

Abstract
Background
Transmissible gastroenteritis (TGE) is a highly contagious intestinal disease caused by transmissible gastroenteritis virus (TGEV). The primary techniques for identifying TGEV involve enzyme-linked immunosorbent assay (ELISA), polymerase chain reaction (PCR), and fluorescent quantitative PCR (qPCR). However, these approaches are complex, demanding specialized tools and significant time. Therefore, a precise, swift, and effective differential diagnosis method is crucial for TGEV prevention. In recent years, clustered regularly interspaced short palindromic repeats (CRISPR) and Cas-associated proteins have become popular for their high specificity, unique cleavage activity, and ease of detection. CRISPR-Cas12a, a novel RNA-guided nucleic acid endonuclease, is emerging as a powerful molecular scissor.
Results
In this study, we designed three pairs of crRNA targeting the N gene of TGEV. Following the selection of the most appropriate crRNA, we established the loop-mediated isothermal (LAMP) amplification method with a sensitivity of 102 copies/µL. And based on this, we established the CRISPR-Cas12a fluorescence assay with a sensitivity of 100 copies/µL. Furthermore, we established a CRISPR/Cas12a lateral-flow dipstick assay with a sensitivity of 102 copies/µL. Importantly, none of these methods exhibited cross-reactivity with other related viruses, enabling quicker and more straightforward observation of experimental results.
Conclusions
We have successfully developed a CRISPR-Cas12a fluorescence assay and a CRISPR/Cas12a lateral-flow dipstick assay for clinical TGEV detection. Overall, we created a portable, quick, and sensitive TGEV assay with strong specificity utilizing the CRISPR-Cas12a system.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12917-025-04711-1.
Keywords: LAMP, CRISPR-Cas12a, TGEV, Detection
Background
TGE is a highly contagious intestinal disease caused by TGEV [1]. This virus induces acute gastrointestinal symptoms in swine globally, and the current TGEV outbreak presents a considerable threat to the swine industry in China and worldwide due to the added risk of cross-species transmission [2]. TGEV was initially documented in the United States in 1946 and has since been detected in Europe, Asia, Africa, and South America [3]. It is a coronavirus with a positive-stranded RNA genome of about 28.5 kb [4]. Three open reading frames (ORF1, ORF3, and ORF7) mainly encode the translation of nonstructural proteins, whereas structural proteins mainly include: spike (S or E2), small membrane (sM or E), membrane (M or E1), and nucleocapsid (N) proteins [5–7]. N proteins are phosphorylated acidic proteins encoded by the N gene that, together with M proteins, form the internal core of the virus [8]. TGEV can infect swine of all ages and breeds, with younger swine having shorter incubation periods and faster outbreaks [9]. Piglets infected with TGEV within 2 weeks mainly show symptoms such as depression, watery diarrhea, severe vomiting and usually die within 1 week [10].
Therefore, there is a pressing requirement to create straightforward yet efficient TGEV tests. Swift and precise identification of TGEV is crucial for disease prevention and management [11]. Presently, key techniques for detecting TGEV comprise electron microscopy, indirect immunofluorescence, ELISA, PCR, and qPCR [12, 13]. ELISA is a time-intensive approach for TGEV detection, known for its low sensitivity and susceptibility to false positives [14, 15]. Furthermore, LAMP and RT-PCR are viable for TGEV identification due to their high sensitivity; however, they mandate specialized facilities, are unwieldy, and are unsuitable for application in swine farms [16, 17]. In recent years, CRISPR and Cas-associated proteins have gained popularity due to their high specificity, unique cleavage activity, and easy detection [18]. Their application has made them an ideal tool for next-generation pathogen diagnostics and gene editing [19]. CRISPR-Cas12a, a novel RNA-guided nucleic acid endonuclease, is emerging as a potent molecular scissor with potential for genome editing [20]. The integration of the CRISPR-Cas platform with lateral-flow system enables rapid, sensitive, highly specific, cost-effective, and reliable pathogen diagnosis [21].
In this study, CRISPR-Cas12a fluorescence assay and CRISPR/Cas12a lateral-flow dipstick assay were developed using LAMP amplification for the conserved N gene of TGEV. The former achieved sensitivity, accuracy, and visualization through efficient LAMP amplification under Cas12a cleavage, but it required specific fluorescence equipment to view the results. Based on the CRISPR-Cas12a fluorescence assay, in combination with lateral-flow dipstick, can address this limitation and enable accurate detection of low-concentration templates without the need for expensive equipment. It offers easy operation and results in about 1 h, providing a new approach for the prevention and clinical detection of TGEV.
Results
Evaluation of the optimal crRNA
The ranscription-purified crRNA was utilized in the CRISPR-Cas12a fluorescence assay established in this research, and the products were assessed under UV light after the reaction was completed. The three sets of crRNA exhibited distinct green fluorescence under UV light (Fig. 1A), which were quantified through fluorescence PCR and numerically analyzed using GraphPad Prism 8 software. The first pair of crRNAs demonstrated the highest fluorescence signal values, with clearer results visible to the naked eye (Fig. 1B). Consequently, this set was chosen for subsequent experiments.
Fig. 1.

Results of crRNA screening. (A) Visual inspection under UV light. (B) The values were read by fluorescent quantitative PCR and then analyzed numerically by GraphPad Prism 8
Establishment of the LAMP system and assessment of its sensitivity
Six pairs of primers were designed for LAMP detection at both ends of the PAM site, split into two groups. Lane 1 was indistinct, while the amplification effects of lanes 2 and 3 were similar (Fig. 2A). Lanes 1 and 3 proved less effective than lane 2 (Fig. 2B). Gel electrophoresis examination of the amplified products indicated a sensitivity of 102 copies/µL (Fig. 3). Despite the LAMP assay’s high sensitivity of 102 copies/µL, its experimental procedures are laborious and demand costly equipment.
Fig. 2.

LAMP primer screening. (A) Marker: Marker; from TGEV-J1 to TGEV-J3 are LAMP primer set I; from TGEV-J1-NC to TGEV-J3-NC are negative control with ddH2O (B) Marker: Marker; from TGEV-W1 to TGEV-W3 are LAMP primer set II; from TGEV-W1-NC to TGEV-W3-NC are negative control with ddH2O
Fig. 3.
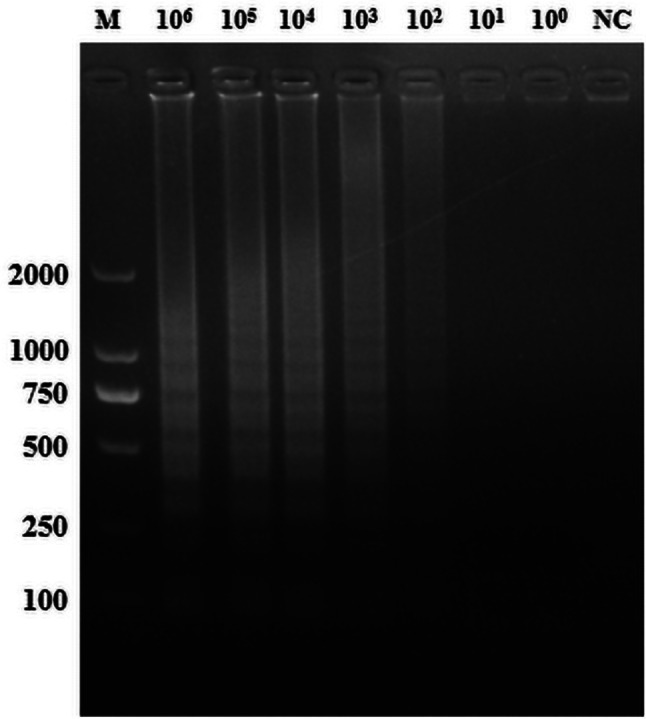
TGEV LAMP sensitivity test results. M: Marker; 106 to 100 indicate tests with varying concentrations of pCI-neo-N recombinant plasmid; NC: negative control with ddH2O
Establishment and optimization of the TGEV detection method by the CRISPR-Cas12a fluorescence assay
Stronger green fluorescence under UV light indicated higher measured fluorescence values and improved detection efficiency. At a reaction temperature of 37 °C, fluorescence values stabilized after 30 min (Fig. 4A). Thus, we determined 30 min as the ideal reaction time. Analysis of fluorescence values during this period revealed the optimal signal at 39 °C (Fig. 4B). Considering the fluorescence signal intensity, subsequent experiments were conducted with an optimal detection condition of 30 min of amplification at 39 °C.
Fig. 4.

Optimization of reaction time and temperature in the CRISPR-Cas12a fluorescence assay. (A) From left to right, the reaction times were 40 min, 35 min, 30 min, 25 min, 20 min, 15 min, and 10 min; NC: negative control of RNase-free ddH2O. (B) From left to right, the reaction temperatures were 43 ℃, 41 ℃, 39 ℃, 37 ℃, 35 ℃, 33 ℃, and 31 ℃; NC: negative control of RNase-free ddH2O
Sensitivity assessment of the CRISPR-Cas12a fluorescence assay
With a 10-fold dilution of the recombinant plasmid pCI-neo-N as the detection template, amplified via LAMP, and ddH2O as the negative control, we observed a gradual decrease in fluorescence value with increasing dilution. The sensitivity of the CRISPR-Cas12a fluorescence assay was determined to be 100 copies/µL (Fig. 5A, B).
Fig. 5.

Sensitivity of the CRISPR-Cas12a fluorescence assay. (A) Sensitivity assessment using various concentrations of pCI-neo-N. (B) End-point fluorescence intensity to determine sensitivity
Specificity assessment of CRISPR-Cas12a fluorescence assay
LAMP-amplified pCI-neo-N was employed as a template under the optimal CRISPR-Cas12a amplification conditions. Only the TGEV group displayed green fluorescence, demonstrating that the TGEV detection through the CRISPR-Cas12a fluorescence assay did not show cross-reactivity with PCV2, PCV3, PPV, PCLV, PRRSV and PEDV (Fig. 6A, B).
Fig. 6.

CRISPR-Cas12a fluorescence assay specificity test results. (A) From 1 to 8 are the experimental groups of TGEV, PCV2, PCV3, PCLV, PRRSV, PPV and PEDV respectively. (B) Determine the endpoint fluorescence intensity for specificity
Establishment and optimization of the CRISPR/Cas12a lateral-flow dipstick assay
To enhance the convenience and speed of TGEV detection, we developed the CRISPR/Cas12a lateral-flow dipstick assay, building upon the CRISPR-Cas12a fluorescence assay. To prevent false positives in the dipstick test from probe concentration issues, we varied probe concentrations for fine-tuning. Through visual inspection, we confirmed the elimination of false positives at a final probe concentration of 150 nM (Fig. 7). To balance cost-effectiveness and crRNA cleavage efficiency, we determined 30 nM as the optimal probe concentration for the assay (Fig. 7).
Fig. 7.
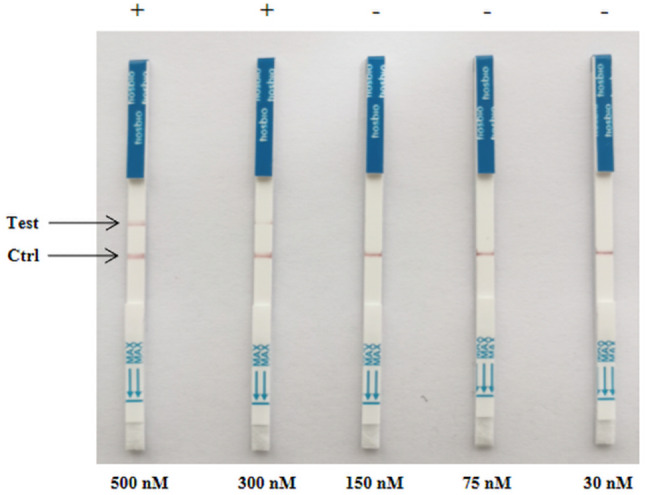
CRISPR-Cas12a lateral-flow dipstick probe concentration optimization results. 500 nM to 30 nM indicate tests with varying concentrations of the probe
Sensitivity and specificity assessment of the CRISPR/Cas12a lateral-flow dipstick assay
The CRISPR/Cas12a lateral-flow dipstick assay achieved a minimum detection limit of 102 copies/µL through LAMP amplification of the recombinant plasmid pCI-neo-N, with ddH2O serving as a negative control (Fig. 8). There was no cross-reactivity observed when LAMP-amplified pCI-neo-N was simultaneously tested as a template with PCV2, PCV3, PPV, PCLV, PRRSV and PEDV (Fig. 9).
Fig. 8.

CRISPR-Cas12a lateral-flow dipstick sensitivity test results. 106 to 100 indicate tests with varying concentrations of pCI-neo-N recombinant plasmid as a template; NC: negative control of RNase-free ddH2O
Fig. 9.
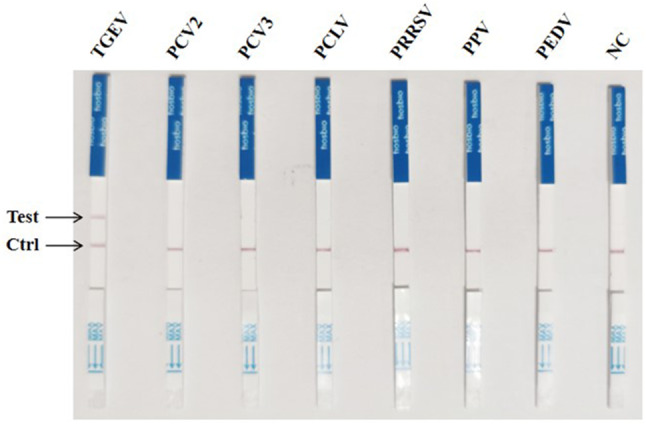
CRISPR-Cas12a lateral-flow dipstick specificity test results. From left to right, the experimental groups were TGEV, PCV2, PCV3, PCLV, PRRSV, PPV and PEDV; NC: negative control of RNase-free ddH2O
Performance of the CRISPR/Cas12a fluorescence and lateral-flow dipstick assay for testing TGEV clinical samples
13 samples were tested using the established CRISPR-Cas12a fluorescence assay, CRISPR/Cas12a lateral-flow dipstick assay, and RT-PCR assay. 13 clinical samples showed positive results for CRISPR-Cas12a fluorescence assay (Fig. 10A, B) and lateral-flow dipstick assay (Fig. 11A), with a 100% concordance rate with RT-PCR data (Fig. 11B). These findings suggest that both the CRISPR-Cas12a fluorescence assay and the CRISPR/Cas12a lateral-flow dipstick assay developed in this study are suitable for clinical tests and other applications.
Fig. 10.

Test results of clinical samples for the CRISPR-Cas12a fluorescence assay. (A) Results of the CRISPR-Cas12a fluorescence assay. (B) End-point fluorescence. From 1 to 13 are 13 clinical samples. NC: negative control of RNase-free ddH2O
Fig. 11.

Results of clinical sample testing by CRISPR-Cas12a lateral-flow dipstick assay and qPCR. (A) Results of the CRISPR/Cas12a lateral-flow dipstick assay. (B) Results of the q-PCR
Discussion
TGEV is a coronavirus that incites severe diarrhea, vomiting, and dehydration in swine, leading to significant mortality rates in piglets under 2 weeks old, causing economic setbacks in the swine industry [22]. It lingers in swine for extended durations, primarily replicating in the respiratory and intestinal tracts, resulting in issues such as villous atrophy. Afflicted swine can harbor the virus, which disperses through feces for approximately 4 weeks [23]. Early identification of TGEV is crucial for curtailing its transmission. However, current detection methods are laborious and time-intensive, necessitating specialized laboratory settings. Hence, there’s a necessity for an assay that is highly visible, straightforward, swift, sensitive, and user-friendly.
There are numerous techniques to detect TGEV, such as real-time reverse transcription loop-mediated isothermal amplification, nanoparticle-associated PCR assay, multiplexed real-time RT-qPCR assay with TaqMan probes, and multiplexed immunoassay [24]. The introduction and advancement of the CRISPR-Cas system represent a significant milestone in 21st-century molecular biology, unlocking numerous possibilities in genetic engineering, genome editing, and beyond [25]. Leveraging the CRISPR-Cas system has enabled the creation of a swift and precise TGEV detection approach. Presently, combining the CRISPR-Cas system with PRA, ERA, and LAMP allows for the detection of various pathogens rapidly, with high sensitivity. For instance, the SFTSV detection method employs recombinase polymerase amplification and the CRISPR-Cas12a system [26]. Furthermore, the integration of reverse transcription-enzymatic recombinase amplification (RT-ERA) and CRISPR-Cas12a technologies offers a straightforward, convenient, and sensitive method for detecting GAstV [27]. The fusion of isothermal amplification and CRISPR-Cas12 DETECTR technology has resulted in a rapid (30–40 min) technique for identifying SARS-CoV-2 in clinical samples [28]. The CRISPR-Cas12a assay stands out due to its reduced time and equipment expenses when compared to conventional PCR and LAMP assays [29].
In this study, we identified optimal crRNA and developed a CRISPR/Cas12a fluorescence assay based on the conserved N gene of TGEV using LAMP amplification. By fine-tuning the reaction conditions to amplify at 39 °C for 30 min, this assay has a minimum detection limit of 100 copies/µL and shows no cross-reactivity with other related viruses. Additionally, we devised a CRISPR/Cas12a lateral-flow dipstick assay with a probe concentration of 30 nM to prevent false-positive outcomes. This assay exhibits a detection sensitivity of 102 copies/µL and, like the fluorescence assay, displays no cross-reactivity with other similar viruses. These advancements offer a more efficient and rapid method for TGEV detection. Our validation in 13 clinical samples using the CRISPR-Cas12a fluorescence assay, lateral-flow dipstick assay, and RT-PCR yielded consistent results, affirming the accuracy and practicality of the TGEV detection method outlined in this study. However, the CRISPR-Cas12a fluorescence assay requires specific equipment to observe results, and have discrepancies between naked eye observations and instrument measurements. In this study, the CRISPR/Cas12 lateral-flow dipstick assay was not optimized for reaction conditions. A fully optimized reaction system could theoretically enhance assay sensitivity. Overall, the CRISPR/Cas12a assay is highly efficient, fast, and sensitive. Combining it with LAMP amplification technology and lateral-flow dipstick simplifies TGEV detection equipment, making it more affordable, user-friendly, and providing valuable support for TGEV prevention and control in swine industry.
Conclusions
In summary, this study successfully established an intuitive, rapid, sensitive, highly specific, and easy-to-operate method for detecting TGEV.
Methods
Material sources and nucleic acid extraction
The recombinant plasmid (pCI-neo-N), an eukaryotic expression vector for the N gene of TGEV, was generously provided by another laboratory [30]. Porcine epidemic diarrhea virus (PEDV), porcine circovirus type 2 (PCV2), porcine circovirus type 3 (PCV3), porcine circovirus-like virus (PCLV), porcine reproductive and respiratory syndrome virus (PRRSV), and porcine poliovirus (PPV) were maintained in our lab [31]. 13 clinical samples were obtained from fecal matter of swine farms suspected of TGEV infection in Anhui Province and were identified as positive by RT-PCR. Viral nucleic acids were extracted using the TIANamp viral DNA/RNA kit (TIANGEN, Beijing, China). Viral nucleic acids were eluted in 25 µl of nuclease-free water and stored at -20 °C until needed. The viral RNA nucleic acids were reverse transcribed with a reverse transcription kit (Yeasen, Shanghai, China). Plasmids were extracted with the Plasmid Extraction Kit (TIANGEN, Beijing, China) following the manufacturer’s instructions.
Design of crRNA sequence, primers and probes
The conserved N gene of TGEV was chosen as the detection target. Three different PAM sites of 5´-TTTN on the N gene were identified for the CRISPR-Cas12a system detection target. Three sets of crRNA were designed based on the gene sequences of the corresponding PAM sites using the Cpf1 primer design website (http://bioinfolab.miamioh.edu/CRISPR-DT/interface/Cpf1_main.php) (Table 1). Six pairs of LAMP primers (Table 2) were designed at either end of the PAM sites using the LAMP PrimerExplorer design software. Tsingke (Nanjing, China) designed the crRNA, primers and probes. For the conserved N gene of TGEV, a highly conserved region of the target gene sequence was chosen, leading to the design of five sets of qPCR primers using Prime Premier 5.0 (Table 3). General Biosystems (Anhui, China) synthesized the primers.
Table 1.
crRNA sequences and probe of TGEV
| Primer Name | Primer Sequence (5΄-3΄) |
|---|---|
| TGEV-crRNA-1 | GAAATTAATACGACTCACTATAGGGTAATTTCTACTAAGTGTAGATCCTGCAGTTCTCTTCCAGGT |
| TGEV-crRNA-1 | ACCTGGAAGAGAACTGCAGGATCTACACTTAGTAGAAATTACCCTATAGTGAGTCGTATTAATTTC |
| TGEV-crRNA-2 | GAAATTAATACGACTCACTATAGGGTAATTTCTACTAAGTGTAGATTTCATGGCACCATCCTTGGC |
| TGEV-crRNA-2 | GCCAAGGATGGTGCCATGAAATCTACACTTAGTAGAAATTACCCT ATAGTGAGTCGTATTAATTTC |
| TGEV-crRNA-3 | GAAATTAATACGACTCACTATAGGGTAATTTCTACTAAGTGTAGATGGTACAAAGTCTCTCGGACA |
| TGEV-crRNA-3 | TGTCCGAGAGACTTTGTACCATCTACACTTAGTAGAAATTACCCT ATAGTGAGTVGTATTAATTTC |
| ssDNA reporter-F | 6-FAM-TTTTTT-BHQ1 |
| ssDNA reporter-L | 6-FAM-TTTTTTTATTTTTTT-Biotin |
Table 2.
Sequence of LAMP primers for the N gene of TGEV
| Primer Name | Primer sequence(5΄-3΄) |
|---|---|
| TGEV-J1-F3 | CTAAGAATGAAAACAAACACACC |
| TGEV-J1-B3 | TCCTTTGAAGTCCAATAGCT |
| TGEV-J1-FIP | AATTGGCTGAACTGCTTCTAGCGAGAACTGCAGGTAAAGGTG |
| TGEV-J1-BIP | GGAGCAGTGCCAAGCATTACCCAAACAGAATGCTAGACAC |
| TGEV-J2-F3 | AACAACAGCAACGCTCTC |
| TGEV-J2-B3 | GGGTAATGCTTGGCACTG |
| TGEV-J2-FIP | CCAGGTGTGTTTGTTTTCATTCTTTAAAGAACGTAGTAACTCTAAGACA |
| TGEV-J2-BIP |
TGCAGGTAAAGGTGATGTGAC AACATTGGCAACGAGGTCAG |
| TGEV-J3-F3 | AGTCACGTTCACACACAA |
| TGEV-J3-B3 | TGTGTCATCAAACACATCTG |
| TGEV-J3-FIP | ACGAGCATAGGCATTAATCTGCACTTGCCAAAGGATGATCC |
| TGEV-J3-BIP | CAGAAGTGGCAAAAGAACAGAGAAAAATGCATCAGGTACCACAT |
| TGEV-W1-F3 | TGACAAGATTTTATGGAGCTAGA |
| TGEV-W1-R3 | TGGTATTTGTGTGTGAACGT |
| TGEV-W1-FIP | GTAATGCTTGGCACTGCTCCGCAGTTCAGCCAATTTTGG |
| TGEV-W1-BIP | CTGAATGTGTTCCATCTGTGTCTGACTTCTATCTGGTCGCC |
| TGEV-W2-F3 | GAGCAGTGCCAAGCATTA |
| TGEV-W2-B3 | GGCATTAATCTGCTGAAGG |
| TGEV-W2-FIP | TGAAGTCCAATAGCTTCCAAACAGCCCACAACTGGCTGAATG |
| TGEV-W2-BIP | AGGAAGATGGCGACCAGATAGAATTGTCCAGTCTTAGGATCA |
| TGEV-W3-F3 | AACAACAGCAACGCTCTC |
| TGEV-W3-B3 | GGGTAATGCTTGGCACTG |
| TGEV-W3-FIP | CCAGGTGTGTTTGTTTTCATTCTTTAAAGAACGTAGTAACTCTAAGACA |
| TGEV-W3-BIP | TGCAGGTAAAGGTGATGTGACAACATTGGCAACGAGGTCAG |
Table 3.
N gene qPCR identification primers for TGEV
| Primer Name | Primer Sequence |
|---|---|
| TGEV-qPCR-F1 | CCATAACCCTCCAGCAAG |
| TGEV-qPCR -R1 | AGCACCACGACTACCAAG |
| TGEV-qPCR -F2 | AGGTAATAGGGACCAACA |
| TGEVq-PCR-R2 | CAGTGCCTAAGTAGTAGAAGA |
| TGEV-qPCR -R3 | TATCTGGTCGCCATCTTC |
| TGEV-qPCR -F4 | CCCCATAACCCTCCAGCAA |
| TGEV-qPCR -R4 | TGGCAACCCAGACAACTCC |
| TGEV-qPCR -F5 | AGCAGTGCCAAGCATTACC |
| TGEV-qPCR -R5 | ATCTGGTCGCCATCTTCCT |
Primer screening and sensitivity detection for LAMP
Six pairs of artificial LAMP primers were evaluated following the guidelines of the LAMP Kit from Sangon Biotech (Shanghai, China). The reaction occurred at 65 ℃ for 1 h, followed by enzyme deactivation at 80 ℃. Amplification outcomes were assessed via agarose gel electrophoresis, identifying the most effective primer pair. To decrease the concentration of the recombinant plasmid pCI-neo-N, we diluted it by 10-fold serial to reach 109-100 copies/µL and then applied the optimized LAMP conditions for amplification. Post-reaction, we analyzed the amplification outcomes through agarose gel electrophoresis to establish the LAMP amplification’s minimum detection threshold.
Optimization of reaction time and temperature in the CRISPR-Cas12a fluorescence assay
To develop a CRISPR-Cas12a fluorescence assay for detecting TGEV, the N gene sequences of 30 TGEV strains were downloaded from GenBank. Afterward, Meglign software was utilized to analyze these sequences for comparison, and the most conserved regions of the N gene sequences were selected as the detection targets. The LAMP amplification products were used as templates, ddH2O was used as negative control. The reaction setup included: 2 µL NEB Buffer 2.1, 1 µL RNA inhibitor, 1 µL target gene, 1 µL crRNA (500 nM), 13 µL RNase-Free ddH2O, 1 µL probe (250 nM), and 1 µL Cas12a (250 nM). A gradient from 10 to 40 min was applied, with increments every 5 min, and a temperature range of 31–43 °C, with increments every 2 °C. The reaction took place in a water bath, the reaction products were photographed under a UV lamp to record the results. Subsequently, the results underwent fluorescence quantitative PCR instrument (Thermo Fisher Scientific, Shanghai, China) analysis for determining the optimal reaction time.
Sensitivity of the CRISPR-Cas12a fluorescence assay
Recombinant plasmid pCI-neo-N was 10-fold isodiluted to 109-100 copies/µL, followed by LAMP amplification and cleavage using the optimized CRISPR-Cas12a fluorescence assay. After the reaction, fluorescence intensity was visualized under UV light and quantified using a fluorescence measurement device to determine the minimum detection limit of the assay.
Optimization of probe concentrations in the CRISPR/Cas12a lateral-flow dipstick assay
The lateral-flow dipsticks used in the assay were procured from Tiosbio (Beijing, China). Probe concentrations were fine-tuned to establish the probes’ minimum detection threshold. The reaction system comprised various probe final concentrations: 1000 nM, 300 nM, 150 nM, 75 nM, and 30 nM, respectively. To create a 20 µL reaction product with RNase-Free ddH2O, 750 nM Cas12a, 1 nM crRNA, 2 µL NEB Buffer 2.1, 0.5 µL RNA inhibitor, and 2 µL LAMP product were employed. Post-reaction, 5 µL of the product was taken, brought to 50 µL with RNase-Free ddH2O, and thoroughly mixed. Utilizing test strips, results were determined following the same method as our previously established approach [32].
Sensitivity of the CRISPR/Cas12a lateral-flow dipstick assay
The plasmid pCI-neo-N recombinante was isodiluted 10-fold to 109-100 copies/µL and subjected to LAMP amplification. Later, 5 µL of the product was diluted to 50 µL with ddH2O and loaded into a lateral-flow dipsticks. The results were then observed after a few minutes to establish the lowest detection limit of the CRISPR/Cas12a lateral-flow dipsticks assay.
Specificity of CRISPR/Cas12a lateral-flow dipstick assay and CRISPR-Cas12a fluorescence assay
pCI-neo-N, PEDV, PCV2, PCV3, PCLV, PRRSV, and PPV (amplified through PCR and confirmed positive by sequencing with sensitivity falling within the detection range) were employed as test samples for specificity assessment, ddH2O was used as a negative control. Amplification was conducted following the optimized CRISPR-Cas12a fluorescence assay, with fluorescence intensity inspection under UV light post-reaction. Fluorescence values were measured using fluorescence quantification equipment to ascertain the specificity of the CRISPR-Cas12a fluorescence assay. The specificity of the CRISPR/Cas12a lateral-flow dipstick assay was also validated by evaluating the Colours development on test strips.
Evaluation of the CRISPR-Cas12a fluorescence assay and lateral-flow dipstick assay using clinical samples
We assessed the effectiveness of the CRISPR-Cas12a fluorescence assay and the lateral flow dipstick assay by isolating viral nucleic acids from 13 fecal samples. The samples were diluted in PBS, washed, and then transferred to a sterile EP tube followed by centrifugation at 8500 rpm for 3 min, the supernatant was taken. Subsequently, 200 µL of the supernatant was mixed with 20 µL of Proteinase K and 200 µL of Carrier RNA, and then vortexed for 15 s. After adding 250 µL of anhydrous ethanol and leaving the mixture at room temperature for 5 min, the solution was transferred into an RNase-Free column, followed by centrifugation and discarding the waste solution. Buffer GD and rinsing solution PW were added, and after centrifugation and discarding the waste solutions, anhydrous ethanol was added and the process was repeated. Lastly, the adsorbent column was moved to a clean RNase-Free centrifuge tube, and the viral RNA were collected at 12,000 r/min. Clinical samples were then tested using the CRISPR-Cas12a fluorescence assay, the CRISPR/Cas12a lateral-flow dipstick assay, and RT-PCR.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
Not applicable.
Author contributions
XJS, JT, and ZQ contributed to conception and design of the study. JLW and ZJH conducted experiments. HYW, JLW, ZJH, LTL, ZC, GJW, ZYW and YS analyzed data. HYW and XJS wrote the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This work was supported by grants the University Synergy Innovation Program of Anhui Province (GXXT-2022-057), Anhui Provincial University Excellent Young Talents Support Program (YQYB2023001) and Research Funds of Joint Research Center for Food Nutrition and Health of IHM (2023SJY01).
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Haiyang Wang and Zhao Qi contributed equally to this work.
References
- 1.Zhao S, Gao Q, Qin T, Yin Y, Lin J, Yu Q, Yang Q. Effects of virulent and attenuated transmissible gastroenteritis virus on the ability of Porcine dendritic cells to sample and present antigen. Vet Microbiol. 2014;171(1–2):74–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turlewicz-Podbielska H, Pomorska-Mól M. Porcine coronaviruses: overview of the state of the Art. Virol Sin. 2021;36(5):833–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng S, Wu H, Chen Z. Evolution of transmissible gastroenteritis virus (TGEV): A codon usage perspective. Int J Mol Sci 2020, 21(21). [DOI] [PMC free article] [PubMed]
- 4.Izeta A, Smerdou C, Alonso S, Penzes Z, Mendez A, Plana-Durán J, Enjuanes L. Replication and packaging of transmissible gastroenteritis coronavirus-derived synthetic minigenomes. J Virol. 1999;73(2):1535–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Charley B, Laude H. Induction of alpha interferon by transmissible gastroenteritis coronavirus: role of transmembrane glycoprotein E1. J Virol. 1988;62(1):8–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Delmas B, Laude H. Assembly of coronavirus Spike protein into trimers and its role in epitope expression. J Virol. 1990;64(11):5367–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ji Z, Dong H, Jiao R, Zhu X, Shi H, Chen J, Shi D, Liu J, Jing Z, Zhang J, et al. The TGEV membrane protein interacts with HSC70 to direct virus internalization through Clathrin-Mediated endocytosis. J Virol. 2023;97(4):e0012823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Risco C, Antón IM, Enjuanes L, Carrascosa JL. The transmissible gastroenteritis coronavirus contains a spherical core shell consisting of M and N proteins. J Virol. 1996;70(7):4773–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chai W, Burwinkel M, Wang Z, Palissa C, Esch B, Twardziok S, Rieger J, Wrede P, Schmidt MF. Antiviral effects of a probiotic Enterococcus faecium strain against transmissible gastroenteritis coronavirus. Arch Virol. 2013;158(4):799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liang X, Wang P, Lian K, Han F, Tang Y, Zhang S, Zhang W. APB-13 improves the adverse outcomes caused by TGEV infection by correcting the intestinal microbial disorders in piglets. J Anim Physiol Anim Nutr. 2022;106(1):69–77. [DOI] [PubMed] [Google Scholar]
- 11.Li C, Liang J, Yang D, Zhang Q, Miao D, He X, Du Y, Zhang W, Ni J, Zhao K. Visual and Rapid Detection of Porcine Epidemic Diarrhea Virus (PEDV) Using Reverse Transcription Loop-Mediated Isothermal Amplification Method. Animals: an open access journal from MDPI 2022, 12(19). [DOI] [PMC free article] [PubMed]
- 12.Ding G, Fu Y, Li B, Chen J, Wang J, Yin B, Sha W, Liu G. Development of a multiplex RT-PCR for the detection of major diarrhoeal viruses in pig herds in China. Transbound Emerg Dis. 2020;67(2):678–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Q, Liu X, Fang Y, Zhou P, Wang Y, Zhang Y. Detection and phylogenetic analyses of Spike genes in Porcine epidemic diarrhea virus strains Circulating in China in 2016–2017. Virol J. 2017;14(1):194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Callebaut P, Pensaert MB, Hooyberghs J. A competitive Inhibition ELISA for the differentiation of serum antibodies from pigs infected with transmissible gastroenteritis virus (TGEV) or with the TGEV-related Porcine respiratory coronavirus. Vet Microbiol. 1989;20(1):9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paton DJ, Brown IH, Vaz EK. An ELISA for the detection of serum antibodies to both transmissible gastroenteritis virus and Porcine respiratory coronavirus. Br Vet J. 1991;147(4):370–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li P, Ren X. Reverse transcription loop-mediated isothermal amplification for rapid detection of transmissible gastroenteritis virus. Curr Microbiol. 2011;62(3):1074–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Niu JW, Li JH, Guan JL, Deng KH, Wang XW, Li G, Zhou X, Xu MS, Chen RA, Zhai SL, et al. Development of a multiplex RT-PCR method for the detection of four Porcine enteric coronaviruses. Front Veterinary Sci. 2022;9:1033864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rajan A, Shrivastava S, Janhawi, Kumar A, Singh AK, Arora PK. CRISPR-Cas system: from diagnostic tool to potential antiviral treatment. Appl Microbiol Biotechnol. 2022;106(18):5863–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paul B, Montoya G. CRISPR-Cas12a: functional overview and applications. Biomedical J. 2020;43(1):8–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chertow DS. Next-generation diagnostics with CRISPR. Sci (New York NY). 2018;360(6387):381–2. [DOI] [PubMed] [Google Scholar]
- 21.Khambhati K, Bhattacharjee G, Singh V. Current progress in CRISPR-based diagnostic platforms. J Cell Biochem. 2019;120(3):2721–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xia L, Yang Y, Wang J, Jing Y, Yang Q. Impact of TGEV infection on the pig small intestine. Virol J. 2018;15(1):102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paul PS, Halbur P, Janke B, Joo H, Nawagitgul P, Singh J, Sorden S. Exogenous Porcine viruses. Curr Top Microbiol Immunol. 2003;278:125–83. [DOI] [PubMed] [Google Scholar]
- 24.Liu Q, Wang HY. Porcine enteric coronaviruses: an updated overview of the pathogenesis, prevalence, and diagnosis. Vet Res Commun. 2021;45(2–3):75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perez Rojo F, Nyman RKM, Johnson AAT, Navarro MP, Ryan MH, Erskine W, Kaur P. CRISPR-Cas systems: ushering in the new genome editing era. Bioengineered. 2018;9(1):214–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang M, Liu S, Xu Y, Li A, Wu W, Liang M, Niu G, Wang Z, Wang T. CRISPR/Cas12a technology combined with RPA for rapid and portable SFTSV detection. Front Microbiol. 2022;13:754995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang K, Zhang W, Xu L, Liu Q, Song X, Shao Y, Tu J, Qi K. Facile, ultrasensitive, and highly specific diagnosis of Goose astrovirus via reverse transcription-enzymatic recombinase amplification coupled with a CRISPR-Cas12a system detection. Poult Sci. 2022;101(12):102208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Broughton JP, Deng X, Yu G, Fasching CL, Servellita V, Singh J, Miao X, Streithorst JA, Granados A, Sotomayor-Gonzalez A, et al. CRISPR-Cas12-based detection of SARS-CoV-2. Nat Biotechnol. 2020;38(7):870–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu H, Hu X, Zeng H, He C, Cheng F, Tang X, Wang J. A rapid and high-throughput system for the detection of Transgenic products based on LAMP-CRISPR-Cas12a. Curr Res Food Sci. 2023;7:100605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ding L, Huang Y, Du Q, Dong F, Zhao X, Zhang W, Xu X, Tong D. TGEV nucleocapsid protein induces cell cycle arrest and apoptosis through activation of p53 signaling. Biochem Biophys Res Commun. 2014;445(2):497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang W, Xu L, Liu Q, Cao Y, Yang K, Song X, Shao Y, Tu J, Qi K. Enzymatic recombinase amplification coupled with CRISPR-Cas12a for ultrasensitive, rapid, and specific Porcine circovirus 3 detection. Mol Cell Probes. 2021;59:101763. [DOI] [PubMed] [Google Scholar]
- 32.Wei J, Li Y, Cao Y, Liu Q, Yang K, Song X, Shao Y, Qi K, Tu J. Rapid and visual detection of Porcine parvovirus using an ERA-CRISPR/Cas12a system combined with lateral flow dipstick assay. Front Cell Infect Microbiol. 2022;12:879887. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files.