

Summary
Enteropathogenic infections cause pathophysiological changes in the host but their effects beyond the gastrointestinal tract are undefined. Here, using Citrobacter rodentium infection in mouse, which mimics human diarrheal enteropathogenic Escherichia coli, we show that gastrointestinal infection negatively affects bone remodeling, leading to compromised bone architecture. Transmission of infection through fecal-oral route from Citrobacter rodentium-infected to non-infected mice caused bone loss in non-infected cage mates. Mice with B cell deficiency (Igh6−/− mice) failed to clear C. rodentium infection and exhibited more severe and long-term bone loss compared to WT mice. Unbiased cytokine profiling showed an increase in circulating tumor necrosis factor α (TNFα) levels following Citrobacter rodentium infection, and immunoneutralization of TNFα prevented infection-induced bone loss completely in WT and immunocompromised mice. These findings reveal rapid, relaying, and modifiable effects of enteropathogenic infections on an extraintestinal organ—bone, and provide insights into the mechanism(s) through which these infections affect extraintestinal organ homeostasis.
Subject areas: Gastroenterology, Molecular physiology, Immune response
Graphical abstract
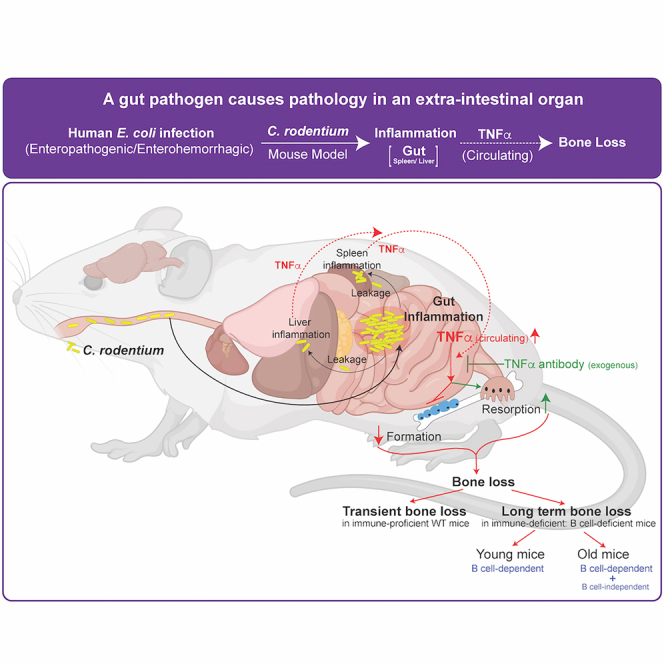
Highlights
-
•
Enteropathogenic E. coli infection in a mouse model damages bone, a gut-distant organ
-
•
Transmission of infection causes bone loss in non-infected cage mates
-
•
B cell-deficient mice unable to clear infection have more severe and sustained bone loss
-
•
Infection-induced increase in circulating TNFα levels contribute to bone loss
Gastroenterology; Molecular physiology; Immune response
Introduction
Escherichia coli (E. coli) is the predominant gram-negative anaerobic bacterium in human colonic flora. It typically colonizes the gastrointestinal (GI) tract of infants within hours of the inception of life and, thereafter, the bacterium and the host remain in a mutually beneficial relationship.1 The pathogenic strains of E. coli, when acquired, attack and invade the intestinal mucosa and secrete toxins, leading to exudation of water and electrolytes, causing enteritis and resulting in ∼1,000,000 deaths per year.2,3 Immunocompetent hosts are capable of clearing the infection within few days or weeks.2 Transmission of pathogenic E. coli to other individuals, often via the fecal-oral route or unhygienic food preparations, leads to deadly outbreaks, particularly in children, the elderly, and immunocompromised individuals.2,4 Human mucosal infections with enteropathogenic and enterohemorrhagic E. coli (EPEC/EHEC) are effectively modeled in mice using a bacterial pathogen, Citrobacter rodentium (C. rodentium).5,6,7 Similar to EPEC and EHEC, C. rodentium infection progresses through colonization, symptomatic, and convalescent phases.8 At the cellular level, cells of the adaptive immune system, such as B lymphocytes, make important contributions in protecting against C. rodentium infection, and mice depleted of B lymphocytes have an impaired ability to clear the infection.9,10,11,12 The pathophysiological consequences of C. rodentium infection beyond the gut remain poorly understood.
In humans, changes in the gastrointestinal microbiome, such as those seen in antibiotic challenges or colitis, are associated with low bone density.13,14 Bones are an integral component of the vertebrate organ system that perform many functions, from supporting soft organs to affecting functions of other organs.15,16,17 To perform these functions, bones are constantly renewed through the process of bone remodeling (BR), which comprises bone resorption by osteoclasts followed by bone formation by osteoblasts.18,19,20,21,22,23 Any dysregulation in BR that occurs as part of normative aging or due to other physiological perturbations leads to osteoporosis, a generalized decrease in bone density resulting in an increased risk of fractures, especially in the elderly.18,19,20,21,22,23,24 While nutritional and genetic aspects of osteoporosis have been well described, it is increasingly becoming apparent that environmental risk factors, immune dysregulation, and gastrointestinal perturbations play an important role in the pathogenesis of osteoporosis.25,26,27,28 The role(s) of host-pathogen interactions, such as those elicited by C. rodentium, and associated mechanisms, in regulating bone density are unknown.
During the process of bacterial clearance from the gastrointestinal tract, immune cells produce cytokines, such as tumor necrosis factor α (TNFα) and CCL2, and lipids, such as PGE2, which are crucial for microbial clearance in a number of mucosal and systemic models, including infection with C. rodentium.29,30 TNFα is an established immune modulator in normal and chronic inflammatory situations, including gastrointestinal inflammation.31 In certain inflammatory conditions, TNFα has been shown to induce bone loss through a decrease in the number and function of osteoblasts, and causes an increase in the differentiation and function of osteoclasts.32,33,34,35,36 Thus, TNFα blockade can potentially limit or reverse bone loss under inflammatory conditions.37,38,39 The effect of enteropathogenic infections on peripheral organ homeostasis, such as bone, under healthy or immunocompromised conditions, and the significance of TNFα and B lymphocytes in this process is undefined.
Using a mouse model of C. rodentium that mimics EPEC/EHEC infections, we show that this infection leads to rapid bone loss, and transmission of infection to uninfected cage-mates decreases their bone volume, as well. Furthermore, our results show that immunocompromised B cell-deficient mice have more severe and sustained bone loss following infection. Through immunoneutralization studies, we illustrate a critical role of circulating TNFα in infection-induced bone loss.
Results
C. rodentium infection causes bone loss in wild-type mice
To investigate the plausible role of host-microbe interactions in the pathogenesis of enteropathogenic infections beyond the gut, we used a mouse model of C. rodentium infection, which mimics enteropathogenic and enterohemorrhagic E. coli infections observed in humans.8 To assess the effect of C. rodentium infection on bone health, 6 weeks-old adult C57Bl/6J wild-type (WT) mice were orally inoculated with a single dose (200 μL) of vehicle or C. rodentium suspension (Figure 1A). We first confirmed C. rodentium infection dynamics by analyzing salient features, viz. fecal bacterial load, gastrointestinal pathology, and peripheral organ colonization (for a detailed description of C. rodentium infection dynamics and colon histopathology, please see previous studies by others and us9,40). Bacterial shedding curve analysis in the feces estimated using the C. rodentium colony forming assay revealed that bacterial numbers peaked within 1 week of infection (1.0 E+06 to 1.0 E+08 CFU/mL of feces), the levels remained elevated till the second week, and declined thereafter to their lowest levels by the third week (Figure 1B). Although C. rodentium was not detectable in the bone and blood at any stage post inoculation, it was detectable, albeit at levels >100,000-fold lower compared with those in the colon, in the spleen and liver, collected on D12 from infected mice (Figure 1C). Four weeks post infection, bacteria were not detected in the colon or in any other peripheral organs tested (Figure 1C). We confirmed that changes in bacterial colonization of the gut also reflected changes in colon weight and colonic hyperplasia, which is known to be associated with C. rodentium infection (Figures 1D and S1A). Notably, C. rodentium-infected mice had no explicit differences in the terminal body weight (BW) acquisition (2.4 ± 0.2 g versus 2.2 ± 0.4 g) and appetite (on day 14) (3.1 ± 0.5 g versus 2.9 ± 0.2 g) with respect to that in vehicle controls. These results confirmed that C. rodentium inoculation in WT mice leads to rapid intestinal colonization within few days; some bacteria at the peak of infection leak to other peripheral organs, such as the liver and spleen, but not to the bone, and infection is cleared by 4 weeks post-inoculation in WT mice.
Figure 1.

Citrobacter rodentium infection of the gut causes bone loss in wild-type mice
(A) Schematic representation of the experimental regimen.
(B) Bacterial shedding analysis using the colony forming unit (CFU) assay in feces at different time points post inoculation of vehicle/C. rodentium in wild-type (WT) mice.
(C) The CFU assay in feces and blood collected on day 12, and in the spleen, liver, and bone collected on day 12 and 28 from WT mice inoculated with vehicle/C. rodentium. Scale bar, 20 mm.
(D) Representative images showing the colon histology, 2 weeks post vehicle/C. rodentium inoculation in WT mice and colonic hyperplasia in infected mice. Scale bar, 100 μm.
(E and F) Representative images of lumbar 4 vertebra, with quantification of bone volume to total volume percentage (BV/TV%) in female (E) and male (F) WT mice, 4 weeks post vehicle/C. rodentium inoculation. Scale bar, 1 mm.
(G–I) Toluidine blue staining of osteoblasts with quantification of Ob.N./T.Ar. and Ob.S./BS showing reduced osteoblast numbers and surface on bone (G), calcein double labeling detecting bone formation fronts, and quantification of bone formation rate and mineral apposition rates (H), and tartrate-resistant alkaline phosphatase-stained osteoclasts (red), with quantification of osteoclast numbers per bone perimeter, osteoclast surface per bone surface (Oc.S./BS), and serum deoxypyridinoline levels (I) in bones of female mice, 4 weeks post vehicle or C. rodentium inoculation. Osteoblasts are marked with arrows, and osteoclast surface with red line on the bone surface (dotted lines). Scale bar, 20 μm (G). Scale bar, 50 μm (H). Scale bar, 20 μm (I). Values are mean ± SEM. n for each group is indicated within each panel. ∗p < 0.05.
We next analyzed the bone architecture in C. rodentium-infected and control WT mice, 4 weeks post-oral inoculation. Bone histological and histomorphometric analysis of trabeculae-rich bones (vertebra) revealed a significant decrease in trabecular bone volume in C. rodentium-infected female mice compared with that in controls (Figure 1E). A similar decrease in trabecular bone volume was noted in male mice after infection (Figure 1F). Analysis of long bone architecture in infected mice using the micro-computed tomography (μCT) analysis revealed a decrease in trabecular bone volume (BV/TV%), characterized by a decrease in trabecular number (Tb.N.), trabecular thickness (Tb.Th.), and an increase in trabecular separation (Figure S1B). Analysis of cortical thickness (Ct.Th.) in the femur revealed a decrease in the infected mice compared with that in controls (Figure S1B). To identify changes in the bone cell types (osteoblasts, bone forming cells; osteoclasts, bone resorbing cells) involved in bone loss following C. rodentium infection, we performed histological analysis of trabecular bone in vertebrae. Analysis of osteoblast parameters in the bones of infected mice showed >1.6-fold decrease in osteoblast numbers per trabecular area (Ob.N./T.Ar.), and osteoblast surface per bone surface (Ob.S./T.Ar.) compared with that in controls (Figure 1G). Accordingly, bone formation (BFR) and mineral apposition rates (MAR), which are measures of the activity of osteoblasts, were decreased in infected mice compared with that in controls (Figure 1H). C. rodentium infection also affected bone resorption parameters because osteoclast numbers and surface per bone surface as well as serum levels of deoxypyridinoline (Dpd) were higher in the infected mice (Figure 1I). Together, the results of bone histological analysis suggest that C. rodentium infection causes bone loss, in both sexes, through a concomitant decrease in bone formation and an increase in bone resorption parameters.
Transmission of C. rodentium infection causes bone loss in initially uninfected WT cage mates
In human pathogenic strains of E. coli are transmitted from infected to uninfected humans through unhygienic food preparations. In mice, enteric bacterial infections are rapidly transmitted to non-infected cage mates due to their coprophagic behavior. We, therefore next asked whether cohousing C. rodentium-inoculated mice with non-inoculated mice causes bone loss in cage-mates not initially infected with C. rodentium. To address this question, we inoculated two mice with C. rodentium culture and two other mice in the same cage with vehicle (Figure 2A). Seven such cages, with a total of 14 mice initially inoculated with C. rodentium culture and 14 mice inoculated with vehicle were used. Colony forming assays using feces samples showed a rapid increase in C. rodentium in the feces of C. rodentium-inoculated mice, which as expected, proceeded to a delayed increase in C. rodentium in the feces of vehicle-inoculated cage mates (Figures 2B and 2C). Body weight was not significantly affected in C. rodentium-infected mouse groups compared with that in non-infected controls (Figure S2A). Bone histomorphometric analysis, 4 weeks post-oral inoculation, revealed that trabecular bone volume measured using histology (Figure 2D), and trabecular and cortical parameters of long bone measured using μCT (Figure 2E) were decreased in both C. rodentium- and vehicle-inoculated cage mates that subsequently contracted the infection compared with those in vehicle-treated non-cage mate controls. Histological and histomorphometric analyses of vertebrae revealed a decline in osteoblast parameters (osteoblast number, bone formation, and MAR; Figures 2F and 2G), and an increase in osteoclast parameters (Figure 2H). These results show that contraction of C. rodentium infection leads to bone loss even in cage mates that were not originally infected with C. rodentium.
Figure 2.

Transmission of Citrobacter rodentium infection causes low bone volume in initially uninfected cage-mates
(A) Schematic representation of the experimental regimen used.
(B and C) Bacterial shedding analysis using the colony forming unit (CFU) assay in feces at different time points (B), and bacterial CFU analysis at day 16 in feces (C) post vehicle/C. rodentium inoculation in infected and uninfected cage mates. Scale bar, 20 mm.
(D) Histological and histomorphometric analysis of lumbar 4 vertebra, with quantification of bone volume over total volume % (BV/TV%). Scale bar, 1 mm
(E) 3D μCT images of proximal tibia, with quantification of bone volume over total volume % (BV/TV%), trabecular numbers (Tb.N.), trabecular thickness (Tb.Th.), trabecular separation (Tb.Sp.), and cortical thickness (Ct.Th.), 4-week post vehicle/C. rodentium inoculation in infected and uninfected cage mates. Scale bar, 1 mm.
(F–H) Representative photomicrographs of toluidine blue-stained osteoblasts and quantification of osteoblast numbers per trabecular area (Ob.N./T.Ar.) (F), bone formation rate (BFR) and mineral apposition rate (MAR) (G), and osteoclast surface per bone surface % (OcS/BS%) (H) 4-week post vehicle or C. rodentium inoculation in infected and initially uninfected cage mates. Scale bar, 20 μm (F). Scale bar, 50 μm (G). Scale bar, 20 μm (H). Values are mean ± SEM. n for each group is indicated within each panel. ∗p < 0.05.
Immunocompromised Igh6−/− mice that lack B cells show a more severe and sustained bone loss following C. rodentium infection
The lymphocytic host response to C. rodentium infection is characterized by mucosal infiltration of immune cells, which is followed by an inflammatory response and clearance of bacteria from the lumen, approximately 4 weeks post infection. B lymphocytes have been shown to be responsible for the clearance of C. rodentium from the intestinal lumen approximately 4 weeks post challenge in the WT mice, and in the absence of B cells, as in Igh6−/− mice, bacterial infection is sustained at least up to 8 weeks.9,41 We, therefore, used B cell-deficient mice to investigate whether these mice would exhibit a more severe bone loss at 4 weeks following C. rodentium infection and whether this effect would be sustained for a longer time compared with that seen in the WT mice.
WT and Igh6−/− mice were inoculated with vehicle or C. rodentium cultures and sacrificed at 4- and 7-week post infection (Figure 3A). We first measured salient features of infection susceptibility of Igh6−/− mice using CFU analysis in the feces, liver, spleen, and bone at 1-, 3-, and 7-week following oral challenge. A detailed description of infection susceptibility of the GI tract and other peripheral organs to C. rodentium infection of Igh6−/− mice has been previously provided by others and us.9,10 The results showed that B cell deficiency led to a sustained infection, and that fecal bacterial load remained high at 4- and 7-week post inoculation, time points at which infected WT mice had the lowest bacterial load (Figures 3A and 3B, and S3A–S3D). This analysis also showed that, following inoculation, bacterial load was comparable between WT and Igh6−/− mice in the feces, but was much higher in the spleen and liver of Igh6−/− mice compared with that in the WT mice (Figures 3B and S3A–S3C). No bacteria were detected at any time point post inoculation in the bone tissue (Figures 3B and S3D).
Figure 3.

Immunocompromised mice that lack B cells (Igh6−/−) show more severe and sustained bone loss than wild-type (WT) mice following Citrobacter rodentium infection
(A) Schematic representation of the experimental regimen used and bacterial shedding analysis using the colony forming unit (CFU) assay in feces at different time points in WT (uninfected), WT (infected), Igh6−/− (uninfected), and Igh6−/− (infected) mice.
(B) Colony forming unit (CFU) assay in feces, bone, spleen, and liver at day 42 post vehicle or C. rodentium inoculation in WT (uninfected), WT (infected), Igh6−/− (uninfected), and Igh6−/− (infected) mice. Scale bar, 20 mm.
(C and D) 3D μCT images of proximal tibia, with quantification of bone volume over total volume percentage (BV/TV%) and cortical thickness (Ct. Th.) 4- (C) and 7-week (D) post vehicle/C. rodentium inoculation in WT or Igh6−/− mice. Scale bar, 1 mm.
(E) Histological and histomorphometric analysis of lumbar 4 vertebra (Scale bar, 1 mm), with quantification of bone volume over total volume % (BV/TV%), osteoblast numbers per trabecular area (Ob.N./T.Ar. Scale bar, 20 μm), bone formation rate (BFR; Scale bar, 50 μm), mineral apposition rate (MAR), and osteoclast surface per bone surface % (OcS/BS%; Scale bar, 20 μm) 7-week post vehicle/C. rodentium inoculation in WT or Igh6−/− mice. Representative micrographs for different histological analysis are shown in panel E. Values are mean ± SEM. n for each group is indicated within each panel. ∗p < 0.05.
After confirming that the absence of B cells caused impaired clearance of bacteria in Igh6−/− mice, we compared the bone volume and architecture in Igh6−/− mice with that in WT mice at 4- and 7-week post C. rodentium infection. At 4-week post infection, analysis of long bone architecture using μCT revealed that vehicle-administered B cell-deficient mice have a lower BV/TV% and Ct. Th. as compared with WT litter-mate controls (Figure 3B)—a result consistent with a previous report.42 At 4 weeks post infection, Igh6−/− mice had a more profound decrease in the BV/TV% compared with that in WT infected mice (Figure 3C). Due to sustained infection, Igh6−/− mice, even after 7-week post C. rodentium inoculation—a time point at which WT infected mice had no fecal bacterial load, and comparable bone parameters to vehicle inoculated WT controls—had compromised bone architecture parameters (Figures 3A, 3D, and S3A–S3D). Bone histological analysis of the vertebrae showed that low bone architecture of Igh6−/− mice affected trabecular bone volume and was caused by a decrease in osteoblast parameters (osteoblast numbers, bone formation, MAR), and an increase in osteoclast parameters (Figure 3E). Bone formation decreased further in C. rodentium-infected B cell-deficient mice compared with that in vehicle-inoculated B cell-deficient mice at 7 weeks (Figure 3E). The presence of higher bacterial load following infection in extraintestinal organs, together with more drastic bone loss in Igh6−/− compared with that in WT mice, suggests that inflammation of other organs could contribute more to the C. rodentium infection-induced bone loss in Igh6−/− mice relative to that seen in WT mice following infection (Figures 3B and S3A–S3D). We note here that trabecular bone loss in the long bones (Figure 3D) are much more pronounced compared to what is seen in the vertebra (Figure 3E).
C. rodentium infection-induced bone loss is associated with increased levels of TNFα
Our analysis indicated that in C. rodentium-infected mice, at least during the initial stages of infection, colon environment secretes molecule(s) in circulation that cause bone loss. To identify the proinflammatory or other cytokines that are associated with infection-induced bone loss, we performed longitudinal cytokine profiling of sera collected from control and C. rodentium-infected mice at 1-, 2-, and 4-week post infection (Figure 4A). The results showed that C. rodentium infection caused a significant increase in serum levels of TNFα and a decrease in RANTES levels at 7 days post infection (Figure 4B), when infection was at its peak, as shown in Figure 1B. Serum levels of TNFα peaked within 1-week post-infection and declined gradually, thereafter (Figure 4C), suggesting that TNFα could be one of the cytokines involved in bone loss observed post C. rodentium infection. We reasoned that if TNFα is involved in this process, then Igh6−/− mice that show more sustained bone loss up to 7 weeks would show higher TNFα levels at that time point. Conversely, WT mice that reverted back to normal gut flora and bone architecture by 7 weeks would show normal TNFα levels. Indeed, TNFα levels in serum and the expression of TNFα gene in the colon showed a sustained increase in B cell-deficient mice at 4- and 7-week post infection, whereas the levels in WT infected mice were equivalent to those in uninfected WT controls (Figures 4D and S4A).
Figure 4.

Tumor necrosis factor α (TNFα) mediates Citrobacter rodentium infection-induced bone loss
(A) Schematic representation of the experimental regimen and heatmap showing levels of various cytokines in wild-type (WT) mice at 1-, 2-, and 4-week post vehicle/C. rodentium inoculation.
(B) Bubble plot showing levels of cytokines in WT mice at 1 week post vehicle/C. rodentium inoculation.
(C) TNFα levels in WT mice at 1-, 2-, and 4-week post vehicle/C. rodentium inoculation.
(D) TNFα levels in WT or Igh6−/− mice at different time points post vehicle or C. rodentium inoculation.
(E and F) Histological and histomorphometric analysis of lumbar 4 vertebra (Scale bar, 1 mm), with quantification of BV/TV%, Ob.N/T.Ar, Oc.S/BS%, and serum osteocalcin levels (E), and 3D μCT images of proximal tibia, with quantification of BV/TV% and Ct. Th. (F) 4 weeks post vehicle/C. rodentium inoculation in WT mice with or without weekly intravenous injections of TNFα neutralizing antibody. Scale bar, 1 mm.
(G) Disease activity index [DAI] score during the course of a noninfectious colitis inducing chemical dextran sodium sulfate (DSS; 2 weeks), and following recovery period of 6 weeks.
(H) 3D μCT images of proximal tibia, with quantification of BV/TV% and Ct.Th. at 2 and 8 weeks in the DSS-induced colitis model. Scale bar, 1 mm.
(I) Serum TNFα levels at 2 and 8 weeks in vehicle [V] or DSS [D] challenged mice in DSS-induced colitis model.
(J) Histological analysis of lumbar 4 vertebra, with quantification of BV/TV%, 3 weeks post vehicle/DSS administration in WT mice with or without weekly intravenous injections of TNFα neutralizing antibody. Values are mean ± SEM. n for each group is indicated within each panel. ∗p < 0.05; ∗∗p < 0.01.
These findings suggest a plausible model in which spiked levels of TNFα secreted by the immune cells in the colon environment, and possibly in other organs, could contribute toward Citrobacter infection-induced bone loss.
A TNFα neutralizing antibody prevents C. rodentium infection-induced bone loss in WT and B cell-deficient mice
Does increased circulating levels of TNFα contribute toward bone loss following C. rodentium infection of the gastrointestinal tract? To address this question, we administered a neutralizing antibody against TNFα through intravenous route a day before the C. rodentium inoculation, and weekly thereafter until the mice were sacrificed at 4 weeks. Administration of TNFα neutralizing antibody to WT or Igh6−/− uninfected mice did not affect their trabecular bone volume in long bones (μCT analysis) compared with that in controls (Figure S4B). In addition, cohousing Igh6−/− with WT mice for 4 weeks prior to C. rodentium infection did not affect the magnitude of bone loss, indicating that basal changes in the microbial population did not affect the response of Igh6−/− mice to C. rodentium infection-induced decrease in bone architecture (Figure S4C). Moreover, colon pathology and serum cytokine profile in Igh6−/− mice under baseline uninfected conditions were similar to that seen in WT mice.9 Analysis of fecal bacterial load on day 14 in WT infected mice that received either TNFα antibody or vehicle showed that TNFα neutralization did not affect the infection dynamics (Figure S4D); this observation suggests that clinical course of the disease is not affected by the anti-TNFα treatment, and is consistent with the findings in a previous study.43 Bone histomorphometric analysis of vertebrae, 4 weeks post infection, showed decreased trabecular bone volume in vehicle-injected C. rodentium-infected mice (Figure 4E). In contrast, mice that were inoculated with C. rodentium and received weekly TNFα neutralizing antibody had a bone architecture similar to that in the uninfected WT controls (Figure 4E). This prevention of bone loss in TNFα neutralizing antibody-treated mice was associated with a normalization of osteoblast and osteoclast parameters (Figures 4E and S4E). Micro-computed tomography analysis of the tibia revealed that administration of TNFα neutralizing antibody prevented bone loss in the appendicular skeleton, as well (Figure 4F). Administration of TNFα neutralizing antibody in the WT uninfected mice did not affect bone mass in the appendicular skeleton (Figure S4F).
We next investigated whether TNFα mediates bone pathology in a non-infectious colitis model. For this purpose, we treated mice with dextran sodium sulfate (DSS), a molecule that severely compromises intestinal barrier leading to “systemic inflammation” and colitis.44,45 DSS treatment for two weeks rapidly decreased body weight, and increased disease activity index (DAI) score reflected in increased colon damage, epithelial disintegration, and inflammatory cell infiltration compared to controls (Figures 4G and S4G). The μCT analysis of long bones and vertebra histology revealed that trabecular bone volume was reduced in DSS-treated mice (Figures 4H and S4H). Surprisingly, however, cortical thickness was not affected 2 weeks post DSS, and was only decreased at 8 weeks in the DSS recovered mice (Figures 4H and S4I). Even after 4 weeks of DSS challenge only a decrease in trabecular bone volume and not cortical thickness was seen (Figure S4J). A comparison of bone pathology caused by DSS-induced barrier damage and C. rodentium mucosal infections in the intestine showed that trabecular bone volume was more severely decreased by DSS treatment, and cortical bone loss was more rapid following C. rodentium infection compared with that in the DSS-induced colitis model (Figures S4I, S4H, 4E, and 4F). Serum levels of TNFα were increased after 2 weeks of DSS treatment, as was observed after C. rodentium infection, and the levels returned back to normal after 6 weeks of recovery period (Figure 4I). Neutralization of circulating TNFα, however, did not prevent DSS-induced trabecular bone loss (Figure 4J). The latter result when viewed in the context of differences in bone architectural changes and a major decrease in body weight in DSS versus C. rodentium infection (Figures 4H, S4H, 4E, and 4F) indicates that other endocrine or local factors, in addition to TNFα, are involved in DSS-induced bone pathology.
We next investigated whether the observed increase in circulating levels of TNFα in B cell-deficient mice following C. rodentium infection contributes to accelerated bone loss in these mice. Analysis of bones from 16-week-old Igh6−/− mice treated with either a TNFα neutralizing antibody or vehicle, before the C. rodentium infection showed that treatment with the neutralizing antibody decreased infection-induced bone loss in the Igh6−/− mice (Figure S4K). However, the bone volume in antibody-treated infected Igh6−/− mice were similar to vehicle-injected uninfected Igh6−/− mice, and the bone volume remained significantly lower than the vehicle-injected uninfected WT mice (Figure S4K). When viewed in conjunction with no baseline changes in gastrointestinal inflammation in Igh6−/− mice, and considering the fact that B cells are produced and home in bone marrow,46 these findings suggested that the baseline decrease in trabecular bone volume and cortical thickness following B cell deficiency and prior to infection could be originating within the bones, and that this process may be independent of gut inflammation. Further supporting the role of B cells within the bone was the fact that B cells produce osteoprotegerin (OPG),47 a secreted factor that potently inhibits bone resorption.48 We measured OPG mRNA levels in the bone marrow (BM) cells collected from WT and Igh6−/− mice. Results showed that OPG mRNA levels were decreased by >70% in the whole BM collected from 16-week-old Igh6−/− mice compared with those in WT controls (Figure S4L). These results when viewed in context of significant loss of bone density in OPG heterozygous mice49 suggested that OPG insufficiency could contribute to bone loss in 16-week-old Igh6−/− mice. To determine the specific contribution of compromised production of B cell-derived OPG in low bone volume in 16-week-old Igh6−/− mice, we performed a rescue experiment in which 6-week-old Igh6−/− mice that are deficient in B cells were replenished with WT B cells through adoptive transfer. Analysis of Igh6−/− mice 8 weeks after B cell replenishment revealed that the B cell transfer normalized OPG levels and rescued bone volume in Igh6−/− mice to the levels seen in controls (Figures S4L and S4M).
The previously analysis shows that the low baseline bone density seen in the B cell deficient Igh6−/− mice is caused by an impaired production of B cell-derived OPG within the bones. However, the low bone density observed at the baseline in 16-week-old B cell-deficient mice was a confounding factor and limited the assessment of specific contribution of TNFα in causing bone loss in Igh6−/− mice following C. rodentium infection. To circumvent this problem, we utilized younger 6-week-old B cell-deficient mice, which did not show a compromised bone architecture (Figure S4N), for TNFα immunoneutralization following infection. C. rodentium infection induced a decline in bone volume in vehicle-injected Igh6−/− mice, but animals administered TNFα antibody showed bone volumes similar to that in uninfected WT controls (Figure S4N). Together, these results show that the observed rise in circulating TNFα levels post C. rodentium infection is one of the key events that is responsible for the decrease in bone architecture in WT and young B cell-deficient mice. In the older B cell-deficient mice although the infection-induced bone loss is recovered through TNFα immunoneutralization the baseline bone loss caused by a B cell dependent changes in OPG production remains. Together, the results in young and old B cell deficient mice illustrate importance of B cells in intestinal infection-induced bone loss during rapid growth phase of bone as in young mice and in more homeostatic changes in bone as seen in older B cell-deficient mice.
Discussion
The main findings of the present study are that C. rodentium infection causes bone loss in WT mice, and that the loss is more severe and long-term in immunocompromised B cell-deficient mice. C. rodentium infection is restricted mostly to the gut; however, some bacteria, albeit very less, do leak at the peak of infection to other organs, but not to the bones. Infection-induced changes in bone architecture were observed much later after the infection had been completely cleared from the system in the WT mice, suggesting that the effect of a gastrointestinal pathogen lasts much longer after the pathogen has been cleared from the host. Cytokine and cellular phenotyping analyses showed that circulating TNFα plays an important role in infection-induced changes in bone architecture in WT mice, and due to compromised bacterial clearance, the infection causes more severe and sustained bone loss in immunocompromised conditions.
The beneficial effects of intestinal symbiotic microflora on BR have been known for some time50,51; however, to our knowledge this is the first report that shows an effect of a gastrointestinal pathogen on the bone. Our results show that during the initial stages of C. rodentium colonization, infection is restricted to the gut lumen (days 2–3). However, at later stages of infection, some bacteria cross the gut barrier and colonize, although minimally, the liver and spleen, but not the bone (day 14 post infection). In the B cell-deficient genetic mouse model, the gut barrier is further compromised, and bacteria affect systemic tissues more prominently, albeit at 3- to 4-fold lower levels, and are detectable in the liver, spleen, and lymph nodes, but not in the bone. Therefore, an increase in inflammation of other peripheral organs, viz., the liver and spleen, could contribute to the C. rodentium infection-induced inflammation culminating in bone loss in mice. We note that we only analyzed long bones for bacterial load, and it is possible that other bones could have C. rodentium colonization. Moreover, owing to technical limitations in obtaining a large amount of blood from WT infected mice, it is very much possible that some bacteria could be present in the circulation.
Analysis of bone loss in non-infectious DSS-induced colitis model reveals that the structural and molecular basis of bone pathology in this model is different from that seen after mucosal C. rodentium infection. First, DSS model severely compromises the intestinal barrier and leads to a major decrease in body weight, which is not affected by C. rodentium infection. Second, DSS treatment induced a decrease only in trabecular but both trabecular and cortical bones were rapidly decreased following C. rodentium infection. Third, although TNFα neutralization completely prevented bone loss in the C. rodentium infection model, it did not affect bone loss caused by DSS-induced colitis, suggesting that in the DSS-induced bone loss model other yet to be identified components, such as immune cells, pro-inflammatory cytokines, or nutritional axes might contribute to bone loss. For instance, DSS-induced gut inflammation also drives, in the gut-associated lymphoid tissues,52,53 the differentiation of Th17 cells, which have been shown to contribute to bone loss in the murine periodontitis model54 and human.55,56 Malabsorption of some nutrients, such as vitamin B12 from ileum,57 a part of small intestine affected by DSS,58 that positively regulate bone architecture,59 may further compound bone regulation in the DSS model. We, however, acknowledge the fact that these differences noted between C. rodentium- and DSS-induced colitis models may differ in other models of non-infectious colitis. These questions need to be addressed in future studies.
How does infection restricted to non-skeletal organs affect the bone volume? Our unbiased cytokine profiling showed that spiked levels of TNFα in the blood contribute to bone loss following infectious colitis. Given that some bacteria are present in other organs, such as the liver and spleen, TNFα secreted from these organs, at the peak of infection, could potentially contribute to bone loss. These changes in cytokine levels are in concordance with previous reports of a post infection increase in serum TNFα levels.60 However, our studies do not rule out that other circulating molecules besides TNFα could also contribute to bone loss in C. rodentium-induced colitis models because in this model, the beneficial effects exerted by endogenous gut microbiota, such as serotonin, and IGF-1 synthesis, could also be compromised61,62,51,.63 Although we did not determine the cellular source of TNFα in this study, previous studies have shown that TNFα+ T cells and Th17 cells can affect bone volume under certain conditions, such as gonadal failure.28 It is also possible that colon-resident immune cells or those that cause an inflammatory response in other organs, such as the spleen and liver, during infectious colitis can increase circulating TNFα levels that could contribute to bone loss.60 Future studies are, thus, needed to identify the immune cell repertoire that secretes TNFα in the circulation, in different organs, and possibly in the bone marrow, as it may provide means to neutralize spiked TNFα levels without compromising infection clearance and would restrict extraintestinal damages caused by infectious colitis.
Although our data show that TNFα levels are increased in parallel with bone loss induced by C. rodentium in B cell-deficient mice up to 7-week post infection, TNFα alone is not entirely responsible for the sustained infection-induced bone loss in 16-week-old B cell-deficient mice because the administration of TNFα-neutralizing antibodies only partially protected the mutant mice from infection-induced bone loss. Indeed, our analysis further highlights that B cell deficiency regulates BR locally in the older mice through a compromised synthesis of OPG, a factor secreted by B cells. The beneficial role of B cells in the BR during normative aging in the present and previous studies42 is in contrast to their detrimental roles in cell functions in some other organ systems.64 However, in younger 6-week-old mutant mice, OPG produced by the B cells does not appear to contribute in any significant way to BR, and thus the young mutant mice have normal bone volume at the baseline. Importantly, younger 6-week-old mutant mice also showed accelerated bone loss following C. rodentium infection, and TNFα neutralization completely prevented bone loss caused by infection suggesting that impaired clearance of bacteria following B cell deficiency causes long-term bone loss and that increased TNFα levels mediate bone loss in this model as well. Our findings of increased susceptibility of bone tissue to C. rodentium infection in an immunocompromised condition associated with elevated TNFα levels may be relevant for clinical conditions, such as infections in immune-compromised patients, infants, and young animals who are unable to clear the infections.65,66,67
The cellular organization of an organism is complex and interorgan communications affect their (patho)physiology. Using C. rodentium infection in mice as a model, we illustrate that gastrointestinal infection affects extra-intestinal organ physiology, such as bone, long after the infection has been cleared from the host. With the newfound understanding that pathogenic C. rodentium infections affect the bone volume in a mouse model, it will now be important to analyze the changes in BR following E. coli infections in humans, especially under immunocompromised conditions. Furthermore, it will now be crucial to identify genes and pathways, the contribution of other organs, if any, and other endocrine factors besides TNFα, that underlie bone loss induced by C. rodentium. Additionally, future studies need to address whether other enteropathogenic infections have similar deleterious effects on bone, and whether immunoneutralization of TNFα in a curative regimen will treat extraintestinal pathologies caused by these pathogens. Our study highlights the need to look at other organs beyond the bone that may be affected as a result of gastrointestinal infections despite not showing the presence of bacteria.
Limitations of the study
In this work, we have provided evidence that gastrointestinal infection by C. rodentium, which is restricted to the gut and mimics human diarrheal enteropathogenic E. coli, causes deleterious changes in bone, an extraintestinal organ. We also provide insights on how infection leads to secretion of inflammatory cytokine TNFα that contributes to bone loss, and suggest TNFα neutralization as a potential therapeutic target in controlling extraintestinal pathologies caused by such infections. Three of the limitations that will be addressed in the immediate future are: how TNFα action on bone cells underlies infection-induced bone loss? Through the use of mouse genetic models of TNFα action it needs to be determined whether TNFα is solely responsible for infection induced bone loss or there are other factors that contribute to this process, and lastly what is the evidence that gastrointestinal infections may affect extraintestinal organs in humans? More work is therefore needed to address these important questions and our future studies will be focused on these issues.
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Vijay K. Yadav (vy78@njms.rutgers.edu).
Materials availability
This study did not generate new unique reagents or mouse lines.
Data and code availability
-
•
All data reported in this article will be shared by the lead contact upon request.
-
•
This article does not report the original code.
-
•
Any additional information required to reanalyze the data reported in this article is available from the lead contact upon request.
Acknowledgments
This work was supported by the Wellcome Trust grant 098051 (V.K.Y. and G.D.), NIH Grants HD107574 (V.K.Y.), AG079980 (V.K.Y.), Health@InnoHK (G.D.), Innovation and Technology Commission Funding (G.D.), DK098378 (I.I.I.), and AI144808 (I.I.I.). We apologize to numerous investigators whose work has not been cited due to space constraints. We thank the Molecular Pathology Shared Resource of the Herbert Irving Comprehensive Cancer Center at Columbia University for imaging, supported by NIH grant #P30 CA013696. We thank the Animal Research Support Facility staff at Columbia University, Wellcome Sanger Institute, National Institute of Immunology and National Center for Cell Science for assistance with animal experiments.
Author contributions
Conceptualization: V.K.Y.; methodology: V.K.Y., G.D., X.E.G., and S.C.; investigation: K.S., C.B., N.H., S.R.M., M.E.E., A.M., M.A.Y., S.S.K., A.S., X.S.L., A.D., and D.P.; supervision: V.K.Y., G.D., U.R.M., G.L., I.I.I., D.P., and S.C.; funding acquisition: V.K.Y. and G.D.; writing—original draft: V.K.Y., P.S., A.G., A.S., M.A.Y., and K.S.; writing—review and editing: V.K.Y., M.A.Y., G.L., and K.S.
Declaration of interests
All the authors declared no competing interests.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-TNFα antibody | Biolegend, San Diego, USA | MP6-XT22 |
| Bacterial and virus strains | ||
| Citrobacter rodentium | Pr. Frankel, Wiles et al.40 | ICC180 |
| Chemicals, peptides, and recombinant proteins | ||
| DSS MW 36–50 kDa | MP Biomedicals, Santa Ana, CA | 9011-18-1 |
| Calcein | Sigma | C0875 |
| Alizarin Red S | Sigma | A3882 |
| TRIzol | ThermoFisher | 15596026 |
| Critical commercial assays | ||
| Bio-plex Pro Mouse Cytokine Multiplex Assay | BioRad Inc., Hercules, CA | |
| TNFα ELISA kit | Millipore Inc. | EZMTNFA |
| Ocn Mouse ELISA Kit | Abcam Inc | ab285236 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | Jackson Laboratory | JAX: 000664, RRID: IMSR_JAX:000664 |
| Mouse: Igh6−/− in C57BL/6N background | Jackson Laboratory | |
| Oligonucleotides | ||
| TNFα primers Forward-AAGCCTGTAGCCCACGTCGTA Reverse-GGCACCACTAGTTGGTTGTCTTTG |
Sigma | N/A |
| OPG primers Forward- ACAGTTTGCCTGGGACCAAA Reverse- TCACAGAGGTCAATGTCTTGGA |
Sigma | |
| Software and algorithms | ||
| Osteo measure software | OsteoMetrics, Inc., GA, USA | N/A |
| GraphPad Prism Version 8 | GraphPad | N/A |
| ImageJ | NIH | N/A |
| NRecon | Bruker | N/A |
| CTan | Bruker | N/A |
| mCT Vol | Bruker | N/A |
Experimental model and study participant details
Animals and ethics approval
Six weeks-old C57BL/6J female and male mice obtained from the Wellcome Trust Sanger Institute breeding colonies or from The Jackson Laboratory (Bar Harbor, Maine) were used for the studies. B cell-deficient mice on a C57BL/6N background, homozygous for a targeted mutation in the gene for immunoglobulin heavy chain 6 (Igh6−/− mice),68 were bred at the Wellcome Trust Sanger Institute from breeder mice originally obtained from The Jackson Laboratory. For experiments performed at the Columbia University, Igh6−/− mice and controls were purchased directly from The Jackson Laboratory, acclimatized to the housing conditions for at least 1 week before being used for experiments. At no point, mice from different groups were cohoused. All animals had ad libitum access to food and water. Animal husbandry and experimental procedures were conducted according to the United Kingdom Animals (Scientific Procedures) Act of 1986. All procedures involving live mice were approved by the United Kingdom Home Office under the United Kingdom Animal (Scientific) Procedures Act and by institutional ethical committees of Wellcome Sanger Institute (PPL80/2479), Columbia University (AABO2550), and National Center for Cell Science (NCCS/IAEC/B358).
Method details
Bacterial infection of mice
C. rodentium strain ICC180 was used in this study.40 Bacterial inoculums were prepared by culturing bacteria overnight at 37°C in 100 mL of Luria Bertani (LB) broth supplemented with nalidixic acid (100 μg/mL) on a bacterial shaker (220 rpm). Cultures were harvested by centrifugation and resuspended in a 1:10 volume of Dulbecco's phosphate-buffered saline (D-PBS). Mice were orally inoculated under anesthesia using a gavage needle with 200 μL of overnight LB-grown C. rodentium suspension, 10X concentrated in PBS (∼5 × 109 cfu) or an equal volume of PBS. The viable count of the inoculum was determined by retrospective plating on LB agar supplemented with nalidixic acid (100 μg/mL). Infection was monitored through biweekly/weekly feces collection, which were homogenized in PBS, serially diluted, and plated onto nalidixic acid and kanamycin containing agar plates to ensure that the colonies only resembled C. rodentium. These colonies were then counted to generate a shedding curve.
Measurement of the C. rodentium burden
At regular time points post infection, fecal samples from individual mice were collected in separate sterile micro-centrifuge tubes. Fecal samples were weighed, and for every 0.01 g of feces, 100 μL of sterile PBS was added (for example, for 0.02 g of feces, 200 μL PBS was added). For the analysis, the liver, spleen, and bone samples were weighed and homogenized using a similar scheme as used for feces. Between different organ homogenizations, hand gloves were changed and the homogenizer probe was sanitized to avoid cross contamination. The fecal, liver, spleen, and bone samples were homogenized on a vortex and serially diluted. The number of viable bacteria was determined based on viable count on LB agar containing nalidixic acid (100 μg/mL).
TNFα neutralizing antibody treatment
Wild type or Igh6−/− mice were administered 100 μg (2 × 104 units) of neutralizing anti-TNFα antibody (MP6-XT22; Biolegend, San Diego, USA), diluted in 0.05 mL of sterile 1X PBS, through the intravenous route (tail), a day before the C. rodentium inoculation and weekly, thereafter. The same experimental regimen was followed as described above for assessing the basal effect of neutralizing anti-TNFα antibody administration on the bone in WT or Igh6−/− mice.
Dextran sodium sulfate-induced acute colitis in mice
Acute colitis was induced in WT C57BL/6J mice by administering DSS through the drinking water. Mice were treated with 2% (w/v) DSS (MW 36–50 kDa; MP Biomedicals, Santa Ana, CA) for a week, followed by 1% DSS for another week. Mice were grouped into four groups as follows: Control-8 weeks, DSS treatment followed by 6 weeks recovery, Control-2 weeks, and DSS treatment for 2 weeks. At no point, mice from different groups were cohoused. Development, progression, and recovery of colitis were monitored daily by measuring weight loss, stool consistency, and bloody diarrhea as reported previously by us.69 Mice were clinically scored based on the disease activity index (DAI) score used to evaluate the DSS-induced colitis. The DAI was calculated as the total score (body weight decrease + stool consistency + rectal bleeding) divided by 12.
Colon histology
Colon was collected from uninfected and infected mice at indicated timepoints, cleared of feces through flushing with 1X PBS, and weighed with a sensitive balance to measure changes in colon weight. The terminal 0.5 cm of the colon was removed aseptically, placed in 5% formaldehyde, and incubated overnight at room temperature for subsequent histological analysis. Tissues were processed using a Shandon excelsior tissue processor (Thermo Fisher Scientific) and then embedded in paraffin wax. Five-micrometer sections were cut using a Leica RM2125 microtome and transferred to Superfrost Plus slides (VWR International). The sections were deparaffinized by incubating them twice in Histoclear II solution for 10 min. The sections were then rehydrated by incubation in 100%, 90%, and 70% ethanol for 5 min each, followed by two 5 min washes in PBS. The sections were stained first in Mayer’s hematoxylin and then in eosin and dehydrated and mounted before assessment.
Cytokine profiling using the multiplex cytokine assay
A multiplex biometric enzyme-linked immunosorbent assay (ELISA)-based immunoassay, containing dyed microspheres conjugated with a monoclonal antibody specific for a target protein, was used according to the manufacturer’s instructions (Bio-plex Pro Mouse Cytokine Multiplex Assay; BioRad Inc., Hercules, CA). The cytokines assayed were granulocyte colony stimulating factor (G-CSF), granulocyte-monocyte colony stimulating factor (GM-CSF), IFN-γ, IL1-α, IL1-β, IL2, IL4, IL5, IL6, IL7, IL9, IL10, IL12 (p40), IL12 (p70), IL13, IL15, IL17, IP10, KC, CCL2 (monocyte chemoattractant protein-170), CCL4 (macrophage inflammatory protein [MIP]-1β), MIP-1α, MIP2, RANTES, and TNFα. Briefly, serum samples were diluted 1:4 and incubated with antibody-coupled beads for 2 h at room temperature, with shaking. The immune complexes were washed, then incubated with biotinylated detection antibody for 1 h at room temperature and, finally, incubated for 30 min with streptavidin–phycoerythrin prior to assessing the cytokine concentrations. The concentrated recombinant cytokines were provided by the vendor (BioRad, Inc.). A broad range (1.95–32,000 pg/mL) of standards was used to establish standard curves to maximize the sensitivity and dynamic range of the assay. Cytokine levels were determined using a Bio-Plex array reader (an automated flow-based microfluidics device that uses a dual-laser fluorescent detector with real-time digital signal processing for quantitation; Luminex, Austin, TX). This instrument quantitates multiplex immunoassays in a 96-well format on very small fluid volumes. The concentrations of analytes in these assays were calculated using a standard curve with the software provided by the manufacturer.
Bioassays
Serum was prepared using BD Vacutainer SST, snap frozen in liquid nitrogen, and stored at −80°C, until analyzed. Serum TNFα levels were measured using a Mouse TNFα ELISA kit (Millipore Inc. or Biolegend) and osteocalcin (Ocn) was measured using an Ocn Mouse ELISA Kit (Abcam Inc.).
Bone histology and histomorphometry
Skeletal processing and histological and histomorphometric analyses were performed as described previously.71 For histological analysis, mice were euthanized at indicated ages between 1100 and 1400 h. Briefly, for assessment of dynamic histomorphometric indices, mice were injected with calcein, 2 and 4 days prior to sacrifice, according to the standard calcein double-labeling procedure.59,72,73 After sacrifice, internal organs were removed from the animals and whole skeleton was pinned to a thermocol board and immersed in 4% neutral buffered formalin for 12–14 h at room temperature. After fixation, the skeleton was cut into the vertebral column (lumbar vertebrae 1 to 5, with associated muscles) or long bone (with associated muscles) for processing and embedding. Undecalcified bones were dehydrated in a graded series of ethanol, embedded in methyl methacrylate, and 5 μm sections were prepared on a rotary microtome (Leica Inc.), as described previously.72,73 The sections were stained with 1% toluidine blue (osteoblast parameters), Von Kossa/Van Gieson reagent (stains mineralized bone matrix in black and non-mineralized bone matrix in red), or tartrate-resistant alkaline phosphatase (TRAP, osteoclast parameters) stain and visualized using a Zeiss microscope (Carl Zeiss, Jena, Germany). Histomorphometric analysis was performed on tibiae and vertebrae according to the American Society for Bone and Mineral Research (ASBMR) standards74 using the OsteoMeasure Analysis System (Osteometrix, Atlanta, GA). For BV/TV% analysis, Von Kossa/Van Gieson-stained sections were imaged at 5× magnification and analyzed using the ImageJ software. For osteoblast parameters, toluidine blue-stained sections were visualized at 20× magnification, and analyses were performed in the secondary spongiosa in at least 15 fields per vertebra. Osteoblasts were counted only when they were in a group of three or more on the bone surface. For osteoclast parameters, TRAP-stained sections were visualized at 20× magnification and analyses were performed in the secondary spongiosa in at least 15 fields per vertebra or long bone section. For bone formation analysis, sections were cleared in xylene, mounted in DPX, and visualized under ultraviolet light at 40× magnification, and analysis was performed to detect bone formation fronts.
Micro-computed tomography (μCT) analysis
Trabecular bone and cortical architecture of the proximal tibia were assessed using a μCT system (Skyscan 1172), as described previously.72,73 The tibia bone specimen was stabilized with gauze in a 2 mL centrifuge tube filled with 70% ethanol and fastened in the specimen holder of the μCT scanner. One-hundred μCT slices, corresponding to a 1.05 mm region distal from the growth plate, were acquired at an isotropic spatial resolution of 10.5 μm. A global thresholding technique was applied to binarize gray-scale μCT images where the minimum between the bone and bone marrow peaks in the voxel gray value histogram was chosen as the threshold value. The trabecular bone compartment was segmented using a semiautomatic contouring method and subjected to a model-independent morphological analysis72,73 using the standard software provided by the manufacturer of the μCT scanner. 3D morphological parameters included model-independent measures by distance transformation (DT) of bone volume fraction (BV/TV), Tb.Th∗ (trabecular thickness), Tb.N∗ (trabecular number), and Tb.Sp∗ (trabecular separation).75,76
Quantification and statistical analysis
Results are given as mean ± standard error. Statistical analysis was performed using the Student’s t test (2-tailed) or Chi-Square test (see figure legends). For all panels in Figures 1, 2, 3, 4, and S1–S4, #p < 0.05 and ∗p < 0.01 are versus WT or control. A p value < 0.05 was considered significant.
Published: January 16, 2025
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2025.111802.
Supplemental information
References
- 1.Martinson J.N.V., Walk S.T. Escherichia coli Residency in the Gut of Healthy Human Adults. EcoSal Plus. 2020;9:1–19. doi: 10.1128/ecosalplus.ESP-0003-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nataro J.P., Kaper J.B. Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 1998;11:142–201. doi: 10.1128/cmr.11.1.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ikuta K.S., Swetschinski L.R., Robles Aguilar G., Sharara F., Mestrovic T., Gray A.P., Davis Weaver N., Wool E.E., Han C., Gershberg Hayoon A., et al. Global mortality associated with 33 bacterial pathogens in 2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet. 2022;400:2221–2248. doi: 10.1016/S0140-6736(22)02185-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramakrishna B.S. Prevalence of intestinal pathogens in HIV patients with diarrhea: implications for treatment. Indian J. Pediatr. 1999;66:85–91. doi: 10.1007/BF02752359. [DOI] [PubMed] [Google Scholar]
- 5.Borenshtein D., McBee M.E., Schauer D.B. Utility of the Citrobacter rodentium infection model in laboratory mice. Curr. Opin. Gastroenterol. 2008;24:32–37. doi: 10.1097/MOG.0b013e3282f2b0fb. [DOI] [PubMed] [Google Scholar]
- 6.Simmons C.P., Goncalves N.S., Ghaem-Maghami M., Bajaj-Elliott M., Clare S., Neves B., Frankel G., Dougan G., MacDonald T.T. Impaired resistance and enhanced pathology during infection with a noninvasive, attaching-effacing enteric bacterial pathogen, Citrobacter rodentium, in mice lacking IL-12 or IFN-gamma. J. Immunol. 2002;168:1804–1812. doi: 10.4049/jimmunol.168.4.1804. [DOI] [PubMed] [Google Scholar]
- 7.Bhinder G., Sham H.P., Chan J.M., Morampudi V., Jacobson K., Vallance B.A. The Citrobacter rodentium mouse model: studying pathogen and host contributions to infectious colitis. J. Vis. Exp. 2013;72 doi: 10.3791/50222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collins J.W., Keeney K.M., Crepin V.F., Rathinam V.A.K., Fitzgerald K.A., Finlay B.B., Frankel G. Citrobacter rodentium: infection, inflammation and the microbiota. Nat. Rev. Microbiol. 2014;12:612–623. doi: 10.1038/nrmicro3315. [DOI] [PubMed] [Google Scholar]
- 9.Maaser C., Housley M.P., Iimura M., Smith J.R., Vallance B.A., Finlay B.B., Schreiber J.R., Varki N.M., Kagnoff M.F., Eckmann L. Clearance of Citrobacter rodentium requires B cells but not secretory immunoglobulin A (IgA) or IgM antibodies. Infect. Immun. 2004;72:3315–3324. doi: 10.1128/IAI.72.6.3315-3324.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simmons C.P., Clare S., Ghaem-Maghami M., Uren T.K., Rankin J., Huett A., Goldin R., Lewis D.J., MacDonald T.T., Strugnell R.A., et al. Central role for B lymphocytes and CD4+ T cells in immunity to infection by the attaching and effacing pathogen Citrobacter rodentium. Infect. Immun. 2003;71:5077–5086. doi: 10.1128/iai.71.9.5077-5086.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chaplin D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010;125:S3–S23. doi: 10.1016/j.jaci.2009.12.980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shlomchik M.J., Craft J.E., Mamula M.J. From T to B and back again: positive feedback in systemic autoimmune disease. Nat. Rev. Immunol. 2001;1:147–153. doi: 10.1038/35100573. [DOI] [PubMed] [Google Scholar]
- 13.Ke K., Arra M., Abu-Amer Y. Mechanisms Underlying Bone Loss Associated with Gut Inflammation. Int. J. Mol. Sci. 2019;20 doi: 10.3390/ijms20246323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bundela R.P.S., Ashdhir P., Narayan K.S., Jain M., Pokharna R.K., Nijhawan S. Prevalence and risk factors for low bone mineral density in ulcerative colitis. Indian J. Gastroenterol. 2017;36:193–196. doi: 10.1007/s12664-017-0758-0. [DOI] [PubMed] [Google Scholar]
- 15.Rodan G.A., Martin T.J. Therapeutic approaches to bone diseases. Science. 2000;289:1508–1514. doi: 10.1126/science.289.5484.1508. [DOI] [PubMed] [Google Scholar]
- 16.Clemens T.L., Karsenty G. The osteoblast: an insulin target cell controlling glucose homeostasis. J. Bone Miner. Res. 2011;26:677–680. doi: 10.1002/jbmr.321. [DOI] [PubMed] [Google Scholar]
- 17.Fukumoto S., Yamashita T. FGF23 is a hormone-regulating phosphate metabolism--unique biological characteristics of FGF23. Bone. 2007;40:1190–1195. doi: 10.1016/j.bone.2006.12.062. [DOI] [PubMed] [Google Scholar]
- 18.Manolagas S.C. Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr. Rev. 2000;21:115–137. doi: 10.1210/edrv.21.2.0395. [DOI] [PubMed] [Google Scholar]
- 19.Zaidi M. Skeletal remodeling in health and disease. Nat. Med. 2007;13:791–801. doi: 10.1038/nm1593. [DOI] [PubMed] [Google Scholar]
- 20.Kobayashi T., Kronenberg H.M. Overview of skeletal development. Methods Mol. Biol. 2014;1130:3–12. doi: 10.1007/978-1-62703-989-5_1. [DOI] [PubMed] [Google Scholar]
- 21.Berendsen A.D., Olsen B.R. Bone development. Bone. 2015;80:14–18. doi: 10.1016/j.bone.2015.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niedzwiedzki T., Filipowska J. Bone remodeling in the context of cellular and systemic regulation: the role of osteocytes and the nervous system. J. Mol. Endocrinol. 2015;55:R23–R36. doi: 10.1530/JME-15-0067. [DOI] [PubMed] [Google Scholar]
- 23.Sharan K., Yadav V.K. Hypothalamic control of bone metabolism. Best Pract. Res. Clin. Endocrinol. Metabol. 2014;28:713–723. doi: 10.1016/j.beem.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 24.Khosla S. Personalising osteoporosis treatment for patients at high risk of fracture. Lancet Diabetes Endocrinol. 2019;7:739–741. doi: 10.1016/S2213-8587(19)30266-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iseme R.A., McEvoy M., Kelly B., Agnew L., Walker F.R., Attia J. Is osteoporosis an autoimmune mediated disorder? Bone Rep. 2017;7:121–131. doi: 10.1016/j.bonr.2017.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Southerland J.C., Valentine J.F. Osteopenia and osteoporosis in gastrointestinal diseases: diagnosis and treatment. Curr. Gastroenterol. Rep. 2001;3:399–407. doi: 10.1007/s11894-001-0082-8. [DOI] [PubMed] [Google Scholar]
- 27.Tyagi A.M., Darby T.M., Hsu E., Yu M., Pal S., Dar H., Li J.Y., Adams J., Jones R.M., Pacifici R. The gut microbiota is a transmissible determinant of skeletal maturation. Elife. 2021;10 doi: 10.7554/eLife.64237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu M., Pal S., Paterson C.W., Li J.Y., Tyagi A.M., Adams J., Coopersmith C.M., Weitzmann M.N., Pacifici R. Ovariectomy induces bone loss via microbial-dependent trafficking of intestinal TNF+ T cells and Th17 cells. J. Clin. Invest. 2021;131 doi: 10.1172/JCI143137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim Y.G., Kamada N., Shaw M.H., Warner N., Chen G.Y., Franchi L., Núñez G. The Nod2 sensor promotes intestinal pathogen eradication via the chemokine CCL2-dependent recruitment of inflammatory monocytes. Immunity. 2011;34:769–780. doi: 10.1016/j.immuni.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grainger J.R., Wohlfert E.A., Fuss I.J., Bouladoux N., Askenase M.H., Legrand F., Koo L.Y., Brenchley J.M., Fraser I.D.C., Belkaid Y. Inflammatory monocytes regulate pathologic responses to commensals during acute gastrointestinal infection. Nat. Med. 2013;19:713–721. doi: 10.1038/nm.3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beutler B., Cerami A. The biology of cachectin/TNF--a primary mediator of the host response. Annu. Rev. Immunol. 1989;7:625–655. doi: 10.1146/annurev.iy.07.040189.003205. [DOI] [PubMed] [Google Scholar]
- 32.Takayanagi H. Osteoimmunology and the effects of the immune system on bone. Nat. Rev. Rheumatol. 2009;5:667–676. doi: 10.1038/nrrheum.2009.217. [DOI] [PubMed] [Google Scholar]
- 33.Daoussis D., Andonopoulos A.P., Liossis S.N.C. Wnt pathway and IL-17: novel regulators of joint remodeling in rheumatic diseases. Looking beyond the RANK-RANKL-OPG axis. Semin. Arthritis Rheum. 2010;39:369–383. doi: 10.1016/j.semarthrit.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 34.Kobayashi K., Takahashi N., Jimi E., Udagawa N., Takami M., Kotake S., Nakagawa N., Kinosaki M., Yamaguchi K., Shima N., et al. Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J. Exp. Med. 2000;191:275–286. doi: 10.1084/jem.191.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pascher E., Perniok A., Becker A., Feldkamp J. Effect of 1alpha,25(OH)2-vitamin D3 on TNF alpha-mediated apoptosis of human primary osteoblast-like cells in vitro. Horm. Metab. Res. 1999;31:653–656. doi: 10.1055/s-2007-978815. [DOI] [PubMed] [Google Scholar]
- 36.Gilbert L., He X., Farmer P., Boden S., Kozlowski M., Rubin J., Nanes M.S. Inhibition of osteoblast differentiation by tumor necrosis factor-alpha. Endocrinology. 2000;141:3956–3964. doi: 10.1210/endo.141.11.7739. [DOI] [PubMed] [Google Scholar]
- 37.Lee C.K., Lee E.Y., Chung S.M., Mun S.H., Yoo B., Moon H.B. Effects of disease-modifying antirheumatic drugs and antiinflammatory cytokines on human osteoclastogenesis through interaction with receptor activator of nuclear factor kappaB, osteoprotegerin, and receptor activator of nuclear factor kappaB ligand. Arthritis Rheum. 2004;50:3831–3843. doi: 10.1002/art.20637. [DOI] [PubMed] [Google Scholar]
- 38.Redlich K., Görtz B., Hayer S., Zwerina J., Doerr N., Kostenuik P., Bergmeister H., Kollias G., Steiner G., Smolen J.S., Schett G. Repair of local bone erosions and reversal of systemic bone loss upon therapy with anti-tumor necrosis factor in combination with osteoprotegerin or parathyroid hormone in tumor necrosis factor-mediated arthritis. Am. J. Pathol. 2004;164:543–555. doi: 10.1016/S0002-9440(10)63144-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saito H., Kojima T., Takahashi M., Horne W.C., Baron R., Amagasa T., Ohya K., Aoki K. A tumor necrosis factor receptor loop peptide mimic inhibits bone destruction to the same extent as anti-tumor necrosis factor monoclonal antibody in murine collagen-induced arthritis. Arthritis Rheum. 2007;56:1164–1174. doi: 10.1002/art.22495. [DOI] [PubMed] [Google Scholar]
- 40.Wiles S., Clare S., Harker J., Huett A., Young D., Dougan G., Frankel G. Organ specificity, colonization and clearance dynamics in vivo following oral challenges with the murine pathogen Citrobacter rodentium. Cell Microbiol. 2004;6:963–972. doi: 10.1111/j.1462-5822.2004.00414.x. [DOI] [PubMed] [Google Scholar]
- 41.Vallance B.A., Deng W., Knodler L.A., Finlay B.B. Mice lacking T and B lymphocytes develop transient colitis and crypt hyperplasia yet suffer impaired bacterial clearance during Citrobacter rodentium infection. Infect. Immun. 2002;70:2070–2081. doi: 10.1128/iai.70.4.2070-2081.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Y., Toraldo G., Li A., Yang X., Zhang H., Qian W.P., Weitzmann M.N. B cells and T cells are critical for the preservation of bone homeostasis and attainment of peak bone mass in vivo. Blood. 2007;109:3839–3848. doi: 10.1182/blood-2006-07-037994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bennett K.M., Parnell E.A., Sanscartier C., Parks S., Chen G., Nair M.G., Lo D.D. Induction of Colonic M Cells during Intestinal Inflammation. Am. J. Pathol. 2016;186:1166–1179. doi: 10.1016/j.ajpath.2015.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Okayasu I., Hatakeyama S., Yamada M., Ohkusa T., Inagaki Y., Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98:694–702. doi: 10.1016/0016-5085(90)90290-h. [DOI] [PubMed] [Google Scholar]
- 45.Hamdani G., Gabet Y., Rachmilewitz D., Karmeli F., Bab I., Dresner-Pollak R. Dextran sodium sulfate-induced colitis causes rapid bone loss in mice. Bone. 2008;43:945–950. doi: 10.1016/j.bone.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 46.Nunez C., Nishimoto N., Gartland G.L., Billips L.G., Burrows P.D., Kubagawa H., Cooper M.D. B cells are generated throughout life in humans. J. Immunol. 1996;156:866–872. [PubMed] [Google Scholar]
- 47.Yun T.J., Chaudhary P.M., Shu G.L., Frazer J.K., Ewings M.K., Schwartz S.M., Pascual V., Hood L.E., Clark E.A. OPG/FDCR-1, a TNF receptor family member, is expressed in lymphoid cells and is up-regulated by ligating CD40. J. Immunol. 1998;161:6113–6121. [PubMed] [Google Scholar]
- 48.Simonet W.S., Lacey D.L., Dunstan C.R., Kelley M., Chang M.S., Lüthy R., Nguyen H.Q., Wooden S., Bennett L., Boone T., et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–319. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 49.Bucay N., Sarosi I., Dunstan C.R., Morony S., Tarpley J., Capparelli C., Scully S., Tan H.L., Xu W., Lacey D.L., et al. osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998;12:1260–1268. doi: 10.1101/gad.12.9.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sjogren K., Engdahl C., Henning P., Lerner U.H., Tremaroli V., Lagerquist M.K., Backhed F., Ohlsson C. The gut microbiota regulates bone mass in mice. J. Bone Miner. Res. 2012;27:1357–1367. doi: 10.1002/jbmr.1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Britton R.A., Irwin R., Quach D., Schaefer L., Zhang J., Lee T., Parameswaran N., McCabe L.R. Probiotic L. reuteri treatment prevents bone loss in a menopausal ovariectomized mouse model. J. Cell. Physiol. 2014;229:1822–1830. doi: 10.1002/jcp.24636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kulkarni N., Meitei H.T., Sonar S.A., Sharma P.K., Mujeeb V.R., Srivastava S., Boppana R., Lal G. CCR6 signaling inhibits suppressor function of induced-Treg during gut inflammation. J. Autoimmun. 2018;88:121–130. doi: 10.1016/j.jaut.2017.10.013. [DOI] [PubMed] [Google Scholar]
- 53.Kulkarni N., Pathak M., Lal G. Role of chemokine receptors and intestinal epithelial cells in the mucosal inflammation and tolerance. J. Leukoc. Biol. 2017;101:377–394. doi: 10.1189/jlb.1RU0716-327R. [DOI] [PubMed] [Google Scholar]
- 54.Zhang J., Wang A.X., Wu Y., Zhang S. IL-1 receptor antagonist (IL-1RA) suppresses a hyper-IL-17 response-mediated bone loss in a murine experimental periodontitis. Arch. Oral Biol. 2022;144 doi: 10.1016/j.archoralbio.2022.105555. [DOI] [PubMed] [Google Scholar]
- 55.Ikeuchi T., Moutsopoulos N.M. Osteoimmunology in periodontitis; a paradigm for Th17/IL-17 inflammatory bone loss. Bone. 2022;163 doi: 10.1016/j.bone.2022.116500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cheng W.C., Hughes F.J., Taams L.S. The presence, function and regulation of IL-17 and Th17 cells in periodontitis. J. Clin. Periodontol. 2014;41:541–549. doi: 10.1111/jcpe.12238. [DOI] [PubMed] [Google Scholar]
- 57.Booth C.C., Mollin D.L. The site of absorption of vitamin B12 in man. Lancet. 1959;1:18–21. doi: 10.1016/s0140-6736(59)90979-1. [DOI] [PubMed] [Google Scholar]
- 58.Geier M.S., Smith C.L., Butler R.N., Howarth G.S. Small-intestinal manifestations of dextran sulfate sodium consumption in rats and assessment of the effects of Lactobacillus fermentum BR11. Dig. Dis. Sci. 2009;54:1222–1228. doi: 10.1007/s10620-008-0495-4. [DOI] [PubMed] [Google Scholar]
- 59.Singh P., Telnova S., Zhou B., Mohamed A.D., Mello V.D., Wackerhage H., Guo X.E., Panda A.K., Yadav V.K. Maternal vitamin B12 in mice positively regulates bone, but not muscle mass and strength in post-weaning and mature offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2021;320:R984–R993. doi: 10.1152/ajpregu.00355.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goncalves N.S., Ghaem-Maghami M., Monteleone G., Frankel G., Dougan G., Lewis D.J., Simmons C.P., MacDonald T.T. Critical role for tumor necrosis factor alpha in controlling the number of lumenal pathogenic bacteria and immunopathology in infectious colitis. Infect. Immun. 2001;69:6651–6659. doi: 10.1128/IAI.69.11.6651-6659.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang J., Motyl K.J., Irwin R., MacDougald O.A., Britton R.A., McCabe L.R. Loss of Bone and Wnt10b Expression in Male Type 1 Diabetic Mice Is Blocked by the Probiotic Lactobacillus reuteri. Endocrinology. 2015;156:3169–3182. doi: 10.1210/EN.2015-1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sugisawa E., Takayama Y., Takemura N., Kondo T., Hatakeyama S., Kumagai Y., Sunagawa M., Tominaga M., Maruyama K. RNA Sensing by Gut Piezo1 Is Essential for Systemic Serotonin Synthesis. Cell. 2020;182:609–624.e21. doi: 10.1016/j.cell.2020.06.022. [DOI] [PubMed] [Google Scholar]
- 63.Ohlsson C., Engdahl C., Fåk F., Andersson A., Windahl S.H., Farman H.H., Movérare-Skrtic S., Islander U., Sjögren K. Probiotics protect mice from ovariectomy-induced cortical bone loss. PLoS One. 2014;9 doi: 10.1371/journal.pone.0092368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Khan S., Chakraborty M., Wu F., Chen N., Wang T., Chan Y.T., Sayad A., Vásquez J.D.S., Kotlyar M., Nguyen K., et al. B Cells Promote T Cell Immunosenescence and Mammalian Aging Parameters. bioRxiv. 2023 doi: 10.1101/2023.09.12.556363. Preprint at. [DOI] [Google Scholar]
- 65.Fagundes-Neto U., De Martini-Costa S., Pedroso M.Z., Scaletsky I.C. Studies of the small bowel surface by scanning electron microscopy in infants with persistent diarrhea. Braz. J. Med. Biol. Res. 2000;33:1437–1442. doi: 10.1590/s0100-879x2000001200006. [DOI] [PubMed] [Google Scholar]
- 66.Fagundes-Neto U., Scaletsky I.C. The gut at war: the consequences of enteropathogenic Escherichia coli infection as a factor of diarrhea and malnutrition. Sao Paulo Med. J. 2000;118:21–29. doi: 10.1590/s1516-31802000000100006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Blanco M., Blanco J.E., Blanco J., de Carvalho V.M., Onuma D.L., Pestana de Castro A.F. Typing of intimin (eae) genes in attaching and effacing Escherichia coli strains from monkeys. J. Clin. Microbiol. 2004;42:1382–1383. doi: 10.1128/jcm.42.3.1382-1383.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kitamura D., Roes J., Kühn R., Rajewsky K. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature. 1991;350:423–426. doi: 10.1038/350423a0. [DOI] [PubMed] [Google Scholar]
- 69.Pathak M., Padghan P., Halder N., Lal G., Kulkarni N., Kulkarni N., Sonar S.A. CCR9 signaling in dendritic cells drives the differentiation of Foxp3(+) Tregs and suppresses the allergic IgE response in the gut. Eur. J. Immunol. 2020;50:404–417. doi: 10.1002/eji.201948327. [DOI] [PubMed] [Google Scholar]
- 70.Verlaan S., Aspray T.J., Bauer J.M., Cederholm T., Hemsworth J., Hill T.R., McPhee J.S., Piasecki M., Seal C., Sieber C.C., et al. Nutritional status, body composition, and quality of life in community-dwelling sarcopenic and non-sarcopenic older adults: A case-control study. Clin. Nutr. 2017;36:267–274. doi: 10.1016/j.clnu.2015.11.013. [DOI] [PubMed] [Google Scholar]
- 71.Singh P., Gollapalli K., Mangiola S., Schranner D., Yusuf M.A., Chamoli M., Shi S.L., Lopes Bastos B., Nair T., Riermeier A., et al. Taurine deficiency as a driver of aging. Science. 2023;380 doi: 10.1126/science.abn9257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sharan K., Lewis K., Furukawa T., Yadav V.K. Regulation of bone mass through pineal-derived melatonin-MT2 receptor pathway. J. Pineal Res. 2017;63 doi: 10.1111/jpi.12423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lewis K.E., Sharan K., Takumi T., Yadav V.K. Skeletal Site-specific Changes in Bone Mass in a Genetic Mouse Model for Human 15q11-13 Duplication Seen in Autism. Sci. Rep. 2017;7:9902. doi: 10.1038/s41598-017-09921-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Parfitt A.M., Drezner M.K., Glorieux F.H., Kanis J.A., Malluche H., Meunier P.J., Ott S.M., Recker R.R. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J. Bone Miner. Res. 1987;2:595–610. doi: 10.1002/jbmr.5650020617. [DOI] [PubMed] [Google Scholar]
- 75.Feldkamp L.A., Goldstein S.A., Parfitt A.M., Jesion G., Kleerekoper M. The direct examination of three-dimensional bone architecture in vitro by computed tomography. J. Bone Miner. Res. 1989;4:3–11. doi: 10.1002/jbmr.5650040103. [DOI] [PubMed] [Google Scholar]
- 76.Gundersen H.J., Boyce R.W., Nyengaard J.R., Odgaard A. The Conneulor: unbiased estimation of connectivity using physical disectors under projection. Bone. 1993;14:217–222. doi: 10.1016/8756-3282(93)90144-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this article will be shared by the lead contact upon request.
-
•
This article does not report the original code.
-
•
Any additional information required to reanalyze the data reported in this article is available from the lead contact upon request.