

Abstract
Cuproptosis, a recently identified copper-dependent cell death mechanism, remains poorly unexplored in ovarian cancer (OC). This study systematically evaluates clinically significant cuproptosis-related genes (CRGs) as potential prognostic biomarkers in OC. Cox regression analysis and LASSO algorithms were used to develop a prognostic risk model incorporating 5 CRGs (CD8B2, GJB2, GRIP2, MELK, and PLA2G2D) within the TCGA cohort. This model stratified OC patients into high-risk and low-risk groups, with the high-risk group exhibiting significantly shorter overall survival compared to the low-risk group. The model's predictive accuracy for prognosis in OC patients was validated in the TCGA training cohort, TCGA testing cohort, and ICGC external validation cohorts. Among these 5 signature genes, the number of cuproptosis genes associated with GJB2 is the largest, so we selected GJB2 for further validation. qPCR revealed that GJB2 was highly expressed in OC cells and tumor tissues. The high expression of GJB2 was closely associated with poor prognosis in OC patients. Functionally, GJB2 silencing suppressed OC cell proliferation and migration while its overexpression promoted malignant progression and EMT. Furthermore, GJB2 regulated copper homeostasis and reduced cuproptosis sensitivity, while also facilitating immune escape by inhibiting CD8+ T cell infiltration and cytokine secretion, revealing its multiple roles in OC progression. In conclusion, we established a novel prognostic model incorporating 5 CRGs that effectively predicts clinical outcomes and characterizes the immune microenvironment in OC. Our findings particularly highlight GJB2 as a key regulator of cuproptosis with significant potential as both a prognostic biomarker and therapeutic target for OC management.
Supplementary Information
The online version contains supplementary material available at 10.1007/s10495-025-02119-8.
Keywords: Ovarian cancer, Prognostic signature, GJB2, Proliferation and migration, Cuproptosis, Immune infiltration
Introduction
Ovarian cancer (OC) is the second highest cause of mortality in gynecological malignancies worldwide, and its high mortality is mainly related to the difficulty of early diagnosis, high aggressiveness, and the development of chemotherapy resistance [1]. Due to the absence of distinctive symptoms and reliable biomarkers, most patients are diagnosed at advanced stages [2]. Despite advances in treatments such as targeted therapy and immunotherapy, the 5-year overall survival rate remains low, largely due to recurrence, drug resistance, and therapeutic uncertainty [3–5]. In clinical applications, the accurate assessment of prognosis in cancer patients relies on molecular biomarkers, which can help identify clinical risk groups and guide treatment decisions, making them a crucial component of personalized therapy [6].
Emerging evidence demonstrates that dysregulated copper homeostasis plays a pivotal role in cancer pathogenesis across multiple malignancies [7, 8]. Some studies have shown that decreased serum copper concentrations are linked to the development of endometrial cancer [9], and an imbalance in copper homeostasis has also been observed in the progression of head and neck cancers [10]. Recently, a groundbreaking study by Tsvetkov et al. identified a novel form of copper-dependent cell death, termed cuproptosis [11]. This mechanism, previously unidentified, has significant implications for understanding new regulatory pathways in cancer. While this breakthrough has opened new avenues for exploring copper-mediated regulatory networks in cancer biology, the potential connection between cuproptosis and OC pathogenesis remains largely speculative, with no definitive mechanistic evidence currently available to substantiate this relationship in OC models or clinical samples.
Gap Junction Protein Beta 2 (GJB2), also known as connexin 26 (Cx26), is a member of the connexin family and plays a key role in the formation of hemichannels and gap junctions. The opening of hemichannels facilitates the release of signaling molecules, such as ATP and glutamate, into the extracellular space [12]. Emerging evidence demonstrates that aberrant GJB2 expression disrupts gap junction intercellular communication (GJIC) across multiple malignancies, including breast, colorectal, and non-small cell lung cancers [13–15]. Beyond its canonical role in GJIC, connexins have been shown to orchestrate cancer progression through hemichannel-mediated modulation of autocrine/paracrine signaling networks [16]. Previous studies have shown that GJIC mediated by Cx26 promotes tumorigenesis by regulating cell proliferation and differentiation, while also enhancing cancer cell migration by reducing tumor cell adhesion [17, 18]. Teleki et al. [19] reported that GJB2 expression was downregulated after chemotherapy in breast cancer patients, suggesting an association between GJB2 expression and chemotherapy response, and highlighting its potential as an anti-cancer drug target. Although significant progress has been made in understanding GJB2 functions in other cancer types, its pathophysiological roles in OC, particularly the specific mechanisms underlying therapy resistance, metastasis, and tumor microenvironment modulation, remain completely uncharted territory.
This study aimed to identify predictive cuproptosis-related genes (CRGs) in OC that can not only provide valuable insight into the molecular networks, the signaling pathway, and the tumor immune infiltration that is related to cuproptosis in OC but can also be used to identify OC patients at high risk of poor survival. In our research, we identified 5 CRGs (CD8B2, GJB2, GRIP2, MELK, and PLA2G2D) and developed a prognostic model. Based on the CRG risk score, we classified the TCGA-OC patients into a high-risk group and a low-risk group. The functional role of CRGs in OC was further explored through gene set enrichment analysis (GSEA), assessing their potential for predicting prognosis, diagnosis, tumor immune infiltration, chemotherapeutic drug sensitivity, and immunotherapy efficacy, with both internal and external validations. Among these 5 signature genes, GJB2 was associated with the largest number of cuproptosis genes, so we selected the key gene GJB2 for further validation. To elucidate the oncogenic properties of GJB2, OC cells were transfected with GJB2-related plasmid, then performed CCK-8, colony formation assay, wound healing assay, and Transwell migration assay to evaluate the impact of CJB2 on the proliferation and migration of cells. To further characterize the molecular mechanisms underlying GJB2's oncogenic effects, we employed qPCR and Western blotting analyses to systematically evaluate its impact on the Epithelial-Mesenchymal Transition (EMT) signaling pathway activation. Simultaneously, through GO enrichment analysis of GJB2-correlated genes, we identified significant involvement in copper ion binding pathways, which was subsequently validated by qPCR showing marked alterations in key cuproptosis regulators following GJB2 overexpression. Later, cell viability assays also confirmed GJB2's regulatory impact on cuproptosis sensitivity. Finally, Transwell migration and flow cytometric analyses revealed that GJB2 inhibits CD8+ T cell infiltration and suppresses cytokine secretion, indicative of its role in immune evasion. Collectively, these findings establish GJB2 as a multifaceted oncogene in OC pathogenesis, driving tumor progression through proliferative, migratory, EMT-promoting, cuproptosis-modulating, and immunoevasive mechanisms, while highlighting its potential as a prognostic biomarker and therapeutic target.
Materials and methods
Data collection
The flowchart of the study design is illustrated in Fig. 1. A total of 379 OC patients and 88 normal ovarian samples were involved in the present study. The transcriptome data of TCGA-OC was downloaded from the UCSC Xena server (https://xenabrowser.net/) [20]. A total of 88 normal ovarian tissue mRNA expression samples were downloaded from the Genotype-Tissue Expression (GTEx) database. Then converted and normalized the FPKM values of TCGA-OC (379 OC patients) and GTEx-ovary (88 normal samples) to TPM values, and finally obtained an integrated expression matrix. The "NormalizeBetweenArrays" instruction of R package "limma" was used to remove batch-specific effects in the process. The clinical information of OC cases was obtained from The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/) [21, 22], including OS, survival status, age, grade, and stage. The summarized clinical data of the OC patients are shown in Table S1. The concrete clinical information for TCGA whole dataset OC patients is in Table S2. Study participants with incomplete clinical information were excluded. OC data for Ovarian cancer-Australia OV-AU items were collected from the International Cancer Genome Consortium (ICGC, https://icgc.org) database. A total of 85 samples were included in the column as an independent verification cohort for analysis. GSE38666, GSE105437, and GSE63885 were derived from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/). These datasets were achieved from common databases, and strict guidelines were obeyed according to these databases. The 97 cuproptosis genes (CGs) were extracted from a previous study for subsequent bioinformatics analysis [11, 23–25], listed in Table S3.
Fig. 1.

Flowchart of the Study
Mutation, correlation analysis, and differential analysis of DECGs
Differential expression genes (DEGs) in the TCGA-OC samples and normal ovarian samples were analyzed by the “limma” package in RStudio (|log2FC|> 1, p < 0.05). Venn diagrams were used to identify TCGA-DEGs that intersected with CGs, and the overlapping genes were defined as differentially expressed cuproptosis genes (DECGs). To identify the expression differences of DECGs between the normal and tumor samples, the R “limma” and “ggpubr” packages were used to generate the boxplots for comparison. We then utilized the “maftools” [26] R package to generate a waterfall plot that allowed us to examine the mutation frequency and mutation type of DECGs in each sample. Finally, the correlation analysis of 18 DECRs was drawn with the "corrplot" R package.
Construction and validation of cuproptosis-related prognosis model
To identify cuproptosis-related genes (CRGs), we used the "limma" package to calculate the correlation between DECGs and CRGs [27]. Using the correlation coefficient|R|> 0.4 and p < 0.001 as the screening criteria, a total of 1435 CRGs with expression values were obtained. We further screened the differentially expressed CRGs (|log2FC|> 3, p < 0.05) and finally obtained 71 differentially expressed CRGs. Then used univariate Cox regression analysis to obtain CRGs related to the prognosis of OC patients, and we carried out multivariate Cox regression analysis and least absolute shrinkage and selection operator (LASSO) Cox regression to construct the CRGs predictive signature for OC patients. Finally, the prognostic risk model was constructed by 5 CRGs (CD8B2, GJB2, GRIP2, MELK, and PLA2G2D). For each patient, the risk score was calculated based on the following formula: Cuproptosis risk score = ∑(Expi × Coefi). (Expi denotes each signature gene's expression level, and Coefi denotes the corresponding coefficient).
The reliability of the prognostic signature was validated using internal validation (TCGA-OC) and external validation groups (ICGC-OC). We randomly divided all patients from the TCGA-OC database into the training and testing groups. Based on the median value of the risk score, the training group, the testing group, and all patients were classified into high-risk and low-risk groups, respectively. In both training and validation groups, the signature’s predictive capability was assessed by Kaplan–Meier (KM) survival analysis and ROC curves using the “timeROC”, “survival”, and “survminer” R packages. Additionally, we also created a nomogram using the "survival", “rms”, and “regplot” R packages to predict the 1-, 3-, and 5-year survival rates and calibrated the signature to evaluate its consistency with practice. To measure the nomogram's capacity to differentiate and predict, the concordance index (C-index) was also determined. The C-index ranged from 0.5 to 1.0, with a higher C-index indicating stronger differentiating ability of the predictive model. Additionally, we utilized the “ggplot2” R package to perform PCA analysis, which allowed us to observe the distribution of samples in the high-risk and low-risk groups.
Functional enrichment analysis and immune cell infiltration analysis
The biological processes (BP), molecular functions (MF), and cellular components (CC) of cuproptosis-related genes and GJB2-correlated genes were determined using Gene Ontology (GO) analysis [28] by using the clusterProfiler package [29] and org.Hs.eg.db package. Additionally, the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis [30] and Gene set variation analysis (GSVA) [31] were used to investigate the pathways involved in differentially expressed genes in various risk groups. The gene sets of hallmarkers were obtained from the Molecular Signatures Database (MSigDB, http://software.broadinstitute.org/gsea/msigdb/index.jsp) for GSVA analysis.
Correlation of the prognostic signature with TME and immune checkpoints
"Reshape2", "tidyverse", "ggplot2", "ggpubr" and "ggExtra" R packages were used to analyze the correlation of risk score with immune cells and plot a heatmap depicting the relationship of signature genes with immune cells. Additionally, we compared the Tumor Microenvironment (TME) scores between high-risk and low-risk groups using the "reshape2" and "ggpubr" R packages. To further investigate the signature's predictive capability in immunotherapy response, we analyzed the expression of immune checkpoints between high-risk and low-risk groups using "limma", "ggplot2", "ggpubr" and "ggExtra" R packages [32].
Drug sensitivity analyses
Half-maximal inhibitory concentration (IC50) data for anticancer drugs were obtained from the Genomics of Drug Sensitivity in Cancer (GDSC) database (https://www.cancerrxgene.org/) [33, 34]. The drug distribution in the high-risk and low-risk score groups was analyzed and visualized using the "pRRophetic" [35] R package. The Wilcoxon sign rank test was used to compare IC50 differences between common anti-tumor drugs in the high-risk and low-risk groups. Differences in common anti-tumor drugs between high-expression and low-expression groups of GJB2 are also analyzed using the same methods.
Protein expression in the HPA database
The Human Protein Atlas (HPA) database (https://www.proteinatlas.org/) was used for the comparison of protein expression of GJB2 between normal ovarian tissue and OC tissue samples by immunohistochemistry (IHC) [36].
Cell culture and transfection
All cell lines used in this study included human normal ovarian surface epithelial cells (Immortalized Ovarian Surface Epithelial, IOSE) and OC cell lines, HO 8910, A2780, OC314, and SKOV3 (STR identification), were cultured in RPMI-1640 (BI) replenished with 10% fetal bovine serum (FBS) (BI), 100 µg/ml penicillin (Sigma) and 100 µg/ml streptomycin (Sigma). All cells were cultured at 37 °C in a humidified 5% CO2 incubator. Cells were collected for the experiment at the indicated time. 5 × 105 °C cells were seeded in a six-well plate and then reached 60–70% fusion degree per well, jetPRIME was applied for transient transfection plasmid and/or siRNA in vitro according to manufacturer's instructions. These cells were digested and collected after 48 h of transfection for the subsequent assays. All of the sequences of primers are listed in Table S4.
RNA isolation and qPCR
To detect the gene expression of GJB2 and the changes resulting from GJB2 si/OE transfection, we performed qRT-PCR analysis. Total RNA was isolated using Trizol reagent (Vazyme, Nanjing, China). Complementary DNA (cDNA) was synthesized using the TransScript Uni All-in-One First-Strand cDNA Synthesis SuperMix for qPCR (One-Step gDNA Removal) (TransGen Biotech, AU341-02-V2). qPCR was performed by the Applied Biosystems 7500 Real-Time PCR System and the ChamQ Universal SYBR qPCR Master Mix (Vazyme, Q711-02). We chose glyceraldehyde-3-phosphate dehydrogenase (GAPDH) to normalize GJB2, CDH1, CDH2, VIM, FDX1, LIAS, DLAT, and PDHB expression levels. Relative RNA abundances were calculated by the standard 2−ΔΔCt method. The sequences of all qPCR primers are listed in Table S4.
Cytokine secretion assay
OC cells were seeded at a density of 1 × 105 cells per well in 12-well plates and allowed to adhere overnight. Following adhesion, cells were transfected with either GJB2-overexpression plasmid (GJB2-Flag) or empty vector control (Flag). 24 h post-transfection, OTK3-activated T cells were added to the transfected tumor cells at a 5:1 effector-to-target ratio (T cells: tumor cells). The co-culture system was maintained for 12 h, after which 500 µl of BFA/monensin solution (1 × working concentration) was added to block cytokine secretion. Following 6 h of incubation, all T cells were harvested for subsequent intracellular cytokine analysis. For cell surface and intracellular staining, cells were first incubated with APC-anti human CD8 antibody at 4 °C for 30 min in the dark for extracellular staining. Following washing, cells were fixed with 100 µl of fixation buffer and incubated at 4 °C for 30 min protected from light. Permeabilization was then performed by adding 200 µl of 1 × Perm wash buffer and incubating at 4 °C for 30 min or overnight. For intracellular staining, an antibody cocktail containing PerCP-anti human GZMB and PE-anti human IFN-γ was prepared, and 50 µl of the premixed antibodies was added to each sample, followed by incubation at room temperature for 30 min in the dark. The reaction was stopped by adding 150 µl of FCM buffer to each well. Finally, cells were resuspended in 300 µl of 1 × PBS, filtered through a cell strainer, and transferred to flow cytometry tubes for acquisition.
Transwell assay
Cell migration was measured by Transwell assays using the 24-well transwell chambers with 8 μm polycarbonate membranes (Corning Incorporated, USA). For the assay of GJB2-regulated tumor cell migration ability, OC cells were collected after transfection with GJB2 overexpression plasmid or siRNA for 24 h. A total of 2 × 105 cells in 200 µl serum-free medium were added to the upper chamber. The lower chamber contained 800 µl medium with 20% FBS as a chemoattractant. For the assay of GJB2's ability to attract CD8+ T cells, OC cells overexpressing GJB2 were seeded into the lower chamber. 24 h after transfection, a total of 1 × 106 CD8+ T cells were added to the upper chamber in 200 µl medium containing 10% FBS. At the same time, 800 µl medium containing 10% FBS was added to the lower chamber. After incubation for 24 h, non-migrating cells were removed with a cotton swab, the remaining cells were subsequently fixed with 4% paraformaldehyde for 30 min and stained with crystal violet for 20 min. The relative cell number was calculated a light microscope (Olympus, Tokyo, Japan).
Wound healing assay
After being transfected with GJB2 siRNA and GJB2-OE for 24 h, OC cells were seeded into a 12-well plate and then scraped with a 200 µl pipette tip. Cell migration images were captured at 0, 24, 36, 48, 60, and 72 h after scratching.
Cell counting kit-8 (CCK-8) assay
The influence of GJB2 siRNA and GJB2-OE on cell proliferation was evaluated by CCK-8 assay. Specifically, OC cells (3 × 103 cells/well) were planted into 96-well plates and cultivated at 37 °C for 12, 24, 48, and 72 h, respectively. At different time points, the cells were incubated with the mixture containing 10 µl CCK-8 and 100 µl fresh medium for 2 h at 37 °C. Finally, absorbance was measured at 450 nm with an enzyme-labeled apparatus.
Colony formation assay
A total of 500 cells per well were spread onto six-well plates and cultured at 37 °C for 2 weeks. The fresh medium was changed every three days. Finally, the colonies were treated with 4% paraformaldehyde fixation for 30 min and crystal violet staining for 20 min. Subsequently, a statistical analysis of the colony number was conducted.
Cell viability
Cells were inoculated into 96-well plates at a rate of 3000 cells/well and allowed to attach for 12 h. Elesclomol and copper chloride (CuCl2) solution were added 1:1 for 24 h. Then 100 µl of fresh medium containing 10% CCK8 solution was added and the 450 nm absorbance was detected following incubation for 2 h at 37 °C using an enzyme-labeled apparatus. As for the chemical rescue assay, copper ion chelator (TTM) was added after plating overnight, then Elesclomol-CuCl2 was added into plates and incubated for 24 h.
Western blotting
Cells were cultured in 60 mm dishes to approximately 80% confluence and then lysed with ice-cold lysis RIPA buffer (P0013B) containing 1% protease inhibitor cocktail and the concentration of the supernatant was determined by BCA. After loading the target proteins onto an SDS poly acrylamide gel, the proteins were transferred from the gel to a PVDF membrane. Subsequently, the membranes were incubated at 4 °C overnight with GJB2 (ER1902-42, HUABIO, 1:1000), E-cadherin (20,874–1-AP, Proteintech, 1:2000), N-cadherin (22,018–1-AP, Proteintech, 1:2000), Vimentin (10,366–1-AP, Proteintech, 1:2000) and GAPDH (Utibody, UM4002, 1:2000) after being blocked with 5% BSA for 2 h. After washed 3 times with TBST, the membranes were incubated with a secondary antibody (Zhengneng Biotechnology) for 90 min at room temperature. Protein bands were detected and analyzed using enhanced ECL chemiluminescence. Digital images were taken by a MiniChemi from SageCreation, Beijing, China.
Statistical analyses
For the data comparison between the two groups, we adopted the t-test for variables with a normal distribution and the Wilcoxon rank sum test for variables conforming to non-normal distribution. The overall survival rates of each group were compared using a KM analysis in conjunction with a log-rank test. Additionally, we developed the prognostic signature using the univariate Cox-LASSO-multivariate Cox regression analysis methods. We performed all statistical analyses using the R software (version 4.2.0) and GraphPad Prism software (version 9.0). Statistical significance is indicated with asterisks (*). A two-sided p value of < 0.05 was considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Results
DECGs and molecular characterization in OC patients
To investigate the correlation between cuproptosis-related genes (CRGs) and OC, we compiled a list of 97 cuproptosis genes (CGs) from published articles[23–25], as shown in Table S3. Gene ontology (GO) pathway enrichment analysis of these CGs revealed significant associations with various copper signaling pathways (Fig. 2A). We then compared normal ovarian and OC samples, identifying a total of 5,723 differentially expressed genes (DEGs) (|Log2FC|> 1, p < 0.05; Fig. 2B). Among the 97 CGs, 18 were found to be differentially expressed in the TCGA cohort (Fig. 2B). Of these, 8 DECGs (differentially expressed cuproptosis genes) were upregulated, while 10 were downregulated, indicating dysregulation of cuproptosis in OC (Fig. 2C, Table S5). Further analysis of the expression correlations among the 18 DECGs revealed highly consistent relationships between the regulators of cuproptosis (Fig. 2D).
Fig. 2.

DECGs and Molecular Characterization in OC Patients. A GO pathway enrichment analysis of CGs involved signaling pathway. B Venn diagram showing the DECGs shared by TCGA-DEGs and CGs, 18 DECGs were found in the TCGA cohort. C Differential expression of 18 DECGs between normal tissue and OC tissues. D The correlations between the expression of 18 DECGs. E Mutation frequencies of 18 DECGs in 462 patients with OC from the TCGA cohort. F Frequency of CNV alterations in 18 DECGs. Red dots represented CNV amplification, while green dots represented CNV deletion. G Location of CNV alterations of 18 DECGs on chromosomes in the TCGA-OC cohort. H Sankey diagram of coexpression between 18 DECGs and 1453 CRGs. I The volcano plot depicted the expression patterns of CRGs in TCGA-OC. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
We next analyzed the somatic mutation frequency of the 18 DECGs and found that 56 out of 462 OC samples (12.12%) harbored mutations in these genes, predominantly in the form of missense mutations (Fig. 2E). We also evaluated somatic copy number variations (CNVs) in the DECGs and observed widespread CNVs across all 18 genes. The frequencies of CNVs in DECGs in OC patients are shown in Fig. 2F, while the chromosomal locations of altered CNVs in these genes are depicted in Fig. 2G. Finally, Pearson correlation analysis revealed significant associations between 1,453 CRGs and the 18 DECGs (|R|> 0.4, p < 0.001), as illustrated in the Sankey diagram (Fig. 2H, Table S6). Among these, 71 differential genes were identified within the 1,453 CRGs, including 30 upregulated and 41 downregulated CRGs in tumor tissues (|Log2FC|> 3, p < 0.05; Fig. 2I, Table S7).
Construction and validation of CRGs signature
Univariate Cox regression analysis of 71 CRGs identified 9 CRGs significantly associated with the prognosis of OC patients (Fig. S1A, Table S8). To minimize overfitting during signature generation, we applied LASSO regression analysis, which revealed 7 significant genes and highlighted the trajectory changes in the regression coefficients of mRNAs, as well as estimated prediction accuracy through 1000 cross-validations (Fig. S1B-C). These high-dimensional data were further reduced using a multifactorial Cox proportional hazards regression model, resulting in the selection of 5 CRGs for the development of a risk score model to predict overall survival (OS) in OC patients. The selected genes included CD8B2, GJB2, GRIP2, MELK, and PLA2G2D (Fig. 3A). A heatmap illustrating the relationship between the 18 DECGs and the 5 CRGs is shown in Fig. 3B. Risk score = 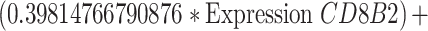
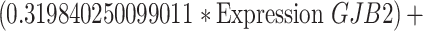

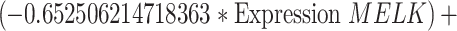
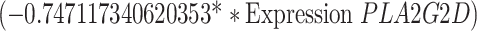 . Based on the median risk score, we classified patients in the TCGA-OC cohort into high-risk and low-risk groups for survival analysis. The KM method was used to compare the OS between the two groups, and the results revealed that the OS of patients in the high-risk group was significantly worse than that of the low-risk group (p = 0.003, Fig. 3C). Given the strong correlation between the risk model and patient outcomes, we further investigated whether the prognostic value of the 5 CRGs could serve as an independent prognostic factor. In the univariate Cox analysis, both age and risk scores showed statistically significant associations with OS (p < 0.001, Fig. 3D). Furthermore, in multivariate Cox regression analysis, both variables retained their prognostic significance for OS (Fig. 3E). Subsequently, OC patients were divided into 3 groups (all, training, and testing) to assess the accuracy and feasibility of the prognostic risk model in predicting OC prognosis. The detailed expression levels of the risk genes, risk scores, and risk group assignments are provided in Table S9. The analysis revealed that, in all 3 groups (all, training, and testing), the expression levels of the 5 key risk-scoring genes differed significantly between the high-risk and low-risk groups (Fig. 3F). Specifically, GRIP2, GJB2, and CD8B2 were more highly expressed in the high-risk group, whereas PLA2G2D and MELK had lower expression levels (Fig. 3F). Scatter plots for the 3 groups (all, training, and testing) demonstrated that the mortality rate of OC patients increased with higher risk scores (Fig. 3F).
. Based on the median risk score, we classified patients in the TCGA-OC cohort into high-risk and low-risk groups for survival analysis. The KM method was used to compare the OS between the two groups, and the results revealed that the OS of patients in the high-risk group was significantly worse than that of the low-risk group (p = 0.003, Fig. 3C). Given the strong correlation between the risk model and patient outcomes, we further investigated whether the prognostic value of the 5 CRGs could serve as an independent prognostic factor. In the univariate Cox analysis, both age and risk scores showed statistically significant associations with OS (p < 0.001, Fig. 3D). Furthermore, in multivariate Cox regression analysis, both variables retained their prognostic significance for OS (Fig. 3E). Subsequently, OC patients were divided into 3 groups (all, training, and testing) to assess the accuracy and feasibility of the prognostic risk model in predicting OC prognosis. The detailed expression levels of the risk genes, risk scores, and risk group assignments are provided in Table S9. The analysis revealed that, in all 3 groups (all, training, and testing), the expression levels of the 5 key risk-scoring genes differed significantly between the high-risk and low-risk groups (Fig. 3F). Specifically, GRIP2, GJB2, and CD8B2 were more highly expressed in the high-risk group, whereas PLA2G2D and MELK had lower expression levels (Fig. 3F). Scatter plots for the 3 groups (all, training, and testing) demonstrated that the mortality rate of OC patients increased with higher risk scores (Fig. 3F).
Fig. 3.

Construction and Validation of CRGs Signature. A The forest plot demonstrated the hazard ratio of 5 CRGs with prognostic values filtered by the univariate Cox regression analysis. B The heatmap of the correlation between 18 DECGs and 5 CRGs. C KM curves for OS of the two risk groups (chi-square test, p = 0.003). D Univariate Cox analysis of the signature with clinical phenotypes. E Multivariate Cox regression analysis of the signature with clinical phenotypes. F The expression heatmap of 5 CRGs in the 3 groups (all, training, and testing). The ranked dot plot indicates the risk score distribution and the scatter plot presents the patients' survival status. G ROC curves to predict the sensitivity and specificity of 1-, 2-, and 3-year survival according to the risk score in the 3 groups (all, training, and testing). (H-J) PCA between the high-risk and low-risk groups based on the 18 DECGs H, All CRGs I, and 5 CRGs risk model J. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
The receiver operating characteristic (ROC) curve analysis revealed that the area under the curve (AUC) values for evaluating survival outcomes in OC patients at 1, 2, and 3 years were all greater than 0.5, demonstrating the high specificity and sensitivity of the prediction model (Fig. 3G). Three-dimensional principal component analysis (PCA) showed that the distribution of high-risk and low-risk patients was indistinguishable based on the 18 DECGs and all CRGs expression sets (Fig. 3H, I). However, the distribution of high-risk and low-risk groups based on the 5 CRGs was most distinct (Fig. 3J). These results indicate that the 5 CRGs used in the risk model effectively distinguish between high-risk and low-risk samples, further supporting the utility of our method in differentiating these populations. Overall, these findings suggest that the prognostic risk model, based on the 5 genes (CD8B2, GJB2, GRIP2, MELK, and PLA2G2D), can serve as a reliable predictor of OC prognosis.
Heatmap and KM curves of clinicopathological variables in the high-risk and low-risk groups
To facilitate the clinical application of the prediction model, we integrated clinical data and gene features from TCGA-OC patient samples. We analyzed the association between the CRGs' prognostic signature and clinicopathological characteristics. The heatmap illustrates the distribution of age, grade, stage, and risk score across all OC patient samples (Fig. S2A). The proportions of clinicopathological variables in the high-risk and low-risk groups are shown in Fig. S2B-D. Patients with missing clinical information were excluded from the analysis. We further explored whether prognosis varied across different clinical subgroups by separating samples based on age (≤ 65 and > 65), tumor grade (2 and 3), and stage (III and IV). Survival rates were worse for high-risk patients compared to low-risk patients in all subgroups (Fig. S2E–G). These results suggest that our risk model can accurately predict the prognosis across various clinical subgroups of OC.
Prognostic analysis of the 5 CRGs risk model in TCGA and ICGC cohorts
The prognostic risk score, combined with age and tumor grade, was used to generate the ROC survival curve. The results indicated that, compared to other clinical traits, the CRGs prognostic risk scoring system accurately predicted the 1-, 3-, and 5-year survival rates of OC samples (Fig. 4A–C, AUC = 0.620, 0.593, and 0.601, respectively). Using the 5 CRGs features, we constructed a prognostic nomogram to predict the 1-, 3-, and 5-year OS probabilities. As shown in Fig. 4D, the score assigned to each factor corresponds to its contribution to survival risk. The accuracy of the nomogram was verified using calibration curves, which demonstrated a high degree of agreement between actual values and predicted values (Fig. 4E). Moreover, the C-index for the risk score was greater than 0.5, indicating that the risk model offers a significant advantage in predicting OS in OC patients (Fig. 4F). To assess the classification ability of the risk model based on the 5 CRGs, we performed PCA (Fig. 4G) and t-distributed stochastic neighbor embedding (t-SNE) (Fig. 4H) on all TCGA datasets. The results showed distinct distributions between the high-risk and low-risk groups, suggesting that the prognostic signature accurately differentiates between them (Fig. 4G, H). Additionally, when we compared our CRGs signature prognostic model with the Tumor Immune Dysfunction and Exclusion (TIDE) model and the Tumor Inflammation Signature (TIS) model, we found that our model had a superior ability to predict the 1-year survival rate of OC samples (Fig. 4I, AUC = 0.620, 0.579, and 0.497, respectively).
Fig. 4.

Prognostic Analysis of the 5 CRGs Risk Model in TCGA and ICGC Cohorts. A–C ROC curve of 1-, 3-, and 5-year OS for multiple prognostic indicators of OC samples. D The nomogram, a quantitative model for predicting clinical prognosis, to predict 1-year, 3-year, and 5-year OS in the OC patients of the TCGA-OC cohort using 3 factors, including Age, Grade, and risk score. E The calibration curves indicated that the nomogram accurately predicted the 1-, 3-, and 5-year OS of OC patients in the TCGA cohort. F A concordance index (C-index) was generated to assess the identification and forecasting capabilities of the nomogram. G The PCA analysis based on the prognostic signature demonstrated that the patients in the different risk score groups were distributed in two directions. Red and blue dots represent the high-risk group and the low-risk group. H t-SNE analysis of risk groups in all TCGA cohorts. I Comparison of the prognostic risk model with TIS models and TIDE models. TIS: Tumor Inflammation Signature; TIDE: Tumor Immune Dysfunction and Exclusion. J KM survival analysis of high and low-risk groups in ICGC cohorts. K 1-, 2-, and 3-year ROC curve analysis in ICGC cohorts. L The distribution of the risk score in the ICGC cohorts. M The correlation of survival time and risk scores in ICGC cohorts. N PCA between low-risk and high-risk groups in ICGC cohorts. O t-SNE analysis of risk groups in the ICGC cohorts. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
In the ICGC cohorts, patients with OC were also divided into high-risk and low-risk groups. KM curves revealed that patients in the low-risk group had a significantly better survival benefit (Fig. 4J). Additionally, the AUC values for 1-, 2-, and 3-year survival in the ICGC cohorts were 0.645, 0.708, and 0.691, respectively (Fig. 4K). The scatter plot further demonstrated that patients in the low-risk group exhibited more favorable prognostic outcomes (Fig. 4L, M). Finally, PCA and t-SNE analyses of the ICGC data sets revealed a clear distinction between high-risk and low-risk components, indicating that the prognostic signature accurately differentiated the two groups (Fig. 4N, O). These results collectively demonstrate that the gene signature performs well in predicting the prognosis of patients in both groups.
Risk score of CRGs predicts the signaling pathways, tumor microenvironment, and immune cell infiltration
We conducted GSEA using both KEGG and GO databases to investigate the differences in biological functions and signaling pathways between the high-risk and low-risk groups defined by the 5 CRG signatures (Fig. 5A, B, Fig. S3A-B). The high-risk group exhibited significant enrichment in pathways such as ECM receptor interaction, focal adhesion, and WNT signaling, while the low-risk group was notably enriched in pathways related to chemokine signaling and primary immunodeficiency (Fig. 5A, B). We also analyzed the immune microenvironment of samples with different risk scores and obtained their immune expression profiles (Fig. S4A). Additionally, GSVA enrichment analysis revealed significant differences between the high-risk and low-risk groups in several immune-related pathways, including Type II IFN response, MHC class I, HLA, cytolytic activity, inflammation promotion, T cell co-stimulation, checkpoint regulation, T cell co-inhibition, and APC co-inhibition (Fig. S4B, Table S10).
Fig. 5.

Risk Score of CRGs Predicts the Signaling Pathways, Tumor Microenvironment, and Immune Cell Infiltration. (A-B) GSEA enrichment analysis (KEGG) between the high-risk A and low-risk B groups. C The abundance of infiltrating immune cell types in the high-risk and low-risk groups. D Comparison of immune-related scores between the high-risk and low-risk groups. E Violin plot illuminating the difference in the exclusion score between the high-risk and low-risk groups. F Survival analysis curves of the high-TMB and low-TMB groups. G Effect of TMB with different risks on the probability of survival. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
We next used CIBERSORT to compare immune cell infiltration between the high-risk and low-risk groups. In the low-risk group, we observed a higher proportion of memory B cells, plasma cells, CD8+ T cells, activated memory CD4+ T cells, follicular helper T cells, activated NK cells, and M1 macrophages. In contrast, the high-risk group had a lower proportion of naive B cells, resting memory CD4+ T cells, monocytes, and activated mast cells (Fig. 5C). Since human leukocyte antigen (HLA) genes are critical for antigen presentation, we analyzed HLA indexes that are closely associated with immune function. Our analysis revealed a significant upregulation of HLA gene expression in the low-risk group compared to the high-risk group, encompassing both classical class I (HLA-A) and class II molecules (HLA-DQA1, HLA-DOB, HLA-DMB), as well as the non-classical HLA-F (Fig. S4C). This elevated HLA expression profile suggests enhanced antigen presentation capacity in low-risk patients, which is generally associated with more robust anti-tumor immune responses. Additionally, we examined the expression of m6A-related genes in the high-risk and low-risk groups. The results showed that only FTO expression differed significantly, with higher expression in the high-risk group compared to the low-risk group (Fig. S4D).
To further evaluate the predictive value of risk scores for immunotherapy, we evaluated immune scores in OC cells using the ESTIMATE algorithm and found that the low-risk group had higher immune scores (Fig. 5D), indicating a more immunologically active tumor microenvironment. Importantly, our analysis of immune exclusion patterns demonstrated a significant positive association with risk scores (Fig. 5E), suggesting that tumors from high-risk patients exhibit characteristics of immunologically "cold" tumors that are less responsive to immune checkpoint blockade (ICI) therapy. Previous studies have shown that Tumor Mutation Burden (TMB) is positively correlated with tumor stage, grade, and immune cell infiltration [37]. We categorized OC patients into "high-TMB" and "low-TMB" groups using median cutoff points and performed survival analyses. Kaplan–Meier analysis revealed that patients in the high-TMB group had significantly better OS compared to those in the low-TMB group (p < 0.001, Fig. 5F). A combined survival analysis of TMB and CRG risk scores further revealed that patients with both high TMB and low-risk scores had the best prognosis, while those with low TMB and high-risk scores had the worst prognosis (Fig. 5G). We conclude that patients in the low-risk group are more likely to benefit from immunotherapy.
Immunotherapy analysis and drug sensitivity prediction in high-risk and low-risk groups
Several studies have shown that combination therapy with anti-CTLA-4 and anti-PD-1 can increase the proportion of activated CD8+ T cells and NK cells while reducing the number of suppressive immune cells in the tumor microenvironment, thereby enhancing the therapeutic efficacy against cancer [38, 39]. Building on this background and our analysis of immune checkpoints, we sought to investigate whether there are differences in immunotherapy response between high-risk and low-risk groups. To do so, we downloaded the OC immunotherapy score data from The Cancer Immunome Atlas (TCIA, https://tcia.at/) and assessed the correlation of immunotherapy response between the two risk groups. The results indicated that patients in the low-risk group had a better response to immunotherapy, whether treated with anti-CTLA-4 and anti-PD-1 alone or in combination (Fig. 6A–D). This finding further supports our hypothesis that the risk score can predict the sensitivity and efficacy of immunotherapy.
Fig. 6.

Immunotherapy Analysis and Drug Sensitivity Prediction in High-risk and Low-risk Groups. A–D Differences in sensitivity to anti-PD-1, anti-CTLA-4, and the combination of these two antibodies in different risk score groups. E, F Boxplot showing the mean differences in estimated IC50 values of 6 representative drugs (Ruxolitinib, Temozolomide, Tubastatin A, Trametinib, Midostaurin, and 17-AAG) between the two risk groups
Chemotherapy resistance remains a significant challenge in the treatment of OC and contributes substantially to cancer-related mortality. Identifying the chemotherapy-sensitive population could optimize the therapeutic efficacy for OC patients, maximizing the benefits of standard chemotherapy protocols. To further investigate the drug resistance potential between high-risk and low-risk groups, we used the "pRRophetic" package to estimate the IC50 values of chemotherapy drugs and inhibitors for both groups. 6 representative drugs are shown in Fig. 6E-F. Our analysis identified Ruxolitinib, Temozolomide, and Tubastatin A as potential candidates for treating patients in the low-risk group, while Trametinib, Midostaurin, and 17-AAG (Tanespimycin) may offer therapeutic benefits for patients in the high-risk group.
Analysis of GJB2 expression and survival prognosis in OC
To further validate the prognostic signature, we conducted functional assays through in vitro experiments. Among these 5 signature genes (CD8B2, GJB2, GRIP2, MELK, and PLA2G2D), GJB2 was associated with the largest number of cuproptosis genes (Table S6) and exhibited strong positive correlations with multiple key tumor-related signaling pathways (Fig. 7A). A literature review showed that GJB2 has not been previously reported in OC, prompting us to include GJB2 for further functional and mechanistic analysis in this study. First, we assessed GJB2 expression in normal ovarian surface epithelial cells (IOSE) and OC cell lines (HO 8910, A2780, OC314, and SKOV3). The qPCR results indicated that GJB2 expression was significantly higher in OC cell lines compared to IOSE cells (Fig. 7B). Subsequently, we analyzed clinical specimens from 42 OC patients and 16 normal ovarian controls obtained from Xiangya Hospital, Central South University. qPCR analysis demonstrated significantly elevated GJB2 expression levels in tumor tissues compared to normal ovarian tissues (Fig. 7C). Additionally, we analyzed GJB2 protein expression patterns in OC through immunohistochemical data from the Human Protein Atlas (HPA, https://www.proteinatlas.org/) database. The IHC results consistently demonstrated upregulated GJB2 protein expression in OC tissues compared to normal ovarian tissue (Fig. 7D), corroborating our qPCR-based observations and further supporting the potential oncogenic role of GJB2 in OC development.
Fig. 7.

Analysis of GJB2 Expression and Survival Prognosis in OC. A GSVA analyzed the signaling pathways involved in 5 characteristic genes (CD8B2, GJB2, GRIP2, MELK, and PLA2G2D). B The expression of GJB2 in normal ovarian surface epithelial cells (IOSE) and OC cell lines (HO 8910, A2780, OC314, and SKOV3) were detected by qPCR. CGJB2 expression levels in OC and normal ovarian tissues were quantified by qPCR. D Representative IHC staining for GJB2 protein in normal ovarian tissue and OC tissue, taken from the HPA with permission on its website. E–G The violin plot depicts the upregulation of GJB2 in OC tumor samples in TCGA + GTEx, GSE38666, and GSE105437. H–J ROC Curves Displaying the Sensitivity and Specificity of GJB2 for the Diagnosis of OC Patients from the TCGA, GSE38666, and GSE105437 Dataset. K KM survival curves for OS in OC patients according to the tumor expression of GJB2. L KM survival curves for PFS in OC patients according to the tumor expression of GJB2. M GEPIA2 online website analysis showed a correlation between GJB2 and the OC stage. N Meta-analysis depicting forest plots of GJB2 expression in GSE63885 datasets as a univariate predictor of OS. O ROC curves display the sensitivity and specificity of GJB2 for the diagnosis of OC patients from the GSE63885 datasets. X-axis indicates false-positive rates, and Y-axis indicates true-positive rates. P Correlation between GJB2 expression and the sensitivity of GDSC drugs (top 30). Positive correlation indicates a higher gene expression may lead to drug resistance. Negative correlation indicates a higher gene expression may make drug-sensitive. In all statistical plots, data are expressed as the mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Through comprehensive bioinformatic analysis of multiple independent cohorts, we consistently observed significant upregulation of GJB2 expression in OC tissues versus normal controls, as evidenced by data from the TCGA + GTEx (Fig. 7E), GSE38666 (Fig. 7F), and GSE105437 (Fig. 7G). Additionally, we assessed the diagnostic performance of GJB2 using the area under the ROC curve (AUC) in the TCGA (Fig. 7H), GSE38666 (Fig. 7I), and GSE105437 (Fig. 7J) datasets. The AUC values were 0.954, 0.744, and 0.918, respectively, indicating that GJB2 has strong diagnostic value in OC patients. Importantly, analysis using the KM Plotter website (https://kmplot.com/analysis/) revealed that high expression of GJB2 was associated with poorer OS outcomes in the TCGA-OC dataset (Fig. 7K). In addition to OS, high expression of GJB2 was also linked to worse progression-free survival (PFS) outcomes (Fig. 7L). Similarly, the GEPIA2 website (http://gepia2.cancer-pku.cn/#index) revealed that GJB2 expression increases with the advancing OC stage (Fig. 7M). To further investigate the relationship between GJB2 expression and OC prognosis, we evaluated its prognostic significance in the GEO dataset cohort (GSE63885). Meta-analysis confirmed that GJB2 is an independent prognostic factor, with high expression associated with poor prognosis in OC patients (Fig. 7N). Additionally, we assessed the diagnostic utility of GJB2 to differentiate between tumor and normal samples in the GSE63885 cohort using ROC analysis. The ROC curve showed that the AUC values for predicting patient survival at 1-, 3-, and 5-years were all greater than 0.5 (Fig. 7O). These findings from independent validation cohorts collectively demonstrate that GJB2 holds significant diagnostic and prognostic value for OC patients.
Subsequently, we investigated the relationship between GJB2 expression and the response to anti-tumor drugs. Gene Set Cancer Analysis (GSCA, http://bioinfo.life.hust.edu.cn/GSCA/#/) indicated a significant correlation between GJB2 expression and drug sensitivity. The results revealed that GJB2 expression was negatively associated with the sensitivity to several chemotherapeutic agents, including Afatinib, Cetuximab, Docetaxel, Gefitinib, and Trametinib (Fig. 7P). To further explore potential therapeutic options, we used the "prophetic" R package to estimate the IC50 values of various chemotherapy agents and inhibitors in groups with high and low GJB2 expression. 8 representative drugs are shown in Fig. S5. The analysis showed that the high GJB2 expression group exhibited greater sensitivity to Paclitaxel, MG-132, Sunitinib, Lapatinib, Dasatinib, Pazopanib, Cyclopamine, and Crizotinib. These findings suggest that OC patients with high GJB2 expression may benefit from treatment with these drugs. Therefore, GJB2 expression could serve as a predictor of chemotherapy sensitivity, enabling the estimation of chemotherapy responses based on GJB2 expression levels in OC patients.
GJB2 modulates malignant phenotypes and EMT in OC cells
In this study, SKOV3 and OC314 cells exhibited higher expression levels of GJB2 compared to A7820 and HO 8910 cells (Fig. 7B). To further investigate the role of GJB2, we selectively knocked down GJB2 in SKOV3 and OC314 cells using siRNA. The siRNA constructs GJB2-si2 and GJB2-si3 were chosen for subsequent assays due to their effective knockdown efficiency (Fig. 8A, S6A). To assess the impact of GJB2 knockdown on cell proliferation, we performed CCK-8 (Fig. 8B, S6B) and colony formation (Fig. 8C, S6C) assays in SKOV3/OC314 cells with or without GJB2 knockdown. The results showed that, following GJB2 knockdown, the proliferation rate in the GJB2-si2 and GJB2-si3 groups was significantly reduced compared to the siNC control group (Fig. 8B-C, S6B-C). We also examined the effect of GJB2 knockdown on cell migration using Transwell (Fig. 8D, S6D) and wound healing (Fig. 8E, S6E) assays. Both assays revealed a marked reduction in the migratory ability of OC cells upon inhibition of GJB2 expression. Collectively, these findings suggest that silencing GJB2 expression inhibits both the proliferation and migration of OC cells.
Fig. 8.

GJB2 Modulates Malignant Phenotypes and EMT in OC Cells. A The transfection efficiency of GJB2 siRNA in SKOV3 was detected by qPCR. B The CCK-8 assay showed that GJB2 interference significantly inhibited SKOV3 cell proliferation at 12, 24, 48, and 72 h. C Knockdown of GJB2 inhibited the proliferation ability of SKOV3 cells and was evaluated by a colony formation assay. D–E Transwell (D) and wound healing (E) assays were performed to determine the effect of GJB2 knockdown on the migration ability of SKOV3 cells. F Validation of GJB2 expression after transfection of GJB2-OE and negative control (NC) plasmids in A2780 cells using qPCR and Western blotting assays. G CCK-8 assay displaying the influence of GJB2-OE on the cell viability. H The colony formation assay displays the effect of GJB2 upregulation on cell proliferation ability. I–J The effects of GJB2-OE on OC cell migration were evaluated through Transwell (I) and wound healing (J) assays. K Bioinformatic analysis of GJB2-associated signaling pathways in OC using GSCA. L–M The mRNA and protein levels of GJB2, E-cadherin, N-cadherin, and Vimentin in A2780 cells after GJB2 overexpression were detected by qPCR (L) and Western blotting (M). In all statistical plots, data are expressed as the mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
We further investigated the potential effects of GJB2 upregulation on OC cells. GJB2 overexpression (GJB2-OE) plasmids were constructed and transfected into HO 8910 and A2780 cells, followed by the detection of GJB2 overexpression using qPCR and Western blotting assays. The results showed that GJB2 was markedly elevated in both HO 8910 and A2780 cells (Fig. 8F, S6F). In both CCK-8 (Fig. 8G, S6G) and colony formation (Fig. 8H, S6H) assays, GJB2-OE promoted OC cell proliferation. Additionally, in comparison to the control group, GJB2-OE significantly increased the relative migration rate of OC cells in both Transwell (Fig. 8I, S6I) and wound healing (Fig. 8J, S6J) assays. These findings suggest that targeting GJB2 could offer potential therapeutic strategies for treating OC.
After determining GJB2's critical role in regulating the malignant progression of OC, we next investigated its potential involvement in EMT. Bioinformatics analysis using the GSCA platform (Gene Set Cancer Analysis, http://bioinfo.life.hust.edu.cn/GSCA/#/) revealed significant enrichment of EMT-related pathways in OC samples with high GJB2 expression (Fig. 8K). To validate these findings experimentally, we examined EMT marker expression in GJB2-overexpressing OC cells. Both qPCR (Fig. 8L) and Western blotting (Fig. 8M) analyses demonstrated that GJB2 overexpression significantly downregulated the epithelial marker E-cadherin while upregulating the mesenchymal markers N-cadherin and Vimentin. These results suggest that GJB2 may promote OC metastasis by activating the EMT processes.
GJB2 is associated with copper ionophore-induced cell death
Building upon our investigation of GJB2's functional roles, we further examined its potential involvement in cuproptosis regulation. GO enrichment analysis of GJB2-correlated genes (|R|> 0.45, p < 0.001, Table S11) revealed significant enrichment in copper ion binding pathways (GO:0005507, p = 2.4 × 10–4, Fig. 9A), suggesting GJB2 may modulate cuproptosis through copper-related processes. Subsequent qPCR analysis revealed that overexpression of GJB2 in OC cells led to significant downregulation of key cuproptosis regulators (FDX1, DLAT, LIAS, and PDHB) (Fig. 9B). These findings collectively indicate that GJB2 participates in cuproptosis regulation by modulating the expression of core pathway components.
Fig. 9.

GJB2 is Related to Copper Ionophore-induced Cell Death. A GO enrichment analysis of signaling pathways associated with GJB2-correlated genes (|R|> 0.45, p < 0.001). B Analysis of GJB2, FDX1, DLAT, LIAS, and PDHB expression in GJB2-overexpressing OC cells by qPCR. C–D HO 8910 and A2780 cells were exposed to different doses of Elesclomol-CuCl2 for 24 h and detected by CCK-8 reagent. E Representative images of OC cells treated with Elesclomol-CuCl2 with or without TTM for 24 h. Scale bars represent 200 μm. F The rescue effect of TTM in HO 8910 and A2780 treated with Elesclomol-CuCl2 was explored through CCK-8 assay. G–J HO 8910 and A2780 cells transfected with GJB2-OE were treated with various concentrations of elesclomol for 24 h. Cell viability was evaluated by CCK-8 assay. In all statistical plots, data are expressed as the mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Accumulating evidence suggests that Elesclomol is a copper-binding small molecule and Elesclomol-induced cell death results from the accumulation of intracellular copper rather than from the direct effects of Elesclomol itself [40, 41]. However, the mechanisms underlying copper-induced cytotoxicity in OC cells remain unclear. To investigate this, we first assessed the IC50 values of HO 8910 and A2780 cells treated with Elesclomol-CuCl2 (1:1) for 24 h. The IC50 values were found to be 30 nM and 15 nM, respectively (Fig. 9C, D). We then constructed a cuproptosis induction model, which included 3 experimental groups: negative control (NC), Elesclomol-CuCl2, and TTM-Elesclomol-CuCl2. In the Elesclomol-CuCl2 group, HO 8910 cells were treated with 30 nM Elesclomol-CuCl2 (1:1, Elesclomol is a potent copper ionophore which induces cuproptosis) for 24 h. In the TTM-Elesclomol-CuCl2 group, HO 8910 cells were pretreated overnight with 20 μM tetrathiomolybdate (TTM, a copper ion chelator that inhibits cuproptosis), and then treated with 30 nM Elesclomol-CuCl2 for 24 h. The results showed that TTM rescued the viability of HO 8910 cells under Elesclomol-CuCl2-induced conditions (Fig. 9E, F). Similar results were observed in another OC cell line A2780 (Fig. 9E, F). These findings collectively suggest that Elesclomol-induced OC cell death is dependent on copper accumulation. To further elucidate the role of copper in OC cells and clarify the function of GJB2 in cuproptosis, we assessed the recovery of OC cell viability in the Vector and GJB2-OE groups following treatment with Elesclomol-CuCl2. Both groups were treated with varying concentrations of Elesclomol-CuCl2 for 24 h, and cell viability was measured using the CCK-8 assay. The results indicated that GJB2-OE significantly increased the IC50 value (Fig. 9G, H). Additionally, the bar chart revealed a significant difference in cell viability between the two groups after 24 h of Elesclomol-CuCl2 treatment, with the GJB2-OE group showing markedly higher viability than the Vector group (Fig. 9I, J). This suggests that GJB2 overexpression enables OC cells to recover viability more rapidly after Elesclomol-CuCl2 treatment. In conclusion, GJB2-OE mitigates copper-induced cell death in OC cells treated with Elesclomol-CuCl2.
GJB2 inhibits the infiltration of CD8+ T cells and promotes immune escape
The tumor immune microenvironment (TIME) has emerged as a critical focus in cancer research, with growing evidence highlighting its role in tumor progression and therapeutic response. While our previous studies have established that abnormal GJB2 expression can regulate the malignant progression in OC, its specific impact on immune regulation remains poorly understood. To investigate GJB2's immunomodulatory role, we first applied the TIDE algorithm to assess the potential efficacy of immunotherapy across OC subgroups stratified by GJB2 expression levels. Given that higher TIDE scores correlate with increased immune evasion and reduced likelihood of benefiting from ICI therapy [42, 43], our analysis revealed that elevated GJB2 expression in the TCGA cohort was associated with significantly higher TIDE, Dysfunction, and Exclusion scores (Fig. 10A, C). These findings collectively indicate that GJB2 overexpression may contribute to an immunosuppressive TIME, potentially diminishing immunotherapy responsiveness in OC patients. To further investigate the immunomodulatory role of GJB2, we examined its correlation with immunosuppressive components in the tumor microenvironment, including regulatory T cell marker FOXP3, M2 macrophage marker CD163, and immune checkpoint molecules (TIGIT, CTLA4, and CD274). Strikingly, GJB2 expression showed significant positive correlations with all these immunosuppressive markers (FOXP3, CD163, TIGIT, CTLA4, and CD274) (Fig. 10D), strongly suggesting that GJB2 overexpression contributes to establishing an immunosuppressive tumor microenvironment in OC.
Fig. 10.

GJB2 Inhibits the Infiltration of CD8+ T Cells and Promotes Immune Escape. (A-C) Violin plot illuminating the difference of the TIDE score A, Dysfunction score B, and Exclusion score C between the GJB2 high- and low-expression groups. D Box plot displaying differential expression of immune checkpoints between GJB2 high- and low-expression groups. E CD8+ T cell migration to lower chambers was quantified following GJB2 overexpression in OC cells. F Flow cytometry analysis of Granzyme B (GZMB) and IFN-γ secretion by CD8+ T cells co-cultured with GJB2-overexpressing OC cells (E: T ratio = 5:1). In all statistical plots, data are expressed as the mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
To elucidate the mechanism by which GJB2 modulates immune cell function, we performed Transwell migration assays which revealed that GJB2-overexpressing OC cells exhibited significantly reduced chemotactic attraction of CD8+ T cells compared to controls (Fig. 10E), consistent with its established pro-tumorigenic role. This impaired recruitment capacity suggests that GJB2 may facilitate immune evasion by inhibiting CD8+ T cell infiltration. Furthermore, in functional co-culture experiments using OTK3-activated primary human CD8+ T cells (effector: target ratio = 5:1), flow cytometric analysis demonstrated that GJB2 overexpression in tumor cells substantially suppressed the production of key cytokines (Granzyme B and IFN-γ) compared to control cells after 24 h of co-culture (Fig. 10F). These findings collectively demonstrate that GJB2 not only inhibits CD8+ T cell migration but also functionally impairs their cytotoxic potential, ultimately fostering an immune-evasive tumor microenvironment.
Discussion
Ovarian cancer (OC) is one of the most common malignancies of the genital tract, with high mortality rates and a significant impact on the health of women worldwide [44]. The conventional therapeutic approach for OC typically consists of cytoreductive surgery combined with platinum-based chemotherapy [45]. Nevertheless, a substantial proportion of patients ultimately develop platinum resistance, leading to disease recurrence [46]. Despite advances in characterizing molecular prognostic factors for OC, the field still lacks definitive markers that reliably predict tumor development and serve as clinically useful biomarkers [47]. Therefore, it is crucial to identify promising biomarkers for OC prognosis prediction and to develop effective therapeutic strategies.
In recent years, researchers have focused on developing therapies that induce cancer cell death, as triggering the death of malignant cells is a viable strategy for cancer treatment [48, 49]. Based on different signaling pathways, there are many types of cell death, such as apoptosis, necroptosis, pyroptosis, ferroptosis, and autophagy-dependent cell death [50]. A recent study by Tsvetkov et al. introduced a novel form of copper-induced programmed cell death, termed cuproptosis, which operates through mechanisms distinct from those of classical cell death [11]. Copper, as an essential cofactor for a variety of metalloenzymes that contribute to tumor initiation, invasion, and metastasis [7, 51, 52], has recently emerged as a promising target for developing antitumor therapies. Emerging evidence highlights the critical roles of cuproptosis genes in tumor progression. For example, LIPT1 significantly influences hepatocellular carcinoma cell proliferation and invasion [53], and FDX1 induces cuproptosis and regulates anti-tumor immunity in colon cancer [54]. Recent mechanistic studies reveal that copper ions (Cu⁺) directly bind to lipoylated DLAT, triggering protein aggregation that generates toxic stress and ultimately leads to cuproptosis. Notably, TCA cycle inhibition causes acetyl-CoA accumulation, which competitively suppresses DLAT activity and consequently inhibits cuproptosis through feedback regulation [11, 55, 56]. Similarly, LIAS gene knockout has been shown to attenuate copper-induced cell death [11]. Likewise, studies in clear cell renal cell carcinoma demonstrate that reduced expression of PDHB, a key cuproptosis regulator, is associated with diminished copper-dependent cytotoxicity [57]. Although there is growing interest in the potential connection between cuproptosis and OC, there is currently no direct experimental or clinical evidence to support such a link.
In this work, we enrolled more CRGs to separate OC samples according to the CGs expression levels from public datasets. Using the relevant data of OC patients in TCGA, we constructed a CRG prognosis prediction model based on the regulation of the cuproptosis process. In this research, 5 CRGs (CD8B2, GJB2, GRIP2, MELK, and PLA2G2D) were chosen to construct a prognostic signature through LASSO Cox regression analysis. OC patients were stratified into high-risk and low-risk groups based on the risk score. The ROC curve demonstrated the strong predictive performance of the signature. A predictive nomogram was then created by integrating the risk score with clinical factors such as age, grade, and stage, thereby enhancing the model's utility and ease of application. External validation using ICGC cohorts confirmed the robustness of the signature's predictive ability. Functional analyses revealed that CRGs associated with the low-risk and high-risk groups were linked to immune-related pathways. A comparison of immune cell infiltration and activated pathways between the two groups showed that the high-risk group exhibited generally lower levels of infiltrating immune cells and reduced activity of immune-related pathways compared to the low-risk group. Among these 5 signature genes, GJB2 was associated with the largest number of CGs and showed significant positive associations with critical oncogenic pathways. Studies demonstrate that GJB2 is highly expressed in various cancers including hepatocellular carcinoma (HCC) [58], non-small cell lung cancer (NSCLC) [59], and breast carcinomas [60], showing significant correlation with poor patient prognosis. Its oncogenic mechanisms involve activation of the NF-κB/HIF-1α/GLUT1/PD-L1 signaling axis, glycolytic regulation, and immune microenvironment remodeling. For instance, in HCC, GJB2 promotes immune suppression by translocating to the cytoplasm and activating the NF-κB pathway [58], while in NSCLC, its expression can be upregulated by CAR10 [59]. However, the specific functions and mechanisms of GJB2 in OC remain unclear and warrant further investigation.
Based on preliminary findings, we prioritized GJB2 for comprehensive in vitro characterization. Both qPCR assays and bioinformatics analyses revealed that GJB2 was highly expressed in OC cells and tumor samples, with its elevated expression correlating positively with stage and worse prognosis. Our findings demonstrate that GJB2 exerts an oncogenic function in OC progression. Silencing GJB2 markedly suppressed OC cell proliferation and migration, whereas GJB2 overexpression enhanced these malignant phenotypes and triggered the EMT. Simultaneously, GO enrichment analysis revealed a significant association between GJB2 and the copper ion binding pathway, with key cuproptosis regulators changed significantly after GJB2 overexpression. Furthermore, overexpression of GJB2 weakened the sensitivity of OC cells to cuproptosis. Finally, we demonstrated that GJB2 markedly suppresses CD8+ T cell infiltration and reduces the secretion of key cytokines (GZMB and IFN-γ), revealing its critical role in promoting immune escape. Our findings establish GJB2 as a multifunctional oncogenic regulator that may shape the immunosuppressive tumor microenvironment through three distinct mechanisms: promoting regulatory T cell recruitment and M2 macrophage polarization, inhibiting CD8+ T cell infiltration and function, and upregulating immune checkpoint molecule expression. While previous studies have highlighted the role of GJB2 in cancer progression [13, 14, 61], our study provides new evidence for its specific involvement in OC cuproptosis. GJB2 was found to interact with the expression of numerous CRGs in OC, both positively and negatively, conjecture that GJB2 may regulate the cuproptosis process through multiple mechanisms. Nonetheless, additional research is needed to determine the specific regulatory mechanisms by which GJB2 modulates OC cuproptosis.
These findings suggest the potential clinical application of GJB2 in the following aspects. Compared to BRCA status (primarily reflecting homologous recombination deficiency) [62], GJB2 better characterizes tumor proliferative and metastatic potential. Combined analysis of both markers could provide a more comprehensive evaluation of tumor biological characteristics and treatment sensitivity. In contrast to CA-125 (mainly used for clinical diagnosis and treatment monitoring) [63], GJB2 expression levels objectively indicate tumor malignancy. Integrating both markers significantly improves predictive accuracy for long-term prognosis and therapeutic response. Regarding immunotherapy prediction, high GJB2 expression shows a significant association with an immunosuppressive microenvironment, suggesting its potential as a predictive biomarker for patient response to immune checkpoint inhibitors.
There may be some possible limitations in the current study. First, the major part of the cohort in this study is derived from the TCGA, GEO, and ICGC databases, which are retrospective studies and need to be validated by prospective analysis. Second, while this study has elucidated the oncogenic mechanisms of GJB2, its clinical application requires further validation through large-scale clinical cohorts to verify its predictive value, along with prospective studies to evaluate treatment efficacy based on GJB2 stratification. Third, none of the anti-GJB2 targeting drugs have been tested in clinical trials to date. As such, the potential immunotherapeutic effects of anti-GJB2 treatment remain speculative. In the future, there is a critical need to develop and evaluate anti-tumor immunotherapeutic agents targeting GJB2.
In conclusion, our study elucidates the regulatory mechanisms of cuproptosis in OC, laying the theoretical foundation for developing novel therapies targeting this pathway. Significantly, we not only developed a cuproptosis-related prognostic model for OC but also pinpointed GJB2 as a key molecular target, a discovery that opens new avenues for targeted therapy and immunotherapy development in OC treatment.
Conclusions
Overall, the prognostic model established in this study holds significant clinical value for guiding targeted therapy and immunotherapy in OC. Our findings identify GJB2 as a pivotal CRG, whose elevated expression correlates strongly with poor patient outcomes. Functionally, GJB2 not only promotes OC cell proliferation, migration, and EMT, but also modulates cuproptosis by reducing cellular sensibility to copper-induced cell death. In terms of immune regulation, GJB2 fosters an immunosuppressive tumor microenvironment by suppressing CD8+ T cell infiltration and cytokine secretion. These discoveries provide novel insights into the regulatory mechanisms of cuproptosis in OC and open new avenues for developing GJB2-targeted therapeutic strategies.
Supplementary Information
Below is the link to the electronic supplementary material.
Table S1 The clinical characteristics of OC patients in the TCGA dataset. Table S2 The concrete clinical information for TCGA whole dataset OC patients. Table S3 The cuproptosis genes extracted from the previous research. Table S4 Sequences of primers. Table S5 Differential expression of CGs. Table S6 Correlation and regulation of DECGs and CRGs. Table S7 Differential expression of CRGs. Table S8 Univariate Cox analysis of identifying 9 CRGs associated with OS. Table S9 The detailed risk genes expression, risk score, and risk group. Table S10 The GSVA functional enrichment analysis. Table S11 GJB2-correlated genes.
Supplementary file 2 (ZIP 7234 kb)
Acknowledgements
We sincerely thank the public databases, including TCGA, GTEx, ICGC, GEO, HPA, GEPIA, and KM plotter, for providing open access.
Abbreviations
- OC
Ovarian cancer
- TCGA
The cancer genome atlas
- GTEx
Genotype tissue expression
- ICGC
International cancer genome consortium
- CGs
Cuproptosis genes
- DEGs
Differentially expressed genes
- DECGs
Differentially expressed cuproptosis genes
- CRGs
Cuproptosis-related genes
- CNVs
Copy number variations
- LASSO
Least absolute shrinkage and selection operator
- OS
Overall survival
- KM
Kaplan–Meier
- ROC
Receiver operating characteristic
- AUC
Areas under the curve
- PCA
Principal component analysis
- t-SNE
T-distributed Stochastic Neighbor Embedding
- C-index
Concordance index
- IC50
Half-maximal inhibitory concentration
- TIDE
Tumor immune dysfunction and exclusion
- ICI
Immune checkpoint inhibitor
- TIS
Tumor inflammation signature
- CSCs
Cancer stem cells
- GO
Gene ontology
- GSVA
Gene set variation analysis
- GDSC
Genomics of drug sensitivity in cancer
- GSCA
Gene set cancer analysis
- TCIA
The cancer immunome atlas
- IOSE
Immortalized ovarian surface epithelial
- OE
Overexpression
- NC
Negative control
- GJB2
Gap junction protein beta-2
- Cx26
Connexin 26
- TMB
Tumor mutation burden
- TME
Tumor microenvironment
- TIME
Tumor immune microenvironment
- EMT
Epithelial-mesenchymal transition
Authors contributions
H.L. and G.Y. conceived and guided the study. H.L. mainly took charge of searching the literature, conducting the data analysis and experiments, producing the figures, and writing. G.S. revised the manuscript and conducted the experiments. K.G. downloaded the data from the databases. M.W. and Y.L. organized the data. Z.T., Q.P., and G.Y. revised the manuscript. All authors have read and approved the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China [No. 82173376, No. 82372711], and the Natural Science Foundation of Hunan Province [2022JJ70079].
Data availability
Data is provided within the manuscript or supplementary information files.
Declarations
Competing interests
The authors declare no competing interests.
Ethics approval
The study was reviewed and approved by the Ethics Committee of Xiangya Hospital Central South University, approval number 202204081. All clinical samples were collected with informed consent from patients. We declare that all methods are reported in accordance with the declaration of Helsinki.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Zhihui Tan, Email: tanzhihui@csu.edu.cn.
Qiong Pan, Email: 1517682924@qq.com.
Gang Yin, Email: gangyin@csu.edu.cn.
References
- 1.Siegel RL, Giaquinto AN, Jemal A (2024) Cancer statistics, 2024. CA Cancer J Clin 74:12–49. 10.3322/caac.21820 [DOI] [PubMed] [Google Scholar]
- 2.Kuroki L, Guntupalli SR (2020) Treatment of epithelial ovarian cancer. BMJ 371:m3773. 10.1136/bmj.m3773 [DOI] [PubMed] [Google Scholar]
- 3.Wilson MK, Pujade-Lauraine E, Aoki D et al (2017) Fifth ovarian cancer consensus conference of the gynecologic cancer InterGroup: recurrent disease. Ann Oncol 28:727–732. 10.1093/annonc/mdw663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang L, Zhao W, Huang J et al (2022) Development of a dendritic cell/tumor cell fusion cell membrane nano-vaccine for the treatment of ovarian cancer. Front Immunol 13:828263. 10.3389/fimmu.2022.828263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marrelli D, Ansaloni L, Federici O et al (2022) Cytoreductive surgery (CRS) and HIPEC for advanced ovarian cancer with peritoneal metastases: Italian PSM oncoteam evidence and study purposes. Cancers (Basel). 10.3390/cancers14236010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu F, Quan F, Xu J et al (2019) Breast cancer prognosis signature: linking risk stratification to disease subtypes. Brief Bioinform 20:2130–2140. 10.1093/bib/bby073 [DOI] [PubMed] [Google Scholar]
- 7.Ge EJ, Bush AI, Casini A et al (2022) Connecting copper and cancer: from transition metal signalling to metalloplasia. Nat Rev Cancer 22:102–113. 10.1038/s41568-021-00417-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y, Zhang L, Zhou F (2022) Cuproptosis: a new form of programmed cell death. Cell Mol Immunol 19:867–868. 10.1038/s41423-022-00866-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Atakul T, Altinkaya SO, Abas BI et al (2020) Serum copper and zinc levels in patients with endometrial cancer. Biol Trace Elem Res 195:46–54. 10.1007/s12011-019-01844-x [DOI] [PubMed] [Google Scholar]
- 10.Ressnerova A, Raudenska M, Holubova M et al (2016) Zinc and copper homeostasis in head and neck cancer: review and meta-analysis. Curr Med Chem 23:1304–1330. 10.2174/0929867323666160405111543 [DOI] [PubMed] [Google Scholar]
- 11.Tsvetkov P, Coy S, Petrova B et al (2022) Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 375:1254–1261. 10.1126/science.abf0529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Omori Y, Duflot-Dancer A, Mesnil M et al (1998) Role of connexin (gap junction) genes in cell growth control: approach with site-directed mutagenesis and dominant-negative effects. Toxicol Lett 96–97:105–110. 10.1016/s0378-4274(98)00056-3 [DOI] [PubMed] [Google Scholar]
- 13.Naoi Y, Miyoshi Y, Taguchi T et al (2007) Connexin26 expression is associated with lymphatic vessel invasion and poor prognosis in human breast cancer. Breast Cancer Res Treat 106:11–17. 10.1007/s10549-006-9465-8 [DOI] [PubMed] [Google Scholar]
- 14.Ezumi K, Yamamoto H, Murata K et al (2008) Aberrant expression of connexin 26 is associated with lung metastasis of colorectal cancer. Clin Cancer Res 14:677–684. 10.1158/1078-0432.Ccr-07-1184 [DOI] [PubMed] [Google Scholar]
- 15.Yang J, Qin G, Luo M et al (2015) Reciprocal positive regulation between Cx26 and PI3K/Akt pathway confers acquired gefitinib resistance in NSCLC cells via GJIC-independent induction of EMT. Cell Death Dis 6:e1829. 10.1038/cddis.2015.197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Campenhout R, Gomes AR, De Groof TWM et al (2021) Mechanisms underlying connexin hemichannel activation in disease. Int J Mol Sci. 10.3390/ijms22073503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu JI, Wang LH (2019) Emerging roles of gap junction proteins connexins in cancer metastasis, chemoresistance and clinical application. J Biomed Sci 26:8. 10.1186/s12929-019-0497-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Radman BA, Alhameed AMM, Shu G et al (2024) Cellular elasticity in cancer: a review of altered biomechanical features. J Mater Chem B 12:5299–5324. 10.1039/d4tb00328d [DOI] [PubMed] [Google Scholar]
- 19.Teleki I, Krenacs T, Szasz MA et al (2013) The potential prognostic value of connexin 26 and 46 expression in neoadjuvant-treated breast cancer. BMC Cancer 13:50. 10.1186/1471-2407-13-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldman MJ, Craft B, Hastie M et al (2020) Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol 38:675–678. 10.1038/s41587-020-0546-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hutter C, Zenklusen JC (2018) The cancer genome atlas: creating lasting value beyond its data. Cell 173:283–285. 10.1016/j.cell.2018.03.042 [DOI] [PubMed] [Google Scholar]
- 22.Wang Z, Jensen MA, Zenklusen JC (2016) A practical guide to the cancer genome atlas (TCGA). Methods Mol Biol 1418:111–141. 10.1007/978-1-4939-3578-9_6 [DOI] [PubMed] [Google Scholar]
- 23.Ma C, Li F, Gu Z et al (2023) A novel defined risk signature of cuproptosis-related long non-coding RNA for predicting prognosis, immune infiltration, and immunotherapy response in lung adenocarcinoma. Front Pharmacol 14:1146840. 10.3389/fphar.2023.1146840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yan T, Yang H, Meng Y et al (2023) Targeting copper death genotyping associated gene RARRES2 suppresses glioblastoma progression and macrophages infiltration. Cancer Cell Int 23:105. 10.1186/s12935-023-02950-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang Y, Yin D, Wu L (2022) Identification of cuproptosis-related subtypes and development of a prognostic signature in colorectal cancer. Sci Rep 12:17348. 10.1038/s41598-022-22300-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mayakonda A, Lin DC, Assenov Y et al (2018) Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res 28:1747–1756. 10.1101/gr.239244.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ritchie ME, Phipson B, Wu D et al (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucl Acids Res 43:e47. 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ashburner M, Ball CA, Blake JA et al (2000) Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat Genet 25:25–29. 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu G, Wang LG, Han Y et al (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16:284–287. 10.1089/omi.2011.0118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ogata H, Goto S, Sato K et al (1999) KEGG: kyoto encyclopedia of genes and genomes. Nucl Acids Res 27:29–34. 10.1093/nar/27.1.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hänzelmann S, Castelo R, Guinney J (2013) GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf 14:7. 10.1186/1471-2105-14-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pardoll DM (2012) The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12:252–264. 10.1038/nrc3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Geeleher P, Cox NJ, Huang RS (2014) Clinical drug response can be predicted using baseline gene expression levels and in vitro drug sensitivity in cell lines. Genome Biol 15:R47. 10.1186/gb-2014-15-3-r47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang W, Soares J, Greninger P et al (2013) Genomics of drug sensitivity in cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucl Acids Res 41:D955-961. 10.1093/nar/gks1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Geeleher P, Cox N, Huang RS (2014) pRRophetic: an R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS ONE 9:e107468. 10.1371/journal.pone.0107468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Asplund A, Edqvist PH, Schwenk JM et al (2012) Antibodies for profiling the human proteome-the human protein atlas as a resource for cancer research. Proteomics 12:2067–2077. 10.1002/pmic.201100504 [DOI] [PubMed] [Google Scholar]
- 37.Bi F, Chen Y, Yang Q (2020) Significance of tumor mutation burden combined with immune infiltrates in the progression and prognosis of ovarian cancer. Cancer Cell Int 20:373. 10.1186/s12935-020-01472-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reardon DA, Gokhale PC, Klein SR et al (2016) Glioblastoma eradication following immune checkpoint blockade in an orthotopic, immunocompetent model. Cancer Immunol Res 4:124–135. 10.1158/2326-6066.Cir-15-0151 [DOI] [PubMed] [Google Scholar]
- 39.Rotte A (2019) Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J Exp Clin Cancer Res 38:255. 10.1186/s13046-019-1259-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buccarelli M, D’Alessandris QG, Matarrese P et al (2021) Elesclomol-induced increase of mitochondrial reactive oxygen species impairs glioblastoma stem-like cell survival and tumor growth. J Exp Clin Cancer Res 40:228. 10.1186/s13046-021-02031-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao W, Huang Z, Duan J et al (2021) Elesclomol induces copper-dependent ferroptosis in colorectal cancer cells via degradation of ATP7A. Mol Oncol 15:3527–3544. 10.1002/1878-0261.13079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen Y, Li ZY, Zhou GQ et al (2021) An immune-related gene prognostic index for head and neck squamous cell carcinoma. Clin Cancer Res 27:330–341. 10.1158/1078-0432.Ccr-20-2166 [DOI] [PubMed] [Google Scholar]
- 43.Wu J, Li L, Zhang H et al (2021) A risk model developed based on tumor microenvironment predicts overall survival and associates with tumor immunity of patients with lung adenocarcinoma. Oncogene 40:4413–4424. 10.1038/s41388-021-01853-y [DOI] [PubMed] [Google Scholar]
- 44.Bray F, Laversanne M, Sung H et al (2024) Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 74:229–263. 10.3322/caac.21834 [DOI] [PubMed] [Google Scholar]
- 45.Battistini C, Cavallaro U (2023) Patient-derived in vitro models of ovarian cancer: powerful tools to explore the biology of the disease and develop personalized treatments. Cancers (Basel). 10.3390/cancers15020368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Zyl B, Tang D, Bowden NA (2018) Biomarkers of platinum resistance in ovarian cancer: what can we use to improve treatment. Endocr Relat Cancer 25:R303-r318. 10.1530/erc-17-0336 [DOI] [PubMed] [Google Scholar]
- 47.Radu MR, Prădatu A, Duică F et al (2021) Ovarian cancer: biomarkers and targeted therapy. Biomedicines. 10.3390/biomedicines9060693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Strasser A, Vaux DL (2020) Cell death in the origin and treatment of cancer. Mol Cell 78:1045–1054. 10.1016/j.molcel.2020.05.014 [DOI] [PubMed] [Google Scholar]
- 49.Zhang Y, Tang J, Jiang C et al (2025) Metabolic reprogramming in cancer and senescence. MedComm (2026) 6:e70055. 10.1002/mco2.70055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qi X, Li Q, Che X et al (2022) Application of regulatory cell death in cancer: based on targeted therapy and immunotherapy. Front Immunol 13:837293. 10.3389/fimmu.2022.837293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fouani L, Menezes SV, Paulson M et al (2017) Metals and metastasis: exploiting the role of metals in cancer metastasis to develop novel anti-metastatic agents. Pharmacol Res 115:275–287. 10.1016/j.phrs.2016.12.001 [DOI] [PubMed] [Google Scholar]
- 52.Shanbhag VC, Gudekar N, Jasmer K et al (2021) Copper metabolism as a unique vulnerability in cancer. Biochim Biophys Acta Mol Cell Res 1868:118893. 10.1016/j.bbamcr.2020.118893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yan C, Niu Y, Ma L et al (2022) System analysis based on the cuproptosis-related genes identifies LIPT1 as a novel therapy target for liver hepatocellular carcinoma. J Transl Med 20:452. 10.1186/s12967-022-03630-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang L, Cao Y, Guo W et al (2023) High expression of cuproptosis-related gene FDX1 in relation to good prognosis and immune cells infiltration in colon adenocarcinoma (COAD). J Cancer Res Clin Oncol 149:15–24. 10.1007/s00432-022-04382-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peng X, Zhao H, Ye L et al (2024) Biomarker identification and risk prediction model development for differentiated thyroid carcinoma lung metastasis based on primary lesion proteomics. Clin Cancer Res 30:3059–3072. 10.1158/1078-0432.Ccr-23-3806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xie J, Yang Y, Gao Y et al (2023) Cuproptosis: mechanisms and links with cancers. Mol Cancer 22:46. 10.1186/s12943-023-01732-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang H, Yang Z, He X et al (2023) Cuproptosis related gene PDHB is identified as a biomarker inversely associated with the progression of clear cell renal cell carcinoma. BMC Cancer 23:804. 10.1186/s12885-023-11324-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu H, Li X, Zhang C et al (2024) GJB2 promotes HCC progression by activating glycolysis through cytoplasmic translocation and generating a suppressive tumor microenvironment based on single cell RNA sequencing. Adv Sci (Weinh) 11:e2402115. 10.1002/advs.202402115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li S, Liu Y, Qiu G et al (2021) Long non-coding RNA CAR10 facilitates non-small cell lung cancer cell migration and invasion by modulating the miR-892a/GJB2 pathway. Cancer Manag Res 13:1967–1979. 10.2147/cmar.S287386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Castellana B, Escuin D, Peiró G et al (2012) ASPN and GJB2 are implicated in the mechanisms of invasion of ductal breast carcinomas. J Cancer 3:175–183. 10.7150/jca.4120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu X, Furuya T, Li D et al (2010) Connexin 26 expression correlates with less aggressive phenotype of intestinal type-gastric carcinomas. Int J Mol Med 25:709–716. 10.3892/ijmm_00000395 [DOI] [PubMed] [Google Scholar]
- 62.Ramus SJ, Gayther SA (2009) The contribution of BRCA1 and BRCA2 to ovarian cancer. Mol Oncol 3:138–150. 10.1016/j.molonc.2009.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Krivak TC, Tian C, Rose GS et al (2009) A gynecologic oncology group study of serum CA-125 levels in patients with stage III optimally debulked ovarian cancer treated with intraperitoneal compared to intravenous chemotherapy: an analysis of patients enrolled in GOG 172. Gynecol Oncol 115:81–85. 10.1016/j.ygyno.2009.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 The clinical characteristics of OC patients in the TCGA dataset. Table S2 The concrete clinical information for TCGA whole dataset OC patients. Table S3 The cuproptosis genes extracted from the previous research. Table S4 Sequences of primers. Table S5 Differential expression of CGs. Table S6 Correlation and regulation of DECGs and CRGs. Table S7 Differential expression of CRGs. Table S8 Univariate Cox analysis of identifying 9 CRGs associated with OS. Table S9 The detailed risk genes expression, risk score, and risk group. Table S10 The GSVA functional enrichment analysis. Table S11 GJB2-correlated genes.
Supplementary file 2 (ZIP 7234 kb)
Data Availability Statement
Data is provided within the manuscript or supplementary information files.