. Instead, hN1 folds outward to form the paddle structure. Both structures underline the newly gained flexibility of the N-loop that is required for interaction with Fe protein (see below). In the complex with Fe protein and MoFe protein, this paddle structure is retained but, as hN1 returns to its position at the [2Fe:2S] cluster, the entire N-loop is tightly bound to the FeSII core, and the interaction of R92 with the C-terminal carboxylate is no longer possible. Instead, this carboxylate then can occupy a position directly above the labile [4Fe:4S] cluster of the Fe protein (see below).
Interactions of FeSII with nitrogenase Fe protein
On analytical SEC, FeSII interacted readily with the Fe protein NifH2, but not with MoFe protein in the absence of NifH2 (Fig. 2h), establishing that the FeSII2–NifH2 complex forms first. In particular, the substoichiometric provision of MoFe protein led to an accumulation of FeSII:NifH complexes of variable stoichiometry (Fig. 2h and Extended Data Fig. 1d,e), while all MoFe protein was found in a set of different complexes (Extended Data Fig. 1f,g). Under all conditions tested, the FeSII protein formed a stable dimer but its initial interaction with dimeric NifH2 was through one protomer only. In the protected complex, FeSII2 inserted itself between NifH2 and NifD2K2 (Fig. 3a), covering the functional interface of the two nitrogenase components (Extended Data Fig. 6). In the complex, each FeSII monomer had a NifH2 homodimer bound (Fig. 3b). The FeSII2 dimer interface was identical to the one observed in the uncomplexed structures of the ferredoxin (Fig. 2a–c), and the N-loop was in the locked conformation (Fig. 2d, Extended Data Fig. 3e), giving the protomer a globular shape (Fig. 3c and Extended Data Fig. 4b). As FeSII2 interacted with Fe protein, the initial point of contact is probably through the N-loop paddle of activated FeSII in its oxidized state (Fig. 2c). As the N-loop retracts, it pulls both proteins closely together into a tight complex, with the [2Fe:2S] cluster of FeSII close to the [4Fe:4S] cluster of NifH2. The minimal distance of the clusters of 12.5 Å allows for efficient electron transfer. The interaction of FeSII2 and NifH2 was asymmetric, and the N-loop interacted predominantly with one monomer (Fig. 3d). In detail, the N-loop formed two new salt bridges to NifH2, R72:E69H (superscript denotes residues of NifH) and K76:E112H, as well as hydrogen bonds from N73 to E69H and from N108H to the backbone carbonyl of K76 (Fig. 3e). The sum of these interactions constitutes a significant binding strength for complex formation. Residues R72, K76 and N73 are all part of helix hN1 in the N-loop. Note that the NifH2 dimer that binds to one FeSII protomer does not form any interactions with the second protomer. The second interface of interaction within the complex not only brings the two metal clusters of the protein partners close together, but it also places the C-terminal amino acid A122 of FeSII in a position directly above the most sensitive spot of NifH2—the bridging [4Fe:4S] cluster (Fig. 3f). In the reduced structure of FeSII2, this terminus was fixed by the other protomer (Fig. 2a), but it gained flexibility in the open state (Fig. 2c). Once the complex locks in place, A122 is again well defined and stabilized by a hydrogen bond to residue E41 in the cluster-binding loop. The C-terminal carboxylate at A122 is also hydrogen-bonded to residue E118 of FeSII that in turn forms a salt bridge to R101H (Fig. 3f). Interestingly, in the NifH2D2K2 complex structures locked in place with either ADP–AlF4− or AMPPCP17,31, the arginine residues R101H form the only salt bridges in the interface of the component proteins, making them a main contributor to complex stability (Extended Data Fig. 6a–c). FeSII thus stably binds to Fe protein, preventing its interaction with MoFe protein and protecting the sensitive [4Fe:4S] cluster of Fe protein. Characterized by numerous, strong salt bridges and supported by further hydrogen-bonding interactions, this first association is strong, but a 3D variability analysis of the cryo-EM maps shows that the NifH dimers bound to FeSII in the complex retain significant flexibility (Supplementary Video 1). Note also that we observed in vitro that FeSII alone did not prevent oxidative damage to Fe protein in the absence of MoFe protein5. Although the rapid interaction of FeSII with Fe protein is the key initiation event of complex formation, the subsequent binding of MoFe protein to this core unit (Fig. 3b) is indispensable for attaining its protective function.
Fig. 3. Interactions of FeSII with the Fe protein NifH.

a, Cryo-EM map highlighting FeSII (blue), which bridges and separates a MoFe protein heterotetramer and a Fe protein dimer. b, In the complex, FeSII acts as a dimer and interacts with two NifH2 dimers. Top, EM map. Bottom, cartoon representation. c, The interaction of a FeSII monomer with a NifH2 dimer is asymmetric, and FeSII fully covers the surface area above the O2-sensitive [4Fe:4S] cluster of NifH. d, A detailed view of the interface shows that interactions are focused on two areas. One NifH monomer interacts with helix hN1 of the N-loop, and the core of FeSII resides close to the [4Fe:4S] cluster of the Fe protein. e, Key interactions of helix hN1 with NifH are two salt bridges (R72:E69H, K76:E112H) and hydrogen bonds involving N73 and N108H. f, The C terminus of FeSII is placed immediately above the [4Fe:4S] cluster of NifH, and E41 and E118 interact with the Fe protein. The interaction blocks R101H in both NifH monomers.
Complex formation with MoFe protein
Tight binding to Fe protein has left the activated FeSII with its N-loops retracted, in a conformation that is probably close to the locked state. Through this it gains affinity to MoFe protein, allowing for the association of the larger nitrogenase component to bind, thereby providing a starting point for filament formation. FeSII interacts mostly with the surface of NifD (Fig. 4a), covering a patch above the FeMo cofactor that was suggested to serve as one entry point for the protons required for N2 reduction, terminating in residue H196D (Fig. 4b)8. In addition, K63 of FeSII forms a salt bridge to D204D of MoFe protein, and the negative end of the helix dipole of hN2 points directly at the FeMo cofactor (Fig. 4b). The only interaction with NifK occurs through a short, protruding loop of the subunit, 119TEDAAVFG126, which contains the highly conserved F125K (in bold) that is also found as F125D in NifD (Fig. 4c). Due to the asymmetric binding of FeSII2 onto the NifDK heterodimer, this second phenylalanine is not in contact with any other component of the complex (Fig. 4d). The two F125 residues are conserved in this position in all known structures of nitrogenases, where they symmetrically support the formation of the complex with Fe protein. There, the phenylalanine resides in a pocket on the surface of Fe protein, whereas in the FeSII protein interaction it packs into a groove directly adjacent to the [2Fe:2S] cluster (Fig. 4c,d). In its asymmetric position on NifD, FeSII does not cover the surface position above P-cluster, that is, the twofold pseudosymmetry axis of the NifDK heterodimer. However, it extends residue R24 to form a hydrogen bond to the backbone carbonyl of L158D, so that the positively charged guanidinium moiety of the arginine side chain is situated almost precisely on this pseudo-twofold axis (Fig. 4f). The small ferredoxin thus specifically occupies the interaction interface of the two nitrogenase components, separating them while shielding access to their oxygen-sensitive metal clusters. Key to the interaction with MoFe protein is the conformation of helix hN2 in the locked state (Fig. 2c). Although this helix interacts with NifD only at its C-terminal end, its arrangement in the N-loop paddle (Fig. 2b) rather than in the open conformation of the reduced state of FeSII (Fig. 2a) is a strict prerequisite for the observed mode of complex formation.
Fig. 4. Binding of FeSII to NifDK.

a, The FeSII dimer (blue/purple) in contact with two copies of MoFe protein. The two bound Fe proteins are omitted for clarity (Fig. 1c). b, FeSII binds to NifD (yellow) above FeMo cofactor, with a salt bridge forming between K63 of the ferredoxin and D204D of the enzyme. FeSII also covers H196D—a proposed entry point for H+ to the active site. c, On the other side of FeSII, the [2Fe:2S] cluster binds close to F125K. d, This residue is one of a conserved pair of phenylalanines that are crucial for interaction with Fe protein (Extended Data Fig. 5c). In the complex, the Fe protein is cradled in a deep pocket in FeSII. e, The pair of phenylalanines in NifD and NifK and their relative position to the P-cluster. f, View onto the surface of MoFe protein along the pseudo-twofold axis relating NifD and NifK. FeSII binds largely on NifD but extends the side chain of R24 to reside precisely on the NifDK interface, above the P-cluster.
Intermolecular electron transfer
Our structural and biochemical data show that FeSII first associates with Fe protein—the most sensitive component of the nitrogenase system—and that this core complex then recruits MoFe protein, as detailed above. Upon complex formation, the [2Fe:2S] cluster of FeSII is in its oxidized state, whereas under diazotrophic conditions its interaction partner, Fe protein, will mostly be in the reduced state. With a midpoint potential of −262 mV versus standard hydrogen electrode26, FeSII can obtain an electron by the more strongly reducing Fe protein NifH (E0′ = −420 mV versus standard hydrogen electrode32), so that the [2Fe:2S] cluster in FeSII is again reduced (Figs. 3b and 5)33. When bound to NifH, the N-loop paddle retains its conformation seen in the oxidized state and, rather than returning to the free reduced and inactive state, FeSII attains its locked conformation (Fig. 2d). Only with the retracted N-loop in the paddle conformation can the FeSII–NifH complex now bind to the MoFe protein, where the same process at the opposite NifDK protomer then leads to filament growth. The bidirectional extension of the protected filaments that these sequential interactions enable assures the most rapid response possible to oxygen stress, with a minimum required amount of FeSII. The two protomers of the FeSII dimer interact independently with two copies of the Fe protein dimer, but we have found earlier that the oxidation of both clusters shows positive cooperativity, providing another means of accelerating complex formation5. There is no indication of a further redox event between FeSII and NifDK. The midpoint potential of reduced FeSII should not be sufficiently negative to reduce the P-cluster of the catalytic dinitrogenase but, while the reverse process is feasible on the basis of potential differences, we did not observe any complex formation of FeSII with NifDK in the absence of NifH (Fig. 2h). Our model implies that the interaction with NifDK requires FeSII to already be in its locked conformation, that is, bound to NifH, and the sequential binding can then be rationalized independent of whether the protective effect of the complex extends to the more stable MoFe protein itself. What remains to be explained is how the complex dissociates once the acute phase of oxidative stress is overcome. All metal clusters in the protected filament are shielded from access to external electron donors. We find that the complex is stabilized in the presence of O2 and dissociates upon addition of dithionite (Fig. 1a and Extended Data Fig. 1), but it is unknown whether the release of the components is a simple first-order reaction or whether more components are involved.
Fig. 5. Sequential formation of the protected FeSII:NifH:NifDK complex.

Conformational protection of Mo-nitrogenase is initiated by the oxidative activation of reduced, dimeric FeSII, which leads to the extension of the N-loops in both protomers. This state interacts only with NifH but not with NifDK. The FeSII dimer first binds one NifH dimer, followed by a second one to form an initiator complex that is able to provide significant protection to the more sensitive Fe protein component5. The initiator complex then successively recruits two NifD2K2 heterotetramers to assemble the core particle analysed in the present study. As the initiator complex can bind to both sides of the MoFe protein heterotetramer, this interaction leads directly to the polymerization into filaments (Fig. 1g).
In summary, the data presented here highlight the remarkable features of the small ferredoxin FeSII that can sense oxidative stress and react by polymerizing the far larger nitrogenase components into a protected filament to overcome short phases of intermittent oxidative stress. The free-living diazotroph A. vinelandii is well adapted to generating intracellular anaerobicity through its high respiration rates, and arguably this situation is of high relevance for the use of a heterologously produced nitrogenase. The most promising strategy for producing functional nitrogenase in food crops focuses on plant mitochondria34,35. As energy-conserving organelles they can provide the ATP required by the enzyme, and the aerobic respiratory chain in their inner membrane renders their inner matrix space the most anoxic environment in a plant cell. In particular at night, when no oxygenic photosynthesis occurs, mitochondria operate on the complete oxidation of sugars or fatty acids and provide a rather constant low-O2 environment were a recombinant nitrogenase can mostly operate safely, whereas the presence of FeSII can provide short-term protection during short oxygen bursts, as it does in its native host A. vinelandii. FeSII production in this context seems feasible in eukaryotic mitochondria that contain the machinery for iron–sulfur cluster biogenesis36,37, making the 1968 discovery of this second ‘Shethna protein’ a valuable addition to the puzzle of nitrogenase bioengineering aimed at alleviating the dependency of food crops on inorganic fertilizer.
Methods
Production of recombinant A.vinelandii FeSII
A codon-optimized gene encoding A. vinelandii FeSII (AVIN_39700) was synthesized (GeneArt) and inserted into the multiple cloning site of the expression vector pET21a (Novagen) by means of Gibson assembly, excluding the hexahistidine affinity tag offered by the vector38. Chemically competent E. coli BL21(DE3) C43 cells39 were transformed with the plasmid40. For protein production, the cells were cultivated at 37 °C in LB medium supplemented with 100 µg ml−1 of ampicillin. Gene expression was induced by the addition of 0.1 mM isopropyl-β-d-1-thiogalactopyranoside at an optical density at 600 nm (OD600nm) of 0.6 and collected by centrifugation after 5 h. The cell pellet was resuspended in 50 mM sodium acetate buffer at pH 5.2 and disrupted by three passages through a microfluidizer at 150 MPa (Maximator HPL6). After readjusting the pH to 5.2, cell debris was removed by centrifugation at 80,000g for 45 min. The supernatant was loaded onto a cation exchange column (5 ml HiTrap SP HP, Cytiva) equilibrated with 50 mM sodium acetate buffer at pH 5.2. After washing, the column was developed with a step gradient from 0 to 100 mM NaCl. Fractions containing FeSII were pooled, concentrated by ultrafiltration and subsequently loaded on a HiLoad Superdex 75 26/600 (Cytiva) size-exclusion column equilibrated with 50 mM HEPES/NaOH buffer at pH 8.0 and 100 mM NaCl. Fractions containing FeSII were pooled, concentrated by ultrafiltration (10 kDa MWCO, Sartorius), flash frozen and stored in liquid nitrogen.
Production of nitrogenase component proteins
A. vinelandii (Lipman 1903, DSM2289)41 was cultivated oxically in modified Burk medium42. A preculture of 100 ml was inoculated with cells from a glycerol stock and grown diazotrophically to an OD600nm of 2.5. The culture was then used to inoculate main cultures of 500 ml, which were grown to an OD600nm of 2.5 and collected by centrifugation. All subsequent steps were performed anoxically in an inert gas chamber (95% N2, 5% H2 atmosphere, Coy Laboratory Products) or by using modified Schlenk techniques. All buffers were degassed by repeated cycles of vacuum and N2 and were supplemented with 2.5 mM Na2S2O4 adjusted to pH 7.5 with Tris base. The cell pellet was resuspended in 50 mM Tris-HCl buffer at pH 7.4 and disrupted by three passages through a microfluidizer at 150 MPa (Maximator HPL6) under an N2-atmosphere. Cell debris was removed by centrifugation at 80,000g for 30 min and the supernatant was loaded on an anion exchange column (50 ml Q Sepharose, GE Healthcare) equilibrated with 50 mM Tris-HCl buffer at pH 7.4. After washing, the protein was eluted with a linear gradient of NaCl. The MoFe protein eluted at a conductivity of 27 mS cm−1, while Fe protein eluted at 42 mS cm−1. The concentrated proteins were further purified separately by SEC (HiLoad Superdex 200 26/600, Cytiva), equilibrated with 20 mM Tris-HCl buffer at pH 7.4 with 200 mM NaCl. The purified proteins were concentrated by ultrafiltration (100 kDa and 30 kDa MWCO, Sartorius), flash frozen and stored in liquid nitrogen.
Activity assays
Nitrogenase activity was monitored by following the non-physiological reduction of acetylene to ethylene that can be quantified straightforwardly by gas chromatography carried out as described elsewhere5. In brief, in a 10 ml Wheaton vial, 1 ml of reaction mixture was prepared anoxically, containing 0.65 µmol of NifD2K2 and 1.3 µmol of NifH2 and, if applicable, 1.3 µmol of FeSII dimer. The mixture further contained 125 mg l−1 of phosphocreatine kinase, 15 mM of phosphocreatine, 2.5 mM ATP, 5 mM MgCl2 and 20 mM Tris-HCl buffer at pH 7.4. Pure O2 gas was added through a gastight syringe to the desired final concentrations and incubated for variable times. After 1 or 5 min, oxygen was removed by flushing the headspace with 100% Ar, and subsequently Na2S2O4 was added to a final concentration of 2.5 mM. For the assay, the respective missing components were added and 1 ml of C2H2 was injected to the headspace with a gastight syringe and the reaction was allowed to proceed for 3 min at 30 °C and then stopped by addition of 250 µl of glacial acetic acid. A sample of 1 ml of headspace was taken and injected into a gas chromatograph for quantification of ethylene.
Complex formation
The following steps were performed anoxically in an inert gas chamber. MoFe protein (NifD2K2), Fe protein (NifH2) and FeSII (dimer)) inert were combined in a molar ratio of 1:3:5 and their buffer solution was exchanged to anoxic 20 mM Tris-HCl buffer at pH 7.4 with 40 mM NaCl, 5 mM MgCl2 and 1 mM Na2S2O4, using a NAP-5 desalting column (Cytiva). The protein mixture was transferred to a 12 ml vial containing a stirrer, sealed with a rubber septum and N2 turnover was initiated by the addition of 2.5 mM ATP under vigorous stirring. After 1 min, 60 µl of the gas phase were exchanged for air, resulting in an oxygen concentration of 0.1%. For assays with alternative nitrogenases, the final oxygen concentration was 1%. After 5 min of incubation and stirring, the protein mixture was loaded onto a Superose 6 Increase 10/300 GL column (Cytiva), equilibrated with anoxic 20 mM Tris-HCl buffer at pH 7.4 with 5 mM MgCl2. Fractions for cryo-EM analysis were collected in anoxic vials under positive N2 pressure. For other experiments, molar ratio of proteins, salt concentration, oxygen concentration and incubation time were the same unless indicated otherwise.
Analytical size-exclusion chromatography
To analyse the potential binary precursor complexes FeSII:NifDK and FeSII:NifH, the following steps were carried out in an inert gas chamber. For FeSII:NifDK, the buffers of oxic FeSII (35 mg ml−1, 1.2 mg) and dithionite-containing NifDK (64 mg ml−1, 3.6 mg) in a molar ratio of 3:1 were exchanged separately to anoxic, dithionite-free 20 mM Tris-HCl buffer at pH 7.4 with 25 mM NaCl, 5 mM MgCl2 using a NAP-5 desalting column. The proteins were collected in the same reaction tube leading to a volume of 500 µL, incubated for 5 min and sterile filtered. A 100 µl sample of the protein mixture was then loaded onto a Superdex 200 Increase 10/300 GL column (Cytiva) equilibrated with anoxic 20 mM Tris-HCl buffer at pH 7.4 with 25 mM NaCl and 5 mM MgCl2 connected to a Viscotek GPCmax (Malvern Panalytical) with a TDA 305 containing a refractive index (RI) detector and a right angle light scattering (RALS) detector for mass determination that was calibrated using bovine serum albumin. We analysed the binary FeSII:NifH complex identically with a molar ratio of 3:1 using FeSII (20 mg ml−1, 2.2 mg) and NifH (42 mg ml−1, 1.8 mg) with a target buffer consisting of 20 mM Tris-HCl buffer at pH 7.4 and 5 mM MgCl2 on a Superose 6 Increase 10/300 GL equilibrated with the same buffer.
Negative staining grid preparation and imaging
For negative staining EM, the ternary complex was obtained as stated above, with the following modifications: a molar component protein ratio of 1:2:1 was used, turnover buffer did not contain NaCl, the protein mixture was exposed to 0.5% oxygen for 15 min and the SEC step was omitted. After incubation with oxygen, 2.5 µl of protein mixture at a concentration of 0.01–0.1 mg ml−1 was applied to a glow-discharged TEM grid (carbon film supported, 300 Mesh, Sigma-Aldrich) for 1 min and then blotted. The grid was washed with two drops of water and stained with 2% uranyl acetate. After drying for 24 h, the grids were analysed on a Hitachi HT7800 transmission electron microscope at 100 kV.
Cryo-EM grid preparation and data collection
The ternary complex was diluted to 0.75 mg ml−1 in an inert gas chamber, transferred into a PCR tube and flash frozen in liquid N2 until further use. All cryo grids were prepared with 2.5 µl of protein sample using a Vitrobot Mark IV (ThermoFisher Scientific). After thawing on ice, protein samples were applied to glow-discharged Quantifoil Au R2/1 M300 grids under constant N2 flow, incubated for 5 s, blotted for 3 s with filter paper and flash frozen in liquid ethane cooled by liquid nitrogen. The dataset was recorded on a 200 kV Glacios cryo-transmission electron microscope (ThermoFischer Scientific) equipped with a Gatan K3 detector with a pixel size of 0.878 Å px−1, 30 frames and an exposure time of 4 s with a total dose of 50.56 e− Å−2.
Single-particle analysis, structural modelling and refinement
Initially, the raw video stacks were motion-corrected with RELION v.3.1 (ref. 43) and per-micrograph defocus values were estimated using CTFFIND v.4.1 (ref. 44). Particle picking was done using Laplacian-of-Gaussian blob detection (Extended Data Fig. 2). The particles were extracted with a box size of 400 pixels and sorted by three rounds of two-dimensional (2D) classification using a mask diameter of 250 Å. The particles were then transferred to CryoSPARC v.4.3 (ref. 45) for ab initio reconstruction. The model was refined to 2.99 Å in several rounds of non-uniform refinement46, CTF refinement47 and heterogeneous refinement implementing C2 symmetry. In a second round, the raw video stacks were motion-corrected followed by per-micrograph defocus value estimation, both with CryoSPARC v.4.3. The final model of the first refinement round was taken as a template for template-based particle picking in CryoSPARC v.4.3. Particles were extracted with two box-sizes: 420 pixels to obtain a high-resolution map for model building and 900 pixels to obtain a low-resolution map to visualize the helical supercomplex (Extended Data Fig. 2d). Particles extracted with a box size of 420 pixels were sorted by 2D classification followed by heterogeneous refinement using the final model of the first round of refinement and two trash classes to further sort bad particles out. The resulting map was refined to 2.89 Å in several rounds of non-uniform refinement, CTF refinement and heterogeneous refinement implementing C2 symmetry. Particles extracted with a box size of 900 pixels were binned to 300 pixels and subjected to two rounds of 2D classification followed by ab initio reconstruction. This model was refined by non-uniform refinement and then taken as a template, together with two trash classes, for heterogeneous refinement on all extracted particles to increase the number of good particles resulting in a map of higher quality. The resulting map was further refined to 5.36 Å, which is the resolution limit for the pixel size used, in several rounds of non-uniform refinement, 3D classification and heterogeneous refinement.
The 2.89 Å resolution map was used for model building. As starting model for MoFe, Protein Data Bank (PDB) entry 3U7Qwas used, for the Fe protein PDB entry 1FP6 and for the FeSII protein a model created by AlphaFold2 (refs. 48,49). All models were rigid-body fitted into the density map using UCSF ChimeraX50, hand-refined using COOT51, applied C2 symmetry and real-space refined in PHENIX52. The quality of the structure was validated by MolProbity53. Data collection and refinement statistics are summarized in Extended Data Table 1. Figures were generated with PyMOL (Schrödinger LLC) or UCSF ChimeraX50.
Crystallization, data collection, structure solution and refinement for reduced FeSII
A. vinelandii FeSII protein was crystallized using the sitting-drop vapour diffusion method in an inert gas chamber using an OryxNano drop dispenser (Douglas Instruments). FeSII protein (46 mg ml−1) was reduced by adding 10 mM Na2S2O4 adjusted to pH 7.5 with Tris base. Large, 3D crystals were obtained by mixing 0.3 µl of protein solution with the same volume of a reservoir solution containing 50 mM glycine at pH 9 and 55 % (v/v) of polyethylene 400 on a 96-well crystallization plate (MRC 2, SwissSci). After 8 days, crystals were collected, mounted on a nylon loop and flash frozen in liquid nitrogen. Diffraction data were collected at the European Synchrotron Radiation Facility on beamline FIP2-BM07 using a Pilatus 6M detector at an X-ray wavelength of 0.9795 Å; 360° of data were collected with 0.2° oscillations per image. Reflections were recorded to below 1.3 Å. Raw data was auto-processed using autoPROC54 and the resolution was cut at 1.45 Å using AIMLESS55 from the CCP4 (ref. 56) suite. The structure was solved by using MOLREP57 with oxidized FeSII (PDB 5FFI) as search model, yielding a solution with one FeSII dimer in the asymmetric unit of the P 41212 unit cell. The model was built with COOT51 and refined using REFMAC5 (ref. 58) and PHENIX52. Data collection and refinement statistics are summarized in Extended Data Table 2. Figures were generated using PyMOL (Schrödinger LLC).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41586-024-08355-3.
Supplementary information
3D variability analysis of the FeSII:Mo-nitrogenase complex structure. A 3D variability analysis carried out using CryoSPARC highlights the flexible binding of the two copies of the Fe protein NifH2 to the dimeric FeSII protein. In contrast, the interaction of the small ferredoxin with the MoFe protein NifD2K2 shows substantially higher rigidity.
Acknowledgements
We thank F. Detemple for support with activity measurements. We thank J. Rudiger, M. Eichhorst and A. Khansary for assistance with experiments, and R. Hinnenberg and M. Rodriguez Franco at the EM facility at the Faculty of Biology, University of Freiburg, S. Steimle at the Cryo-EM Facility, University of Freiburg, and M. Chami at the BioEM laboratory of the Basel Biocenter for excellent assistance with data collection. This work was supported by Deutsche Forschungsgemeinschaft (CRC 1381, project ID 403222702 and project ID 536145634) (O.E.) and the European Research Council (Horizon Europe AdG no. 101141673). High-performance computing resources were available at the bwHPC cluster of the federal state of Baden-Württemberg and Deutsche Forschungsgemeinschaft (INST 35/134-1 FUGG). The Hitachi HT7800 electron microscope used for negative-stain imaging (project ID 426849454) as well as the Cryo-TEM at the Cyro-EM facility of the University of Freiburg (project ID 506518771) were supported by Deutsche Forschungsgemeinschaft and are operated within the Microscopy and Image Analysis Platform (MIAP) and the Life Imaging Centre (LIC), Freiburg.
Extended data figures and tables
Author contributions
P.F. and O.E. designed the experiments. P.F. and S.F. carried out the experiments. P.F., L.Z. and O.E. processed data. P.F. and L.Z. built and refined the structural model. P.F., L.Z. and O.E. wrote the manuscript.
Peer review
Peer review information
Nature thanks Justin North and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Data availability
The atomic coordinates and cryo-EM map of the FeSII–nitrogenase complex have been deposited with the Protein Data Bank at http://www.pdb.org with accession number 8RHP and the Electron Microscopy Data Bank (EMDB) with accession number EMD-19178. Coordinates and structure factors for the reduced form of FeSII have been deposited with the Protein Data Bank at http://www.pdb.org with accession number 8RHO.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
is available for this paper at 10.1038/s41586-024-08355-3.
Supplementary information
The online version contains supplementary material available at 10.1038/s41586-024-08355-3.
References
- 1.Robson, R. L. & Postgate, J. R. Oxygen and hydrogen in biological nitrogen fixation. Annu. Rev. Microbiol.34, 183–207 (1980). [DOI] [PubMed] [Google Scholar]
- 2.Robson, R. L. Characterization of an oxygen-stable nitrogenase complex isolated from Azotobacter chroococcum. Biochem. J.181, 569–575 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shethna, Y. I., DerVartanian, D. V. & Beinert, H. Non heme (iron-sulfur) proteins of Azotobacter vinelandii. Biochem. Biophys. Res. Commun.31, 862–868 (1968). [DOI] [PubMed] [Google Scholar]
- 4.Moshiri, F., Kim, J. W., Fu, C. L. & Maier, R. J. The FeSII protein of Azotobacter vinelandii Is not essential for aerobic nitrogen fixation, but confers significant protection to oxygen-mediated inactivation of nitrogenase in vitro and in vivo. Mol. Microbiol.14, 101–114 (1994). [DOI] [PubMed] [Google Scholar]
- 5.Schlesier, J., Rohde, M., Gerhardt, S. & Einsle, O. A conformational switch triggers nitrogenase protection from oxygen damage by Shethna protein II (FeSII). J. Am. Chem. Soc.138, 239–247 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Ågren, G. I., Wetterstedt, J. Å. M. & Billberger, M. F. K. Nutrient limitation on terrestrial plant growth—modeling the interaction between nitrogen and phosphorus. New Phytol.194, 953–960 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Burris, R. H. & Roberts, G. P. Biological nitrogen-fixation. Annu. Rev. Nutr.13, 317–335 (1993). [DOI] [PubMed] [Google Scholar]
- 8.Einsle, O. & Rees, D. C. Structural enzymology of nitrogenase enzymes. Chem. Rev.120, 4969–5004 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Einsle, O. Catalysis and structure of nitrogenase. Curr. Opin. Struct. Biol.83, 102719 (2023). [DOI] [PubMed] [Google Scholar]
- 10.Rutledge, H. L. & Tezcan, F. A. Electron transfer in nitrogenase. Chem. Rev.120, 5158–5193 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Danyal, K., Dean, D. R., Hoffman, B. M. & Seefeldt, L. C. Electron transfer within nitrogenase: evidence for a deficit-spending mechanism. Biochemistry50, 9255–9263 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seefeldt, L. C. et al. Reduction of substrates by nitrogenases. Chem. Rev.120, 5082–5106 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris, D. F. et al. Mechanism of N2 reduction catalyzed by Fe-nitrogenase involves reductive elimination of H2. Biochemistry57, 701–710 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raugei, S., Seefeldt, L. C. & Hoffman, B. M. Critical computational analysis illuminates the reductive-elimination mechanism that activates nitrogenase for N2 reduction. Proc. Natl Acad. Sci. USA115, E10521–E10530 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trncik, C., Detemple, F. & Einsle, O. Iron-only Fe-nitrogenase underscores common catalytic principles in biological nitrogen fixation. Nat. Catal.6, 415–424 (2023). [Google Scholar]
- 16.Rohde, M., Laun, K., Zebger, I., Stripp, S. T. & Einsle, O. Two ligand-binding sites in CO-reducing V nitrogenase reveal a general mechanistic principle. Sci. Adv.7, eabg4474 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
-
17.Schindelin, H., Kisker, C., Sehlessman, J. L., Howard, J. B. & Rees, D. C. Structure of ADP •
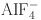 -stabilized nitrogenase complex and its implications for signal transduction. Nature387, 370–376 (1997).
[DOI] [PubMed] [Google Scholar]
-stabilized nitrogenase complex and its implications for signal transduction. Nature387, 370–376 (1997).
[DOI] [PubMed] [Google Scholar] - 18.Dixon, R. & Kahn, D. Genetic regulation of biological nitrogen fixation. Nat. Rev. Microbiol.2, 621–631 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Jacobs, D., Mitchell, D. & Watt, G. D. The concentration of cellular nitrogenase proteins in Azotobacter vinelandii whole cells as determined by activity measurements and electron paramagnetic resonance spectroscopy. Arch. Biochem. Biophys.324, 317–324 (1995). [DOI] [PubMed] [Google Scholar]
- 20.Weiss, M. C. et al. The physiology and habitat of the last universal common ancestor. Nat. Microbiol.1, 16116 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Gallon, J. R. The oxygen sensitivity of nitrogenase—a problem for biochemists and microorganisms. Trends Biochem. Sci6, 19–23 (1981). [Google Scholar]
- 22.Zehr, J. P. Nitrogen fixation by marine cyanobacteria. Trends Microbiol.19, 162–173 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Zhang, L. & Einsle, O. Architecture of the RNF1 complex that drives biological nitrogen fixation. Nat. Chem. Biol.20, 1078–1085 (2024). [DOI] [PubMed] [Google Scholar]
- 24.Dalton, H. & Postgate, J. R. Effect of oxygen on growth of Azotobacter chroococcum in batch and continuous cultures. J. Gen. Microbiol.54, 463–473 (1968). [DOI] [PubMed] [Google Scholar]
- 25.Bulen, W. A. & LeComte, J. R. Nitrogenase complex and its components. Methods Enzymol.24, 456–470 (1972). [DOI] [PubMed] [Google Scholar]
- 26.Moshiri, F., Crouse, B. R., Johnson, M. K. & Maier, R. J. The “nitrogenase-protective” FeSII protein of Azotobacter vinelandii: overexpression, characterization, and crystallization. Biochemistry34, 12973–12982 (1995). [DOI] [PubMed] [Google Scholar]
- 27.Maier, R. J. & Moshiri, F. Role of the Azotobacter vinelandii nitrogenase-protective Shethna protein in preventing oxygen-mediated cell death. J. Bacteriol.182, 3854–3857 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peña, C., Reyes, C., Larralde-Corona, P., Corkidi, G. & Galindo, E. Characterization of Azotobacter vinelandii aggregation in submerged culture by digital image analysis. FEMS Microbiol. Lett.207, 173–177 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Sippel, D. & Einsle, O. The structure of vanadium nitrogenase reveals an unusual bridging ligand. Nat. Chem. Biol.13, 956–960 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kabasakal, B. V. et al. The crystal structure of Shethna protein II (FeSII) from Azotobacter vinelandii suggests a domain swap. Acta Crystallogr. D Struct. Biol.80, 599–604 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tezcan, F. A. et al. Nitrogenase complexes: multiple docking sites for a nucleotide switch protein. Science309, 1377–1380 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Lanzilotta, W. N. & Seefeldt, L. C. Changes in the midpoint potentials of the nitrogenase metal centers as a result of iron protein molybdenum–iron protein complex formation. Biochemistry36, 12976–12983 (1997). [DOI] [PubMed] [Google Scholar]
- 33.Veeger, C. et al. in Nitrogen Fixation Vol. 1 (eds Newton, W. E. & Orme-Johnson, W. H.) 111–137 (University Park Press, 1980).
- 34.del Campo, J. M. S. et al. Overview of physiological, biochemical, and regulatory aspects of nitrogen fixation in Azotobacter vinelandii. Crit. Rev. Biochem. Mol. Biol.57, 492–538 (2023). [DOI] [PubMed] [Google Scholar]
- 35.Buren, S. & Rubio, L. M. State of the art in eukaryotic nitrogenase engineering. FEMS Microbiol. Lett.365, fnx274 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raulfs, E. C., O’Carroll, I. P., Dos Santos, P. C., Unciuleac, M. C. & Dean, D. R. In vivo iron-sulfur cluster formation. Proc. Natl Acad. Sci. USA105, 8591–8596 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Braymer, J. J. & Lill, R. Iron-sulfur cluster biogenesis and trafficking in mitochondria. J. Biol. Chem.292, 12754–12763 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gibson, D. G. et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods6, 343–345 (2009). [DOI] [PubMed] [Google Scholar]
- 39.Miroux, B. & Walker, J. E. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol.260, 289–298 (1996). [DOI] [PubMed] [Google Scholar]
- 40.van Die, I. M., Bergmans, H. E. & Hoekstra, W. P. Transformation in Escherichia coli: studies on the role of the heat shock in induction of competence. J. Gen. Microbiol.129, 663–670 (1983). [DOI] [PubMed] [Google Scholar]
- 41.Lipman, J. G. Experiments on the transformation and fixation of nitrogen by bacteria. Rep. NJ Agric. Exp. Stn 24, 217–285 (1903). [Google Scholar]
- 42.Spatzal, T. et al. Evidence for interstitial carbon in nitrogenase FeMo cofactor. Science334, 940 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scheres, S. H. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol.180, 519–530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol.192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Punjani, A., Zhang, H. & Fleet, D. J. Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat. Methods17, 1214–1221 (2020). [DOI] [PubMed] [Google Scholar]
- 47.Zivanov, J., Nakane, T. & Scheres, S. H. W. Estimation of high-order aberrations and anisotropic magnification from cryo-EM data sets in RELION-3.1. IUCrJ7, 253–267 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Varadi, M. et al. AlphaFold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res.50, D439–D444 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goddard, T. D. et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci.27, 14–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr.60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 52.Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct. Biol.75, 861–877 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr.66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vonrhein, C. et al. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D Biol. Crystallogr.67, 293–302 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Evans, P. R. & Murshudov, G. N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr.69, 1204–1214 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Winn, M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr.67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vagin, A. & Teplyakov, A. MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr.30, 1022–1025 (1997). [Google Scholar]
- 58.Vagin, A. A. et al. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. D Biol. Crystallogr.60, 2184–2195 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
3D variability analysis of the FeSII:Mo-nitrogenase complex structure. A 3D variability analysis carried out using CryoSPARC highlights the flexible binding of the two copies of the Fe protein NifH2 to the dimeric FeSII protein. In contrast, the interaction of the small ferredoxin with the MoFe protein NifD2K2 shows substantially higher rigidity.
Data Availability Statement
The atomic coordinates and cryo-EM map of the FeSII–nitrogenase complex have been deposited with the Protein Data Bank at http://www.pdb.org with accession number 8RHP and the Electron Microscopy Data Bank (EMDB) with accession number EMD-19178. Coordinates and structure factors for the reduced form of FeSII have been deposited with the Protein Data Bank at http://www.pdb.org with accession number 8RHO.