

ABSTRACT
Atopic dermatitis, prurigo nodularis, and chronic spontaneous urticaria are immune‐mediated, inflammatory skin conditions characterized by intense itch and disease‐specific skin lesions. Despite their different clinical presentations, the three diseases are unified by an aberrant type 2 immune response involving type 2 cytokines, immune cells, and sensory nerves that may underlie their shared clinical manifestations of inflammation and pruritus. The chronic nature of these conditions is associated with significant impairment in patients' quality of life and psychological disorders, such as anxiety and depression. This article reviews type 2 inflammation and its role in atopic dermatitis, prurigo nodularis, and chronic spontaneous urticaria, focusing on the pathophysiologic drivers of type 2 inflammation in each dermatologic condition. Understanding the shared immune mechanisms that underlie these seemingly distinct skin diseases and other concomitant inflammatory conditions is critical for applying therapeutic interventions targeting the type 2 immune pathway.
Keywords: atopic dermatitis, chronic spontaneous urticaria, cytokine, prurigo nodularis, type 2 inflammation
1. Introduction
Chronic inflammation is driven by abnormal or dysregulated immune responses, often triggered by complex interactions between intrinsic and environmental factors. Under normal physiological circumstances, the role of type 2 inflammation is to provide defense against multicellular parasitic pathogens and environmental irritants, toxins, and allergens. However, aberrant type 2 immune signaling is associated with several inflammatory conditions, such as atopic dermatitis (AD), asthma, chronic rhinosinusitis with nasal polyposis, eosinophilic esophagitis, and chronic obstructive pulmonary disease. Recently, dysregulated type 2 inflammation has been identified as contributing to other skin conditions beyond AD, specifically prurigo nodularis (PN) and chronic spontaneous urticaria (CSU). In all three skin diseases, an aberrant type 2 immune response leads to intense itch and skin lesions, which are often chronic and can significantly impact the quality of life. However, despite their shared type 2 pathophysiology, the clinical presentations of AD, PN, and CSU diverge, with distinct patterns of skin manifestations and clinical courses.
This review aims to provide an overview of type 2 inflammation and its contribution to AD, PN, and CSU. Specifically, we highlight the role of immune cells and cytokines that contribute to disease activity and progression. We also explore the therapeutic rationale for targeting the type 2 inflammatory axis to modulate the immune dysregulation in AD, PN, and CSU.
2. Overview of the Immune System
The human immune system consists of innate and adaptive responses. The innate immune system recognizes molecular patterns shared by many pathogens. It provides an initial, rapid, non‐specific immune response, whereas the adaptive immune system targets specific invading pathogens and provides long‐term immune protection through immunologic memory [1]. Activation of the adaptive immune system is a highly orchestrated process involving various signals from the innate system to confer the specific host response through clonal selection [2]. While immune responses can be categorized in several ways, they are conventionally grouped into three major modules based on shared activity and functions, distinguished by the involvement of specific innate and adaptive immune cells and cytokines (Figure 1; Supporting Information) [3].
FIGURE 1.
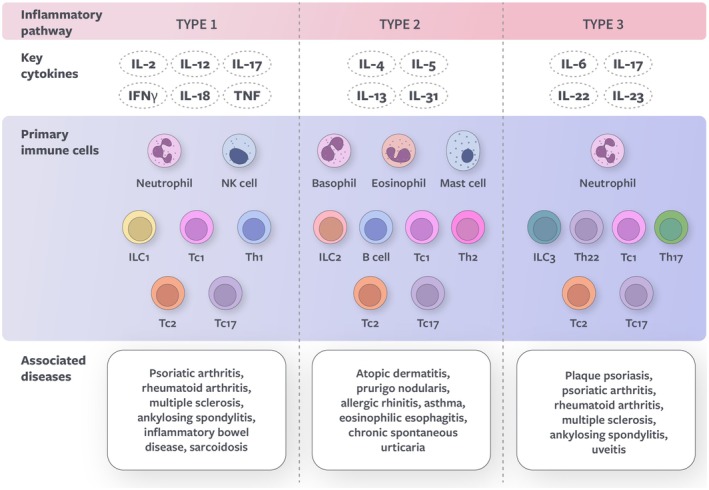
Overview of types 1, 2, and 3 immune responses. IFN, interferon; IL, interleukin; ILC, innate lymphoid cell; NK, natural killer; Tc, T cytotoxic cell; Th, T helper cell; TNF, tumor necrosis factor.
Type 1 immunity predominantly targets intracellular pathogens, including viruses, intracellular bacteria, and transformed cancer cells. It is mediated by T helper (Th) type 1 cells, T cytotoxic (Tc) cells, and group 1 innate lymphoid cells (ILC1s). Infected or malignant cells are eliminated through the release of proinflammatory cytokines, such as interferon (IFN), interleukin (IL)‐12, and tumor necrosis factor (TNF) [4].
Type 2 immunity provides defense against parasites, mainly helminths, and diverse environmental threats, including toxins and irritants. It is driven by Th2 cells, Tc2 cells, group 2 ILCs (ILC2s), and mast cells, which release alarmins and signature cytokines (IL‐4, IL‐5, IL‐13, and IL‐31). In turn, type 2 cytokines recruit effector cells, such as eosinophils and basophils, and promote further Th2 polarization, keratinocyte hyperproliferation, and B cell class switching to immunoglobulin E (IgE), IgG, and IgA. Type 2 immune mechanisms aim to eliminate parasites by triggering expulsion (e.g., the itch/scratch reflex) and fibrosis (e.g., wound repair) [5].
Type 3 immunity mainly protects against extracellular fungi and bacteria. Its main cellular components are group 3 ILCs (ILC3s), Th17, and Tc17 cells, which produce IL‐17 and IL‐22 cytokines. IL‐17 cytokines stimulate the production of a broad array of proinflammatory proteins secreted by keratinocytes, including antimicrobial peptides and IL‐36 [6]. This results in the recruitment of neutrophils and activation of other immune cells to protect against pathogens through phagocytosis and tissue inflammation [3].
Under normal physiologic conditions, the various types of immune responses serve protective roles and have multiple layers of regulatory mechanisms. However, chronic inflammation and inflammatory diseases can arise when immune responses become dysregulated. Dysregulated type 1 immunity contributes to psoriatic arthritis, rheumatoid arthritis, Hashimoto thyroiditis, and multiple sclerosis [3, 7]. Dysregulated type 2 immunity can contribute to various chronic inflammatory diseases, including AD, allergic rhinitis, allergic asthma, PN, CSU, chronic rhinosinusitis with nasal polyposis, and eosinophilic esophagitis [8, 9]. Type 3 diseases include plaque psoriasis and ankylosing spondylitis [6, 10].
3. The Role of IL‐4 and IL‐13 in Type 2 Inflammation
Type 2 immunity is characterized by the expression of type 2 cytokines (IL‐4, IL‐5, IL‐13, and IL‐31), crucial in regulating immune responses to allergens, irritants, and parasites. This occurs in concert with innate cells, adaptive cells, cutaneous neurons, and keratinocytes that perpetuate the polarization of a type 2 immune response. Type 2 cytokines and their receptors are widely expressed among cell types from both the innate and adaptive arms of the immune system [11, 12, 13, 14, 15]. In AD, PN, and CSU, type 2 cytokines are critical mediators of inflammation at both the systemic and local tissue levels and employ the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway to exert their effect through direct receptor binding (see Supporting Information) [16].
IL‐4 and IL‐13 are the two primary effector cytokines in type 2 immunity. They share some functions but are not identical or interchangeable. Both IL‐4 and IL‐13 promote IgE production, mast cell and basophil activation and degranulation, fibrotic tissue remodeling, skin barrier dysfunction, and microbiome imbalance [17, 18, 19, 20, 21]. In addition, IL‐4 and IL‐13 act on receptors in sensory neurons, increasing their sensitivity to several pruritogens [22]. IL‐4, but not IL‐13, is essential for Th2 cell differentiation and polarization, further amplifying the systemic inflammatory response through the release of IL‐4, IL‐5, IL‐13, and IL‐31 [19, 20, 23, 24]. In contrast, IL‐13 has a role in peripheral immunity as an effector cytokine in end‐organ tissues [25]. It contributes to skin pathology by promoting collagen deposition [17, 21]. IL‐4, IL‐13, and IL‐5 control eosinophil recruitment and trafficking into tissue, whereas IL‐5 also controls eosinophil survival and differentiation [19, 20]. IL‐31 is primarily produced by differentiated T lymphocytes and mediates itch sensations upon binding to its receptor IL‐31RA, expressed on various cell types, including sensory neurons, keratinocytes, and immune cells [24, 26]. In addition, IL‐31 may contribute to inflammation, skin barrier dysfunction, and fibrosis [26, 27].
Activating immune cells and enhancing immune cell trafficking to tissues amplify type 2 inflammation and establish a chronic and persistent disease phenotype [8, 18]. Continued activation of type 2 immunity may lead to the emergence of resident memory T lymphocytes and long‐term changes in the immune response.
4. Neuroimmune Interactions
Immune cells, neurons, and cytokines work together to perpetuate the chronic inflammatory state at the tissue level. In the localized tissue microenvironment, the proximity of immune cells and sensory neurons fosters dynamic crosstalk, enabling reciprocal stimulation and modulation of inflammatory pathways. The interaction between nerves, mast cells, T cells, eosinophils, and other immune cells triggers the release of proinflammatory mediators such as histamines and cytokines, promoting itch, pain, and inflammation. Additionally, sensory neurons can be activated by alarmins (IL‐33, thymic stromal lymphopoietin; see Supporting Information) produced by keratinocytes. Activated sensory neurons release neuropeptides, such as substance P (see Supporting Information), which further activate immune cells and promote chronic inflammation [8, 9, 20, 28, 29].
Recent therapeutic strategies have revealed that targeting mediators of type 2 inflammation can alleviate various dermatologic conditions previously thought to have different disease drivers. AD, PN, and CSU are distinct skin conditions with disease‐specific signs and symptoms, but they share an intricate crosstalk between sensory cells and type 2 immune cells. It is now appreciated that sensory neurons play a crucial role in relaying signals, generating their molecular messengers that impact surrounding immune components in the skin. However, this shared pathophysiology manifests uniquely in each disorder. In AD and PN, the itch is persistent, with primary skin lesions reflective of the itch‐scratch cycle. In contrast, itch and pain in CSU fluctuate with the transient nature of urticarial lesions and leave no residual skin changes upon resolution. Unraveling the intricacies of type 2 inflammation in AD, PN, and CSU enhances our understanding of the immune response and holds promise for therapeutic interventions targeting their common elements.
5. Atopic Dermatitis
AD is a debilitating systemic disease characterized by recurrent eczematous lesions, skin barrier disruption, and intense pruritus [30]. AD usually develops in childhood and often persists into adulthood [31], with an estimated prevalence of 11.8% in the US pediatric population [32] and 7.3% in the US adult population [33]. AD pathogenesis involves a complex interplay of genetic and environmental factors. Genetic predisposition to AD can derive from mutations in genes related to skin barrier function (e.g., FLG and LOR) and immune response regulation (e.g., CARD14 and TLR genes) [34]; such genetic variations may increase susceptibility to environmental triggers such as irritants, allergens, and pathogens [35, 36].
Patients with AD experience a multidimensional disease burden that can have a significant impact on quality of life. The persistent itch and pain can lead to sleep disturbances, reduced productivity, and absenteeism from work and school [37, 38, 39]. These effects, together with the distress caused by visible skin lesions, can strongly contribute to mental health disorders such as anxiety and depression [37]. Moreover, AD is often associated with comorbidities such as allergic conditions, skin infections, and cardiovascular diseases [40].
Type 2 cytokines are key drivers of the pathophysiology of AD (Figure 2). Disruption of the epithelial barrier triggers the release of alarmins (see Supporting Information), which act as early soluble signals to stimulate the polarization of a type 2 immune response [42, 43]. Type 2 inflammation leads to the activation of immune effector cells, which infiltrate the skin and perpetuate the inflammatory response. Elevated Th2 cytokines result in the downregulation of antimicrobial peptides and barrier proteins in keratinocytes, weakening the skin defenses and increasing the risk of infections [44, 45]. IL‐4 and IL‐13 have sensitized neurons to a range of pruritogens (e.g., IL‐31 and histamine), resulting in chronic itch [22, 46]. Furthermore, a feedback loop occurs where IL‐4, produced by Th2 cells and potentially mast cells and basophils, acts on Th2 cells to trigger the production of IL‐31, along with more IL‐4 and IL‐13 [19, 23, 47]. Scratching can also further disrupt the epithelial barrier and lead to a continued increase in alarmin production by keratinocytes, thereby promoting disease chronicity in the skin [47].
FIGURE 2.
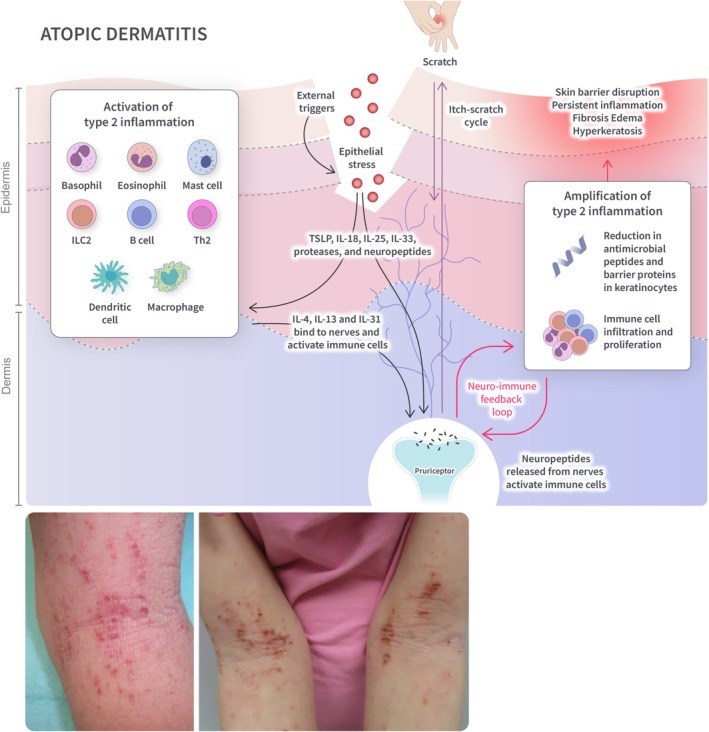
Overview of type 2 inflammation in AD. Skin barrier disruption triggers the release of alarmins (IL‐25, IL‐33, TLSP), which stimulate a type 2 immune response. Type 2 cytokines (IL‐4 and IL‐13) activate immune cells and sensitize neurons to pruritogens, leading to an itch‐scratch cycle. A neuro‐immune feedback loop results in the amplification of type 2 inflammation and further skin barrier disruption through scratching. IL, interleukin; ILC, innate lymphoid cell; Th, T helper cell; TSLP, thymic stromal lymphopoietin. Source: Siegfried et al. [41].
The chronic inflammatory signaling driven by type 2 cytokines and immune effector cells can lead to maladaptive responses. Persistent scratching can give rise to epidermal thickening (lichenification) and excoriations [42]. Skin barrier function is further disrupted, resulting in a vicious cycle of increased susceptibility to irritants and allergens, inflammation, and skin barrier dysfunction [30]. In addition, a dysfunctional skin barrier is often associated with alterations in the skin microbiome and increased colonization by Staphylococcus aureus [48]. The overgrowth of S. aureus can trigger inflammatory responses in keratinocytes through the activation of pattern recognition receptors and promote the release of toxins, further aggravating the epidermal barrier disruption, lesion chronicity, and susceptibility to infections [48, 49].
Emollients and moisturizers are pivotal in managing AD to restore skin barrier function and thereby reduce inflammatory signals produced by activated keratinocytes [50, 51]. Topical corticosteroids, available in various potency levels, may further alleviate inflammation and pruritus by modulating immune responses in the skin. However, prolonged use can lead to skin atrophy, so their application requires noncontinuous treatment and monitoring [51, 52]. Non‐steroidal anti‐inflammatories such as topical calcineurin inhibitors (e.g., tacrolimus and pimecrolimus), topical JAK inhibitors (e.g., ruxolitinib), topical phosphodiesterase 4 inhibitors (e.g., crisaborole and roflumilast) and aryl hydrocarbon receptor modulators (e.g., tapinarof) offer an alternative for sensitive areas such as the face, neck, and genitals [50, 51, 52, 53, 54, 55]. In cases of moderate‐to‐severe AD unresponsive to topical treatments, phototherapy or systemic medications, such as oral immunosuppressants, may be considered. Traditional immunosuppressive agents used to treat AD include azathioprine, cyclosporine, and methotrexate; however, these medications require close laboratory monitoring due to their narrow therapeutic index, multiple drug–drug interactions, and the potential for serious side effects, including kidney or liver injury, bone marrow suppression, pulmonary fibrosis, and hypertension [51, 56, 57].
More recently, injectable biologics (e.g., dupilumab, tralokinumab, lebrikizumab, and nemolizumab) have demonstrated efficacy and safety in the treatment of moderate‐to‐severe AD [51, 57, 58, 59, 60, 61, 62]. Dupilumab, the first biologic approved for the treatment of AD, modulates the type 2 immune response and inflammation through dual inhibition of IL‐4 and IL‐13 by blocking the IL‐4Rα subunit [51, 57]. Tralokinumab and lebrikizumab reduce the inflammatory response associated with AD through inhibition of IL‐13 [58, 59]. In contrast, nemolizumab inhibits the signaling of IL‐31 by blocking IL‐31Rα and is therefore effective at reducing pruritus and inflammation [60, 61, 62]. However, the ability of nemolizumab to adequately suppress type 2 inflammatory circuits in the skin through the inhibition of IL‐31 signaling alone has not been fully elucidated. Additional clinical studies are needed to compare the clinical significance of IL‐31 blockade versus selective blockade of IL‐4 and/or IL‐13 signaling in primary cutaneous conditions.
For patients who are not responsive to biologics and other systemic therapies, oral JAK inhibitors have emerged as an efficacious alternative, though more studies are needed to assess their long‐term safety [63, 64]. JAK inhibitors alleviate AD symptoms by blocking the JAK/STAT signaling pathway, which is involved in intracellular signaling pathways for a broad range of inflammatory cytokines and growth factors [57, 63, 64]. Finally, early clinical studies exploring the OX40 receptor and its ligand (OX40L) as therapeutic targets for AD have recently shown promise [65, 66, 67]. OX40, primarily expressed on the surface of activated T cells, modulates immune responses by promoting T‐cell survival and proliferation [67, 68, 69]. OX40L belongs to the TNF superfamily and is primarily expressed on antigen‐presenting cells, such as dendritic and B cell populations [70]. By targeting the OX40/OX40L pathway, novel drugs such as rocatinlimab (anti‐OX40), telazorlimab (anti‐OX40), and amlitelimab (anti‐OX40L) aim to regulate memory and regulatory T‐cell responses, thereby reducing the inflammation and immune dysfunction observed in AD [65, 66, 67, 69].
6. Prurigo Nodularis
PN is a chronic inflammatory skin condition with an estimated prevalence of 72 out of 100,000 individuals in the U.S. adult population [71]. The diagnosis of PN is based on the following clinical features: firm, itchy lesions that can present as nodules, papules, or plaques, generally with a bilateral distribution on the arms and trunk and sparing areas that are hard to reach; chronic pruritus lasting 6 weeks or more; history and/or signs of repeated scratching, picking, or rubbing [72, 73, 74]. PN primarily affects middle‐aged to older patients (50 years of age and older) [75]. PN signs and symptoms (intense itch, skin lesions, bleeding of excoriated lesions, scars with skin dyspigmentation) [72, 76, 77] can have a severe impact on patients' quality of life and are associated with sleep disturbance, absenteeism from work, symptoms of depression and anxiety, and a feeling of shame and helplessness [72, 78].
The exact pathophysiology of PN is still being elucidated. However, PN is thought to occur due to neuronal dysfunction, sensitization to itch, and the subsequent development of an itch‐scratch cycle [72]. Activation of sensory neurons through neuronal dysfunction and scratching may trigger the release of neuropeptides (see Supporting Information), which activate immune cells (Figure 3). Immune cell activation and the subsequent release of type 2 (IL‐4, IL‐13, and IL‐31) and type 3 (IL‐17) proinflammatory cytokines are likely to promote the development of chronic inflammation [9, 79, 80, 81]. The stimulation of fibroblasts by IL‐4 and IL‐13 may promote dermal fibrosis through extracellular matrix deposition, fibroblast proliferation, and myofibroblast differentiation [82]; furthermore, IL‐4 and IL‐13 are thought to trigger the production of periostin, which enhances itch and contributes to dermal fibrosis [83, 84]. Periostin acts on keratinocytes to induce the production of proinflammatory cytokines, further driving chronicity [83]. In addition, the stimulation of sensory neurons by IL‐4, IL‐13, and IL‐31 induces persistent itch [9, 81]. Chronic neurogenic inflammation and immune cell infiltration may lead to fibrotic nodules and epidermal hyperplasia, further exacerbated by the itch‐scratch cycle [72, 80, 85].
FIGURE 3.
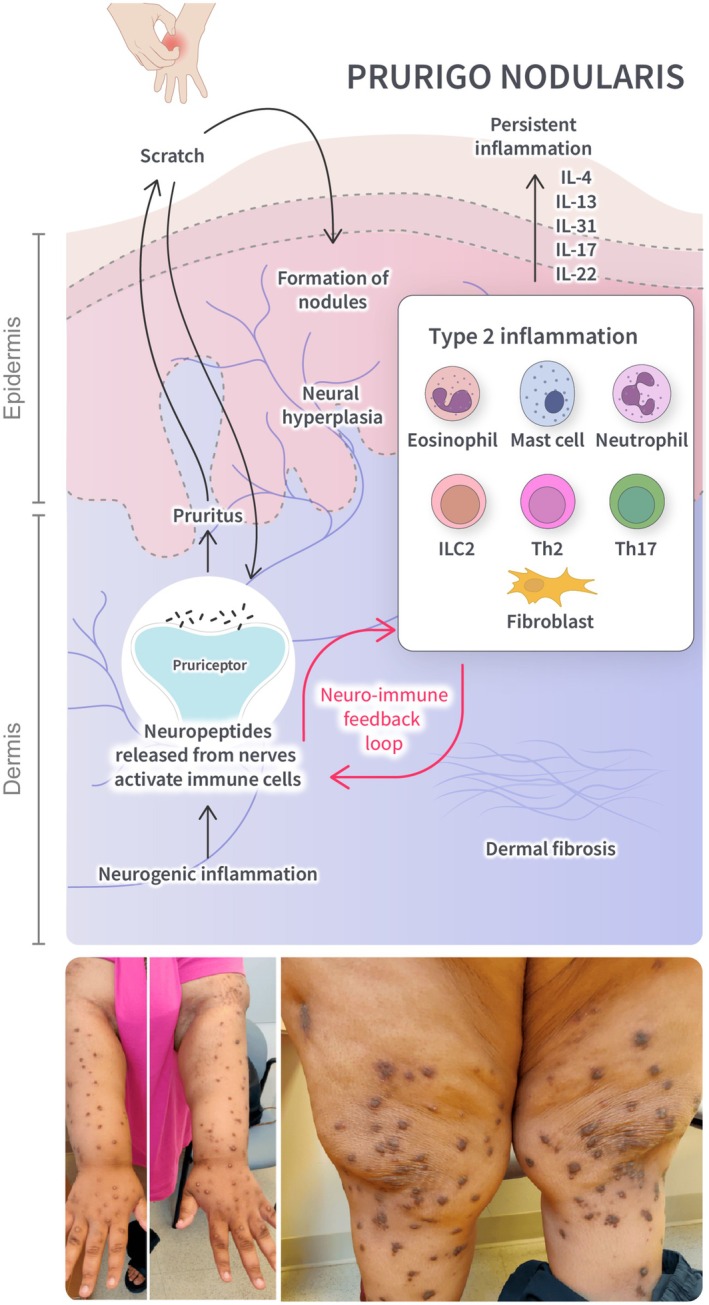
Overview of type 2 inflammation in PN. Neurogenic inflammation triggers the release of neuropeptides, which activate immune cells. This leads to the release of type 2 cytokines, resulting in itch and chronic inflammation. Persistent scratching may lead to the formation of nodules. IL‐4 and IL‐13 stimulate fibroblasts, promoting dermal fibrosis. IL, interleukin; ILC, innate lymphoid cell; Th, T helper cell. Source: Kwon et al. [73].
Treatment options for PN typically involve a multifaceted approach tailored to the patient's symptoms and underlying factors, often combining topical and oral systemic therapies [74]. Topical corticosteroids or calcineurin inhibitors may be applied directly to affected areas to alleviate itching and reduce inflammation [74]. Phototherapy and off‐label systemic medications, such as gabapentinoids and antidepressants, may be considered for more widespread and persistent cases to modulate the neural pathways associated with itch and the development of PN [74, 86, 87]. In more severe cases that are resistant to other treatments, immunosuppressive medications such as oral corticosteroids, methotrexate, cyclosporine, or azathioprine may be considered, although their potential risks and long‐term side effects must be carefully evaluated [86, 87].
Dupilumab, which inhibits the signaling of cytokines IL‐4 and IL‐13, is the first approved systemic therapy for adults with PN [88, 89]. Treatment with dupilumab in two phase 3 trials led to significant improvements in multiple aspects of PN, with a safety profile consistent with its known safety profile [89]. Nemolizumab, also approved for PN, targets the IL‐31 receptor and has been associated with improvements in PN lesions and pruritus in phase 2 and 3 trials [88, 90, 91]. Furthermore, JAK inhibitors abrocitinib and upadacitinib have shown promise for managing PN, offering another targeted approach to interrupt type 2 inflammatory pathways [92, 93, 94]. The potential benefits of selective blockade of IL‐17 or IL‐23 for treating PN are not yet known but may be useful in a subset of PN patients with increased IL‐17 signaling in PN lesions.
7. Chronic Spontaneous Urticaria
CSU is a chronic, heterogeneous, inflammatory condition characterized by recurrent transient, itchy wheals (hives), with or without angioedema, for more than 6 weeks without an identifiable trigger [95, 96, 97]. CSU differs from chronic inducible urticaria, induced by a definite trigger (e.g., cold, pressure, or ultraviolet exposure), and acute urticaria, defined by a duration of less than 6 weeks [97]. CSU and chronic inducible urticaria can occur simultaneously in the same patient.
CSU affects all age groups, with a higher incidence in 20‐ to 40‐year‐olds and a prevalence of 0.08%–0.11% in the United States [97, 98]. In adults, CSU is twice as common in women than in men [98]. Despite the self‐remitting nature of the disease, many patients with CSU experience a prolonged disease course that can last for more than 5 years [99]. The appearance of CSU lesions is unpredictable and associated with anticipatory fear, debilitating itch, impaired sleep, and occasional skin pain or a burning sensation [97, 100]. Furthermore, CSU is often accompanied by comorbid autoimmune diseases (e.g., vitiligo, autoimmune thyroiditis) and, to a lesser extent, atopic conditions (e.g., AD, asthma) [97, 100]. CSU strongly affects patients' quality of life and is linked to psychosocial disorders, such as depression and anxiety [97, 101].
While the pathogenesis of CSU is not fully understood, activation of the mast cell through various autoimmune mechanisms is central to this condition [102, 103]. CSU clinical manifestations are driven by mast cell degranulation and the subsequent release of histamines, followed by leukotrienes, prostaglandins, and proinflammatory cytokines (IL‐4, IL‐13, IL‐31) [102, 103, 104]. Mast cell degranulation in CSU may occur via IgE‐dependent (type I autoimmunity/autoallergic) as well as IgE‐independent pathways (type IIb autoimmunity); in the IgE‐dependent pathway, autoallergens induce cross‐linking of IgE antibodies bound to the high‐affinity IgE receptor (FcεRI) on the surface of mast cells, whereas the IgE‐independent pathway may involve the presence of IgG autoantibodies directed against IgE and FcεRI [102, 103, 104]. The release of inflammatory mediators leads to vasodilation, increased extravasation, activation of sensory skin nerves, and the recruitment of immune cells [103, 104]. In addition to mast cells, the pathogenesis of CSU involves a perivascular infiltrate of inflammatory cells, including T lymphocytes (predominantly Th2), basophils, and eosinophils [102].
Type 2 inflammation is thought to contribute to the pathophysiology of CSU through multiple processes (Figure 4). IL‐4 is a key driver of differentiating naïve T lymphocytes from Th2 cells [19, 103]. IL‐4 and IL‐13 promote B‐cell activation and IgE production and enhance autoantibody‐mediated mechanisms, as well as trafficking of type 2 immune cells to the site of inflammation [19, 102, 103, 104]. At the neuroimmune axis, IL‐4 and IL‐13 activate and sensitize nerves to a range of pruritogens, including histamines and IL‐31 [19, 102, 105]. Released neuropeptides from activated neurons potentially act as ligands by binding to MRGPRX2 receptors on cutaneous mast cells, leading to further activation and release of various inflammatory mediators [97, 102]. Collectively, these effects may lead to mast cell hyperactivity and contribute to a continuous cycle of neuroimmune inflammation [97, 102, 103].
FIGURE 4.
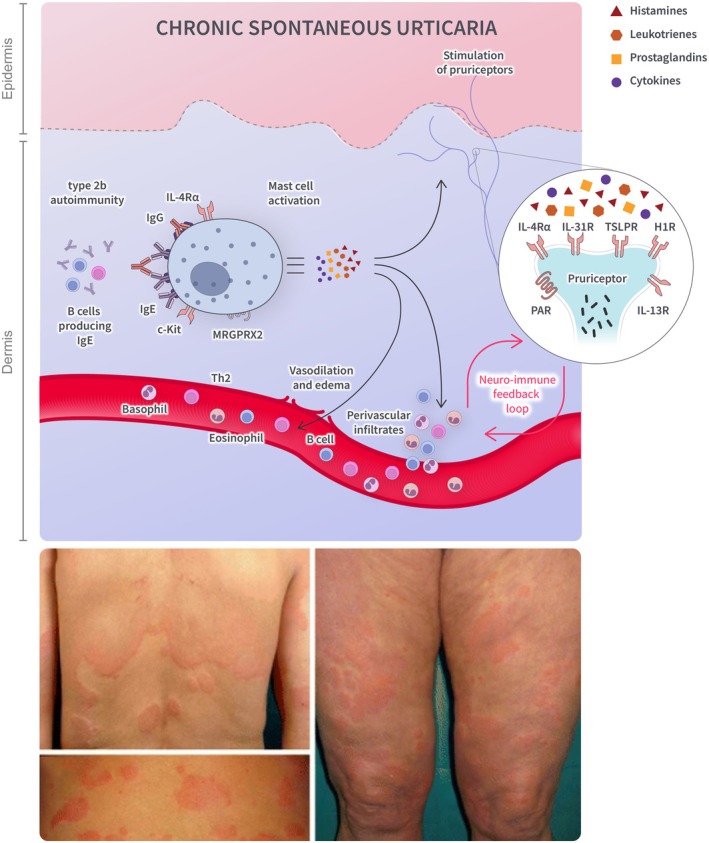
Overview of type 2 inflammation in CSU. Mast cell degranulation is induced by IgE antibodies binding to high‐affinity IgE receptors on the mast cell surface and/or by IgG antibodies targeting IgE and high‐affinity IgE receptors. Mast cell activation leads to the release of histamines, followed by leukotrienes, prostaglandins, and cytokines. The release of inflammatory mediators results in the recruitment of immune cells, vasodilation, and activation of skin nerves. A perivascular infiltrate of inflammatory cells is also thought to drive clinical manifestations of CSU. BTK, Bruton's tyrosine kinase pathway; H, histamine; Ig, immunoglobulin; IL, interleukin; ILC, innate lymphoid cell; MRGPRX2, mas‐related G‐protein coupled receptor member; X2, PAR protease‐activated receptor; R, receptor; SYK, spleen tyrosine kinase pathway; Th, T helper cell, TSLP, thymic stromal lymphopoietin. Source: Marzano et al. [95].
Treatment of CSU can pose a challenge due to the recurrent and unpredictable nature of the disease. CSU treatment options aim to prevent new skin lesions, alleviate symptoms, and improve the patient's quality of life [96, 106, 107]. Nonsedating antihistamines, such as second‐generation H1‐antagonists, are the first‐line treatment for CSU [106, 107, 108]. Many patients, however, do not achieve symptom control with H1 antihistamines, even after up‐dosing [109]. Omalizumab, an anti‐IgE monoclonal antibody, is approved for antihistamine‐refractory cases [110], but approximately 25% of these patients remain symptomatic [111, 112]. Dupilumab has recently been approved for CSU in Japan, Brazil, and the United Arab Emirates and is currently under regulatory review for CSU in the US and EU, following phase 3 clinical trials [112, 113]. Recent clinical studies with Bruton's tyrosine kinase (BTK) inhibitors, such as remibrutinib and rilzabrutinib, have shown promise in patients who are refractory to H1 antihistamines, supporting the rationale for interrupting type 2 inflammatory pathways and mast cell activation, respectively, in the management of CSU [114, 115]. Phase 2 clinical studies evaluating oral remibrutinib showed a rapid onset of action, clinical efficacy, and a favorable safety profile over 52 weeks [114, 116]; these treatment characteristics were validated in the early analysis of phase 3 clinical studies [117, 118]. Oral BTK inhibition as a treatment for CSU will be further explored in planned phase 3 clinical trials for rilzabrutinib. A variety of other novel mechanisms of action, including blockade of c‐KIT and siglec‐6 receptors on mast cells, are currently being evaluated and will potentially expand CSU treatment options.
8. Shared Pathophysiology of Atopic Dermatitis, Prurigo Nodularis, Chronic Spontaneous Urticaria, and Beyond
AD, PN, and CSU are distinct inflammatory conditions with unique clinical presentations. Yet, they are unified by an interplay of cellular and molecular factors that modulate type 2 inflammation and neuroimmune interactions, which lead to their shared clinical manifestations of inflammation, pruritus, epidermal barrier dysfunction, and primary skin lesions. Dysregulation of sensory nerves is implicated in itch and neurogenic inflammation across the three conditions [42, 72, 85, 97, 102]. The interplay of dysregulated modules in neurons, cytokines, mediators, and immune cells at the epidermis and dermis contributes to the unique clinical manifestations observed in each condition.
The shared thread of a skewed, overactive type 2 immune response and neurosensitization may explain the common co‐occurrence of allergic and atopic inflammatory conditions. AD is a prevalent comorbidity in patients with PN [73, 76] and is often associated with a personal or family history of asthma and allergic rhinitis [40]. Furthermore, the predisposition to allergic responses and increased levels of IgE antibodies seen in AD and asthma are also observed in CSU [119, 120].
In addition to AD, PN, and CSU, type 2 immune circuits may contribute to the pathogenesis of other dermatological conditions, such as bullous pemphigoid, lichen planus, Grover's disease, and chronic pruritus of unknown origin. In bullous pemphigoid, type 2 inflammatory cytokines (particularly IL‐4, IL‐5, and IL‐13) are thought to drive the recruitment of mast cells and eosinophils, contributing to blister formation [121]. The immunological pathways underlying lichen planus and Grover's disease are poorly understood, though both are considered mixed inflammatory conditions involving type 1 and 2 immune mechanisms [122, 123, 124]. Several cases of cutaneous lichen planus and Grover disease have been responsive to dupilumab [124, 125, 126, 127, 128], suggesting a potential role of type 2 immune responses in the pathogenesis of both conditions. The number of published cases reporting the potential clinical benefits of type 2 blockade in numerous dermatologic and nondermatologic immune conditions is rapidly growing, with multiple biologic agents available to clinicians. These findings highlight the complex interplay between type 2 immunity and diverse clinical manifestations, underscoring the need for further research into type 2 inflammation across different diseases.
A deeper understanding of the type 2 pathway has led to novel therapies targeting shared mechanisms in various type 2 inflammatory conditions (Table 1). Targeting multiple co‐occurring conditions with a single therapy can simplify treatment regimens and enhance patient outcomes. For instance, dupilumab modulates the immune response by inhibiting IL‐4 and IL‐13 signaling and has demonstrated efficacy in AD, PN, CSU, asthma, eosinophilic esophagitis, chronic obstructive pulmonary disease, and chronic rhinosinusitis with nasal polyps and is currently being evaluated in bullous pemphigoid, chronic pruritus of unknown origin, and pruritus of lichen simplex chronicus [88, 112, 113, 129, 130, 131, 132, 133, 134, 135]. Omalizumab targets IgE and is used for asthma, CSU, chronic rhinosinusitis with nasal polyps, and food allergies [110, 136, 137] but was not successful in AD trials [138]. Similarly, while effective in AD, biologics targeting only IL‐13 (e.g., tralokinumab, lebrikizumab) did not significantly improve severe asthma and chronic obstructive pulmonary disease [139, 140, 141, 142]. Nemolizumab, an anti‐IL‐31R biologic, has demonstrated efficacy in the management of AD and PN but had paradoxical effects of AD and asthma exacerbation in a subset of patients [61, 62, 143], leading to questions about its ability to regulate type 2 inflammation in the skin alone. This highlights the many unanswered questions in elucidating the functions of the different type 2 cytokines and immune cells. For example, are there distinct types of mast cell stimuli that induce various types of mast cell mediators, leading to differences in itch modalities (histaminergic vs. nonhistaminergic)? What are the long‐term implications of underlying systemic type 2 inflammation on human health? Could the regulation of IL‐4 in Th2 polarization and/or the dual inhibition of IL‐4 and IL‐13 influence immune processes locally and systemically, providing a more comprehensive approach to managing type 2 inflammatory conditions? What are the unique immunological and biological functions of IL‐4 alone? Is IL‐4 more relevant than IL‐13 in type 2 inflammation? Future investigations into shared pathways rather than distinct disease states likely hold the key to understanding AD, PN, CSU, and many other type 2 inflammatory conditions.
TABLE 1.
Approved therapeutics targeting type 2 immune responses.
| Drug | Mechanism | Approved indications |
|---|---|---|
| Dupilumab | Anti‐IL‐4/IL‐13 | Atopic dermatitis |
| Prurigo nodularis | ||
| Asthma | ||
| Eosinophilic esophagitis | ||
| Chronic rhinosinusitis with nasal polyps | ||
| Chronic obstructive pulmonary disease | ||
| Omalizumab | Anti‐IgE | Chronic spontaneous urticaria |
| Asthma | ||
| Chronic rhinosinusitis with nasal polyps | ||
| Food allergies | ||
| Tralokinumab, Lebrikizumab | Anti‐IL‐13 | Atopic dermatitis |
| Nemolizumab | Anti‐IL‐31 | Atopic dermatitis |
| Prurigo nodularis | ||
| Abrocitinib, Upadacitinib | JAK1 inhibitor | Atopic dermatitis |
9. Concluding Remarks
Immune dysregulation can contribute to several chronic inflammatory conditions. Although AD, PN, and CSU are distinct clinical entities, they are all characterized by chronic itch and skin lesions that significantly affect quality of life. Unveiling the complexity of type 2 immunity has fueled the clinical development of advanced, targeted therapies that inhibit key elements within this immune pathway. Such therapies have transformed our ability to alleviate the disease burden of several type 2 inflammatory diseases while simultaneously setting new disease clearance and tolerability standards. Current challenges lie in understanding the intricate and non‐linear nature of type 2 immune responses, identifying disease biomarkers that predict clinical responses to specific therapies, developing treatment regimens that restore immune tolerance and/or induce disease remission, and assessing long‐term patient outcomes.
Conflicts of Interest
Raj Chovatiya has served as an advisor, consultant, speaker, and/or investigator for AbbVie, Amgen, Apogee Therapeutics, Arcutis Biotherapeutics, Argenx, Aslan Pharmaceuticals, Beiersdorf, Boehringer Ingelheim, Bristol Myers Squibb, Cara Therapeutics, Dermavant, Eli Lilly, FIDE, Formation Bio, Galderma, Genentech, GSK, Incyte, L'Oréal, LEO Pharma, Nektar Therapeutics, Novartis, Opsidio, Pfizer, RAPT Therapeutics, Regeneron Pharmaceuticals Inc., Sanofi, Sitryx, and UCB. Jason Hawkes has served as an advisor, consultant, speaker, medical board member, and/or counselor for Apogee Therapeutics, Arcutis Biotherapeutics, Boehringer Ingelheim, Blueprint Medicines, Bristol Myers Squibb, Boxer Capital LLC, Galderma, Institute for Systems Biology (ISB), International Psoriasis Council, Janssen, LEO Pharma, National Psoriasis Foundation, Novartis, Pfizer, Regeneron Pharmaceuticals Inc., Sanofi, Sun Pharma, Takeda, and UCB. Douglas DiRuggiero has served as an advisor, consultant, and/or speaker for AbbVie, Amgen, Arcutis Biotherapeutics, Boehringer Ingelheim, Bristol Myers Squibb, Dermavant, Eli Lilly, Galderma, Incyte, Janssen, LEO Pharma, Medscape, National Psoriasis Foundation, Novartis, Pfizer, Regeneron Pharmaceuticals Inc., Sanofi, UCB, and WebMD. Leigh Ann Pansch has served as an advisor, consultant, and/or speaker for AbbVie, Arcutis Biotherapeutics, Beiersdorf, Boehringer Ingelheim, Bristol Myers Squibb, Dermavant, Eli Lilly, Galderma, Johnson & Johnson, LEO Pharma, Novartis, Pfizer, Regeneron Pharmaceuticals Inc., Sanofi, and UCB. Elizabeth Simcox is an employee and shareholder of Regeneron Pharmaceuticals Inc. Tayler Gonzalez is an employee of and may hold stock and/or stock options in Sanofi.
Supporting information
Appendix S1.
Acknowledgments
This research was sponsored by Sanofi and Regeneron Pharmaceuticals Inc. Medical writing/editorial assistance was provided by Alessandra Iannino, PhD, of Excerpta Medica, and was funded by Sanofi and Regeneron Pharmaceuticals Inc., according to the Good Publication Practice guidelines. The authors thank publication managers Robert McDonald of Sanofi and Purvi Smith of Regeneron Pharmaceuticals Inc. for their input and support.
Funding: This work was supported by Sanofi and Regeneron Pharmaceuticals Inc.
Data Availability Statement
Data sharing does not apply to this article, as no datasets were generated or analyzed.
References
- 1. Chaplin D. D., “Overview of the Immune Response,” Journal of Allergy and Clinical Immunology 125, no. 2 Suppl 2 (2010): S3–S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sonnenberg G. F. and Hepworth M. R., “Functional Interactions Between Innate Lymphoid Cells and Adaptive Immunity,” Nature Reviews. Immunology 19, no. 10 (2019): 599–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Annunziato F., Romagnani C., and Romagnani S., “The 3 Major Types of Innate and Adaptive Cell‐Mediated Effector Immunity,” Journal of Allergy and Clinical Immunology 135, no. 3 (2015): 626–635. [DOI] [PubMed] [Google Scholar]
- 4. Kaiko G. E., Horvat J. C., Beagley K. W., and Hansbro P. M., “Immunological Decision‐Making: How Does the Immune System Decide to Mount a Helper T‐Cell Response?,” Immunology 123, no. 3 (2008): 326–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. De Kouchkovsky D. A., Ghosh S., and Rothlin C. V., “Negative Regulation of Type 2 Immunity,” Trends in Immunology 38, no. 3 (2017): 154–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hawkes J. E., Chan T. C., and Krueger J. G., “Psoriasis Pathogenesis and the Development of Novel Targeted Immune Therapies,” Journal of Allergy and Clinical Immunology 140, no. 3 (2017): 645–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Coates L. C., FitzGerald O., Helliwell P. S., and Paul C., “Psoriasis, Psoriatic Arthritis, and Rheumatoid Arthritis: Is all Inflammation the Same?,” Seminars in Arthritis and Rheumatism 46, no. 3 (2016): 291–304. [DOI] [PubMed] [Google Scholar]
- 8. Akdis C. A., Arkwright P. D., Brüggen M. C., et al., “Type 2 Immunity in the Skin and Lungs,” Allergy 75, no. 7 (2020): 1582–1605. [DOI] [PubMed] [Google Scholar]
- 9. Garcovich S., Maurelli M., Gisondi P., Peris K., Yosipovitch G., and Girolomoni G., “Pruritus as a Distinctive Feature of Type 2 Inflammation,” Vaccines (Basel) 9, no. 3 (2021): 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jethwa H. and Bowness P., “The Interleukin (IL)‐23/IL‐17 Axis in Ankylosing Spondylitis: New Advances and Potentials for Treatment,” Clinical and Experimental Immunology 183, no. 1 (2016): 30–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Reinhardt R. L., Liang H. E., and Locksley R. M., “Cytokine‐Secreting Follicular T Cells Shape the Antibody Repertoire,” Nature Immunology 10, no. 4 (2009): 385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bao K. and Rheinardt R. L., “The Differential Expression of IL‐4 and IL‐13 and Its Impact on Type‐2 Immunity,” Cytokine 75, no. 1 (2015): 25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fahy J. V., “Type 2 Inflammation in Asthma–Present in Most, Absent in Many,” Nature Reviews. Immunology 15, no. 1 (2015): 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gauthier M., Ray A., and Wenzel S. E., “Evolving Concepts of Asthma,” American Journal of Respiratory and Critical Care Medicine 192, no. 6 (2015): 660–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gibbs B. F., Patsinakidis N., and Raap U., “Role of Pruritic Cytokine IL‐31 in Autoimmune Skin Diseases,” Frontiers in Immunology 10 (2019): 1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schwartz D. M., Kanno Y., Villarino A., Ward M., Gadina M., and O'Shea J. J., “JAK Inhibition as a Therapeutic Strategy for Immune and Inflammatory Diseases,” Nature Reviews. Drug Discovery 17, no. 1 (2017): 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bieber T., “Interleukin‐13: Targeting an Underestimated Cytokine in Atopic Dermatitis,” Allergy 75, no. 1 (2020): 54–62. [DOI] [PubMed] [Google Scholar]
- 18. Furue M., “Regulation of Skin Barrier Function via Competition Between AHR Axis Versus IL‐13/IL‐4‐JAK‐STAT6/STAT3 Axis: Pathogenic and Therapeutic Implications in Atopic Dermatitis,” Journal of Clinical Medicine 9, no. 11 (2020): 3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gandhi N. A., Bennett B. L., Graham N. M. H., Pirozzi G., Stahl N., and Yancopoulos G. D., “Targeting Key Proximal Drivers of Type 2 Inflammation in Disease,” Nature Reviews. Drug Discovery 15, no. 1 (2016): 35–50. [DOI] [PubMed] [Google Scholar]
- 20. Gandhi N. A., Pirozzi G., and Graham N. M. H., “Commonality of the IL‐4/IL‐13 Pathway in Atopic Diseases,” Expert Review of Clinical Immunology 13, no. 5 (2017): 425–437. [DOI] [PubMed] [Google Scholar]
- 21. Simpson E. L., Guttman‐Yassky E., Eichenfield L. F., et al., “Tralokinumab Therapy for Moderate‐To‐Severe Atopic Dermatitis: Clinical Outcomes With Targeted IL‐13 Inhibition,” Allergy 78, no. 11 (2023): 2875–2891. [DOI] [PubMed] [Google Scholar]
- 22. Oetjen L. K., Mack M. R., Feng J., et al., “Sensory Neurons Co‐opt Classical Immune Signaling Pathways to Mediate Chronic Itch,” Cell 171, no. 1 (2017): 217–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stott B., Lavender P., Lehmann S., Pennino D., Durham S., and Schmidt‐Weber C. B., “Human IL‐31 Is Induced by IL‐4 and Promotes TH2‐Driven Inflammation,” Journal of Allergy and Clinical Immunology 132, no. 2 (2013): 446–454. [DOI] [PubMed] [Google Scholar]
- 24. Furue M., Yamamura K., Kido‐Nakahara M., Nakahara T., and Fukui Y., “Emerging Role of Interleukin‐31 and Interleukin‐31 Receptor in Pruritus in Atopic Dermatitis,” Allergy 73, no. 1 (2018): 29–36. [DOI] [PubMed] [Google Scholar]
- 25. Junttila I. S., Mizukami K., Dickensheets H., et al., “Tuning Sensitivity to IL‐4 and IL‐13: Differential Expression of IL‐4Rα, IL‐13Rα1, and γc Regulates Relative Cytokine Sensitivity,” Journal of Experimental Medicine 205, no. 11 (2008): 2595–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bağci I. S. and Ruzicka T., “IL‐31: A New Key Player in Dermatology and Beyond,” Journal of Allergy and Clinical Immunology 141, no. 3 (2018): 858–866. [DOI] [PubMed] [Google Scholar]
- 27. Nemmer J. M., Kuchner M., Datsi A., et al., “Interleukin‐31 Signaling Bridges the Gap Between Immune Cells, the Nervous System and Epithelial Tissues,” Frontiers in Medicine 8 (2021): 639097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Voisin T. and Chiu I. M., “Mast Cells Get on Your Nerves in Itch,” Immunity 50, no. 5 (2019): 1117–1119. [DOI] [PubMed] [Google Scholar]
- 29. Trier A. M., Mack M. R., and Kim B. S., “The Neuro‐Immune Axis in Skin Sensation, Inflammation, and Immunity,” Journal of Immunology 202, no. 10 (2019): 2829–2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Weidinger S. and Novak N., “Atopic Dermatitis,” Lancet 387, no. 10023 (2016): 1109–1122. [DOI] [PubMed] [Google Scholar]
- 31. Chovatiya R. and Silverberg J. I., “Evaluating the Longitudinal Course of Atopic Dermatitis: Implications for Clinical Practice,” American Journal of Clinical Dermatology 23, no. 4 (2022): 459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wan J., Takeshita J., Shin D. B., and Gelfand J. M., “Mental Health Impairment Among Children With Atopic Dermatitis: A United States Population‐Based Cross‐Sectional Study of the 2013–2017 National Health Interview Survey,” Journal of the American Academy of Dermatology 82, no. 6 (2020): 1368–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chiesa Fuxench Z. C., Block J. K., Boguniewicz M., et al., “Atopic Dermatitis Is America Study: A Cross‐Sectional Study Examining the Prevalence and Disease Burden of Atopic Dermatitis in the US Adult Population,” Journal of Investigative Dermatology 139, no. 3 (2019): 583–590. [DOI] [PubMed] [Google Scholar]
- 34. Bin L. and Leung D. Y. M., “Genetic and Epigenetic Studies of Atopic Dermatitis,” Allergy, Asthma and Clinical Immunology 12 (2016): 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Leung D. Y. M. and Guttman‐Yassky E., “Deciphering the Complexities of Atopic Dermatitis: Shifting Paradigms in Treatment Approaches,” Journal of Allergy and Clinical Immunology 134, no. 4 (2014): 769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hoffjan S. and Stemmler S., “Unravelling the Complex Genetic Background of Atopic Dermatitis: From Genetic Association Results Towards Novel Therapeutic Strategies,” Archives of Dermatological Research 307, no. 8 (2015): 659–670. [DOI] [PubMed] [Google Scholar]
- 37. Simpson E. L., Bieber T., Eckert L., et al., “Patient Burden of Moderate to Severe Atopic Dermatitis (AD): Insights From a Phase 2b Clinical Trial of Dupilumab in Adults,” Journal of the American Academy of Dermatology 74, no. 3 (2016): 491–498. [DOI] [PubMed] [Google Scholar]
- 38. Vakharia P. P., Chopra R., Sacotte R., et al., “Burden of Skin Pain in Atopic Dermatitis,” Annals of Allergy, Asthma & Immunology 119, no. 6 (2017): 548–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zuberbier T., Orlow S. J., Paller A. S., et al., “Patient Perspectives on the Management of Atopic Dermatitis,” Journal of Allergy and Clinical Immunology 118, no. 1 (2006): 226–232. [DOI] [PubMed] [Google Scholar]
- 40. Davis D. M. R., Drucker A. M., Alikhan A., et al., “American Academy of Dermatology Guidelines: Awareness of Comorbidities Associated With Atopic Dermatitis in Adults,” Journal of the American Academy of Dermatology 86, no. 6 (2022): 1335–1336. [DOI] [PubMed] [Google Scholar]
- 41. Siegfried E. C. and Hebert A. A., “Diagnosis of Atopic Dermatitis: Mimics, Overlaps, and Complications,” Journal of Clinical Medicine 4 (2015): 884–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Weidinger S., Beck L. A., Bieber T., Kabashima K., and Irvine A. D., “Atopic Dermatitis,” Nature Reviews. Disease Primers 4, no. 1 (2018): 1. [DOI] [PubMed] [Google Scholar]
- 43. Beck L. A., Cork M. J., and Amagai M., “Type 2 Inflammation Contributes to Skin Barrier Dysfunction in Atopic Dermatitis,” JID Innovations: Skin Science From Molecules to Population Health 2, no. 5 (2022): 100131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Clausen M. L., Slotved H. C., Krogfelt K. A., Andersen P. S., and Agner T., “In Vivo Expression of Antimicrobial Peptides in Atopic Dermatitis,” Experimental Dermatology 25, no. 1 (2016): 3–9. [DOI] [PubMed] [Google Scholar]
- 45. Nguyen H. L. T., Trujillo‐Paez J. V., Umehara Y., et al., “Role of Antimicrobial Peptides in Skin Barrier Repair in Individuals With Atopic Dermatitis,” International Journal of Molecular Sciences 21, no. 20 (2020): 7607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wong L., Yen Y., and Lee C., “The Implications of Pruritogens in the Pathogenesis of Atopic Dermatitis,” International Journal of Molecular Sciences 22, no. 13 (2021): 7227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Haddad E., Cyr S. L., Arima K., McDonald R. A., Levit N. A., and Nestle F. O., “Current and Emerging Strategies to Inhibit Type 2 Inflammation in Atopic Dermatitis,” Dermatology and Therapy 12, no. 7 (2022): 1501–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Williams M. R. and Gallo R. L., “The Role of the Skin Microbiome in Atopic Dermatitis,” Current Allergy and Asthma Reports 15, no. 11 (2015): 65. [DOI] [PubMed] [Google Scholar]
- 49. Simpson E. L., Schlievert P. M., Yoshida T., et al., “Rapid Reduction in Staphylococcus aureus in Atopic Dermatitis Subjects Following Dupilumab Treatment,” Journal of Allergy and Clinical Immunology 152, no. 5 (2023): 1179–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lipman Z. M., Labib A., and Yosipovitch G., “Current Clinical Options for the Management of Itch in Atopic Dermatitis,” Clinical, Cosmetic and Investigational Dermatology 14 (2021): 959–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Goh M. S., Yun J. S., and Su J. C., “Management of Atopic Dermatitis: A Narrative Review,” Medical Journal of Australia 216, no. 11 (2022): 587–593. [DOI] [PubMed] [Google Scholar]
- 52. Frazier W. and Bhardwaj N., “Atopic Dermatitis: Diagnosis and Treatment,” American Family Physician 101, no. 10 (2020): 590–598. [PubMed] [Google Scholar]
- 53. Pinto L. M., Chiricozzi A., Calabrese L., Mannino M., and Peris K., “Novel Therapeutic Strategies in the Topical Treatment of Atopic Dermatitis,” Pharmaceutics 14, no. 12 (2022): 2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Eichenfield L., Boguniewicz M., Simpson E., et al., “Once‐Daily Roflumilast Cream 0.15% for Atopic Dermatitis: Pooled Results: From INTEGUMENT–1/2 Phase 3 Trials,” Annals of Allergy, Asthma & Immunology 131 (2023): S91. [Google Scholar]
- 55. Silverberg J. I., Eichenfield L. F., Hebert A. A., et al., “514 – Tapinarof Cream 1% Once Daily: Significant Efficacy in the Treatment of Atopic Dermatitis in Two Pivotal Phase 3 Trials in Adults and Children Down to 2 Years of Age,” British Journal of Dermatology 190 (2024): ii17–ii18. [DOI] [PubMed] [Google Scholar]
- 56. Johnson B. B., Franco A. I., Beck L. A., and Prezzano J. C., “Treatment‐Resistant Atopic Dermatitis: Challenges and Solutions,” Clinical, Cosmetic and Investigational Dermatology 12 (2019): 181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Davis D. M. R., Drucker A. M., Alikham A., et al., “Guidelines of Care for the Management of Atopic Dermatitis in Adults With Phototherapy and Systemic Therapies,” Journal of the American Academy of Dermatology 90, no. 2 (2024): e43–e56. [DOI] [PubMed] [Google Scholar]
- 58. Wollenberg A., Blauvelt A., Guttman‐Yassky E., et al., “Tralokinumab for Moderate‐To‐Severe Atopic Dermatitis: Results From Two 52‐Week, Randomized, Double‐Blind, Multicentre, Placebo‐Controlled Phase III Trials (ECZTRA 1 and ECZTRA 2),” British Journal of Dermatology 184, no. 3 (2021): 437–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bernardo D., Bieber T., and Torres T., “Lebrikizumab for the Treatment of Moderate‐To‐Severe Atopic Dermatitis,” American Journal of Clinical Dermatology 24, no. 5 (2023): 753–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Silverberg J. I., Pinter A., Pulka G., et al., “Phase 2B Randomized Study of Nemolizumab in Adults With Moderate‐To‐Severe Atopic Dermatitis and Severe Pruritus,” Journal of Allergy and Clinical Immunology 145, no. 1 (2020): 173–182. [DOI] [PubMed] [Google Scholar]
- 61. Orfali R. L. and Aoki V., “Blockage of the IL‐31 Pathway as a Potential Target Therapy for Atopic Dermatitis,” Pharmaceutics 15, no. 2 (2023): 577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Igarashi A., Katsunuma T., Matsumura T., Komazaki H., and Nemolizumab‐JP04 Study Group , “Efficacy and Safety of Nemolizumab in Paediatric Patients Aged 6‐12 Years With Atopic Dermatitis With Moderate‐To‐Severe Pruritus: Results From a Phase III, Randomized, Double‐Blind, Placebo‐Controlled, Multicentre Study,” British Journal of Dermatology 180 (2024): 20–28. [DOI] [PubMed] [Google Scholar]
- 63. Mikhaylov D., Ungar B., Renert‐Yuval Y., and Guttman‐Yassky E., “Oral Janus Kinase Inhibitors for Atopic Dermatitis,” Annals of Allergy, Asthma & Immunology 130, no. 5 (2023): 577–592. [DOI] [PubMed] [Google Scholar]
- 64. Lee K. P., Plante J., Korte J. E., and Eltson D. M., “Oral Janus Kinase Inhibitors in the Treatment of Atopic Dermatitis: A Systematic Review and Meta‐Analysis,” Skin Health and Disease 3, no. 1 (2023): e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Weidinger S., Bieber T., Cork M. J., et al., “Safety and Efficacy of Amlitemab, a Fully Human Nondepleting, Noncytotoxic Anti‐OX40 Ligand Monoclonal Antibody, in Atopic Dermatitis: Results of a Phase IIa Randomized Placebo‐Controlled Trial,” British Journal of Dermatology 189, no. 5 (2023): 531–539. [DOI] [PubMed] [Google Scholar]
- 66. Rewerska B., Sher L. D., Alpizar S., et al., “Phase 2b Randomized Trial of OX40 Inhibitor Telazorlimab for Moderate‐To‐Severe Atopic Dermatitis,” Journal of Allergy and Clinical Immunology 3, no. 1 (2024): 100195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Croft M., Esfandiari E., Chong C., et al., “OX40 in the Pathogenesis of Atopic Dermatitis – A New Therapeutic Target,” American Journal of Clinical Dermatology 25, no. 3 (2024): 447–461. Erratum: American Journal of Clinical Dermatology 25, 3 (2024): 463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Furue M. and Furue M., “OX40L‐OX40 Signaling in Atopic Dermatitis,” Journal of Clinical Medicine 10, no. 12 (2021): 2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lé A. M. and Torres T., “OX40‐OX40L Inhibition for the Treatment of Atopic Dermatitis – Focus on Rocatinlimab and Amlitelimab,” Pharmaceutics 14, no. 12 (2022): 2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Croft M., So T., Duan W., and Soroosh P., “The Significance of OX40 and OX40L to T‐Cell Biology and Immune Disease,” Immunological Reviews 229, no. 1 (2009): 173–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Huang A. H., Canner J. K., Khanna R., Kang S., and Kwatra S. G., “Real‐World Prevalence of Prurigo Nodularis and Burden of Associated Diseases,” Journal of Investigative Dermatology 140, no. 2 (2020): 480–483. [DOI] [PubMed] [Google Scholar]
- 72. Pereira M. P., Steinke S., Zeidler C., et al., “European Academy of Dermatology and Venereology European Prurigo Project: Expert Consensus on the Definition, Classification and Terminology of Chronic Prurigo,” Journal of the European Academy of Dermatology and Venereology 32, no. 7 (2018): 1059–1065. [DOI] [PubMed] [Google Scholar]
- 73. Kwon C. D., Khanna R., Williams K. A., Kwatra M. M., and Kwatra S. G., “Diagnostic Workup and Evaluation of Patients With Prurigo Nodularis,” Medicines (Basel, Switzerland) 6, no. 4 (2019): 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Elmariah S., Kim B., Berger T., et al., “Practical Approaches for Diagnosis and Management of Prurigo Nodularis: United States Expert Panel Consensus,” Journal of the American Academy of Dermatology 84, no. 3 (2021): 747–760. [DOI] [PubMed] [Google Scholar]
- 75. Hughes J. D. M., Woo T. E., Belzberg M., et al., “Association Between Prurigo Nodularis and Etiologies of Peripheral Neuropathy: Suggesting a Role for Neural Dysregulation in Pathogenesis,” Medicines (Basel, Switzerland) 7, no. 1 (2020): 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Iking A., Grundmann S., Chatzigeorgakidis E., Phan N. Q., Klein D., and Ständer S., “Prurigo as a Symptom of Atopic and Non‐Atopic Diseases: Aetiological Survey in a Consecutive Cohort of 108 Patients,” Journal of the European Academy of Dermatology and Venereology 27, no. 5 (2013): 550–557. [DOI] [PubMed] [Google Scholar]
- 77. Pereira M. P., Hoffmann V., Weisshaar E., et al., “Chronic Nodular Prurigo: Clinical Profile and Burden. A European Cross‐Sectional Study,” Journal of the European Academy of Dermatology and Venereology 34, no. 10 (2020): 2373–2383. [DOI] [PubMed] [Google Scholar]
- 78. Jørgensen K. M., Egeberg A., Gislason G. H., Skov L., and Thyssen J. P., “Anxiety, Depression and Suicide in Patients With Prurigo Nodularis,” Journal of the European Academy of Dermatology and Venereology 31, no. 2 (2017): e106–e107. [DOI] [PubMed] [Google Scholar]
- 79. Tsoi L. C., Hacini‐Rachinel F., Fogel P., et al., “Transcriptomic Characterization of Prurigo Nodularis and the Therapeutic Response to Nemolizumab,” Journal of Allergy and Clinical Immunology 149, no. 4 (2022): 1329–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Park K., Mori T., Nakamura M., and Tokura Y., “Increased Expression of mRNAs for IL‐4, IL‐17, IL‐22 and IL‐31 in Skin Lesions of Subacute and Chronic Forms of Prurigo,” European Journal of Dermatology 21, no. 1 (2011): 135–136. [DOI] [PubMed] [Google Scholar]
- 81. Mack M. R. and Kim B. S., “The Itch‐Scratch Cycle: A Neuroimmune Perspective,” Trends in Immunology 39, no. 12 (2018): 980–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Nguyen J. K., Austin E., Huang A., Mamalis A., and Jagdeo J., “The IL‐4/IL‐13 Axis in Skin Fibrosis and Scarring: Mechanistic Concepts and Therapeutic Targets,” Archives of Dermatological Research 312, no. 2 (2020): 81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Masuoka M., Shiraishi H., Ohta S., et al., “Periostin Promotes Chronic Allergic Inflammation in Response to Th2 Cytokines,” Journal of Clinical Investigation 122, no. 7 (2012): 2590–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hashimoto T., Nattkemper L. A., Kim H. S., et al., “Dermal Periostin: A New Player in Itch of Prurigo Nodularis,” Acta Dermato‐Venereologica 101, no. 1 (2021): adv00375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zhong W., Wu X., Zhang W., et al., “Aberrant Expression of Histamine‐Independent Pruritogenic Mediators in Keratinocytes May Be Involved in the Pathogenesis of Prurigo Nodularis,” Acta Dermato‐Venereologica 99, no. 6 (2019): 579–586. [DOI] [PubMed] [Google Scholar]
- 86. Williams K. A., Huang A. H., Belzberg M., and Kwatra S. G., “Prurigo Nodularis: Pathogenesis and Management,” Journal of the American Academy of Dermatology 83, no. 6 (2020): 1567–1575. [DOI] [PubMed] [Google Scholar]
- 87. Labib A., Ju T., Vander Does A., and Yosipovitch G., “Immunotargets and Therapy for Prurigo Nodularis,” ImmunoTargets and Therapy 11 (2022): 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Müller S., Zeidler C., and Ständer S., “Chronic Prurigo Including Prurigo Nodularis: New Insights and Treatments,” American Journal of Clinical Dermatology 25, no. 1 (2024): 15–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Yosipovitch G., Mollanazar N., Ständer S., et al., “Dupilumab in Patients With Prurigo Nodularis: Two Randomized, Double‐Blind, Placebo‐Controlled Phase 3 Trials,” Nature Medicine 29, no. 5 (2023): 1180–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ständer S., Yosipovitch G., Legat F. J., et al., “Trial of Nemolizumab in Moderate‐To‐Severe Prurigo Nodularis,” New England Journal of Medicine 382, no. 8 (2020): 706–716. [DOI] [PubMed] [Google Scholar]
- 91. Ständer S., Yosipovitch G., Legat F. J., et al., “436 Nemolizumab Monotherapy Was Associated With Significant Improvements in Prurigo Activity Score in Adult Patients With Moderate‐To‐Severe Prurigo Nodularis: Results From a Phase 3 Trial (OLYMPIA 2),” British Journal of Dermatology 188, no. Suppl 3 (2023): ljad162.056. [Google Scholar]
- 92. Sun F. and Wu Z., “Successful Treatment of Refractory Prurigo Nodularis With Abrocitinib,” Clinical Case Reports 12, no. 3 (2024): e8606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Fakhraie S. and Chovatiya R., “Janus Kinase Inhibition in the Treatment of Prurigo Nodularis,” Dermatitis 35, no. 3 (2023): 299–300. [DOI] [PubMed] [Google Scholar]
- 94. Muntaner‐Virgili C., Moreno‐Vilchez C., Torrecilla‐Vall‐Llossera C., and Figueras‐Nart I., “Upadacitinib for Prurigo Nodularis,” JAAD Case Reports 48 (2024): 131–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Marzano A. V., Tedeschi A., Menicanti C., Asero R., Crosti C., and Cugno M., “Chronic Spontaneous Urticaria: The Emerging Role of Coagulation,” Current Dermatology Reports 2 (2013): 18–23. [Google Scholar]
- 96. Zuberbier T., Latiff A. H. A., Abuzakouk M., et al., “The International EAACI/GA2LEN/EuroGuiDerm/APAAACI Guideline for the Definition, Classification, Diagnosis and Management of Urticaria,” Allergy 77, no. 3 (2022): 734–766. [DOI] [PubMed] [Google Scholar]
- 97. Kolkhir P., Giménez‐Arnau A. M., Kulthanan K., Peter J., Metz M., and Maurer M., “Urticaria,” Nature Reviews. Disease Primers 8, no. 1 (2022): 61. [DOI] [PubMed] [Google Scholar]
- 98. Maurer M., Weller K., Bindslev‐Jensen C., et al., “Unmet Clinical Needs in Chronic Spontaneous Urticaria. A GA2LEN Task Force Report,” Allergy 66, no. 3 (2011): 317–330. [DOI] [PubMed] [Google Scholar]
- 99. Balp M., Halliday A. C., Severin T., et al., “Clinical Remission of Chronic Spontaneous Urticaria (CSU): A Targeted Literature Review,” Dermatology and Therapy 12, no. 1 (2022): 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Maurer M., Abuzakouk M., Bérard F., et al., “The Burden of Chronic Spontaneous Urticaria Is Substantial: Real‐World Evidence From ASSURE‐CSU,” Allergy 72 (2017): 2005–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Balp M., Vietri J., Tian H., and Isherwood G., “The Impact of Chronic Urticaria From the Patient's Perspective: A Survey in Five European Countries,” Patient 8, no. 6 (2015): 551–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Giménez‐Arnau A. M., DeMontojoye L., Asero R., et al., “The Pathogenesis of Chronic Spontaneous Urticaria: The Role of Infiltrating Cells,” Journal of Allergy and Clinical Immunology: In Practice 9, no. 6 (2021): 2195–2208. [DOI] [PubMed] [Google Scholar]
- 103. Zhou B., Li J., Liu R., Zhu L., and Peng C., “The Role of Crosstalk of Immune Cells in Pathogenesis of Chronic Spontaneous Urticaria,” Frontiers in Immunology 13 (2022): 879754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Dobrican C. T., Muntean I. A., Pintea I., Petricău C., Deleanu D. M., and Filip G. A., “Immunological Signature of Chronic Spontaneous Urticaria (Review),” Experimental and Therapeutic Medicine 23, no. 6 (2022): 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Ingrasci G., Lipman Z. M., Hawash A. A., Girolomoni G., and Yosipovitch G., “The Pruritogenic Role of the Type 2 Immune Response in Diseases Associated With Chronic Itch,” Experimental Dermatology 30, no. 9 (2021): 1208–1217. [DOI] [PubMed] [Google Scholar]
- 106. Johal K. J. and Saini S. S., “Current and Emerging Treatments for Chronic Spontaneous Urticaria,” Annals of Allergy, Asthma & Immunology 125, no. 4 (2020): 380–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Chang J., Cattelan L., Ben‐Shoshan M., Le M., and Netchiporouk E., “Management of Pediatric Chronic Spontaneous Urticaria: A Review of Current Evidence and Guidelines,” Journal of Asthma and Allergy 14 (2021): 187–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Mandel V. D., Alicandro T., Pepe P., et al., “Chronic Spontaneous Urticaria: A Review of Pathological Mechanisms, Diagnosis, Clinical Management, and Treatment,” European Medical Journal 5, no. 1 (2020): 29–39. [Google Scholar]
- 109. Agache I., Rocha C., Pereira A., et al., “Efficacy and Safety of Treatment With Omalizumab for Chronic Spontaneous Urticaria: A Systematic Review for the EAACI Biologicals Guidelines,” Allergy 76, no. 1 (2021): 59–70. [DOI] [PubMed] [Google Scholar]
- 110. Genentech , Xolair (Omalizumab). Prescribing Information (Genentech USA, Inc., 2016). [Google Scholar]
- 111. Tharp M. D., Bernstein J. A., Kavati A., et al., “Benefits and Harms of Omalizumab Treatment in Adolescent and Adult Patients With Chronic Idiopathic (Spontaneous) Urticaria. A Meta‐Analysis of “Real‐World” Evidence,” JAMA Dermatology 155, no. 1 (2019): 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Maurer M., Casale T. B., Saini S. S., et al., “Dupilumab in Patients With Chronic Spontaneous Urticaria (LIBERTY‐CSU CUPID): Two Randomized, Double‐Blind, Placebo‐Controlled, Phase 3 Trials,” Journal of Allergy and Clinical Immunology 154, no. 1 (2024): 184–194. [DOI] [PubMed] [Google Scholar]
- 113. Casale T., Saini S., Bernstein J., et al., “Dupilumab Significantly Improves Itch and Hives in Patients With Chronic Spontaneous Urticaria (CUPID Study C),” Annals of Allergy, Asthma & Immunology 133, no. 6 Suppl 2 (2024): S2. [Google Scholar]
- 114. Maurer M., Berger W., Giménez‐Arnau A., et al., “Remibrutinib, a Novel BTK Inhibitor, Demonstrates Promising Efficacy and Safety in Chronic Spontaneous Urticaria,” Journal of Allergy and Clinical Immunology 150, no. 6 (2022): 1498–1506. [DOI] [PubMed] [Google Scholar]
- 115. Maurer M. M., Gimenez‐Arnau A., Ferrucci S., et al., “Efficacy and Safety of Rilzabrutinib in Patients With Chronic Spontaneous Urticaria: 12‐Weel Results From the RILESCU Phase 2 Dose‐Ranging Study,” Journal of Allergy and Clinical Immunology 153, no. 2 Suppl (2024): AB373. [Google Scholar]
- 116. Jain V., Giménez‐Arnau A., Hayama K., et al., “Remibrutinib Demonstrates Favorable Safety Profile and Sustained Efficacy in Chronic Spontaneous Urticaria Over 52 Weeks,” Journal of Allergy and Clinical Immunology 153, no. 2 (2024): 479–486. [DOI] [PubMed] [Google Scholar]
- 117. Mosnaim G., Gimenez‐Arnau A., Hide M., et al., “Efficacy of Remibrutinib in Patients With Chronic Spontaneous Urticaria With or Without Prior Exposure to Biologics in the Phase 3 REMIX‐1 and REMIX‐2 Studies,” Journal of Allergy and Clinical Immunology 153, no. 2 Suppl (2024): AB369. [Google Scholar]
- 118. Metz M., Gimenez‐Arnau A., Hide M., et al., “Long‐Term Efficacy and Safety of Remibrutinib in Patients With Chronic Spontaneous Urticaria in the Phase 3 REMIX‐1 and REMIX‐2 Studies,” Presented as a Late Oral Abstract Session on Clinical Trials at EAACI, Valencia, Spain, 2024; May 31–June 3 2024.
- 119. Altrichter S., Shen Fok J., Jiao Q., et al., “Total IgE as a Marker for Chronic Spontaneous Urticaria,” Allergy, Asthma & Immunology Research 13, no. 2 (2021): 206–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Papapostolou N., Xepapadaki P., Katoulis A., and Makris M., “Comorbidities of Chronic Urticaria: A Glimpse Into a Complex Relationship,” Frontiers in Allergy 3 (2022): 1008145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Werth V. P., Murrell D. G., Joly P., et al., “Pathophysiology of Bullous Pemphigoid: Role of Type 2 Inflammation and Emerging Treatment Strategies (Narrative Review),” Advances in Therapy 41 (2024): 4418–4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Pietschke K., Holstein J., Meier K., et al., “The Inflammation in Cutaneous Lichen Planus Is Dominated by IFN‐ϒ and IL‐21—A Basis for Therapeutic JAK1 Inhibition,” Experimental Dermatology 30 (2021): 262–270. [DOI] [PubMed] [Google Scholar]
- 123. Boch K., Langan E. A., Kridin K., Zillikens D., Ludwig R. J., and Bieber K., “Lichen Planus,” Frontiers in Medicine 8 (2021): 737813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Butler D. C., Kollhoff A., and Berger T., “Treatment of Grover Disease With Dupilumab,” JAMA Dermatology 157, no. 3 (2021): 353–356. [DOI] [PubMed] [Google Scholar]
- 125. Zhai L. L., Savage K. T., Qiu C. C., Jin A., Valdes‐Rodriguez R., and Mollanazar N. K., “Chronic Pruritus Responding to Dupilumab—A Case Series,” Medicine 6 (2019): 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Pousti B. T., Jin A., Sklovar L., et al., “Dupilumab for the Treatment of Lichen Planus,” Cutis 107, no. 4 (2021): E8–E10. [DOI] [PubMed] [Google Scholar]
- 127. Kazemi S., Murphrey M., and Hawkes J. E., “Rapid Resolution of Widespread Cutaneous Lichen Planus and Generalized Pruritus in an Elderly Patient Following Treatment With Dupilumab,” JAAD Case Reports 30 (2022): 108–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Barei F., Torretta S., Morini N., and Ferrucci S., “A Case of Grover Disease Treated With Dupilumab: Just Serendipity or a Future Perspective?,” Dermatologic Therapy 35 (2022): e15429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Hendricks A. J., Yosipovitch G., and Shi V. Y., “Dupilumab Use in Dermatologic Conditions Beyond Atopic Dermatitis – A Systematic Review,” Journal of Dermatological Treatment 31, no. 1 (2021): 19–28. [DOI] [PubMed] [Google Scholar]
- 130. Zhao L., Wang Q., Liang G., et al., “Evaluation of Dupilumab in Patients With Bullous Pemphigoid,” JAMA Dermatology 159, no. 9 (2023): 953–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Dellon E. S., Rothenberg M. E., Collins M. H., et al., “Dupilumab in Adults and Adolescents With Eosinophilic Esophagitis,” New England Journal of Medicine 387, no. 25 (2022): 2317–2330. [DOI] [PubMed] [Google Scholar]
- 132. Bhatt S. P., Rabe K. F., Hanania N. A., et al., “Dupilumab for COPD With Type 2 Inflammation Indicated by Eosinophil Counts,” New England Journal of Medicine 389, no. 3 (2023): 205–214. [DOI] [PubMed] [Google Scholar]
- 133. Bachert C., Han J. K., Desrosiers M., et al., “Efficacy and Safety of Dupilumab in Patients With Severe Chronic Rhinosinusitis With Nasal Polyps (LIBERTY NP SINUS‐24 and LIBERTY NP SINUS‐52): Results From Two Multicentre, Randomised, Double‐Blind, Placebo‐Controlled, Parallel‐Group Phase 3 Trials,” Lancet 394, no. 10209 (2019): 1638–1650. [DOI] [PubMed] [Google Scholar]
- 134. Jeon J., Wang F., Badic A., and Kim B. S., “Treatment of Patients With Chronic Pruritus of Unknown Origin With Dupilumab,” Journal of Dermatological Treatment 33, no. 3 (2022): 1754–1757. [DOI] [PubMed] [Google Scholar]
- 135. Teresa J. U., Ashley V. D., Mohsin N., and Yosipovitch G., “Lichen Simplex Chronicus Itch: An Update,” Acta Dermato‐Venereologica 102 (2022): 4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Casale T. B., Gimenez‐Arnau A. M., Bernstein J. A., Holden M., Zuberbier T., and Maurer M., “Omalizumab for Patients With Chronic Spontaneous Urticaria: A Narrative Review of Current Status,” Dermatology and Therapy 13, no. 11 (2023): 2573–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Zuberbier T., Wood R. A., Bindslev‐Jensen C., et al., “Omalizumab in IgE‐Mediated Food Allergy: A Systematic Review and Meta‐Analysis,” Journal of Allergy and Clinical Immunology: In Practice 11, no. 4 (2023): 1134–1146. [DOI] [PubMed] [Google Scholar]
- 138. Wang H. H., Li Y. C., and Huang Y. C., “Efficacy of Omalizumab in Patients With Atopic Dermatitis: A Systematic Review and Meta‐Analysis,” Journal of Allergy and Clinical Immunology 138, no. 6 (2016): 1719–1722. [DOI] [PubMed] [Google Scholar]
- 139. Busse W. W., Brusselle G. G., Korn S., et al., “Tralokinumab Did Not Demonstrate Oral Corticosteroid‐Sparing Effects in Severe Asthma,” European Respiratory Journal 53, no. 2 (2019): 1800948. [DOI] [PubMed] [Google Scholar]
- 140. Panettieri R. A., Wang M., Braddock M., Bowen K., and Colice G., “Tralokinumab for the Treatment of Severe, Uncontrolled Asthma: The ATMOSPHERE Clinical Development Program,” Immunotherapy 10, no. 6 (2018): 473–490. [DOI] [PubMed] [Google Scholar]
- 141. Hanania N. A., Korenblat P., Chapman K. R., et al., “Efficacy and Safety of Lebrikizumab in Patients With Uncontrolled Asthma (LAVOLTA I and LAVOLTA II): Replicate, Phase 3, Randomised, Double‐Blind, Placebo‐Controlled Trials,” Lancet Respiratory Medicine 4, no. 10 (2016): 781–796. [DOI] [PubMed] [Google Scholar]
- 142. Cazzola M., Hanania N. A., Page C. P., and Matera M. G., “Novel Anti‐Inflammatory Approaches to COPD,” International Journal of Chronic Obstructive Pulmonary Disease 18 (2023): 1333–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Kim B. S., “Learning From Nemolizumab: A Promising Therapy for Prurigo Nodularis,” Journal of Allergy and Clinical Immunology 153, no. 6 (2024): 1548–1549. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1.
Data Availability Statement
Data sharing does not apply to this article, as no datasets were generated or analyzed.