

Abstract
The US Food and Drug Administration (FDA) performs safety assessments throughout the life cycle of a drug. Postmarketing safety surveillance promotes the identification of adverse events not known at the time of approval. Adverse event reports, also called individual case safety reports (ICSRs), submitted to FDA, are collected and stored in the FDA Adverse Event Reporting System (FAERS). ICSRs stored in FAERS may be reviewed—along with multiple other data sources—to detect potential safety signals and to perform a thorough evaluation to determine if a causal association exists between a drug and an adverse event. Although FAERS is a powerful tool for drug safety surveillance and assessment, understanding the content, application, and proper interpretation of the data contained in FAERS is necessary to reach scientifically and medically accurate conclusions and contextualize findings. This article aims to highlight considerations and explain fundamental concepts of FAERS to promote accurate analyses and appropriate interpretation of the data available.
For the US Food and Drug Administration (FDA) to approve a drug or biological product for human use, an application holder must provide evidence demonstrating that the drug or biological product (henceforth collectively referred to as a drug unless specified otherwise) is both safe and effective under the conditions prescribed, recommended, or suggested in the proposed labeling.1 Although the information obtained in the drug development program must demonstrate a favorable benefit–risk profile for its approved indication, the full spectrum of adverse drug reactions (ADRs) cannot be captured in preapproval safety data.2 Once a drug is approved by the FDA, however, more information regarding its safety becomes available as it is used in a broader population than that studied in preapproval clinical trials.2
New information about a drug’s safety can be obtained from a variety of data sources, including, but not limited to, adverse event (AE) reports, medication error reports, epidemiologic studies, drug utilization studies, postapproval clinical trials, and registries.2 An AE report, also called an individual case safety report (ICSR), is “a description of an adverse drug experience related to an individual patient or subject.”3-6 “Adverse drug experience” is the term used in the Code of Federal Regulations (CFR) 3-6 and is used interchangeably with “adverse event” throughout this article. Although ICSRs can also concern medication errors and product quality problems, for the purposes of this article, we are specifically referring to a drug and AE report when using the term ICSR. Members of the public (e.g., consumers, healthcare professionals, patients, caregivers) may submit ICSRs directly to FDA. Alternatively, an application holder and others, as specified in the CFR, will submit an ICSR to FDA following communication with a patient, consumer, healthcare professional, or anyone else with knowledge of an AE, according to regulatory requirements.3-6 FDA stores ICSRs in specialized AE databases used to monitor the safety of FDA-approved products, which includes the FDA Adverse Event Reporting System (FAERS) for drugs.7 In addition to the AE reporting databases managed by the FDA, international organizations also maintain databases used to report AEs related to devices, drugs, vaccines, and dietary supplements.7-14 While this article focuses specifically on ICSRs contained in FAERS, the principles discussed may apply to other AE databases.15 Regardless of the database used, a detailed understanding of its strengths and limitations is required to determine how that database should be used.
The objective of this article is to describe the content, application, and proper interpretation of the data contained in FAERS and its publicly available counterparts, including the FAERS Public Dashboard. The target audience includes academic researchers, policy makers, application holders, and others who are interested in studying the safety of drugs and who may access and analyze FAERS data, including data obtained from the FAERS Public Dashboard, as part of those efforts.
THE SCIENCE OF PHARMACOVIGILANCE
To understand how data from FAERS are used, it is first necessary to examine the foundational principles of postmarketing drug safety surveillance. The identification and analysis of AEs is part of a field known as pharmacovigilance, which the World Health Organization (WHO) formally defines as “the science and activities relating to the detection, assessment, understanding and prevention of adverse effects or any other medicine or vaccine related problem.”16 In the United States, FDA performs postmarketing pharmacovigilance to monitor drug safety and takes regulatory actions when needed to ensure safe and effective use of drugs during their life cycle.17
In the medical literature, two common terms, which are distinct but are often erroneously used interchangeably, are AE and ADR.2,18 An AE is “any untoward medical occurrence associated with the use of a drug product in humans, whether or not it is considered related to the drug product.”17,19 An AE can occur during the use of a drug in professional practice; from overdose of a drug, whether accidental or intentional; from abuse of the drug; withdrawal of the drug; and from any failure of expected pharmacological action.3-5 Conversely, an ADR represents a subset of AEs in which there is reason to suspect a causal relationship between the drug and the AE.20 Formally, an ADR is defined as “an undesirable effect, reasonably associated with use of a drug, that may occur as part of the pharmacological action of the drug or may be unpredictable in its occurrence.”20 Although the FDA does not generally require that a causal association be established for ICSRs to be submitted and entered into FAERS, establishing a causal relationship is an important step in the safety signal evaluation process.
Two other important regulatory terms are “serious” and “unexpected” AEs. The CFR defines a serious AE as “any adverse drug experience occurring at any dose that results in any of the following outcomes: death, a life-threatening adverse drug experience, inpatient hospitalization or prolongation of existing hospitalization, a persistent or significant disability/incapacity, or a congenital anomaly/birth defect.”3-5 The CFR definition of a serious AE also includes “[i]mportant medical events that, while not resulting in one of these outcomes, may be considered serious based upon appropriate medical judgment, may jeopardize the patient, or may require medical or surgical intervention to prevent one of the outcomes listed in this definition.”3-5 The CFR defines an unexpected AE as “[a]ny adverse drug experience that is not listed in the current labeling for the drug product. This includes events that may be symptomatically and pathophysiologically related to an event listed in the labeling but differ from the event because of greater severity or specificity.”3-5 Additional terms used in AE reporting are provided in Table 1.
Table 1.
Basic adverse event reporting terms
| Term | Definition |
|---|---|
| Adverse drug experience | An adverse drug experience refers to “any adverse event associated with the use of a drug in humans, whether or not considered drug related, including the following: an adverse event occurring in the course of the use of a drug product in professional practice; an adverse event occurring from drug overdose whether accidental or intentional; an adverse event occurring from drug abuse; an adverse event occurring from drug withdrawal; and any failure of expected pharmacological action.”3,4 |
| Adverse experience | An adverse experience refers to “any adverse event associated with the use of a biological product in humans, whether or not considered product related, including the following: an adverse event occurring in the course of the use of a biological product in professional practice; an adverse event occurring from overdose of the product whether accidental or intentional; an adverse event occurring from abuse of the product; an adverse event occurring from withdrawal of the product; and any failure of expected pharmacological action.”5 |
| Adverse drug reaction/Adverse reaction | An adverse drug reaction refers to “an undesirable effect, reasonably associated with use of a drug, that may occur as part of the pharmacological action of the drug or may be unpredictable in its occurrence. This definition does not include all adverse events observed during the use of a drug, only those adverse events for which there is some basis to believe there is a causal relationship between the drug and the occurrence of the adverse event.”20 |
| Adverse event | An adverse event is “any untoward medical occurrence associated with the use of a drug product in humans, whether or not it is considered related to the drug product.”17,19 An adverse event can occur during the use of a drug in professional practice; from overdose of a drug, whether accidental or intentional; from abuse of the drug; withdrawal of the drug; and from any failure of expected pharmacological action.3-5 |
| Causality assessment | The evaluation of the likelihood that a medicine was the causative agent of an observed adverse event in a specific individual. Causality assessment is usually made according to established algorithms.21 |
| Data mining | An approach that uses statistical or mathematical tools to discover patterns of drug-AEs pairs or other associations in large databases17 that may not otherwise be apparent to the observer. The data mining technique most commonly used for surveillance of FAERS relies on disproportionality analysis, which identifies drug-AE combinations that occur more frequently with one drug than with other drugs in the database.22 |
| Individual case safety report (ICSR) | A description of an adverse event related to an individual patient or subject.3-5 These have also been referred to as adverse drug reaction reports or adverse event reports. |
| Medical Dictionary for Regulatory Activities (MedDRA) | MedDRA is a clinically validated medical terminology for regulatory authorities and the regulated pharmaceutical industry for utilization in data entry, retrieval, evaluation, and presentation, in both pre- and post-marketing phases of the regulatory process. It covers diseases, diagnoses, signs, symptoms, therapeutic indications, investigation names, and qualitative results, as well as medical and surgical procedures, medical, social, and family history. MedDRA is one of the standards required for the electronic transmission of ICSRs. Recommendations on the use of MedDRA are set out in an International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use endorsed ‘Points to consider’ document, as updated annually. MedDRA terminology for adverse event reporting is used globally by the biopharmaceutical industry and regulators to promote consistent reporting and data analysis.23 |
| Pharmacovigilance | The science and activities relating to the detection, assessment, understanding, and prevention of adverse effects or any other possible drug-related problems.16 |
| Proportional reporting ratio | The proportion of reports for an event that involves a particular drug compared to the proportion of reports of this event for all drugs in a spontaneous report database. This is expressed as a ratio and reflects the observed/expected values for that event in the database.21 |
| Reporting odds ratio | The odds of finding an adverse event term among all case reports that mention a particular drug divided by the odds of finding the same adverse event term among all other case reports in the spontaneous report database that do not mention this drug.21 |
| Serious adverse event | Any adverse drug experience occurring at any dose that results in any of the following outcomes: Death, a life-threatening adverse drug experience, inpatient hospitalization or prolongation of existing hospitalization, a persistent or significant disability/incapacity, or a congenital anomaly/birth defect. Important medical events that may not result in death, be life-threatening, or require hospitalization may be considered a serious adverse drug experience when, based upon appropriate medical judgment, they may jeopardize the patient or subject and may require medical or surgical intervention to prevent one of the outcomes listed in this definition. Examples of such medical events include allergic bronchospasm requiring intensive treatment in an emergency room or at home, blood dyscrasias or convulsions that do not result in inpatient hospitalization or the development of drug dependency or drug abuse.3-5 |
| Signal | FDA defines a signal as “information that arises from one or multiple sources (including observations and experiments), that suggests a new potentially causal association, or a new aspect of a known association, between an intervention and an event or set of related events, either adverse or beneficial, that is judged to be of sufficient likelihood to justify further action to verify.”17,21 |
| Solicited reports | Reports derived from organized data collection systems, which include clinical trials, registries, post-approval named patient use programs, other patient support and disease management programs, surveys of patients or healthcare professionals, or information gathering on efficacy or patient compliance.2 |
| Spontaneous report | An unsolicited communication by healthcare professionals or consumers to a company, regulatory authority, or other organization that describes one or more suspected adverse drug reactions in a patient who was given one or more medicinal products and which does not derive from a study or any organized data collection scheme.2,21 |
| Stimulated reporting | A form of spontaneous reporting that is prompted by certain situations, including, but not limited to, a notification by a “Dear Health Care Provider” letter; a publication in the press; or questioning of healthcare professionals by company representatives.2 |
As noted above, a fundamental concept in pharmacovigilance is that of a safety signal. There are multiple existing definitions of the term “safety signal” or “signal” in guidance documents.16,21,24-26 In conducting postmarketing safety surveillance, FDA uses a modified version of the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) definition for “signal” and defines a safety signal as “information that arises from one or multiple sources (including observations and experiments) that suggests a new potentially causal association, or a new aspect of a known association, between an intervention and an event or set of related events, either adverse or beneficial, that is judged to be of sufficient likelihood to justify further action to verify.”17,21 For postmarketing pharmacovigilance, FDA focuses on adverse, rather than beneficial, effects.
The importance of postmarketing pharmacovigilance to supplement premarket pharmacovigilance
Identification of ADRs associated with drugs is an important aspect of premarket drug development and is a standard consideration in clinical development programs. The median number of patients evaluated for safety in clinical development programs for new molecular entities and original biological products is over 1000, which is sufficient to capture common ADRs associated with the drug.27 Randomized, double-blind clinical trials also enable researchers to distinguish AEs that may be manifestations of the underlying disease or condition being treated from true ADRs.2 However, premarket clinical trials, by their nature, cannot identify all ADRs associated with a drug.2 First, clinical trials are conducted for a finite time period, whereas postmarketing safety surveillance occurs throughout the life cycle of a drug.17 Second, because clinical trials have specific inclusion and exclusion criteria, trial participants may have fewer comorbidities and use fewer concomitant medications than the broader population that receives the drug once it is marketed.28 Third, certain populations, such as children and older adults, are typically not included in clinical trials, although they may receive the drug in clinical practice.17,29-32 Fourth, once marketed, the drug may be used for unlabeled conditions or at unlabeled dosages.17 Finally, while the sample size of patients in clinical trials may allow detection of common AEs, it is generally insufficient for capturing AEs with a rare or idiosyncratic presentation such as drug-induced liver injury33,34 and Stevens-Johnson Syndrome or toxic epidermal necrolysis.35
Prescription drug labeling must contain “a summary of the essential scientific information needed for the safe and effective use of the drug.”36 Several sections of labeling contain safety-related information, such as information on ADRs, contraindications, and drug–drug interactions. As the use of the drug becomes more widespread, postmarketing safety surveillance may identify potential new safety signals, which if determined to be ADRs after careful evaluation, are then added to the product’s labeling. These updates occur throughout the product’s life cycle. For example, a study of 278 new molecular entities followed for up to 13 years after approval found that 70% had at least one new ADR added to the label.37 Similarly, a study of 61 new therapeutic biological products followed for up to 16 years after approval found that 89% had at least one ADR or other safety-related action during that period.38
Review of ICSRs has been the foundation of the FDA’s pharmacovigilance system for over 50 years. ICSRs form the basis for more than half of the postmarketing safety-related labeling changes in the United States.39,40 However, there are limitations in postmarketing AE reporting data that must be acknowledged. First, because persons at the point of care, such as healthcare professionals, patients, and consumers, are not required to report AEs to either the drug’s application holder or to FDA, FAERS data do not contain all instances of a particular AE.7 Thus, neither the prevalence nor incidence of an AE can be calculated from FAERS data. Second, the quality of information varies. For example, ICSRs may be missing contextual information that is necessary to rule out exacerbation of underlying disease or to assess the causality between a drug-AE pair, such as temporality between drug exposure and AE onset, comorbidities and concomitant medications, diagnostic data, clinical course of the AE, and the outcome of the AE.2 Third, duplicate ICSR submissions inflate the number of reported AEs, thereby affecting subsequent analyses.2 In a study characterizing FDA pharmacovigilance reviews performed in 2016 (n = 69), the number of duplicate ICSRs excluded ranged from 0 to 190.41 Over half of the reviews had at least 10% of ICSRs identified as duplicates and nearly one-quarter had more than 25% identified as duplicates. In the same study, the number of ICSRs retrieved ranged from 4 to 1376, whereas the overall number of ICSRs excluded ranged from 1 to 1057.41
The historical evolution of AE reporting in the United States
Analysis of ICSRs is a core activity of pharmacovigilance.2 The utility of AE reporting was described as early as 1893 in the medical literature when physicians sought to investigate reports of death after chloroform administration.42 As seen in Figure 1, the evolution of modern pharmacovigilance has been shaped by pivotal events and regulations.
Figure 1.

Important time points in the evolution of modern pharmacovigilance. AMA, American Medical Association; FAERS, FDA Adverse Event Reporting System; FD&C Act, Federal Food, Drug, and Cosmetic Act; ICH, International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use; ICSR, Individual Case Safety Report; SRS, Spontaneous Reporting System.3,4,42-64
In 1952, after receiving a concerning report, the FDA requested the National Research Council to evaluate the available data regarding the risk of blood dyscrasias with the use of chloramphenicol; the ad hoc committee of internists and hematologists ultimately identified a causal association between chloramphenicol use and blood dyscrasias leading to a new warning in drug labeling.48 Subsequently, in 1954, the American Medical Association (AMA) started the “AMA Registry on Blood Dyscrasias” in response to dyscrasia cases with chlorpromazine and to evaluate “blood dyscrasias caused by toxic agents”, including aplastic anemia associated with chloramphenicol.49 In 1955, “to test the practicability of a systematic method of drug mortality and morbidity sampling,” the FDA started a pilot program for the “voluntary reporting of adverse reactions to drugs” in conjunction with five hospitals and in coordination with professional associations, including the AMA, the American Association of Medical Librarians, and the American Society of Hospital Pharmacists.50,51 By 1957, the program had expanded to include 11 hospitals, with plans for a national center for the collection and dissemination of the information reported.51 In 1960, the Adverse Reaction Reporting Program (also referred to as the Hospital Reporting Program), a result of the pilot program, was started at FDA with the intention to “make the scientific community aware of the significant adverse effects of drugs in current use” and to serve as an “early warning” surveillance system for healthcare professionals.51-57 At the time, reports were primarily sent to FDA from federal hospitals, including military facilities, the Veterans Administration, and the Public Health Service. Other sources of reports included the “National Library of Medicine [,] the Pharmaceutical Industry, Food and Drug field investigators, and State Departments of Health.”56
Both the AMA and FDA reporting programs expanded to include all ADRs to drugs by the early 1960s.53 Whereas the AMA focused on physicians and smaller hospitals, the FDA focused on larger hospitals, government hospitals, and university-affiliated hospitals.50 Meanwhile, an amendment to the Federal Food, Drug, and Cosmetic Act in 1962 contained a requirement that forms the basis for regulations that require application holders to report AEs to the FDA.54,59,60 In 1967, the FDA started to create a system for organizing the ICSRs it received, and by 1968, it started entering data from what was then FDA Form 1639 Drug Experience Reports into the Spontaneous Reporting System (SRS).54 The AMA and FDA programs coexisted until 1970 when the AMA discontinued its program due to underreporting and duplication of efforts with the FDA program.50,53 The earliest reports in FAERS date back to 1968. ICSRs were initially coded with event terms using the Coding Symbols for a Thesaurus of Adverse Reaction Terms (COSTART) that was developed by FDA.65 COSTART was used in SRS until November 1997 when the FDA replaced SRS with the Adverse Event Reporting System (AERS, the precursor of FAERS) and began using Medical Dictionary for Regulatory Activities (MedDRA) terminology23,65 (see ‘Structured data in FAERS including MedDRA coding and the FAERS product dictionary‘).
In 1993, FDA launched MEDWatch: The FDA Medical Products Reporting Program, through which health professionals and others could use a single, one-page form to report AEs and product quality problems related to drugs and medical devices.61 Today, MedWatch (previously referred to as MEDWatch) continues to accept ICSRs for drugs, which are subsequently stored in FAERS, as well as other FDA-regulated products (e.g., dietary supplements, medical foods, infant formula) which are stored in databases other than FAERS. The submission of ICSRs through the MedWatch platform on FDA Form 3500 (for health professionals) or FDA Form 3500B (for consumers/patients) online via the FDA website or on paper via facsimile transmission or the mail remains a cornerstone of pharmacovigilance in the United States.2,66,67
THE STRUCTURE AND CONTENT OF FAERS
The FAERS database (previously known as AERS) contains reports of AEs, medication errors, and product quality problems for human drugs submitted to the FDA and supports the FDA’s postmarketing safety surveillance program. As of December 31, 2023, FAERS includes more than 28 million reports. After accounting for follow-ups and duplicates, it represents over 20 million unique reports.68
FAERS reports originate from two main sources: application holders in the pharmaceutical industry and members of the public.2 Regardless of whether reports are submitted to the FDA by application holders or directly by the public, the information in nearly all reports originates at the point of patient care.2
In the first pathway, which represents approximately 95% of all ICSR submissions, application holders are required to create and submit ICSRs to the FDA for any AEs of which they become aware. These requirements also apply to any manufacturer, packer, or distributor whose name appears on the label of an approved drug. Application holders and others with regulatory reporting requirements must submit ICSRs that meet the regulatory definitions of both serious and unexpected, whether foreign or domestic, to the FDA within 15 calendar days of receipt of the information. These reports are known as 15-day “Alert” or “expedited” reports.69 Application holders are required to promptly investigate all AEs associated with postmarketing 15-day “Alert reports” and submit follow-up reports within 15 calendar days of receipt of new information or as requested by the FDA.3-5 All other ICSRs, regardless of seriousness or expectedness, must be reported on a quarterly basis for the first 3 years after the US approval date and on an annual basis thereafter.3-5 These reports are commonly referred to as non-expedited reports. Reports of AEs derived from the scientific literature must be reported if the AE is both serious and unexpected. Finally, 15-day “Alert reports” of AEs derived from postmarketing studies must be reported only if the application holder concludes that there is a reasonable possibility that the drug caused the adverse experience.3-5
For the FDA to enter an ICSR into FAERS, a valid ICSR should include at least the following: one identifiable reporter; one identifiable patient; one AE, ADR, or outcome; and one suspect or interacting drug.64 If any of these four pieces of information is missing, the report is considered incomplete, and FDA does not enter it into FAERS; however, application holders are expected to exercise due diligence to collect the missing data elements.70 Additionally, FDA regulations require that application holders include extensive information, when available, about the patient, the AE, the suspected medical products involved, the person reporting the AE, and the drug application holder.3-6 FDA regulations allow application holders to request the FDA to waive, among other things, any of the AE reporting requirements.71 For drugs other than new molecular entities approved for 3 years or less, the FDA generally grants requests to waive the requirement that application holders submit reports of AEs that are both not serious and labeled, provided that the application holder maintains records of these reports, includes them in summary tabulations in their periodic safety reports (PSRs), and provides them to FDA within 5 days of FDA’s request.71-73
ICSRs are generally classified as originating from unsolicited or solicited sources. Reports that come from individuals (e.g., healthcare professionals, consumers and patients, family members of patients), the biomedical literature, and the internet (e.g., social media posts) are generally described as unsolicited reports.70 The most widely described type of ICSR is a spontaneous AE report, which is defined as “an unsolicited communication by healthcare professionals or consumers to a company, regulatory authority[,] or other organization (e.g., [WHO], regional centers, poison control center) that describes one or more ADRs in a patient who was given one or more medicinal products and that does not derive from a study or any organized data collection scheme.”70 Notably, reporters of AEs do not necessarily contact application holders with the specific intent of reporting an AE; however, the application holder must record, review, and submit to the FDA any mention of an AE according to regulations, regardless of whether there was a causal association to the drug.70
ICSRs that originate from “organized data collection systems, which include clinical trials, post-approval named patient use programs, other patient support and disease management programs, surveys of patients or healthcare providers, or information gathering on efficacy or patient compliance” are considered solicited reports and are not considered spontaneous.70 Different AE databases contain different proportions of unsolicited (e.g., spontaneous) and solicited ICSRs. Knowledge of the origin of reports in an AE database can be an important factor affecting the number and temporal pattern of reports captured by the database.2,74,75
In addition to ICSRs that come from industry, FDA also accepts reports directly from the public. These direct reports are unsolicited and differ from solicited reports generated by organized data collection systems. These direct reports represent approximately 5% of all ICSR submissions in FAERS. Members of the public are not required to report AEs to either an application holder or to the FDA. However, any member of the public can report an AE to the FDA via the MedWatch program. Until 2012, all direct reports were submitted using MedWatch Form 3500, which is designed primarily for healthcare professionals. In 2013, the FDA introduced MedWatch Form 3500B, a patient-friendly form that uses a question-and-answer format, which was found to contribute positively to both reporting quality and quantity of consumer reports.76 In 2019, the FDA introduced a Spanish-language version of MedWatch Form 3500B.
Application holders are also required to submit PSRs, which are pharmacovigilance documents that the application holder prepares summarizing the safety information concerning the drug. These documents accompany the submission of the non-expedited ICSRs covered in the report. In the United States, PSR formats specified in regulations include periodic adverse drug experience reports for drugs3 and periodic adverse experience reports for biological products.5 Through a waiver granted by FDA, application holders may submit PSRs in one of two formats specified by ICH, either as a periodic safety update report or as a periodic benefit–risk evaluation report.17
The informatic structure of the FAERS database adheres to the international safety reporting guidance issued by the ICH E2B (R3), Electronic Transmission of Individual Case Safety Reports (ICSRs).77 Application holders are required to submit AE reports to the FDA electronically3,5 as ICSRs either by database-to-database transmission via the Electronic Submission Gateway following the ICH E2B standard or through the FDA’s Safety Reporting Portal. Members of the public can submit ICSRs using paper copies of the MedWatch form through the United States Postal Service or facsimile transmission or electronically via the MedWatch website. FDA converts AE reports submitted through the Safety Reporting Portal or MedWatch into the ICH E2B format to maintain standardization and compliance with international guidelines within the FAERS database.
Structured data in FAERS including MedDRA coding and the FAERS product dictionary
The FAERS database includes both structured and unstructured data. Structured data include the data elements that provide information to recognize at least one identifiable reporter; one identifiable, though ideally anonymized, patient; one AE, ADR, or outcome; and one suspect or interacting drug.64 As previously mentioned, these elements represent the minimal information required for the successful submission of an ICSR to FAERS; consequently, these four data fields are always populated. Examples of additional structured data include the initial receipt date of the AE report and concomitant suspect drugs.77
To incorporate AEs into FAERS in a structured manner, the AE terms in the report are coded using MedDRA terms to provide a uniform vocabulary that enables efficient searching and analysis of the data. MedDRA contains highly specific, standardized hierarchical medical terminology.23,78 The ICH developed MedDRA in the late 1990s to facilitate the sharing of regulatory information internationally for drugs used by humans.23 MedDRA terminology initially incorporated terms from the WHO adverse reaction terminology (WHO-ART), COSTART, and the International Classification of Diseases (ICD).78 To support global harmonization, MedDRA is available in multiple languages.23 MedDRA terminology is updated twice a year and is maintained by the MedDRA Maintenance and Support Services Organization.23
MedDRA is distinct from the ICD, a global standard classification for reporting diseases and health conditions that is developed and maintained by the WHO. Although these two coding systems are different, ICH and WHO have launched standardized mapping to increase interoperability between MedDRA and ICD codes.79 Application holders perform MedDRA coding for the reports that they submit to the FDA, while the FDA performs MedDRA coding for reports received directly from the public via MedWatch.
Suspect products are coded to valid trade names or active ingredients listed in the FAERS Product Dictionary, which the FDA maintains internally. The FAERS Product Dictionary is based on the information in the Structured Product Labeling (SPL), a Health Level Seven standard for the exchange of product information for all marketed US drugs. Product ingredients must be included in the FDA’s Global Substance Registration System, which generates a Unique Ingredient Identifier that is linked to a SPL. The SPL is a data file that is uploaded to an internal database. Application holders should populate the ICSR suspect product(s) with the Proprietary Medicinal Product Name and its active ingredient as per the SPL.80 Foreign products are usually submitted utilizing the WHO’s drug dictionary terms and coded internally to corresponding active ingredient terms in the FAERS Product Dictionary.
Unstructured data and free-text narratives: Quality affects data usefulness
Unstructured data can include free-text narratives, text-based laboratory values, descriptions of imaging studies or other diagnostic tests, past medical history, and other information. In addition, full clinical records, diagnostic images, and other documents can be attached to the ICSR.80
In the free-text narrative section, the reporter can provide the details of the AE as it occurred or was observed. The purpose of the free-text narrative is to provide the essential information needed to contextualize a drug exposure and its potential association with an AE. In general, it is preferred that data elements for structured fields are taken from narrative descriptions of the drug-AE relationship, instead of generating a narrative from the structured data. To that end, FDA has identified the most useful elements of an ICSR:81
Description of the AE(s) or disease experience, including time to onset of signs or symptoms;
Suspected and concomitant product therapy details (i.e., dose, lot number, schedule, dates, duration), including over-the-counter medications, dietary supplements, and recently discontinued medications;
Patient characteristics, including demographic information (e.g., age, race, sex), baseline medical conditions prior to product therapy, comorbid conditions, use of concomitant medications, relevant family history of disease, and presence of other risk factors;
Documentation of the diagnosis of the events, including methods used to make the diagnosis;
Clinical course of the event and patient outcomes (e.g., hospitalization or death);
Relevant therapeutic measures and laboratory data at baseline, during therapy, and subsequent to therapy, including blood levels, as appropriate;
Information about response to drug dechallenge and rechallenge; and
Any other relevant information (e.g., other details relating to the event or information on benefits received by the patient, if important to the assessment of the event).
Some information from the unstructured free-text narrative is abstracted and placed into structured data to make it searchable. For example, AEs are abstracted from the narrative, coded according to MedDRA, and placed into structured data fields. Application holders are responsible for abstracting AE terms and coding them according to MedDRA prior to submission to the FDA. For reports that the public submits directly to the FDA, FDA staff abstract and code AE terms. In some cases, information that should appear in a structured field, such as age, sex, weight, ethnicity, or race, may appear only in the narrative. One study found that 37.2% of ICSRs were missing age in the structured data, although computer-assisted review of the unstructured narrative data using natural language processing (NLP) was able to identify age and reduced the missingness level to 27%.82
The narrative section can vary from one word to several sentences to multiple paragraphs.83 Very short narratives are unlikely to contain sufficient details for a meaningful review. Conversely, long narratives may only be meaningful if the report contains relevant elements related to the drug and AE, such as detailed past medical history, concomitant medications, and laboratory values.
THE FAERS PUBLIC DASHBOARD AND OTHER PUBLICLY AVAILABLE FAERS DATA: BACKGROUND AND APPROPRIATE USES
The FAERS Public Dashboard was launched on September 28, 2017, to increase public access to and transparency of data in the FAERS database as far back as 1968.68,84 The FAERS Public Dashboard is a publicly available version of the FAERS database that is updated quarterly and contains several structured data fields for ICSRs that have been processed and are ready for evaluation. There is substantial public interest in data from AE databases such as FAERS. In 2023, for example, 74,700 users accessed the FAERS Public Dashboard.85
ICSR narratives and certain structured data elements are not included in the FAERS Public Dashboard to protect personally identifiable information about the patient and reporter, as well as the patient’s protected health information.86 The FAERS Public Dashboard allows the public to query a subset of structured FAERS data fields using their web browser. Queries are generally based on a specific “Product” or set of products based on the drug trade name or generic name. Alternatively, queries may include a specific “Reaction Term” or set of reactions. The FAERS Public Dashboard search function is limited to five product terms or five reaction terms (i.e., MedDRA Preferred Terms). The FAERS Public Dashboard provides the literature references for ICSRs for which the application holder has included the literature article as an attachment and reference in the ICSR.
The FAERS Public Dashboard is updated quarterly; thus, it does not represent the most current data included in the internal FAERS database, which is updated daily. Previously, FAERS data were publicly available only in the form of quarterly files, known as FAERS Quarterly Data Extract (QDE) files, provided in American Standard Code for Information Interchange or Extensible Markup Language file formats.87 Use of these files requires knowledge of relational databases and use of analytic tools.87 Launched in 2014, openFDA also provides FAERS QDE files dating from the first quarter of 2004 in the JavaScript Object Notation file format.88 These files may be appropriate for more robust analyses since the user accesses a data file and, unlike with data from the FAERS Public Dashboard, is not limited to a maximum number of products or reaction terms. However, like the FAERS Public Dashboard, the FAERS QDE files do not contain ICSR narratives and certain structured data fields to protect patient and reporter privacy. The FAERS QDE Files and FAERS Public Dashboard contain almost the exact same limited structured data fields since data in the public dashboard are derived from the same files.
Because the FAERS Public Dashboard and FAERS QDE files do not include all structured fields nor the free-text narrative portion of the ICSR, many important clinical details are missing. As a result, these data cannot be used for causality assessments or clinically-related conclusions. The FAERS Public Dashboard is subject to the same limitations (e.g., the inability to calculate incidence rates) associated with ICSRs as the FAERS database. The FAERS Public Dashboard data may be used, however, to generate hypotheses regarding potential safety signals that may need further evaluation. Additionally, data from the FAERS Public Dashboard can be used to describe AE reporting numbers in general, though the presence of duplicate ICSRs allows only for a description of trends and estimates. If an ICSR of interest is identified, individual ICSRs can be requested by the FAERS “Case ID” which is found in the “Listing of Cases” for a suspect drug, through the Freedom of Information Act (FOIA).86 ICSRs released through the FOIA process include the narrative, which are redacted to remove information that may reveal the patient’s identity or other protected information.
THE APPLICATIONS OF FAERS DATA IN PHARMACOVIGILANCE
The FAERS database is routinely used both for surveillance to identify potential new safety signals and for evaluation of an identified safety signal. As shown in Figure 2, FAERS data are one of multiple data sources used by the FDA to establish evidence concerning drug risks and to support regulatory action(s). Safety surveillance includes the screening of FAERS, the biomedical literature, and other information sources to identify potential safety signals. In addition to the information provided, Table 2 describes misconceptions about FAERS data and its use in drug safety analyses.
Figure 2.
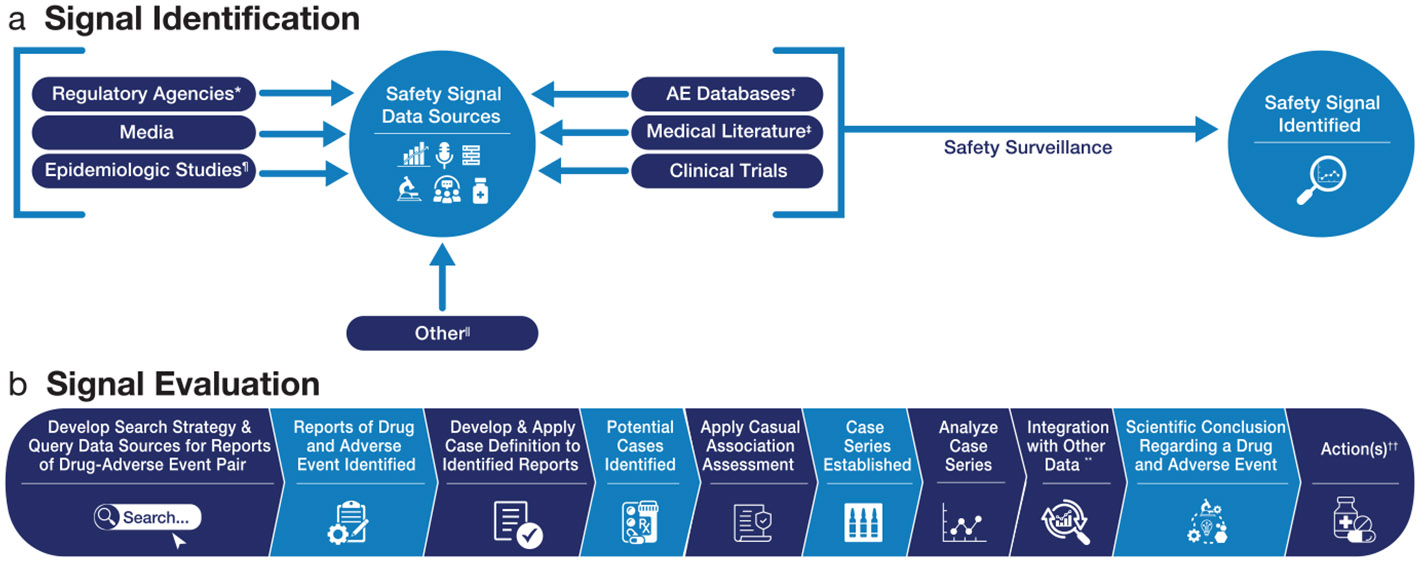
Process map: Safety signal identification and evaluation using postmarketing individual case safety reports.2,17 *For example, notification of a safety signal identified by other drug regulatory agencies. †May contain solicited ICSRs (e.g., from an organized data collection system, such as a registry); unsolicited, non-spontaneous ICSRs (e.g., literature, if the ICSR is derived from a study); unsolicited, spontaneous ICSRs (e.g., from consumers, healthcare professionals, or literature, if derived from a healthcare encounter). Tools such as disproportionality analysis/data mining may be applied in AE databases for safety surveillance and signal detection. ‡Case reports or case series of a drug(s) and adverse event(s). ¶May include studies using electronic healthcare records or in the Sentinel System. ∥For example, poison control center databases, emergency room databases, toxicology databases, and clinical pharmacology studies. **May include other information such as drug utilization, reporting ratios, epidemiologic studies, and clinical pharmacology data. ††For example, require an update to the drug labeling, issue a safety communication, and request additional postmarketing studies. AE, adverse event; ICSR, individual case safety report.
Table 2.
Myth versus fact regarding postmarketing adverse event reporting
| Myth | Fact |
|---|---|
| The MedDRA-coded Preferred Term is a confirmed adverse event | For FDA to enter an ICSR into FAERS, the minimum information for a valid ICSR should include at least the following: one identifiable reporter; one identifiable patient; one AE, ADR, or outcome; and one suspect or interacting drug64; thus, a report may be accepted that does not contain all pertinent details relating to an adverse event. Information submitted to FAERS by reporters is not verified for completeness nor medically confirmed. In addition, the report does not represent an admission from the reporter that the drug caused or contributed to the AE. Reports reflect only the reporter’s observations and opinions and may be incomplete or lack relevant details.89 |
| The MedDRA-coded Preferred Term reported with a drug represents a causal association | AE reports submitted to FAERS are not required to have a causal association between a drug and an AE to be proven nor are the submitted reports reviewed by HCPs for causal association prior to inclusion into the database. The AE reports contain only the reporter’s opinion and observations and may not provide sufficient detail to assess causality. The AE described may be related to other factors, such as underlying disease, another treatment (e.g., other drug, supplement), or other reason.89 |
| FAERS is equally useful for obtaining information about all AEs | While FAERS includes all marketed drug products, uses, and patient populations, it is most useful for AEs that have a rare occurrence and a strong temporal relationship between initial drug exposure and AE.2 |
| The number of cumulative adverse event reports is an incidence | FAERS contains ICSRs from multiple reporting sources, predominantly from application holders and others with regulatory reporting requirements but also includes reports received directly from healthcare professionals and consumers.17 Because of this, duplicate ICSRs occur and are included in crude counts when searching the database for ICSRs containing specific drug-AE combinations. Additionally, the true incidence of AEs may be higher because ADRs are often underreported; one study including data from 12 countries estimated underreporting to adverse event reporting systems of up to 90%.90 Alternatively, stimulated reporting, where publicity about a new important AE/ADR leads to a sudden increase in reporting with a decrease shortly after, can occur.2 |
| Data mining results utilizing MedDRA Preferred Terms and reported drugs represent causal associations | Data mining is a method that generates hypotheses of potential AE-drug combinations based on mathematical calculations to identify patterns or relationships within a large dataset. In FAERS, data mining includes measures of disproportionality.91 Lack of disproportionality does not represent lack of a causal association, absence of a safety signal, or negate a signal detected by other methods.17 |
| Reports coded with a serious outcome of death represent death due to the reported adverse events and drugs | Regulatory definitions for serious outcomes are at the report level and not at the adverse eventlevel for ICSRs submitted in accordance with the ICH E2B(R2) standard.92 As with other adverse events, the report of a death does not necessarily indicate that the drug, or any drug-related adverse event, was the cause of the death. A careful review of the entire ICSR, including the narrative, is necessary to determine whether the drug was involved in the death. The ICH E2B (R3) guidance requires that ICSRs are coded for serious outcomes at the adverse event level. Application holders have until April 1, 2026, to comply.62 |
| AE reports submitted by patients are less informative than reports submitted by healthcare professionals | Some studies demonstrate that patient reports may contain similar or more details than healthcare professional reports and may also provide information to complement healthcare professional reports.2 |
Screening FAERS ICSRs to identify safety signals
The purpose of screening FAERS is to identify potential safety signals, which may involve unlabeled AEs, known AEs reported in an unusual number or with greater severity or specificity, or other new potential safety concerns with the drug. Screening of FAERS involves multiple, methodical, complementary approaches, including targeted report-level review, cumulative screening, and data mining.
Because the number of ICSRs in FAERS is large, a screening approach based on reading each ICSR is not feasible. Instead, ICSR-level screening uses a risk-based approach to identify those ICSRs that will be individually read, taking into consideration factors such as the novelty of the product, its indications, and patient populations.17 To complement ICSR-level screening, cumulative screening of FAERS provides a high-level, aggregate summary of the drug’s postmarketing safety experience based on analyses of all ICSRs for a drug, selected clinically relevant MedDRA terms, serious outcomes, or a specific patient population.17 These complementary approaches may allow more efficient safety signal identification. Other possible sources to identify safety signals include documents submitted by application holders, such as PSRs and risk evaluation and mitigation strategy (REMS) assessment reports, published medical literature, risk management plans, and other information such as data submitted to FDA from international regulatory agencies and the public.
Data mining of FAERS ICSRs using disproportionality analyses and interpretation of results
Data mining of FAERS is an exploratory and hypothesis-generating component of safety surveillance that relies on statistical and mathematical methods to discover patterns of drug–AE pairs or unexpected occurrences that might not otherwise be apparent.17 The data mining technique most commonly used for surveillance of FAERS relies on disproportionality analysis, which identifies drug–AE combinations that occur more frequently with one drug than with other drugs in the database.91 At the FDA, a Multi-item Gamma Poisson Shrinker (MGPS) disproportionality algorithm analyzes the records in FAERS and then quantifies reported associations by producing a set of scores that indicate varying strengths of reporting relationships between drugs and AEs. These scores, known as empirical Bayes geometric mean (EBGM) values, provide a stable estimate of the relative reporting of an AE for a particular drug relative to all other drugs and AEs in FAERS. MGPS also calculates lower and upper 90% confidence limits for EBGM values, denoted as EB05 and EB95, respectively.91 Because EBGM values are based on FAERS data alone, the frequency of occurrence of the AE of interest in the entire database is an important determinant of the EBGM values.91 The same disproportionality analysis performed in another pharmacovigilance database for the same drug–AE pair may yield a different result because the pattern of drug–AE combinations may be different between the two databases.2
For potential safety signals identified through data mining efforts, further analysis of the ICSRs that underlie the disproportionality scores is necessary to determine if an actual safety signal exists that needs evaluation.17 Because disproportionality scores are generated using FAERS data, the general limitations of FAERS data also apply to the results of data mining analyses. Specifically, a disproportionality score is not a measure of causality since that score is based strictly on the numbers of ICSRs, without considering their content. Second, a disproportionality score is not a measure of the incidence of an AE because FAERS data, as noted earlier, are not a full enumeration of all events in a population. Third, disproportionality scores cannot measure the relative risk of an AE across two or more drugs since reporting patterns across multiple drugs may differ and because events are almost always underreported. Fourth, it is important to identify duplicates as they could lead to false-positive signals or masking of a signal (false-negatives), especially in data mining.93-97 Although the software FDA uses for data mining utilizes a dataset that has undergone a deduplication algorithm, some duplicate ICSRs may still remain. Finally, patterns of AE reporting may distort a disproportionality analysis. For example, a study of litigation-associated ICSRs found that large numbers of these types of reports may potentially lead to masking of signals of disproportionate reporting, particularly when the ICSRs are for AEs that are not frequently reported to FAERS.98 For these reasons, disproportionality scores cannot be used to infer causality or frequency of an AE, and drawing definitive causal associations between a drug and AE solely using data mining is a misuse of the technique.2,17,91,99-102
With improved public access to AE databases such as the FAERS Public Dashboard, there has been a corresponding increase in the number of articles published using disproportionality analyses. Pharmacovigilance experts have provided advice on the appropriate use of these data systems, highlighting limitations regarding conclusions that can be drawn from evaluations using these data following the distortion of results from disproportionality analyses in publications.101-106 As early as 2011, de Boer questioned the clinical importance of reporting disproportionality measures, which are derived from AE databases, noting that causality assessments of reports describing specific drug–AE combinations would be more informative. Instead, de Boer recommended that the publication of disproportionality measures from AE databases should be limited to those analyses where techniques to address selective reporting are addressed.99 In another article published in 2011, Montastruc et al. confirmed disproportionality analyses are exploratory in nature and multiple streams of data should be taken into consideration during signal evaluation.100 A decade later, Mouffak et al. evaluated published pharmacovigilance analyses describing disproportionality measures obtained from AE databases and identified misleading data interpretations within the abstracts, main text, and conclusions.107 Over-interpretation was the most frequent type of data misinterpretation, with 40% of abstracts and conclusions inaccurately using causal language.107 More recently, Raschi et al. and Giunchi et al. expressed concerns with the publishing of disproportionality analyses and the quality of such publications.101,102,108 Continued development of statistical data mining methods is also needed.109,110
CONDUCTING A COMPREHENSIVE EVALUATION OF A POTENTIAL SAFETY SIGNAL IDENTIFIED FROM ICSRs
Prioritization of potential safety signals
Once a safety signal is identified, its evaluation must be prioritized, considering safety information including, but not limited to:17
Important potential risks of the product (at the time of or after approval);
AEs for which causal attribution to the drug is biologically plausible rather than a manifestation of the disease being treated;
An increase in the severity or frequency of reporting of a labeled AE, AEs that are unlabeled and serious, reports of reduced effectiveness, and serious AEs with a low background rate in the general population and a high attribution to a drug (e.g., Stevens-Johnson syndrome (SJS), agranulocytosis, Torsade de Pointes);
Deaths, particularly in populations or in patients using the product for indications for which there would not be the expectation of death;
Suspected pharmacodynamic interactions with another drug, a biological product, a dietary supplement, or food; and
Use of a product inconsistent with FDA-approved indications (e.g., off-label use, misuse, abuse).
FDA posts on its website a quarterly report of “any new safety information or potential signal of a serious risk” identified by FAERS within the last quarter, in accordance with 21 U.S.C. 355(k) (5).1,111
Evaluating a safety signal
The risk-based principles guiding FDA’s evaluation of a safety signal are explained in detail in FDA’s Best Practices for FDA Staff in the Postmarketing Safety Surveillance of Human Drug and Biological Products.17 Briefly, safety signal evaluation utilizes a multidisciplinary team to conduct an integrated and comprehensive evaluation of an identified signal using multiple data sources. Evidence from FAERS, the medical literature, and other data sources can be generated by developing a case series, which is a set of selected reports that describe a defined group of patients who received the treatment of interest and experienced the outcome of interest, based on a case definition. To determine if a causal association between the AE and the drug exists, the safety signal evaluation team considers the evidence generated from the case series, along with related information from additional sources, as relevant and available, such as epidemiologic assessments, registry data, clinical trial data, clinical pharmacology studies, drug product utilization analyses, and other data sources (e.g., emergency departments and poison control centers). The data used to inform a safety signal evaluation depends on the nature of the safety issue of concern, and the strengths and limitations of each data source must be considered. Ideally, complementary data would be used to offset the limitations of any individual data source to the maximum extent possible. In some situations, such as when the AE commonly occurs in the population (i.e., has a high background rate) or is a manifestation of the disease being treated, a case series may not be informative. Alternatively, in some situations, a case series generated from FAERS data alone may suffice to provide supporting evidence to result in regulatory action, as described in this section.
Step 1. Developing a case definition.
To identify a case of a particular disease, injury, or health condition, a multidisciplinary team develops a case definition using information from the biomedical literature, clinical guidelines, and expert medical judgment. The case definition typically involves a combination of signs, symptoms, and test results. When uniformly applied, these criteria determine whether a person should be identified as having a particular disease, injury, or other health condition, and thus whether a retrieved ICSR should be included in the case series.
Step 2. Executing a search in FAERS and retrieving ICSRs.
FDA staff do not utilize the FAERS Public Dashboard or the FAERS QDE files to conduct safety signal evaluations because of the limitations previously described. Instead, FDA staff access the entire FAERS database to retrieve ICSRs coded with the MedDRA term signifying the AE term(s) of interest for the safety signal. Depending on the question, the search strategy may be broad to increase sensitivity or narrow to increase specificity. Additionally, the search strategy may focus on all products containing a specific active ingredient, specific formulations of drug products containing the specific active ingredient, all drug products in a specific drug class, and/or a broad spectrum of MedDRA Preferred Terms under the umbrella of Standardised MedDRA Queries,23 which facilitate safety signal analysis and reporting. The search parameters may also specify the population; for example, if the safety question is specific to the pediatric population, the search parameters should be limited to children. Unless there is a specific reason to narrow the search population, however, the search strategy should be broad to capture all potentially relevant ICSRs.
Step 3. Apply a case definition to the identified FAERS ICSRs.
Once the search is completed and ICSRs are retrieved, each ICSR must be individually reviewed to identify those that meet the case definition. This is an essential and at times a laborintensive step for which automated techniques are not yet able to fully replace human review. As a next step, these reports must be reviewed to identify and remove duplicate ICSRs. Duplicate ICSRs refer to separate and unlinked records of the same patient and AE.95 Duplicates occur from various sources reporting on the same patient and AE, such as when a patient is taking multiple drugs and the application holders for each drug submit a highly similar ICSR for their drug to FDA to fulfill reporting requirements; an unlinked follow-up ICSR that is assigned a new Case Identification number in the AE database; or an ICSR of different origin (e.g., a patient and healthcare professional report the same AE).95 Duplicates are identified through matching of both structured (e.g., age, date of birth, country) and unstructured data of the ICSR.95 Deduplication tools are being developed and may aid in this process as part of the Information Visualization Platform (InfoViP).95,112
Step 4. Applying a causality assessment.
Once the case series is established, a causality assessment is conducted for each case. FDA does not endorse a specific causality assessment tool or specific causality categorization terminology, though the categories of probable, possible, unassessable, or unlikely have been previously described.17 The criteria for the causality assessment should be documented, as should the basis for each ICSR’s determination.
The application of a causality assessment requires, among other considerations, a careful evaluation of confounding to determine whether alternative factors, rather than the drug of interest, caused the AE. For example, an analysis of ICSRs describing hypersensitivity reactions following administration of heparin potentially contaminated with oversulfated chondroitin sulfate (OSCS) found that nearly all cases included potentially confounding factors that precluded a determination that death was causally related to the drug.113 Even among the three cases where a probable causal association was identified, factors such as comorbidities were noted to make the causality assessment challenging.113
When performing a causality assessment, one must consider the counterfactual—that is, what would have happened had the patient not taken the drug? For AEs that occur commonly in the absence of drug treatment, such as AEs that resemble manifestations of the underlying condition being treated, it is difficult to determine whether the drug caused the AE on the basis of an ICSR. Instead, AEs that are best suited for causality assessment based on ICSRs are those that have very low background rates, have a short latency between administration of the drug and occurrence of the AE, are not manifestations of the disease or condition being treated, and are usually the result of exposure to a drug or toxin. Such AEs include serious skin reactions (e.g., SJS and toxic epidermal necrolysis), anaphylactic reactions, acute liver injury in the absence of other causes, and progressive multifocal leukoencephalopathy (PML).
The determination of a causal association between a drug and an AE is based on the strength of evidence obtained from the totality of the data available in the postmarketing evaluation. Information from ICSRs alone often does not provide a sufficient basis to suggest a causal relationship between an AE and the use of a drug; however, an ICSR for a rare AE that is typically drug-related or describes a positive rechallenge or positive dechallenge, if well-documented, may provide sufficient evidence to strongly support or determine a causal association. Additionally, because some AEs are rare, other sources of clinical information, such as epidemiological studies, are often not available and ICSRs play a central role in determining the causal association between drug and AE. For example, based on two very well-documented ICSRs that provided sufficient details of the patients’ histories and clinical course, and given that PML only occurs in the setting of immunosuppression, FDA concluded that fingolimod use can lead to reactivation of the John Cunningham virus and development of PML.2,114
Step 5. Characterizing the case series.
Once a causality assessment has been conducted for each ICSR, those ICSRs that meet the case definition but are assessed with an unassessable or unlikely causality are excluded from the case series. The reasons for a determination of an unassessable or unlikely causality should be documented. A case may be considered unassessable if, for example, it meets the case definition but there are confounding factors, such as concomitant drugs or an underlying disease that may have caused or contributed to the AE. A case cannot be causally associated and would be considered unlikely if, for example, the AE meets the case definition, but the AE occurred prior to exposure to the drug since there is an absence of temporality. A careful review, however, is necessary, because a drug may worsen an existing AE. The final case series includes the cases classified as having a probable or possible causal association. A review of these cases can help identify patterns or trends of the AE across the case series. Of note, the final case series with cases categorized as having a probable or possible causal association typically represents a small proportion of the ICSRs retrieved in FAERS.41
Step 6. Evaluating the drug utilization.
Drug utilization analyses can estimate the utilization of a drug and provide additional high-level summary information regarding characteristics of its use, such as descriptive information on the age and sex of recipients, average dose and duration of treatment, and the diseases or conditions for which the drug is prescribed. Within the FDA, drug utilization analyses are generally limited to use in the United States. It is important to understand both the care setting in which the drug is used and the data sources available for these analyses. For prescription drugs used primarily in the outpatient setting, projected national utilization data are generally available. Conversely, for drugs used primarily in hospitals and clinics, national data are generally not available, although data from individual health systems might be available.
Step 7. Determining the reporting ratio for a case series.
A reporting ratio, also called a reporting rate, may provide additional context for the safety signal but should not be considered in isolation. In the context of a case series based on FAERS data, the numerator of the reporting ratio is either the total number of FAERS cases for an AE-drug pair or the number of cases in the final case series, depending on the question to be addressed. The denominator is obtained from a different data source and comes from drug utilization analyses, often the number of prescriptions dispensed or population exposed in the time period corresponding to the time period covered by the FAERS search. When possible, the denominator should approximate the numbers of patients or person-time exposed nationwide as closely as possible and use surrogates of exposure (e.g., prescriptions dispensed, sales data) only if patient-level data is not available.81
The reporting ratio is not an incidence rate because the number of FAERS cases is not a measure of the occurrence of AE in the population.81 The reporting ratio may be contextualized by including an estimate of the background occurrence of an event in the general population, or in a subpopulation with similar characteristics as the exposed population (e.g., pediatric patients) of the reporting ratio.81
Step 8. Assessing data from clinical trials and epidemiologic studies.
Data from clinical trials and epidemiologic studies often inform the safety signal evaluation process. When clinical trials and epidemiologic studies include comparator groups, the ability to infer a causal relationship between an AE and a drug is improved. Post-approval, epidemiologic studies, and postmarketing clinical trials may be conducted to address specific safety issues. Such studies can be based on administrative claims data and electronic health records, on primary data collection, or a combination of the two. Although postmarketing clinical trials are typically conducted to demonstrate efficacy to support additional indications, these clinical trials also collect safety data. In certain circumstances, a clinical trial with the primary aim to study a safety endpoint may be warranted. While the role of postmarketing clinical trials and epidemiologic studies has been discussed extensively elsewhere,115 we provide an example here to demonstrate the importance of multiple data streams.
Within 14 months of approval of the anticoagulant dabigatran, the number of ICSRs received that reported bleeding complications (2347, with 348 coded with a fatal outcome) far exceeded the number of ICSRs for bleeding complications received in the same time period for warfarin (647, with 46 coded with a fatal outcome).116 Based on ICSR counts alone, it might seem as if dabigatran use, in comparison to warfarin use, led to more bleeding AEs in clinical practice, which contrasted with preapproval observations in clinical trials.2 However, a postapproval study using administrative claims data, which included clearly defined numbers of cases and drug utilization in a defined population in a specified period of time, confirmed that the rates of bleeding with dabigatran were not higher than those with warfarin.117
Step 9. Determining the causal association between a drug and AE.
Determining the causal association between a drug and an AE considers the strength of evidence from the totality of all data reviewed during the safety signal evaluation. Considerations include the number of well-documented cases identified in the final case series, consistency of the evidence among different data sources, biological and pharmacologic plausibility, and information about drugs with a similar mechanism of action or within the same class for which a causal association with an AE has already been established. If the evidence to support a causal association is insufficient, the AE may be deemed an AE of interest for continued monitoring. If a causal association between a drug and AE has been determined, regulatory action may be necessary.
Step 10. Strategies to mitigate risk and disseminate safety information.
Available actions to manage the safety risks identified in the safety signal evaluation may include one or more of the following: updates to the drug labeling; communication with consumers or healthcare professionals; further study by FDA; requests for an application holder to conduct studies or trials to obtain additional safety information; requiring a new REMS or modifying an existing REMS; or, on rare occasion, a request for marketing withdrawal.
EMERGING AREAS IN THE USE OF FAERS IN PHARMACOVIGILANCE
AE databases, such as FAERS, can be leveraged to successfully generate safety signals and provide new safety information for a drug–AE combination. Given the large number of AE reports received in FAERS (over 2 million reports yearly) and in an attempt to overcome some of the limitations of FAERS and enhance drug safety surveillance, FDA continues to explore new approaches to improve and complement current pharmacovigilance practices, including research into applying branches of artificial intelligence (AI).83,118-120 Machine learning (ML) and NLP, two types of AI that have been most often applied within the field of pharmacovigilance, have been applied to ICSRs in FAERS to automate tasks such as the identification of potential duplicate ICSRs and the application of a causality assessment.83,121,122
Potential uses of AI may include identification of AEs that may require AE reporting to the FDA, assessment of case validity, case prioritization by determining expectedness and seriousness, report coding, and assessment of report quality.120 AI has also been used to classify the probability of a causal association between an AE and drug.83,121 Tools such as NLP may also aid in assessing FAERS narratives.122 More broadly, AI might assist with the discovery of patterns suggesting drug safety signals in FAERS.123 Additional research is needed to improve the application of AI to pharmacovigilance and to gain acceptance by pharmacovigilance experts.124
CONCLUSION
FAERS is a powerful tool for drug safety surveillance and assessment, as evidenced by the fact that FAERS data are often used to make safety labeling changes.39,40 Understanding the structure, strengths, and limitations of the FAERS database is necessary for drawing scientifically and medically accurate conclusions. It is also imperative to acknowledge and explain any potential limitations of FAERS (and other data sources) to allow clinicians and researchers to appropriately interpret findings. For example, public versions of FAERS data available through the FAERS Public Dashboard and FAERS QDE files do not contain sufficient information to draw causal associations between drug–AE pairings. Similarly, data mining should not be used to draw causal inferences, as causal associations between a drug and AE cannot be established using data mining techniques. Instead, appropriate data sources should be used in a complementary approach during signal detection and signal evaluation, which should typically include the application of a case definition for the AE being addressed followed by an assessment of causality. The considerations and concepts covered in this article should help foster more accurate analyses and interpretations of FAERS data and the principles in the analyses of other AE databases.
ACKNOWLEDGMENTS
The authors would like to thank the following FDA colleagues for their valuable contributions to this manuscript: Robert Ball, Sonja Brajovic, Allen Brinker, Sara Camilli, Vicky Chan, Samantha Cotter, Suranjan De, Ida-Lina Diak, David Moeny, S. Christopher Jones, Douglas Kovich, Maureen Melvin, Monica Muñoz, Tracy Salaam, John Swann, Kaniz Afroz Tanni, Lisa Wolf, Jo Wyeth, and Judith Zander.
FUNDING
No funding was received for this work.
Footnotes
CONFLICT OF INTEREST
The authors declared no competing interests for this work.
DISCLAIMER
The opinions expressed in this manuscript are those of the authors and not necessarily those of the US Food and Drug Administration or the US Government.
Previous presentations: This work has not been previously presented.
References
- 1.U.S. Code. Federal Food, Drug and Cosmetic Act, Subchapter V-Drugs and Devices, Sec. 355-New Drugs, 21 USC § 355 <https://www.govinfo.gov/app/details/USCODE-2023-title21/USCODE-2023-title21-chap9-subchapV-partA-sec355> (2023). Accessed March 25, 2025.
- 2.Dal Pan GJ, Lindquist M & Gelperin K Postmarketing spontaneous pharmacovigilance reporting systems. In Pharmacoepidemiology 6th edn. (eds. Strom BL, Kimmel SE & Hennessy S) 165–201 (Wiley, Hoboken, NJ, 2019). [Google Scholar]
- 3.Code of Federal Regulations. 21 CFR 314.80-Postmarketing reporting of adverse drug experiences <https://www.ecfr.gov/current/title-21/chapter-I/subchapter-D/part-314/subpart-B/section-314.80> (2025). Accessed March 25, 2025.
- 4.Code of Federal Regulations. 21 CFR 310.305-Records and reports concerning adverse drug experiences on marketed prescription drugs for human use without approved new drug applications <https://www.ecfr.gov/current/title-21/chapter-I/subchapter-D/part-310/subpart-D/section-310.305> (2025). Accessed March 25, 2025.
- 5.Code of Federal Regulations. 21 CFR 600.80-Postmarketing reporting of adverse experiences <https://www.ecfr.gov/current/title-21/chapter-I/subchapter-F/part-600/subpart-D/section-600.80> (2025). Accessed March 25, 2025.
- 6.Code of Federal Regulations. 21 CFR 329.100-Postmarketing reporting of adverse drug events under section 760 of the Federal Food, Drug, and Cosmetic Act <https://www.ecfr.gov/current/title-21/chapter-I/subchapter-D/part-329> (2014). Accessed March 25, 2025.
- 7.Food and Drug Administration. FDA Adverse Events Reporting System (FAERS) Public Dashboard: Frequently Asked Questions <https://fis.fda.gov/extensions/FPD-FAQ/FPD-FAQ.html> (2024). Accessed March 25, 2025.
- 8.European Medicines Agency. EudraVigilance <https://www.adrreports.eu/en/index.html> (2024). Accessed March 25, 2025.
- 9.Uppsala Monitoring Centre. VigiBase <https://www.vigiaccess.org/> (2024). Accessed March 25, 2025.
- 10.Health Canada. Canada Vigilance <https://www.canada.ca/en/health-canada/services/drugs-health-products/medeffect-canada/adverse-reaction-database.html> (2025). Accessed March 25, 2025.
- 11.Health Sciences Authority. HSA Adverse Event Online Database <https://www.hsa.gov.sg/adverse-events> (2024). Accessed August 17, 2024.
- 12.Medicines and Healthcare Products Regulatory Agency. Yellow Card <https://yellowcard.mhra.gov.uk/idaps/> (2024). Accessed August 17, 2024.
- 13.Pharmaceuticals and Medical Devices Agency. Japanese Adverse Drug Event Report Database <https://www.pmda.go.jp/> (2024). Accessed August 17, 2024.
- 14.Therapeutic Goods Administration. Database of Adverse Event Notifications <https://www.tga.gov.au/safety/safety/database-adverse-event-notifications-daen-medicines> (2024). Accessed August 17, 2024.
- 15.Varrichio F. et al. Understanding vaccine safety information from the vaccine adverse event reporting system. Pediatr. Infect. Dis. J 23, 287–294 (2004). [DOI] [PubMed] [Google Scholar]
- 16.World Health Organization-Uppsala Monitoring Centre. Pharmacovigilance communications: glossary <https://who-umc.org/pharmacovigilance-communications/glossary/> (2024). Accessed March 25, 2025.
- 17.Food and Drug Administration. Best Practices for FDA Staff in the Postmarketing Safety Surveillance of Human Drug and Biological Products <https://www.fda.gov/media/130216/download?attachment> (2024). Accessed 5 February 2024.
- 18.Nebeker JR, Barach P & Samore MH Clarifying adverse drug events: a clinician’s guide to terminology, documentation, and reporting. Ann. Intern. Med 140, 795–801 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Food and Drug Administration. Guidance for Industry: Adverse Reactions Section of Labeling for Human Prescription Drug and Biological Products – Content and Format <https://www.fda.gov/media/72139/download> (2006). Accessed February 5, 2024.
- 20.Code of Federal Regulations. 21 CFR 201.57-Specific requirements on content and format of labeling for human prescription drug and biological products described in 201.56(b) (1) <https://www.ecfr.gov/current/title-21/chapter-I/subchapter-C/part-201/subpart-B/section-201.57> (2025). Accessed March 25, 2025.
- 21.Council for International Organizations of Medical Sciences (CIOMS). CIOMS Cumulative Glossary with a Focus on Pharmacovigilance <https://cioms.ch/publications/product/cioms-cumulative-glossary-anniversary-edition/> (2024). Accessed May 6, 2025.
- 22.Food and Drug Administration. Data mining at FDA-disproportionality methods <https://www.fda.gov/science-research/data-mining/data-mining-fda-white-paper#disproportionality> (2018). Accessed May 10, 2025.
- 23.Maintenance and Support Services Organization. Medical Dictionary for Regulatory Activities <https://www.meddra.org/> (2024). Accessed August 17, 2024.
- 24.Council for International Organizations of Medical Sciences (CIOMS) Working Group IV. Benefit-risk balance for marketed drugs: evaluating safety signals <https://cioms.ch/publications/product/benefit-risk-balance-for-marketed-drugs-evaluating-safety-signals/> (1998) Accessed April 3, 2024.
- 25.Council for International Organizations of Medical Sciences (CIOMS) Working Group VIII. Practical aspects of signal detection in pharmacovigilance <https://cioms.ch/wp-content/uploads/2018/03/WG8-Signal-Detection.pdf> (2010). Accessed April 3, 2024.
- 26.Council for International Organizations of Medical Sciences (CIOMS). Version 7 (29 October 2024): Glossary of International Council for Harmonization (ICH) terms and definitions <https://cioms.ch/publications/product/glossary-of-ich-terms-and-definitions/> (2024). Accessed October 31, 2024.
- 27.Cherkaoui S. et al. The impact of variability in patient exposure during premarket clinical development on postmarket safety outcomes. Clin. Pharmacol. Ther 110, 1512–1525 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rogers JR, Liu C, Hripcsak G, Cheung YK & Weng C Comparison of clinical characteristics between clinical trial participants and nonparticipants using electronic health record data. JAMA Netw. Open 4, e214732 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Food and Drug Administration. Best Pharmaceuticals for Children Act and Pediatric Research Equity Act: Status Report to Congress July 1, 2015–June 30, 2020 <https://www.fda.gov/media/157840/download?attachment> (2020). Accessed August 17, 2024.
- 30.Zulman DM, Sussman JB, Chen X, Cigolle CT, Blaum CS & Hayward RA Examining the evidence: a systematic review of the inclusion and analysis of older adults in randomized controlled trials. J. Gen. Intern. Med 26, 783–790 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chamberlain C, Kortepeter C & Muñoz M Clinical analysis of adverse drug reactions and pharmacovigilance. In Atkinson’s Principles of Clinical Pharmacology 4th edn. (eds. Huang S-M, Lertora JJL, Vicini P & Atkinson AJ) 499–517 (Academic Press, Cambridge, MA, 2021). [Google Scholar]
- 32.Mohamoud M. et al. Assessment of the impact of mandated postmarketing pediatric-focused safety reviews on safety-related regulatory actions 2013–2019. Clin. Pharmacol. Ther 113, 1368–1377 (2023). [DOI] [PubMed] [Google Scholar]
- 33.Jones SC, Kortepeter C & Brinker AD Postmarketing surveillance of drug-induced liver injury. In Drug-I nduced Liver Injury (eds. Chen M & Will Y) 459–474 (Humana Press, New York, NY, 2018). [Google Scholar]
- 34.Garcia-Cortes M, Robles-Diaz M, Stephens C, Ortega-Alonso A, Lucena MI & Andrade RJ Drug induced liver injury: an update. Arch. Toxicol 94, 3381–3407 (2020). [DOI] [PubMed] [Google Scholar]
- 35.Reyes M, Kortepeter C & Muñoz M Postmarket assessment for drugs and biologics used in dermatology and cutaneous adverse drug reactions. Dermatol. Clin 40, 265–277 (2022). [DOI] [PubMed] [Google Scholar]
- 36.Code of Federal Regulations. 21 CFR 201.56 – Requirements on content and format of labeling for human prescription drug and biological products <https://www.govinfo.gov/app/details/CFR-2024-title21-vol4/CFR-2024-title21-vol4-sec201-56> (2024). Accessed March 25, 2025.
- 37.Pinnow E. et al. Postmarket safety outcomes for new molecular entity (NME) drugs approved by the Food and Drug Administration between 2002 and 2014. Clin. Pharmacol. Ther 104, 390–400 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Bulatao I. et al. Postmarketing safety-related regulatory actions for new therapeutic biologics approved in the United States 2002–2014: similarities and differences with new molecular entities. Clin. Pharmacol. Ther 108, 1243–1253 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lester J, Neyarapally GA, Lipowski E, Graham CF, Hall M & Dal Pan G Evaluation of FDA safety-related drug label changes in 2010. Pharmacoepidemiol. Drug Saf 22, 302–305 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Croteau D, Pinnow E, Wu E, Muñoz M, Bulatao I & Dal Pan G Sources of evidence triggering and supporting safety-related labeling changes: a 10-year longitudinal assessment of 22 new molecular entities approved in 2008 by the US Food and Drug Administration. Drug Saf. 45, 169–180 (2022). [DOI] [PubMed] [Google Scholar]
- 41.Muñoz MA et al. Towards automating adverse event review: a prediction model for case report utility. Drug Saf. 43, 329–338 (2020). [DOI] [PubMed] [Google Scholar]
- 42.The Lancet Commission on Anesthetics. Report of the lancet commission appointed to investigate the subject of the administration of chloroform and other anesthetics from a clinical standpoint. Lancet 141, 629–638 (1893). [Google Scholar]
- 43.DeHovitz RE The 1901 St Louis incident: the first modern medical disaster. Pediatrics 133, 964–965 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Bren L. The road to the biotech revolution–highlights of 100 years of biologics regulation, FDA Consumer Magazine <https://www.fda.gov/files/about%20fda/published/The-Road-to-the-Biotech-Revolution--Highlights-of-100-Years-of-Biologics-Regulation.pdf> (2006). Accessed May 6, 2025. [PubMed]
- 45.Food and Drug Administration. Part I: the 1906 Food and Drugs Act and Its Enforcement <https://www.fda.gov/about-fda/changes-science-law-and-regulatory-authorities/part-i-1906-food-anddrugs-act-and-its-enforcement> (2019). Accessed September 24, 2024.
- 46.Food and Drug Administration. Part II: 1938, Food, Drug, and Cosmetic Act <https://www.fda.gov/about-fda/changes-science-law-and-regulatory-authorities/part-ii-1938-food-drug-cosmetic-act> (2018). Accessed September 24, 2024.
- 47.Wax PM Elixirs, diluents, and the passage of the 1938 Federal Food, Drug and Cosmetic Act. Ann. Intern. Med 122, 456–461 (1995). [DOI] [PubMed] [Google Scholar]
- 48.Lewis CN, Putnam LE, Hendricks FD, Kerlan I & Welch H Chloramphenicol (chloromycetin) in relation to blood dyscrasias with observations on other drugs. Antibiot. Chemother. (Northfield) 2, 601–609 (1952). [PubMed] [Google Scholar]
- 49.De Nosaquo N. Secretary’s introduction. Bull. Am. Med. Assoc. Counc. Pharm. Chem,2 289–291 (1955). [Google Scholar]
- 50.Moser RH Obituary of an idea. JAMA 216, 2135–2136 (1971). [PubMed] [Google Scholar]
- 51.Harvey JL Report from the Food and Drug Administration. Food Drug Cosmet. Law J 12, 71–80 (1957). [Google Scholar]
- 52.Conrad EA Techniques for selecting, evaluating, & compacting the drug literature: American Medical Association. Am. J. Hosp. Pharm 23, 60–63 (1966). [PubMed] [Google Scholar]
- 53.American Medical Association. Reporting adverse drug and medical device events: report of the AMA’s council on ethical and judicial affairs. Food Drug Law J. 49, 359–365 (1994). [Google Scholar]
- 54.Ahmad SR, Goetsch RA & Marks NS Spontaneous reporting in the United States. In Pharmacoepidemiology 4th edn. (eds. Strom BL, Kimmel SE & Hennessy S) 135–159 (Wiley, Hoboken, NJ, 2005). [Google Scholar]
- 55.Kerlan I. Adverse drug reaction reporting: approach to better patient care. Hospitals 34, 65–69 (1960). [PubMed] [Google Scholar]
- 56.Food and Drug Administration. Monthly Report: adverse reactions to drugs and therapeutic devices. August 1965.
- 57.Food and Drug Administration. Reports of suspected adverse reactions to drugs and therapeutic devices: October 1968.
- 58.Food and Drug Administration. Frances Oldham Kelsey: medical reviewer famous for averting a public health tragedy <https://www.fda.gov/about-fda/fda-history-exhibits/frances-oldham-kelsey-medical-reviewer-famous-averting-public-health-tragedy> (2018). sAccessed September 24, 2024.
- 59.Food and Drug Administration. Part III: drugs and foods under the 1938 Act and its amendments <https://www.fda.gov/about-fda/changes-science-law-and-regulatory-authorities/part-iii-drugs-and-foods-under-1938-act-and-its-amendments> (2018). Accessed September 24, 2024.
- 60.United States Code. Federal Food, Drug, and Cosmetic Act: Drug Amendments of 1962 (Kefauver-Harris Act [Public Law 87–781]) <https://www.govinfo.gov/content/pkg/STATUTE-76/pdf/STATUTE-76-Pg780.pdf> (1962). Accessed September 24, 2024.
- 61.Kessler DA Introducing MEDWatch. A new approach to reporting medication and device adverse effects and product problems. JAMA 269, 2765–2768 (1993). [DOI] [PubMed] [Google Scholar]
- 62.Food and Drug Administration. FDA Adverse Event Reporting System (FAERS) Electronic Submissions <https://www.fda.gov/drugs/questions-and-answers-fdas-adverse-event-reporting-system-faers/fda-adverse-event-reporting-system-faers-electronic-submissions> (2024). Accessed September 27, 2024.
- 63.U.S. Congress. 21st Century Cures Act <https://www.congress.gov/114/plaws/publ255/PLAW-114publ255.pdf> (2016). Accessed September 27, 2024.
- 64.Food and Drug Administration. Guidance for Industry: E2B(R3) Electronic Transmission of Individual Case Safety Reports (ICSRs) Implementation Guide – Data Elements and Message Specification <https://www.fda.gov/media/81904/download> (2022). Accessed September 27, 2024.
- 65.National Library of Medicine. Unified Medical Language System (UMLS): CST (COSTART-Synopsis) <https://www.nlm.nih.gov/research/umls/sourcereleasedocs/current/CST/index.html#:~:text=The%20Coding%20Symbols%20for%20a,drug%20and%20biologic%20experience%20reports> (2024). Accessed April 12, 2024.
- 66.Food and Drug Administration. MedWatch Online Voluntary Report Form <https://www.accessdata.fda.gov/scripts/medwatch/index.cfm> (2024). Accessed August 17, 2024.
- 67.Food and Drug Administration. MedWatch Forms for FDA Safety Reporting <https://www.fda.gov/safety/medical-product-safety-information/medwatch-forms-fda-safety-reporting> (2024). Accessed August 17, 2024.
- 68.Food and Drug Administration. FAERS Public Dashboard <https://fis.fda.gov/sense/app/95239e26-e0be-42d9-a960-9a5f7f1c25ee/sheet/7a47a261-d58b-4203-a8aa-6d3021737452/state/analysis> (2024). Accessed September 27, 2024.
- 69.Food and Drug Administration. Expedited Safety Reporting Requirements for Human Drug and Biological Products <https://www.fda.gov/science-research/clinical-trials-and-human-subject-protection/expedited-safety-reporting-requirements-human-drug-and-biological-products> (1997). Accessed December 20, 2024.
- 70.International Council on Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. Post-Approval Safety Data Management: Definitions and Standards for Expedited Reporting E2D: ICH Harmonised Tripartite Guideline < https://www.ema.europa.eu/en/documents/scientific-guideline/international-conference-harmonisation-technical-requirements-registration-pharmaceuticals-human-use-topic-e-2-d-postapproval-safety-data-management-step-5_en.pdf> (2003). Accessed 14 January 2024.
- 71.Food and Drug Administration. Center for Drug Evaluation and Research, Office of Surveillance and Epidemiology. Responding to Requests for Waivers of Postmarketing Safety Reporting Requirements under 21 CFR 314.80 (NDAs), 314.98 (ANDAs), and 600.80 (BLAs) <https://www.fda.gov/media/72450/download> (2022). Accessed August 17, 2024.
- 72.Food and Drug Administration. Guidance for Industry: Postmarketing Safety Reporting for Human Drug and Biological Products Including Vaccines (Draft Guidance) <https://www.fda.gov/media/73593/download> (2001). Accessed August 17, 2024.
- 73.Code of Federal Regulations. 21 CFR 314.90-Applications for FDA Approval to Market a New Drug, Subpart B-Applications, Sec. 314.90, Waivers <https://www.ecfr.gov/current/title-21/chapter-I/subchapter-D/part-314/subpart-B/section-314.90> (2025). Accessed March 25, 2025.
- 74.Harinstein L, Kalra D, Kortepeter CM, Muñoz MA, Wu E & Dal Pan GJ Evaluation of postmarketing reports from industry-sponsored programs in drug safety surveillance. Drug Saf. 42, 649–655 (2019). [DOI] [PubMed] [Google Scholar]
- 75.Marwitz K, Jones SC, Kortepeter CM, Dal Pan GJ & Muñoz M An evaluation of postmarketing reports with an outcome of death in the US FDA adverse event reporting system. Drug Saf. 43, 457–465 (2020). [DOI] [PubMed] [Google Scholar]
- 76.Muñoz MA et al. Impact of a new consumer form on the quantity and quality of adverse event reports submitted to the United States Food and Drug Administration. Pharmacotherapy 39, 1042–1052 (2019). [DOI] [PubMed] [Google Scholar]
- 77.Food and Drug Administration. Guidance Document: FDA Regional Implementation Guide for E2B(R3) Electronic Transmission of Individual Case Safety Reports for Drug and Biological Products <https://www.fda.gov/regulatory-information/search-fda-guidance-documents/fda-regional-implementation-guide-e2br3-electronic-transmission-individual-case-safety-reports-drug> (2024). Accessed August 17, 2024.
- 78.Brown EG, Wood L & Wood S The medical dictionary for regulatory activities (MedDRA). Drug Saf. 20, 109–117 (1999). [DOI] [PubMed] [Google Scholar]
- 79.Medical Dictionary for Regulatory Activities. Mapping <https://www.meddra.org/mapping> (2025). Accessed January 20, 2025.
- 80.Food and Drug Administration. Specifications for preparing and submitting electronic ICSRs and ICSR attachments <https://www.fda.gov/media/132096/download> (2020). Accessed August 17, 2024.
- 81.Food and Drug Administration. Guidance for Industry: Good Pharmacovigilance Practices and Pharmacoepidemiologic Assessment <https://www.fda.gov/regulatory-information/search-fda-guidance-documents/good-pharmacovigilance-practices-and-pharmacoepidemiologic-assessment> (2005). Accessed May 6, 2025.
- 82.Pham P. et al. Leveraging case narratives to enhance patient age ascertainment from adverse event reports. Pharmaceut. Med 35, 307–316 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Han L, Ball R, Pamer CA, Altman RB & Proestel S Development of an automated assessment tool for MedWatch reports in the FDA adverse event reporting system. J. Am. Med. Inform. Assoc 24, 913–920 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Food and Drug Administration. FDA improves accessed to reports of adverse drug reactions <https://www.fda.gov/news-events/press-announcements/fda-improves-access-reports-adverse-drug-reactions> (2017). Accessed August 17, 2024.
- 85.Food and Drug Administration. Office of Surveillance and Epidemiology 2023 Annual Report <https://www.fda.gov/media/180347/download?attachment> (2023). Accessed October 18, 2024.
- 86.Food and Drug Administration. Instructions for requesting individual case reports <https://www.fda.gov/drugs/questions-and-answers-fdas-adverse-event-reporting-system-faers/instr uctions-requesting-individual-case-reports> (2020). Accessed August 17, 2024.
- 87.Food and Drug Administration. FDA Adverse Event Reporting System (FAERS) Quarterly Data Extract Files <https://fis.fda.gov/extensions/FPD-QDE-FAERS/FPD-QDE-FAERS.html> (2024). Accessed August 17, 2024.
- 88.Food and Drug Administration. openFDA <https://open.fda.gov/> (2024). Accessed August 17, 2024.
- 89.Food and Drug Administration. FDA Adverse Event Reporting System (FAERS) Public Dashboard: Frequently Asked Questions (FAQs) <https://fis.fda.gov/extensions/FPD-FAQ/FPD-FAQ.html#_Toc514144622> (2023). Accessed May 6, 2025.
- 90.Hazell L & Shakir SA Under-reporting of adverse drug reactions: a systematic review. Drug Saf. 29, 385–396 (2006). [DOI] [PubMed] [Google Scholar]
- 91.Duggirala HJ et al. Data mining at FDA—white paper <https://www.fda.gov/media/91848/download> (2016). Accessed May 6, 2025. [Google Scholar]
- 92.Food and Drug Administration. Guidance for industry: appendix I (B) to the ICH E2B(R3) ICSRs implementation guide: backwards and forwards compatibility <https://www.fda.gov/media/81913/download> (2022). Accessed September 27, 2024.
- 93.Hauben M, Reich L, DeMicco J & Kim K Extreme duplication in the US FDA adverse events reporting system database. Drug Saf. 30, 551–554 (2007). [DOI] [PubMed] [Google Scholar]
- 94.Hauben M, Zou C, Bright S & Hung E More extreme duplication in the U.S. FDA FAERS database and a suggested check point for disproportionality analysis. Pharmacoepidemiol. Drug Saf 32, 387–391 (2021). [DOI] [PubMed] [Google Scholar]
- 95.Kreimeyer K. et al. Increased confidence in deduplication of drug safety reports with natural language processing of narratives at the US Food and Drug Administration. Front. Drug. Saf 2, 28293864 (2022). [Google Scholar]
- 96.Hung E, Hauben M, Essex H, Zou C & Bright S More extreme duplication in FDA adverse event reporting system detected by literature reference normalization and fuzzy string matching. Pharmacoepidemiol. Drug Saf 32, 387–391 (2023). [DOI] [PubMed] [Google Scholar]
- 97.Kiguba R. et al. Navigating duplication in pharmacovigilance databases: a scoping review. BMJ Open 14, e081990 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Muñoz M & Dal Pan GJ The impact of litigation-associated reports on signal identification in the US FDA’s adverse event reporting system. Drug Saf. 42, 1119–1201 (2019). [DOI] [PubMed] [Google Scholar]
- 99.de Boer A. When to publish measures of disproportionality derived from spontaneous reporting databases? Br. J. Clin. Pharmacol 72, 909–911 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Montastruc J-L, Sommet A, Bagheri H & Lapeyre-Mestre M Benefits and strengths of the disproportionality analysis for identification of adverse drug reactions in a pharmacovigilance database. Br. J. Clin. Pharmacol 72, 905–908 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Raschi E, Salvo F & Khouri C Conceiving, conducting, reporting, interpreting, and publishing disproportionality analyses: a call to action. Br. J. Clin. Pharmacol 88, 3535–3536 (2022). [DOI] [PubMed] [Google Scholar]
- 102.Raschi E. et al. Peer review in pharmacovigilance: lens on disproportionality analysis. Drug Saf. 47, 601–605 (2024). [DOI] [PubMed] [Google Scholar]
- 103.Postigo R. et al. EudraVigilance medicines safety database: publicly accessible data for research and public health protection. Drug Saf. 41, 665–675 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Uppsala Monitoring Centre. Guidelines for using VigiBase data in studies <https://who-umc.org/media/05kldqpj/guidelineusingvigibaseinstudies.pdf> (2021). Accessed April 5, 2024.
- 105.Fusaroli M. et al. The REporting of A Disproportionality analysis for drUg Safety signal detection using individual case safety reports in PharmacoVigilance (READUS-PV): development and statement. Drug Saf. 47, 575–584 (2024a). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fusaroli M. et al. The REporting of A Disproportionality analysis for drUg Safety signal detection using individual case safety reports in PharmacoVigilance (READUS-PV): explanation and elaboration. Drug Saf. 47, 585–599 (2024b). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mouffak A. et al. High prevalence of spin was found in pharmacovigilance studies using disproportionality analyses to detect safety signals: a meta-epidemiological study. J. Clin. Epidemiol 138, 73–79 (2021). [DOI] [PubMed] [Google Scholar]
- 108.Giunchi V, Fusaroli M, Hauben M, Raschi E & Poluzzi E Challenges and opportunities in accessing and analysing FAERS data: a call towards a collaborative approach. Drug Saf. 46, 921–926 (2023). [DOI] [PubMed] [Google Scholar]
- 109.Chakraboty S, Liu A, Ball R & Markatou M On the use of the likelihood ratio test methodology in pharmacovigilance. Stat. Med 41, 5395–5420 (2022). [DOI] [PubMed] [Google Scholar]
- 110.Ding Y, Markatou M & Ball R An evaluation of statistical approaches to postmarketing surveillance. Stat. Med 39, 845–874 (2020). [DOI] [PubMed] [Google Scholar]
- 111.Food and Drug Administration. Potential Signals of Serious Risks/New Safety Information Identified from the FDA Adverse Event Reporting System (FAERS) <https://www.fda.gov/drugs/questions-and-answers-fdas-adverse-event-reporting-system-faers/potential-signals-serious-risksnew-safety-information-identified-fda-adverse-event-reporting-system> (2024). Accessed August 17, 2024.
- 112.Spiker J. et al. Information visualization platform for Postmarket surveillance decision support. Drug Saf. 43, 905–915 (2020). [DOI] [PubMed] [Google Scholar]
- 113.McMahon AW et al. Description of hypersensitivity adverse events following administration of heparin that was potentially contaminated with oversulfated chondroitin sulfate in early 2008. Pharmacoepidemiol. Drug Saf 19, 921–933 (2010). [DOI] [PubMed] [Google Scholar]
- 114.Food and Drug Administration. FDA Drug Safety Communication: FDA investigating rare brain infection in patient taking Gilenya (fingolimod) <https://wayback.archive-it.org/7993/20161022203738/http:/www.fda.gov/Drugs/DrugSafety/ucm366529.htm> (2015). Accessed August 17, 2024.
- 115.Reynolds RF, Lesko SM, Gatto NM, van Staa TP & Mitchell AA The use of randomized controlled trials in pharmacoepidemiology. In Pharmacoepidemiology 6th edn. (eds. Strom BL, Kimmel SE & Hennessy S) 792–812 (Wiley, Hoboken, NJ, 2019). [Google Scholar]
- 116.McConeghy KW, Bress A, Qato DM, Wing C & Nutescu EA Evaluation of dabigatran bleeding adverse reaction reports in the FDA adverse event reporting system during the first year of approval. Pharmacotherapy 34, 561–569 (2014). [DOI] [PubMed] [Google Scholar]
- 117.Southworth MR, Reichman ME & Unger EF Dabigatran and postmarketing reports of bleeding. N. Engl. J. Med 4, 1272–1274 (2013). [DOI] [PubMed] [Google Scholar]
- 118.Food and Drug Administration. Using artificial intelligence & machine learning in the development of drug & biological products: discuss paper and request for feedback <https://www.fda.gov/media/167973/download> (2023). Accessed August 17, 2024.
- 119.Food and Drug Administration. CDER Emerging Drug Safety Technology Program (EDSTP) <https://www.fda.gov/drugs/science-and-research-drugs/cder-emerging-drug-safety-technology-program-edstp#:~:text=The%20EDSTP%20is%20specifically%20focused,AI%2C%20can%20best%20be%20used> (2024). Accessed August 17, 2024.
- 120.Bate A & Hobbiger SF Artificial intelligence, real-world automation and the safety of medicines. Drug Saf. 44, 125–132 (2021). [DOI] [PubMed] [Google Scholar]
- 121.Kreimeyer K. et al. Feature engineering and machine learning for causality assessment in pharmacovigilance: lessons learned from application to the FDA adverse event reporting system. Comput. Biol. Med 135, 104517 (2021). [DOI] [PubMed] [Google Scholar]
- 122.Ball R & Dal Pan G Artificial intelligence for pharmacovigilance: ready for prime time? Drug Saf. 45, 429–438 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Markatou M & Ball R A pattern discovery framework for adverse event evaluation and inference in spontaneous reporting system. Wiley Periodicals (Statistical Analysis and Data Mining) (2014). [Google Scholar]
- 124.Ball R, Talal AH, Dang O, Muñoz M & Markatou M Trust but verify: lessons learned for application of artificial intelligence to case-based clinical decision making from post-marketing drug safety assessment at the US Food and Drug Administration. J. Med. Internet Res 6, 50274 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]