

Abstract
Drug drug Interactions (DDI) present considerable challenges in healthcare, often resulting in adverse effects or decreased therapeutic efficacy. This article proposes a novel deep sequential learning architecture called DDINet to predict and classify DDIs between pairs of drugs based on different mechanisms viz., Excretion, Absorption, Metabolism, and Excretion rate (higher serum level) etc. Chemical features such as Hall Smart, Amino Acid count and Carbon types are extracted from each drug (pairs) to apply as an input to the proposed model. Proposed DDINet incorporates attention mechanism and deep sequential learning architectures, such as Long Short-Term Memory and gated recurrent unit. It utilizes the Rcpi toolkit to extract biochemical features of drugs from their chemical composition in Simplified Molecular-Input Line-Entry System format. Experiments are conducted on publicly available DDI datasets from DrugBank and Kaggle. The model’s efficacy in predicting and classifying DDIs is evaluated using various performance measures. The experimental results show that DDINet outperformed eight counterpart techniques achieving  overall accuracy which is also statistically confirmed by Confidence Interval tests and paired t-tests. This architecture may act as an effective computational technique for drug drug interaction with respect to mechanism which may act as a complementary tool to reduce costly wet lab experiments for DDI prediction and classification.
overall accuracy which is also statistically confirmed by Confidence Interval tests and paired t-tests. This architecture may act as an effective computational technique for drug drug interaction with respect to mechanism which may act as a complementary tool to reduce costly wet lab experiments for DDI prediction and classification.
Keywords: Drug drug interaction, Deep learning, Attention mechanism, Recurrent neural network, Gated recurrent unit
Subject terms: Computational biology and bioinformatics, Target identification
Introduction
Drugs are chemical substances that can effect the body function when consumed1. A drug may be classified by the chemical type of the active ingredient or by the way it is used to treat a particular condition. Each drug can be classified into one or more drug classes. Pharmacology, the study of drugs, encompasses all elements of drugs in medicine, such as their mechanisms of action, physical and chemical characteristics, metabolism, therapeutic uses, and potential toxicity2.
Drug drug interactions (DDIs) refer to the undesirable or desirable side effects with respect to certain biological mechanisms that occur when two or more medications are taken simultaneously2–4. Many diseases necessitate multiple medications, which may increases the risk of DDIs that can be dangerous or even fatal if not properly managed. Nevertheless, medications also have therapeutic benefits. Numerous studies have identified these risks, including research focused on determining the safety of taking two or more drugs concurrently. When a doctor or physician prescribes multiple medications to a patient, it can lead to DDI, which can also be considered a medication error. A drug interaction happens when one medication alters the effect of another, potentially causing harmful outcomes or enhancing the effects of the second drug5. There are three types of drug-to-drug interactions: (i) No interaction; (ii) Antagonistic: This type of interaction between multiple drugs produces an adverse effect, potentially harming the patient; and (iii) Synergistic: This occurs when the interaction between multiple drugs enhances their effect on the body4,6,7.
Therefore, understanding DDIs is crucial for enhancing the drug discovery process and improving patient recovery for serious diseases such as cancer, AIDS, and asthma. Identifying DDIs through wet lab experiments demands a considerable amount of time and intensive effort8. The usual aim of drug interaction prediction is to identify possible interactions between medications that could result in negative effects or decreased effectiveness9. Drug combination prediction aims to enhance the effectiveness of treatments, while drug interaction prediction focuses on identifying potential side effects or challenges that may occur when certain drugs are used together10.
Artificial Intelligence (AI) based computational techniques such as Machine Learning and Deep learning (ML and DL) are used to address the mentioned issues in predicting DDIs. Limited progress has been made in recent years using ML and DL to predict DDIs11–20. These approaches focus on uncovering hidden DDIs between combinations of drugs. However, many of these methods only consider drugs as input and focus solely on predicting DDIs.
Traditional methods for predicting DDIs21 involve high computational cost and time with low predictive accuracy leading to less appreciate for real time human health monitoring, industrial usage. Additionally, traditional methods do not consider the DDI prediction with respect to molecular mechanisms. Therefore, there is a need of the time for mechanism wise accurate computational method to predict DDIs. Current computational methods for predicting DDIs often fall short because they rely on statistical correlations rather than underlying molecular mechanisms, missing key interactions driven by specific pathways or receptor bindings. Incorporating mechanistic data could improve the accuracy and understanding of these predictive models. Moreover existing DDI prediction methods try to predict the interactions without considering the individual molecular mechanisms centric effect. However mechanism centric DDI prediction may uncover the underlying effects of the two drugs in meaningful manner. The motivation behind this work to improve drug safety and efficacy by accurately predicting and understanding DDIs, which can significantly impact patient health. Enhanced prediction methods will help healthcare professionals make informed decisions about drug combinations and dosages, ultimately improving patient outcomes. This work aims to predict and categorize DDIs by analyzing their underlying mechanisms, forecasting increases, decreases, or neutral interactions. The model condenses drug pair features into latent representations to accurately predict interaction classes for unknown drug pairs. This work centers on a model for predicting DDIs using structural features. It begins with data preparation involving Drugbank IDs and DDI data, which are split into training, validation, and test sets. The model architecture integrates gated recurrent unit (GRU), Attention, and Long Short Term Memory (LSTM) blocks, processing features from each drug separately and combining them to predict interactions. The model can make two-class or three-class predictions and is trained to optimize its parameters for accurate DDI predictions. Finally, the model is tested on new samples to evaluate its practical effectiveness.
Major contribution of the present work may be summarize as follows. The proposed DDINet introduces a novel attention based deep sequential learning model with parallel connection for predicting DDIs aiming to enhance the prediction accuracy. The model incorporates mechanism-wise predictions by utilizing the structural features of the pair of drugs extracting from the chemical compositions accepting the Simplified Molecular-Input Line-Entry System (SMILES) format of the drug pairs. Mechanism-wise experimental evaluations highlight the effectiveness of DDINet, demonstrating its superior performance in predicting DDIs compared to counter-part methods.
Various computational methods have been developed to predict DDIs, harnessing advancements in bioinformatics, machine learning and pharmacology. These methods are vital in contemporary healthcare for early identification and evaluation of potential interaction risks. A prevalent approach based on network propagation forecasts potential DDIs by utilizing either a network constructed from drug structural data or a network derived from established DDIs22. This method employs network propagation techniques to infer and predict unknown DDIs within the network framework. ML algorithms are also extensively used for DDI predictions12–15. These algorithms analyze large datasets of known drug interactions to detect patterns and forecast potential interactions between new drug pairs. Inputs to these models often include features like chemical structure, pharmacological properties and gene expression profiles.
Several approaches based on DL is also been used for DDI predictions. Some researchers have used various DL methods such as LSTM, Bi-directional GRU (Bi GRU), Convolution Neural Network (CNN), Transformer based approach, BERT etc. for DDI prediction from text16–20 . Efforts have also been made by researchers for DDI prediction by considering structural features of drugs and trained them by various DL methods like CNN, Deep Neural Network (DNN)23, Autoencoder etc.24–28.
Graph Convolution Neural Networks (GNNs) have also been used by several researchers29–32 for predicting DDIs from structural features of drugs. Feng et al.33 introduced DPDDI, a two-stage model for predicting drug pair interactions. It uses a GCN to extract feature vectors by analyzing drug network structures and a DNN for prediction. Nyamabo et al.34proposed Gated Message Passing Neural Network (GMPNN), which learns chemical substructures from the molecular graph representations of drugs for predicting DDIs.
Additionally, matrix factoring serves as a foundational mathematical framework for addressing various challenges in modeling biological information35. This approach involves breaking down the DDI matrix into multiple matrices, extracting potential features, and reconstructing the matrix to uncover new DDIs. Conventional matrix-factoring techniques such as single-value decomposition (SVD)36, non-negative matrix factorization (NMF)37, and probability matrix factorization (PMF)38 have been utilized.
Although various researchers have attempted to predict DDIs, however mechanism-wise DDI prediction has not been explored. Moreover accuracy of the DDI prediction may be improved further as compared to the reported results of the existing literature.
Results
Training and evaluation result
Figure 1 shows training and validation loss curves for the DDINet model across four mechanisms: excretion, absorption, metabolism, and excretion rate (higher serum level). For excretion, both losses decrease steadily, with some fluctuations. For absorption and metabolism, the losses drop quickly and converge well, showing strong and stable model performance. For the excretion rate (higher serum level), losses decline smoothly, with slight divergence after many epochs, indicating stable training. The training loss values after completion of training become 0.1443, 0.1504, 0.4428 and 0.0691 whereas the validation values logged as 0.3276, 0.1503, 0.4600 and 0.0778 for excretion, absorption, metabolism, and excretion rate (higher serum level) respectively.
Fig. 1.

Graphical representation of the training and validation loss over the epochs produced by the proposed DDINet in terms of (a) Excretion mechanism, (b) Absorption mechanism, (c) Metabolism mechanism, and (d) Excretion rate (higher serum level) mechanism.
The test dataset contains  of the non-interaction and interacting (viz., increase and/or decrease) DDI samples. Confusion matrices achieved by the proposed DDINet method for the aforementioned four mechanisms (viz., excretion, Absorption, Metabolism and excretion rate (higher serum level)) on the test set are shown in Fig. 2. Additionally, the experimental results of the proposed method using five different types of mechanisms (viz., antihypertensive activities, Central Nervous System (CNS) depressant activities, excretion rate which could result in a lower serum level and potentially a reduction in efficacy, hypoglycemic activities and hypotensive activities) of DDIs are reported in the supplementary material of this article.
of the non-interaction and interacting (viz., increase and/or decrease) DDI samples. Confusion matrices achieved by the proposed DDINet method for the aforementioned four mechanisms (viz., excretion, Absorption, Metabolism and excretion rate (higher serum level)) on the test set are shown in Fig. 2. Additionally, the experimental results of the proposed method using five different types of mechanisms (viz., antihypertensive activities, Central Nervous System (CNS) depressant activities, excretion rate which could result in a lower serum level and potentially a reduction in efficacy, hypoglycemic activities and hypotensive activities) of DDIs are reported in the supplementary material of this article.
Fig. 2.

Confusion matrices of the classified test samples using proposed DDINet for (a) Excretion mechanism, (b) Absorption mechanism, (c) Metabolism mechanism, and (d) Excretion rate (higher serum level) mechanism.
Figure 3 presents a graphical representation of the original and predicted graph for test drug pairs in terms of different mechanisms. In this figure, the vertices (with red color) represent the drugs and the edges represent the interactions between two drugs. The original DDIs are shown with single-colored edges (yellow) for excretion, absorption and excretion rate (higher serum level) to represent decreasing interaction between two drugs, wherein, metabolism mechanism, yellow and green color edges represent decreasing and increasing interaction respectively. Incorrectly predicted interactions are depicted with blue color edges in the predicted graphs. Average overall accuracy considering all the four mechanisms achieved by the proposed method is 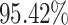 with 0.94, 0.94, 0.95 precision, recall,
with 0.94, 0.94, 0.95 precision, recall,  score respectively as shown in Table 1.
score respectively as shown in Table 1.
Fig. 3.

Graphical representation of original and predicted graph for test drug pairs in terms of different mechanisms.
Table 1.
Results of drug drug interaction prediction (in terms of accuracy, precision, recall and  ) for different mechanisms produced by proposed DDINet.
) for different mechanisms produced by proposed DDINet.
| Mechanism | Accuracy | Precision | Recall |
 Score Score |
|---|---|---|---|---|
| excretion | 94.92% | 0.95 | 0.95 | 0.95 |
| Absorption | 100% | 1.00 | 1.00 | 1.00 |
| Metabolism | 92.15% | 0.92 | 0.92 | 0.92 |
| excretion rate (higher serum level) | 94.61% | 0.95 | 0.95 | 0.95 |
| Overall | 95.42% | 0.95 | 0.96 | 0.95 |
Significant values are shown in bold.
Attention heatmap
Representative test samples (pair of drugs) for each mechanism viz., excretion, absorption, excretion rate (higher serum level) and metabolism for interacting and non-interacting drug pairs are shown respectively in Figs. 4, 5, 6 and 7 in terms of the attention heatmap generated from the attention scores produced by the multi-head attention layer. From the attention heatmap of the excretion mechanism (Fig. 4), it can be observed that latent spaces 7, 20 and 26 are showing high intensity values (shown in light yellowish color) for interacting DDI, whereas same latent spaces are showing low intensity value shown in dark blue color for non-interacting case. Similarly, for absorption, excretion rate (higher serum level) and metabolism mechanisms also we can see the complemented behavior of the attention score’s heatmap. Indicating the fact that the attention mechanism is playing the crucial role in latent space representation for discriminating the interacting (viz., increase/decrease, both) DDIs from non-interacting drug pairs. Thus, attention heatmap of the latent spaces provides an interpretability in support of the DDI prediction.
Fig. 4.

Attention heatmap results of the latent space representation generated by proposed DDINet on excretion mechanism for (a) interacting and (b) non-interacting DDIs.
Fig. 5.

Attention heatmap results of the latent space representation generated by proposed DDINet on absorption mechanism for (a) interacting and (b) non-interacting DDIs.
Fig. 6.

Attention heatmap results of the latent space representation generated by proposed DDINet on excretion rate (higher serum level) mechanism for (a) interacting and (b) non-interacting DDIs.
Fig. 7.

Attention heatmap results of the latent space representation generated by proposed DDINet on metabolism mechanism for (a) increase, (b) decrease and (c) non-interacting DDIs.
Comparative results with other counterpart techniques
Here we present the comparative results of the testing process achieved by the proposed method and the counterpart techniques for the four mechanisms viz., Excretion, Absorption, Metabolism and Excretion rate (higher serum level).
It is worth noting that existing works from the literature so far did not consider the mechanism-wise DDI predictions, rather interaction predictions are done based on the overall mechanisms. However, in our proposed method mechanism wise DDI prediction is done. Therefore, comparisons with existing literature could not be performed. Hence, the results of the proposed DDINet are compared with five popular ML techniques (viz., Multi-Layer Perceptron (MLP), Decision Tree (DT), Random Forest (RF), Naïve Bayes (NB) and Support Vector Machine (SVM)) and three DL methods viz., LSTM, GRU and transformer39 based counterpart DL models40, results of which have been reported in Table 2. From the table it can be clearly observed that the proposed DDINet model outshines other models, as it attained the highest accuracy, precision, recall, and also 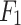 scores. For the excretion mechanism, DDINet is leading with
scores. For the excretion mechanism, DDINet is leading with 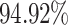 accuracy and an
accuracy and an 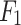 score of 0.95, while LSTM, GRU and transformer based method come next, and NB is the last one (
score of 0.95, while LSTM, GRU and transformer based method come next, and NB is the last one ( accuracy, 0.58
accuracy, 0.58  score). In the Absorption mechanism, the DDINet, LSTM and GRU models were perfect with an accuracy of
score). In the Absorption mechanism, the DDINet, LSTM and GRU models were perfect with an accuracy of 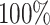 and
and 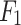 score of 1.0 while NB trailed with an
score of 1.0 while NB trailed with an  score of 0.91. The Metabolism mechanism leads to the following outcome: the best results are for DDINet with
score of 0.91. The Metabolism mechanism leads to the following outcome: the best results are for DDINet with  accuracy and an
accuracy and an  score of 0.92; the second best is LSTM and GRU. NB is significantly underperforming with
score of 0.92; the second best is LSTM and GRU. NB is significantly underperforming with 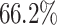 accuracy and an
accuracy and an 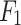 score of 0.22. For the excretion rate (higher serum level) Mechanism, DDINet dominates again with
score of 0.22. For the excretion rate (higher serum level) Mechanism, DDINet dominates again with  accuracy and an
accuracy and an 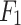 score of 0.95, while LSTM and GRU showed good but relatively lower performance.
score of 0.95, while LSTM and GRU showed good but relatively lower performance.
Table 2.
Summarized results in terms of accuracy, precision, recall and  score achieve by DDI prediction by the proposed DDINet and other compared techniques for (a) Excretion mechanism, (b) Absorption mechanism, (c) Metabolism mechanism and (d) Excretion rate (higher serum level) mechanism.
score achieve by DDI prediction by the proposed DDINet and other compared techniques for (a) Excretion mechanism, (b) Absorption mechanism, (c) Metabolism mechanism and (d) Excretion rate (higher serum level) mechanism.
| Mechanism | Method | Accuracy | Precision | Recall |
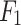 Score Score |
|---|---|---|---|---|---|
| (a) Excretion Mechanism | DDINet | 94.92% | 0.95 | 0.95 | 0.95 |
| LSTM | 87.19% | 0.87 | 0.87 | 0.87 | |
| GRU | 87.85% | 0.88 | 0.88 | 0.88 | |
| Transformer | 81.01% | 0.81 | 0.81 | 0.81 | |
| MLP | 84.98% | 0.85 | 0.85 | 0.85 | |
| RF | 93% | 0.93 | 0.93 | 0.93 | |
| DT | 90% | 0.90 | 0.90 | 0.90 | |
| SVM | 82% | 0.82 | 0.82 | 0.82 | |
| NB | 65% | 0.77 | 0.65 | 0.58 | |
| (b) Absorption Mechanism | DDINet | 100% | 1.0 | 1.0 | 1.0 |
| LSTM | 100% | 1.0 | 1.0 | 1.0 | |
| GRU | 100% | 1.0 | 1.0 | 1.0 | |
| Transformer | 84.61% | 0.88 | 0.86 | 0.85 | |
| MLP | 100% | 1.0 | 1.0 | 1.0 | |
| RF | 92% | 0.93 | 0.92 | 0.91 | |
| DT | 92% | 0.93 | 0.92 | 0.91 | |
| SVM | 100% | 1.0 | 1.0 | 1.0 | |
| NB | 92% | 0.93 | 0.92 | 0.91 | |
| (c) Metabolism Mechanism | DDINet | 92.15% | 0.92 | 0.92 | 0.92 |
| LSTM | 78.67% | 0.79 | 0.79 | 0.74 | |
| GRU | 81.60% | 0.80 | 0.82 | 0.78 | |
| Transformer | 73.85% | 0.70 | 0.47 | 0.48 | |
| MLP | 79.23% | 0.78 | 0.79 | 0.75 | |
| RF | 85% | 0.85 | 0.85 | 0.84 | |
| DT | 78% | 0.78 | 0.78 | 0.78 | |
| SVM | 71% | 0.74 | 0.71 | 0.61 | |
| NB | 26% | 0.62 | 0.26 | 0.22 | |
| (d) Excretion rate (higher serum level) Mechanism | DDINet | 94.61% | 0.95 | 0.95 | 0.95 |
| LSTM | 80.58% | 0.80 | 0.81 | 0.80 | |
| GRU | 84.26% | 0.84 | 0.84 | 0.84 | |
| Transformer | 70.32% | 0.69 | 0.67 | 0.68 | |
| MLP | 79.16% | 0.79 | 0.79 | 0.79 | |
| RF | 90% | 0.91 | 0.90 | 0.90 | |
| DT | 82% | 0.82 | 0.82 | 0.82 | |
| SVM | 72% | 0.72 | 0.72 | 0.71 | |
| NB | 65% | 0.64 | 0.65 | 0.57 |
Overall, DDINet tends to obtain top performance for all evaluation steps, followed by LSTM and GRU, while NB and SVM systematically underperform over most mechanisms.
DDINet outperforms baseline models due to its advanced architecture, which incorporates GRU and LSTM-based feature extraction for capturing both short-to-medium and long-range sequential dependencies40, while attention mechanisms try to extract the importance of the latent space features in the proposed model. The GRU block processes molecular properties of drug chemical features by preserving short-to-medium range dependencies40 while incorporating dropout layers to prevent overfitting, to ensure better generalization. The LSTM block improves memory retention for long-range dependencies40. Additionally, the use of concatenated attention block ensures the model to find important latent space features resulting better classification ability of the model. While the final dense and softmax layers ensure precise classification. Therefore, in summary, the ability to integrate GRU and LSTM layers with attention mechanisms make this model more adaptive with different sequence lengths and varied conditions of data and more robust compared to the other state-of-the-art baseline models.
Confidence interval
A confidence interval (CI)41 acts as a measure of the statistical significance of the results, capturing both error rates and margins of error. A narrow CI, reflecting low error rates and small error margins, indicates precise estimate based on the sample data. In contrast, a wider CI suggests greater uncertainty and reduced precision. The following equation is used to calculate CI41.
 |
1 |
The observed sample error within a sample set s, consisting of n independently drawn examples based on discrete-valued hypothesis h, is represented by the function  . The CI constant, denoted by
. The CI constant, denoted by 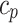 . The CI constants are set at 1.64, 1.96, and 2.58 for confidence levels (CL) of
. The CI constants are set at 1.64, 1.96, and 2.58 for confidence levels (CL) of  ,
, 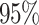 , and
, and 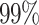 , respectively. Table 3 presents the CI error rates and error margin for the
, respectively. Table 3 presents the CI error rates and error margin for the  ,
, 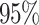 , and
, and  confidence levels, comparing the proposed method with other counterpart models. From the summarized results shown in the Table 3, it is observed that the proposed method has produced lower error rate and narrower error margin than all other counterpart techniques compared for all the mechanisms. It is worthy to note that, four compared methods (i.e,. LSTM, GRU, MLP and SVM) have produced lowest error rates and error margins (0.00) alongwith the proposed DDINet method for absorption mechanism. For all other cases proposed DDINet’s error rate and error margins are lower than other counterpart methods.
confidence levels, comparing the proposed method with other counterpart models. From the summarized results shown in the Table 3, it is observed that the proposed method has produced lower error rate and narrower error margin than all other counterpart techniques compared for all the mechanisms. It is worthy to note that, four compared methods (i.e,. LSTM, GRU, MLP and SVM) have produced lowest error rates and error margins (0.00) alongwith the proposed DDINet method for absorption mechanism. For all other cases proposed DDINet’s error rate and error margins are lower than other counterpart methods.
Table 3.
Results of the Confidence Interval tests in terms of error rate and error bounds of the proposed DDINet model along with other compared methods for (a) Excretion mechanism, (b) Absorption mechanism, (c) Metabolism mechanism, and (d) Excretion rate (higher serum level) mechanism.
| Mechanism | Model | Error Rate | Error Margin | ||
|---|---|---|---|---|---|
| CL 90% | CL 95% | CL 99% | |||
| (a) Excretion Mechanism | DDINet | 0.0508 | 0.0169 | 0.0202 | 0.0266 |
| LSTM | 0.1281 | 0.0257 | 0.0307 | 0.0405 | |
| GRU | 0.1215 | 0.0251 | 0.0300 | 0.0396 | |
| Transformer | 0.1899 | 0.0302 | 0.0361 | 0.0475 | |
| MLP | 0.1502 | 0.0275 | 0.0329 | 0.0433 | |
| RF | 0.0700 | 0.0196 | 0.0234 | 0.0309 | |
| DT | 0.1000 | 0.0231 | 0.0276 | 0.0363 | |
| SVM | 0.1800 | 0.0296 | 0.0353 | 0.0465 | |
| NB | 0.3500 | 0.0367 | 0.0439 | 0.0578 | |
| (b) Absorption Mechanism | DDINet | 0.00 | 0.0000 | 0.0000 | 0.000 |
| LSTM | 0.00 | 0.0000 | 0.0000 | 0.000 | |
| GRU | 0.00 | 0.0000 | 0.0000 | 0.000 | |
| Transformer | 0.1539 | 0.1708 | 0.2042 | 0.2688 | |
| MLP | 0.00 | 0.0000 | 0.0000 | 0.000 | |
| RF | 0.08 | 0.1284 | 0.1534 | 0.202 | |
| DT | 0.08 | 0.1284 | 0.1534 | 0.202 | |
| SVM | 0.00 | 0.0000 | 0.0000 | 0.000 | |
| NB | 0.08 | 0.1284 | 0.1534 | 0.202 | |
| (c) Metabolism Mechanism | DDINet | 0.0785 | 0.0036 | 0.0043 | 0.0057 |
| LSTM | 0.2133 | 0.0055 | 0.0066 | 0.0087 | |
| GRU | 0.1840 | 0.0052 | 0.0063 | 0.0082 | |
| Transformer | 0.2615 | 0.0058 | 0.0069 | 0.0091 | |
| MLP | 0.2077 | 0.0055 | 0.0065 | 0.0086 | |
| RF | 0.1500 | 0.0048 | 0.0058 | 0.0076 | |
| DT | 0.2200 | 0.0056 | 0.0067 | 0.0088 | |
| SVM | 0.2900 | 0.0061 | 0.0073 | 0.0097 | |
| NB | 0.7400 | 0.0059 | 0.0071 | 0.0093 | |
| (d) Excretion Rate (Higher Serum Level) Mechanism | DDINet | 0.0539 | 0.0048 | 0.0057 | 0.0075 |
| LSTM | 0.1281 | 0.0257 | 0.0307 | 0.0405 | |
| GRU | 0.1215 | 0.0251 | 0.0300 | 0.0396 | |
| Transformer | 0.2968 | 0.0096 | 0.0115 | 0.0152 | |
| MLP | 0.1502 | 0.0275 | 0.0329 | 0.0433 | |
| RF | 0.0700 | 0.0196 | 0.0234 | 0.0309 | |
| DT | 0.1000 | 0.0231 | 0.0276 | 0.0363 | |
| SVM | 0.1800 | 0.0296 | 0.0353 | 0.0465 | |
| NB | 0.3500 | 0.0367 | 0.0439 | 0.0578 | |
Paired t-test
The percentage accuracies achieved by the proposed DDINet method, in comparison to other methods, have been statistically validated using a paired t-test42 at a  significance level. Statistically significant results are highlighted in bold in Table 4 for which the p-value is
significance level. Statistically significant results are highlighted in bold in Table 4 for which the p-value is 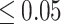 . It is evident from paired t-test results that the DDI prediction accuracy of the proposed method is statistically significant for all the cases, suggesting clear supremacy of the proposed DDINet over its counterpart techniques.
. It is evident from paired t-test results that the DDI prediction accuracy of the proposed method is statistically significant for all the cases, suggesting clear supremacy of the proposed DDINet over its counterpart techniques.
Table 4.
Result of paired t-test of the proposed DDINet model along with other ML-DL based (viz., LSTM, GRU, Transformer, MLP, RF, DT, SVM and NB) methods.
| DDINet Vs. Compared method | Paired t-test in terms of p score for different mechanisms | |||
|---|---|---|---|---|
| Excretion | Absorption | Excretion rate (higher serum level) | Metabolism | |
| LSTM | 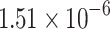 |
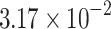 |
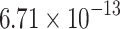 |
 |
| GRU | 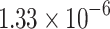 |
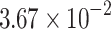 |
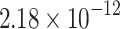 |
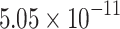 |
| Transformer | 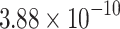 |
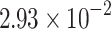 |
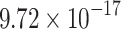 |
 |
| MLP | 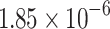 |
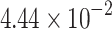 |
 |
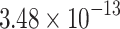 |
| RF |  |
 |
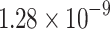 |
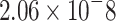 |
| DT | 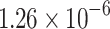 |
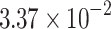 |
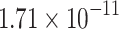 |
 |
| SVM | 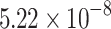 |
 |
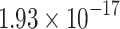 |
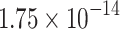 |
| NB | 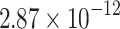 |
 |
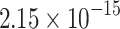 |
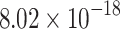 |
Discussion
DDI prediction along with the mechanism classification is an important and challenging area for the researchers of clinical, pharmaceutical and computational biologists. However, literature review suggests that DDI prediction with respect to mechanism has not been carried out so far to the best of the authors’ knowledge. In this respect, this article propose a novel deep sequential learning architecture with attention mechanism called DDINet for predicting DDIs with respect to different mechanisms. The proposed DDINet utilizes the multiple GRU layers with attention mechanism. The method takes the chemical sequence of the pair of drugs and extracts structural features of the drugs (from there SMILES sequences) which are passed to the DDINet as an input. The proposed method is tested on Drugbank and Kaggle dataset. Experimental results suggest that the proposed method can effectively predict the DDIs with respect to various mechanisms such as Excretion, Absorption, Metabolism and Excretion rate (higher serum level) achieving the accuracy 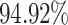 ,
, 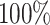 ,
,  and
and  respectively. Since the proposed method is the first of its kind to predict DDIs with respect to individual mechanisms therefore, comparisons are done with respect to baseline counterpart methods such as LSTM, GRU, MLP, RF, DT, SVM and NB from DL and ML techniques. Comparative results demonstrate the superiority of the proposed DDINet over eight popular ML-DL techniques. This architecture can serve as an efficient computational method for predicting drug drug interactions and mechanisms, offering a complementary tool to lessen the reliance on costly wet lab experiments in DDI prediction with respect to different mechanism.
respectively. Since the proposed method is the first of its kind to predict DDIs with respect to individual mechanisms therefore, comparisons are done with respect to baseline counterpart methods such as LSTM, GRU, MLP, RF, DT, SVM and NB from DL and ML techniques. Comparative results demonstrate the superiority of the proposed DDINet over eight popular ML-DL techniques. This architecture can serve as an efficient computational method for predicting drug drug interactions and mechanisms, offering a complementary tool to lessen the reliance on costly wet lab experiments in DDI prediction with respect to different mechanism.
Methods
Dataset and data preprocessing
Initially, drug ID’s (Drugbank ID) along with drug drug interaction information are taken as input. Feature generation for individual drugs are then carried out in the data preparation phase (to be discussed in the subsequent paragraph). Data is subdivided into train, validation and test sets when the data preparation phase is over. After that, the data is passed through the proposed drug drug interaction network (DDINet) architecture. As the model is designed to predict the interaction between two drugs, extracted features of individual drugs (viz., Drug A and Drug B) should be fed into the model as inputs.
The DDI interaction dataset (from Kaggle) consists of two main files drug_interaction.csv and drug_information_1258.csv The first file consists of interacting drug names, actions and mechanisms. The Action column denotes the label for each interaction i.e. increase, decrease. Interactions reported in this dataset are based on a 1258 number of unique drugs. Each unique drug names are represented by unique Drugbank IDs mentioned in the drug_information_1258.csv. To ensure the correctness of the drug interactions given in the Kaggle dataset43. The Drugbank dataset provides various drug-related details such as drug name, drug ID, SMILES (Simplified Molecular-Input Line-Entry System )44 sequence and UniProt ID for 12, 695 unique drugs. The dataset available in Kaggle includes 161, 771 interactions, covering different interaction mechanisms for drug pairs. Each drug pair is associated with its interaction mechanism type (viz., risk or severity of bleeding and hemorrhage, anticoagulant activities, antihypertensive activities, therapeutic efficacy) and corresponding action (increase or decrease). There are 91 different interaction mechanisms in this dataset, involving interactions among 1, 258 unique drugs. The dataset acquired from the Drugbank site contains various drug-related information like drug name, drug ID, SMILES sequence, uniport ID, etc. for a 12695 unique number of drugs. Regarding the dataset available in Kaggle, we have verified the interactions from the Drugbank interaction checker API (available at https://go.drugbank.com/drug-interaction-checker) and found that the interactions given in the Kaggle dataset are correct. The DDI dataset has 91 different types of mechanisms out of which four mechanisms viz., Excretion, Metabolism, Absorption and Excretion (higher serum level) are considered in this study to find out the presence or absence of the interactions with respect to that particular mechanism. Different mechanisms are having information about either increase or decrease or both actions present in DDI. For example, excretion has only decrease action, whereas the antihypertensive activities has both increased and decreased action. Table 5 summarizes the number of DDIs present against four mechanisms viz., Excretion, Absorption, Metabolism and Excretion rate (higher serum level).
Table 5.
Total number of both interaction of four different mechanism.
| Mechanism | Increase | Decrease |
|---|---|---|
| The excretion | N/A | 1185 |
| The absorption | N/A | 29 |
| The metabolism | 13290 | 50454 |
| The excretion rate (higher serum level) | N/A | 18686 |
The selection of these four mechanisms is based on multiple factors, including the number of available samples-ranging from very low (e.g., absorption with only 29 samples) to moderate (e.g., excretion) to high (e.g., metabolism and excretion rate (higher serum levels)). This diversity helps to evaluate the model’s effectiveness and robustness across different data distributions.
Additionally, these mechanisms represent both binary classification task (increase or decrease interactions for absorption, excretion and excretion rate (higher serum level)) and multi-class (3-class) scenarios (increase interaction, decrease interaction, and non-interaction for metabolism), ensuring a comprehensive assessment of the model’s performance across varying classification challenges.
In addition to these four mechanisms, further results on five more mechanisms (such as antihypertensive activities, CNS depressant activities, excretion rate which could result in a lower serum level and potentially a reduction in efficacy, hypoglycemic activities and hypotensive activities) are also reported in the Supplementary 1.
Since predicting the DDI, requires having the non-interacting drugs also (along with the interacting ones) to train any supervised model, therefore, we have randomly selected a pair of drugs (among the total 1258 drugs) that does not participate in the interactions process against a particular mechanisms. By doing so, for instance, a 1100 pair of non-interacting drugs are added against the excretion mechanism. Similarly, for the metabolism, absorption and excretion rate (higher serum level) mechanism, a 10632, 35 and 11878 pair of non-interacting drugs are added respectively.
Each unique Drugbank ID present in the Kaggle dataset43 is then searched from the Drugbank45 database to find its SMILES sequence in order to generate the chemical structural features such as Hall Smart, Amino Acid count, Carbon types extracted using Rcpi toolkit46. These structural features thus represent a unique drug feature vector with lengths 79, 20 and 9 for Hall Smart, Amino Acid count and Carbon types respectively.
The chemical features (viz., Hall Smart, Amino Acid Count, and Carbon Types) play a crucial role in enhancing the predictive power of the model by capturing structural and compositional information for drug pairs required for predicting interactions. Their relevance can be justified based on the following aspects:
The selection of these features provides a high-dimensional representation of chemical compounds, allowing the model to learn more nuanced differences between classes. The inclusion of a larger number of descriptive attributes generated through Hall Smart, Amino Acid Count, and Carbon Types ensure better characterization of molecular structures, contributing to improved classification accuracy as described next. Hall Smart descriptors47,48 encode topological and connectivity-based features of molecular structures, which are crucial for understanding molecular interactions. They help in capturing substructure patterns that are often correlated with biological activity or functional properties of molecules. The amino acid composition directly impacts drug / protein structure and function49,50. By incorporating amino acid count as a feature, the model can identify sequence-based variations that influence molecular behavior, particularly in biomolecular interactions49,50. Carbon atoms form the backbone of most chemical molecules, and their hybridization states, connectivity, and distribution provide critical structural information. The model benefits from this feature by learning patterns related to reactivity, stability, and functional group presence, which are essential for class differentiation51,52. Moreover, the latent space representation generated by these features has been found to positively impact class separation, as evidenced by Principal Component Analysis (PCA) plots as shown in Fig. S2.1 of Supplementary 2. This indicates that these features encapsulate meaningful patterns and variations that help the model distinguish between different classes effectively. Considering the above aspects Hall Smart, Amino Acid Count and Carbon Types have been selected as input features.
The class labels are assigned to the DDI dataset against the feature vectors of a pair of drugs (say, Drug A and Drug B) based on the increase and/or decrease, and neutral action for a particular mechanism. The feature vector for a pair of drugs (Drug A and Drug B) along with their interaction type (represented as the class label) makes the final feature vector which is to be supplied to the model. Both Drug A and Drug B, having the feature vector of size 108 each are concatenated with their corresponding class label which make the final feature vector of size 217 ( ). This is done for all the interactive pair of drugs which have been considered for this proposed work. As this step becoming the final preprocessing step, the preprocessed data now becomes suitable for training the proposed model.
). This is done for all the interactive pair of drugs which have been considered for this proposed work. As this step becoming the final preprocessing step, the preprocessed data now becomes suitable for training the proposed model.
Model design
The schematic representation of the proposed model for predicting DDI from structural features is illustrated in Fig. 8. Initially, drug ID’s (Drugbank ID) along with drug drug interaction information are taken as input. When the data preparation phase is over, data is subdivided into train, validation and test sets. After that, the data is passed through the proposed Durg Drug Interaction Network (DDINet) architecture.
Fig. 8.

Block diagram of proposed DDINet model.
The overall DDINet architecture can be viewed as a combination (fusion) of the GRU block, Attention block and LSTM block. It is worthy to note that different combinations of recurrent neural networks (RNN) and attention mechanisms have been tried in different layers of the hybrid architecture and finalized the model for which it produces the optimum performance. Here, in the proposed model, GRU and LSTM are preferred over RNN because of their ability to handle the vanishing / exploding gradient problem, and attention mechanisms in DDINet are carefully designed to leverage their unique strengths in finding the importance of the latent space features in modeling DDIs.
The GRU block is used for initial feature extraction from different molecular / structural features representations of pair of drugs (say, Drug A and Drug B). GRUs are chosen over LSTMs at this stage due to their computational efficiency and ability to capture short-to medium-term dependencies40, which are crucial for learning meaningful representations from various feature types. To capture the features’ importance, we incorporated attention mechanisms-both single attention and multi-head attention. This step is essential for effectively integrating diverse molecular features with their relative importance in terms of attention scores before passing them to the next stage. Finally, LSTM block is used after the attention mechanism to capture long-term dependencies40 and complex interactions between drug pairs. LSTMs were preferred here because they are well-suited for modeling sequential relationships, which are crucial for understanding cumulative effects in DDIs.
In summary, this hybrid approach ensures that the model benefits from the efficiency of GRU to capture short-to-medium term dependencies, the interpretability of attention using the single and multihead attention mechanism, and the ability of LSTM to retain long-term dependencies, leading to a robust and effective DDI prediction framework.
As the model is designed to predict the interaction between two drugs, extracted features of individual drugs (viz., Drug A and Drug B) should be fed into the model as inputs.
The model initially takes the structural features of Drug A and Drug B, then those are passed through the independent GRU block each for Drug A and Drug B where each GRU block processes each structural feature of individual drug alone. Responses coming out from these last layered GRUs are then concatenated for individual drugs and passed through the attention block. In the attention block, attention scores are calculated for the individual drug and further concatenated in the subsequent phase. These concatenated feature maps of two different drugs are then undergone through a multi-head attention layer. Again these feature maps are subjected to several consecutive LSTMs followed by one dense layer to predict the DDI. Based on the mechanisms DDI may have two class predictions (interacting or non-interacting pair of drugs) or three class predictions (increase, decrease or non-interaction). After completion of the training phase, the model is supplied with test samples for predicting the unknown interactions between a pair of drugs. The detailed architecture of the proposed model is shown in Fig. 9.
Fig. 9.

Detailed architecture of proposed DDINet model.
The extracted structural chemical features from a pair of drugs are passed through the proposed DDINet architecture consisting of a feature extraction phase followed by the classification phase. Feature extraction and classification phases are elaborated in the following subsection.
GRU Block The detailed architecture is shown in Fig. 9 which consists of the GRU blocks each processing three different feature maps of two individual drugs in a very similar fashion. For all the drugs, three different structural features (Hall Smart, Amino Acid count and Carbon types) have been extracted in the dataset preparation block. These features are represented by (
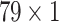 ), (
), (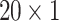 ) and (
) and ( ) for Hall Smart, Amino Acid count and Carbon types respectively. Primarily, three GRUs of two individual drugs accept three aforementioned feature maps. The first GRU takes the feature vector of Amino acid count having the size of (
) for Hall Smart, Amino Acid count and Carbon types respectively. Primarily, three GRUs of two individual drugs accept three aforementioned feature maps. The first GRU takes the feature vector of Amino acid count having the size of (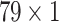 ) and passes through a GRU layer consisting of 50 hidden neurons. A dropout of
) and passes through a GRU layer consisting of 50 hidden neurons. A dropout of 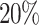 has been applied afterward. The response coming out after the dropout operation is then passed through two sequential GRUs, having the hidden neurons of 30 and 5 respectively. Similarly, two other feature vectors of Hall Smart, Carbon types having the size of
has been applied afterward. The response coming out after the dropout operation is then passed through two sequential GRUs, having the hidden neurons of 30 and 5 respectively. Similarly, two other feature vectors of Hall Smart, Carbon types having the size of  and
and 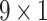 respectively are passed through three GRUs again. Here, the configuration of two sequential GRUs are 15, 10, 5 and 7, 6 and 5 respectively for processing Hall Smart and Carbon types feature vector.
respectively are passed through three GRUs again. Here, the configuration of two sequential GRUs are 15, 10, 5 and 7, 6 and 5 respectively for processing Hall Smart and Carbon types feature vector.Concatenated Attention Block The latent space feature vectors generated by the GRU Block for Drug A and Drug B are passed to the Concatenated Attention Block in the second phase. At the outset, the concatenation operations are applied individually on the responses coming out from the GRU Block for Drug A and Drug B. Then the output of the concatenation layers are passed to the two single-head attention layers having
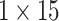 dimension each. Responses generated by the two single-head attention layers are concatenated to produce
dimension each. Responses generated by the two single-head attention layers are concatenated to produce 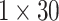 dimensional feature vector and passed to the multi-head attention layers with two heads each with dimension
dimensional feature vector and passed to the multi-head attention layers with two heads each with dimension 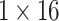 to generate the conjugate latent space representation of dimension
to generate the conjugate latent space representation of dimension  for the pair of drugs Drug A and Drug B.
for the pair of drugs Drug A and Drug B.LSTM Block The third block in the feature extraction phase of the proposed model is the LSTM block where the feature maps coming out from the Concatenated Attention Block are passed through the sequence of three LSTM units as follows. The responses of the Concatenated Attention Block are subjected through the
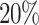 dropout operation to apply to the first LSTM unit generating
dropout operation to apply to the first LSTM unit generating  dimensional output which are again passed to the second LSTM unit with output neurons 16. Next, the third LSTM unit is applied to generate a
dimensional output which are again passed to the second LSTM unit with output neurons 16. Next, the third LSTM unit is applied to generate a  dimensional output vector using the sigmoid activation function. This final block of the feature extraction phase thereby generates the latent space feature representation with dimension (
dimensional output vector using the sigmoid activation function. This final block of the feature extraction phase thereby generates the latent space feature representation with dimension ( ) of the two pairs of drugs to the classification phase.
) of the two pairs of drugs to the classification phase.
Classification phase
The classification Phase comprises one dense layer followed by a softmax activation function which receives the input feature maps of two pairs of drugs extracted in the entire Feature Extraction Phase. This dense layer (with softmax activation) accepts a 5 dimensional feature vector produced by the LSTM block and maps it to the corresponding number of class levels for the prediction. It is worth noting that, the proposed model will act as a binary classifier when the mechanism has only interacting and non-interacting pairs of drugs present; whereas the model will act as multi-class classifier when the mechanism has increasing interaction, decreasing interaction and non-interacting pair of drugs present. Therefore, the output of the dense layer will have two neurons and three neurons present respectively for the case of binary mechanisms and multi-class mechanisms (three).
Validity measures
Here, four performance evaluation measures viz., percentage accuracy, precision, recall, micro-averaged 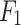 measure are applied for evaluating the overall performance of the proposed DDINet method53.
measure are applied for evaluating the overall performance of the proposed DDINet method53.
Parameters setting for training DDINet
In the article, Rcpi toolkit46 is used for extracting the features of each drug using their SMILES sequence. For classifying the interaction between the pair of drugs, the Google Colab Cloud platform is used. The implementation of the proposed algorithm is performed in TensorFlow along with 1.21.6 version of numpy and 1.3.5 version of pandas.
The proposed model is trained for the absorption mechanism for 250 epochs with a learning rate of 0.001 and for the remaining three mechanisms of the proposed model are trained for 500 epochs with the same learning rate and a dropout rate of 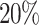 . The proposed DDINet architecture uses Mean Squared Error (MSE) and Categorical Cross-Entropy loss40 as the loss function, for binary class and three class classifiers respectively and Adaptive Momentum (Adam) optimizer40 is employed.
. The proposed DDINet architecture uses Mean Squared Error (MSE) and Categorical Cross-Entropy loss40 as the loss function, for binary class and three class classifiers respectively and Adaptive Momentum (Adam) optimizer40 is employed.
Supplementary Information
Author contributions
A.H. conceived the idea and model. B.S., S.M. and M.R. conducted the experiment. A.H., S.M. and M.R. analyzed the results. A.H., B.S. and S.M. drafted the manuscript. All the authors have reviewed and approved the final manuscript. Additionally, they each confirm that they have contributed significantly to the work and can publicly take responsibility for its content.
Data availability
Data used in this article are already publicly available on Drugbank45 (https://go.drugbank.com/releases/latest) and Kaggle43 (https://www.kaggle.com/datasets/smohsensadeghi/datasetname?resource=download&select=drug_interaction.csv).
Declarations
Competing interests
The authors have no relevant financial or non-financial interests to disclose.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Moumita Roy and Sukanta Majumder contributed equally to this work.
Contributor Information
Anindya Halder, Email: anindya.halder@nehu.ac.in, Email: anindya.halder@gmail.com.
Biswanath Saha, Email: biswanathsaha@nehu.ac.in.
Moumita Roy, Email: moumita.roy.7feb@gmail.com.
Sukanta Majumder, Email: majumder.sukanta85@gmail.com.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-025-93952-z.
References
- 1.World Health Organization. Drug use and health consequences. https://www.who.int/news-room/fact-sheets/detail/drug-use-and-health-consequences, Accessed: 2024-06-10.
- 2.Snyder, I. S. et al.Drug (Encyclopedia Britannica, 2024). https://www.britannica.com/science/drug-chemical-agent.
- 3.Lazarou, J., Pomeranz, B. & Corey, P. Incidence of adverse drug reactions in hospitalized patients: A meta-analysis of prospective studies. Jama279, 1200–1205 (1998). [DOI] [PubMed] [Google Scholar]
- 4.Kusuhara, H. How far should we go? perspective of drug-drug interaction studies in drug development. Drug Metab. Pharmacokinet.29, 227–228 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Dijk, K. V., de VRIES, C. S., van den Berg, P. B., Brouwers, J. & den BERG, L. T. W. J. D. V. Occurrence of potential drug-drug interactions in nursing home residents. Int. J. Pharm. Pract.9, 45–52 (2001).
- 6.Liu, S., Tang, B., Chen, Q. & Wang, X. Drug-drug interaction extraction via convolutional neural networks. Comput. Math. Methods Med.2016, 145 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Percha, B. & Altman, R. Informatics confronts drug-drug interactions. Trends Pharmacol. Sci.34, 178–184 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bjornsson, T. et al. Pharmaceutical research and manufacturers of america (PhRMA) drug metabolism/clinical pharmacology technical working group; fda center for drug evaluation and research (CDER). the conduct of in vitro and in vivo drug-drug interaction studies: A pharmaceutical research and manufacturers of america (PhRMA) perspective. Drug Metab. Dispos.31, 815–832 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Li, T., Wang, C., Zhang, L. & Chen, X. SNRMPACDC: Computational model focused on siamese network and random matrix projection for anticancer synergistic drug combination prediction. Brief. Bioinform.24, bbac503 (2023). [DOI] [PubMed] [Google Scholar]
- 10.Chen, X. et al. NLLSS: Predicting synergistic drug combinations based on semi-supervised learning. PLoS Comput. Biol.12, e1004975 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jang, H. et al. Machine learning-based quantitative prediction of drug exposure in drug-drug interactions using drug label information. NPJ Digit. Med.5, 88 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hung, T. et al. An ai-based prediction model for drug-drug interactions in osteoporosis and paget’s diseases from smiles. Mol. Inform.41, 2100264 (2022). [DOI] [PubMed] [Google Scholar]
- 13.Dang, L. H. et al. Machine learning-based prediction of drug-drug interactions for histamine antagonist using hybrid chemical features. Cells10, 3092 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mei, S. & Zhang, K. A machine learning framework for predicting drug-drug interactions. Sci. Rep.11, 17619 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang, H., Lian, D., Zhang, Y., Qin, L. & Lin, X. Gognn: Graph of graphs neural network for predicting structured entity interactions. arXiv preprintarXiv:2005.05537 (2020).
- 16.Zhu, Y., Li, L., Lu, H., Zhou, A. & Qin, X. Extracting drug-drug interactions from texts with BioBERT and multiple entity-aware attentions. J. Biomed. Inform.106, 103451 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Liu, N., Chen, C. & Kumara, S. Semi-supervised learning algorithm for identifying high-priority drug-drug interactions through adverse event reports. IEEE J. Biomed. Health Inform.24, 57–68 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Zaikis, D. & Vlahavas, I. Drug-drug interaction classification using attention based neural networks. In 11th Hellenic Conference on Artificial Intelligence, 34–40 (2020).
- 19.Warikoo, N., Chang, Y. & Hsu, W. LBERT: Lexically aware transformer-based bidirectional encoder representation model for learning universal bio-entity relations. Bioinformatics37, 404–412 (2021). [DOI] [PubMed] [Google Scholar]
- 20.Fatehifar, M. & Karshenas, H. Drug-drug interaction extraction using a position and similarity fusion-based attention mechanism. J. Biomed. Inform.115, 103707 (2021). [DOI] [PubMed] [Google Scholar]
- 21.Hanton, G. Preclinical cardiac safety assessment of drugs. Drugs R & D8, 213–228 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Shahreza, M., Ghadiri, N., Mousavi, S., Varshosaz, J. & Green, J. A review of network-based approaches to drug repositioning. Brief. Bioinform.19, 878–892 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Ryu, J. Y., Kim, H. U. & Lee, S. Y. Deep learning improves prediction of drug-drug and drug-food interactions. Proc. Natl. Acad. Sci.115, E4304–E4311 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vo, T., Nguyen, N., Kha, Q. & Le, N. On the road to explainable ai in drug-drug interactions prediction: A systematic review. Comput. Struct. Biotechnol. J.20, 2112–2123 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shukla, P. et al. Efficient prediction of drug-drug interaction using deep learning models. IET Syst. Biol.14, 211–216 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu, T., Cui, J., Zhuang, H. & Wang, H. Modeling polypharmacy effects with heterogeneous signed graph convolutional networks. Appl. Intell.51, 8316–8333 (2021). [Google Scholar]
- 27.Masumshah, R., Aghdam, R. & Eslahchi, C. A neural network-based method for polypharmacy side effects prediction. BMC Bioinform.22, 1–17 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin, S. et al. MDF-SA-DDI: Predicting drug-drug interaction events based on multi-source drug fusion, multi-source feature fusion and transformer self-attention mechanism. Brief. Bioinform.23, bbab421 (2022). [DOI] [PubMed] [Google Scholar]
- 29.Luo, Q. et al. Novel deep learning-based transcriptome data analysis for drug-drug interaction prediction with an application in diabetes. BMC Bioinform.22, 1–15 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang, Z., Zhong, W., Lv, Q. & Chen, C.Y.-C. Learning size-adaptive molecular substructures for explainable drug-drug interaction prediction by substructure-aware graph neural network. Chem. Sci.13, 8693–8703 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nyamabo, A. K., Yu, H. & Shi, J.-Y. SSI-DDI: Substructure-substructure interactions for drug-drug interaction prediction. Brief. Bioinform.22, bbab133 (2021). [DOI] [PubMed] [Google Scholar]
- 32.Li, Z. et al. DSN-DDI: An accurate and generalized framework for drug-drug interaction prediction by dual-view representation learning. Brief. Bioinform.24, bbac597 (2023). [DOI] [PubMed] [Google Scholar]
- 33.Feng, Y. H., Zhang, S. W. & Shi, J. Y. DPDDI: A deep predictor for drug-drug interactions. BMC Bioinform.21, 419 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nyamabo, A. K., Yu, H., Liu, Z. & Shi, J.-Y. Drug-drug interaction prediction with learnable size-adaptive molecular substructures. Brief. Bioinform.23, bbab441 (2022). [DOI] [PubMed] [Google Scholar]
- 35.Stražar, M., Žitnik, M., Zupan, B., Ule, J. & Curk, T. Orthogonal matrix factorization enables integrative analysis of multiple RNA binding proteins. Bioinformatics32, 1527–1535 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sarwar, B., Karypis, G., Konstan, J. & Riedl, J. Application of dimensionality reduction in recommender system: A case study. ACM SIGKDD Explor. Newslett.2, 82–86 (2000). [Google Scholar]
- 37.Lee, D. D. & Seung, H. S. Learning the parts of objects by non-negative matrix factorization. Nature401, 788–791 (1999). [DOI] [PubMed] [Google Scholar]
- 38.Mnih, A. & Salakhutdinov, R. Probabilistic matrix factorization. Adv. Neural Inf. Process. Syst.20 (2007).
- 39.Vaswani, A. et al. Attention is all you need. In Advances in Neural Information Processing Systems (NeurIPS) vol. 30 (2017).
- 40.Goodfellow, I., Bengio, Y. & Courville, A. Deep Learning (MIT Press, 2016). [Google Scholar]
- 41.Jordan, M. I. & Mitchell, T. M. Machine learning: Trends, perspectives, and prospects. Science349, 255–260 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Erwin, K. Introductory Mathematical Statistics : Principles and Methods (Wiley, 1970). https://catalog.loc.gov/vwebv/search?earchCode=LCCN&searchArg=70107583 &searchType=1 &permalink=y.
- 43.Sadeghi, S. M. Drug interaction dataset. https://www.kaggle.com/datasets/smohsensadeghi/datasetname?resource=download&select=drug_interaction.csv, Accessed on: 2024-06-18.
- 44.Anderson, E., Veith, G. & Weininger, D. SMILES, a Line Notation and Computerized Interpreter for Chemical Structures (US Environmental Protection Agency, Environmental Research Laboratory, 1987). [Google Scholar]
- 45.Knox, C. et al. Drugbank 6.0: The drugbank knowledgebase for 2024. Nucleic Acids Res.52, D1265–D1275 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cao, D.-S., Xiao, N., Xu, Q.-S. & Chen, A. F. Rcpi: R/bioconductor package to generate various descriptors of proteins, compounds and their interactions. Bioinformatics31, 279–281 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Hall, L. H. & Kier, L. B. The molecular connectivity chi indexes and kappa shape indexes in structure-property modeling. Rev. Comput. Chem. 367–422 (1991).
- 48.Randic, M. On molecular identification numbers. J. Chem. Inf. Comput. Sci.24, 164–175 (1984). [Google Scholar]
- 49.Kyte, J. & Doolittle, R. F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol.157, 105–132 (1982). [DOI] [PubMed] [Google Scholar]
- 50.Fauchere, J.-L. & Pliska, V. Hydrophobic parameters pi of amino-acid side chains from the partitioning of n-acetyl-amino amides. Eur. J. Med. Chem. (1983).
- 51.Hanch, C. & Leo, A. Substituent constants for correlation analysis in chemistry and biology, wiley-interscience. J. Med. Chem. (1979). [DOI] [PubMed]
- 52.Todeschini, R. & Consonni, V. Handbook of Molecular Descriptors (John Wiley & Sons, 2008). [Google Scholar]
- 53.Salton, G. & McGill, M. J. Introduction to Modern Information Retrieval (McGraw-Hill, 1983). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data used in this article are already publicly available on Drugbank45 (https://go.drugbank.com/releases/latest) and Kaggle43 (https://www.kaggle.com/datasets/smohsensadeghi/datasetname?resource=download&select=drug_interaction.csv).