

Abstract
Mastocytosis is characterized by the clonal infiltration and proliferation of neoplastic mast cells into target organs. Clinical features of mastocytosis are based in large part on dysregulated mast cell mediator release. Affected individuals may present with isolated skin involvement or multisystemic disease with a spectrum of symptoms including anaphylaxis, pathologic fractures, and chronic gastrointestinal, neurocognitive, musculoskeletal, and constitutional symptoms. The term “mastocytosis in the skin” refers to individuals with cutaneous infiltration and encompasses both localized and systemic forms of disease. Cutaneous involvement is further categorized into cutaneous mastocytoma, diffuse cutaneous mastocytosis, and maculopapular cutaneous mastocytosis based on morphology. In ~95% of patients with systemic mastocytosis, the disease is driven by the KIT D816V somatic variant. The aim of this clinical review is to highlight the diagnostic considerations, management complexities, and evolving treatment landscape that must be considered when evaluating a patient presenting with mastocytosis in their skin. Clinical manifestations, histopathology, and laboratory parameters are essential to diagnosis and determining the disease burden in those with known or suspected systemic mastocytosis. Once appropriately staged, both skin-directed therapy as well as novel systemic treatment options, including selective tyrosine kinase inhibitors, can be considered with the potential to improve patient outcomes.
Key Points
| This clinical review presents an updated approach to the diagnosis, evaluation, and management of patients presenting with mastocytosis in the skin. |
| As systemic and prognostic implications differ between adult and pediatric populations, careful attention to the unique features that impact an evaluation in these groups is imperative and is outlined within. |
Introduction
Mastocytosis is a clonal mast cell (MC) disease driven by mutations in the KIT gene in the majority of cases [1, 2]. It is characterized by MC proliferation and activation in one or more of the following organs: the bone marrow (BM), liver, spleen, lymph nodes, gastrointestinal tract, and skin. Clinically, the disease may manifest with recurrent pruritus, flushing, urtication, abdominal pain, nausea/vomiting, diarrhea, musculoskeletal pain, vascular instability, headache, and neuropsychiatric difficulties [3]. It is estimated to affect 1 in 10,000 individuals [4], although the precise incidence in the USA remains uncertain and limited by under-diagnosis, which is at least partially attributed to a lack of recognition and awareness of mastocytosis. Mastocytosis may present at any age without sex predilection and familial occurrence may occur but is uncommon [4–6].
Mastocytosis represents a spectrum of phenotypic expression with pathologic and genetic findings affecting presentation and prognosis. In children, the skin is the most common organ involved and, in the vast majority of cases, is the only manifestation of the disease. Pediatric-onset mastocytosis occurs as early as birth, with 85% presenting by age 2 years [7]. Children may be diagnosed with all mastocytosis variants, though the clinical presentation and underlying pathogenesis may differ from adults. For example, most children lack the KIT c.2447 C>T (p.D816V) exon 17 activating mutation often found in adults. Additional distinct KIT genetic alterations have been found in skin biopsies of children with skin-limited disease (cutaneous mastocytosis [CM]), including alterations in exons 8 and 9, partially explaining the differences in the disease course [8]. Although systemic mastocytosis (SM) rarely presents during childhood, recognition is paramount to improving outcomes for these patients. In contrast to children, the vast majority of adults and late adolescents presenting with abnormal MC proliferation in the skin have concurrent SM characterized by extracutaneous neoplastic MC hyperplasia [9]. For this reason, adults found to have cutaneous disease are termed to have “mastocytosis in the skin (MIS)” until staging is complete as cutaneous-limited disease (CM) cannot be definitively diagnosed in this population otherwise.
Across all ages, SM is diagnosed based on BM and extracutaneous organ histopathologic changes as outlined by the World Health Organization and the International Consensus Classification of Myeloid Neoplasms and Acute Leukemias [10–13]. A BM biopsy should therefore be considered in all adult-onset cases and in children demonstrating features suggestive of systemic involvement. Mastocytosis is classified into different subtypes [11]. Bone marrow mastocytosis, indolent SM (ISM), and smoldering SM comprise non-advanced SM, whereas aggressive SM, SM with an associated myeloid hematologic neoplasm, and MC leukemia comprise advanced SM [14]. Indolent SM is the most common form of SM and is diagnosed when criteria for SM are met without evidence of an associated clonal hematologic disorder or MC-related end-organ damage such as severe liver disease, hypersplenism, or significant lymphadenopathy. Patients with CM and ISM have survival rates similar to the general population, whereas survival in patients with advanced SM is reduced owing to a higher disease burden, an associated hematologic neoplasm, or MC-related end-organ damage [15]. Approximately 50% of patients with SM with an associated myeloid hematologic neoplasm and aggressive SM do not display typical cutaneous lesions [9], nor are typical skin lesions common in MC leukemia. Bone marrow mastocytosis, often associated with venom-induced anaphylaxis, also lacks typical skin lesions but has more favorable survival [16]. Well-differentiated SM, when seen in a setting of ISM, may present with a familial pattern and variable cutaneous manifestations [17]. A summary of mastocytosis subtypes is presented in Table 1.
Table 1.
Mastocytosis types and subtypes
| WHO 5th Editiona | International Consensus Classification |
|---|---|
| Cutaneous mastocytosis | |
| Subtypes | |
| Maculopapular cutaneous mastocytosis (urticaria pigmentosa) | Maculopapular cutaneous mastocytosis (urticaria pigmentosa) |
| Monomorphic | Diffuse cutaneous mastocytosis |
| Polymorphic | Mastocytoma of skin |
| Diffuse cutaneous mastocytosis | |
| Cutaneous mastocytoma | |
| Isolated mastocytoma | |
| Multi-localized mastocytoma | |
| Systemic mastocytosis (SM) | |
| Subtypes | |
| Bone marrow mastocytosisb | |
| Indolent SM | |
| Smoldering SM | |
| Aggressive SM | |
| SM with an associated hematologic neoplasmc | |
| Mast cell leukemia | |
| Mast cell sarcoma | |
SM systemic mastocytosis, WHO World Health Organization
aWell-differentiated SM is a morphologic variant present in all subtypes of SM
bBone marrow is now considered a separate subtype of SM in the WHO 5th ed classification of hematolymphoid tumors characterized by no mastocytosis skin lesions, no B-findings, and a basal serum total tryptase level <125 ng/mL, whereas in the International Consensus Classification it is considered a clinicopathologic variant of indolent SM
cIn the International Consensus Classification, SM with an associated hematologic neoplasm has been modified such that the associated hematologic neoplasm is defined as an associated myeloid neoplasm only
Adapted from Arber et al. Blood. 2022;140(11):1200–8 [10]; Khoury et al. Leukemia. 2022;36:1703–19 [11]; National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology. Systemic mastocytosis. 2024; version 3.2024 [14]
Presentation
Morphology
Classically, affected individuals present with red-brown macules, papules, nodules, and/or plaques that produce dermal edema and erythema upon mechanical stimulation (Darier’s sign) [18, 19]. To perform this test accurately, the lesion is stroked five times with moderate pressure using a tongue depressor. Within several minutes, erythema and edema will develop that will be absent in surrounding unaffected skin (thus differentiating this from dermatographism) [20]. The presence of a positive Darier’s sign is particularly useful when establishing a diagnosis in the setting of classic morphology, although in cases of diagnostic uncertainty, histopathology with appropriate staining will be more definitive. Notably, a positive Darier’s sign is more likely to be absent in adults, in patients concurrently receiving antihistamines, and if performed incorrectly [20]. Testing too aggressively may also result in false-positive results or rarely in systemic symptoms—flushing, wheezing, or hypotension—in children with nodular skin lesions.
Atypical presentations require a high index of suspicion and expertise. Skin lesions may have atypical distribution—intertriginous [21, 22], acral [23], unilateral localization [24]—and additional cutaneous features may be present, including pseudoxanthomatous/xanthelasmoid morphology [25–27]. Secondary changes such as acquired cutis laxa and leonine facies are seldomly observed [28, 29]. Non-specific features, including flushing and pruritus, are frequently present and may be the most prominent features of the disease, particularly when the burden of fixed skin lesions is low.
Historically, morphologic classification centered around terms such as urticaria pigmentosa and telangiectasia macularis eruptiva perstans. Recent reclassification has challenged the validity of these as unique variants and instead favors the encompassing term “maculopapular cutaneous mastocytosis” (MPCM). Current guidelines recommend dividing the cutaneous manifestations of mastocytosis into three subtypes, namely, cutaneous mastocytoma, diffuse cutaneous mastocytosis (DCM), and MPCM; this terminology will be utilized throughout this article [20].
Cutaneous mastocytomas are characterized by solitary or a few raised, yellow, red, or brown rubbery ovoid nodules or plaques that may develop secondary blistering and frequently demonstrate a positive Darier’s sign. This form of disease presents in early childhood, is defined by remission during adolescence, and lacks an association with systemic disease [20, 30].
Diffuse cutaneous mastocytosis also generally presents in early childhood. Children with DCM often lack individual lesions and instead manifest with generalized skin thickening (pachydermia) with alteration of skin color. Mechanical irritation frequently produces erythema and cutaneous edema and may evoke blistering. Anaphylaxis may also occur [31]. There is often an elevated serum tryptase level at presentation, but most children lack internal organ involvement. A tendency toward prolonged cutaneous bleeding may be present in the setting of local heparin release, but blood parameters are characteristically normal. Reassuringly, DCM also classically resolves by adolescence [20, 31, 32].
Maculopapular cutaneous mastocytosis represents the most heterogeneous subtype. A consensus statement from the European Competence Network on Mastocytosis, the American Academy of Allergy, Asthma, & Immunology, and the European Academy of Allergology and Clinical Immunology recommended further division of MPCM into polymorphic and monomorphic categories [20]. The polymorphic form predominates in children and is defined by findings of red-brown lesions of varying sizes (Fig. 1). These are often fewer in number compared with monomorphic MPCM, with a predilection for the head and neck in addition to the trunk and extremities [20]. As with cutaneous mastocytoma and DCM, polymorphic MPCM is usually temporary and generally lacks an association with systemic disease. Rarely, adults may also present with polymorphic MPCM or skin lesions consistent with DCM. These individuals are more likely to have an earlier age at onset, BM features of well-differentiated SM, and present with non-D816V KIT mutations [17].
Fig. 1.

Polymorphic maculopapular cutaneous mastocytosis and monomorphic maculopapular cutaneous mastocytosis in children of varied skin phototypes
Monomorphic MPCM is characterized by small uniform red-brown macules and papules with a predilection for the trunk and proximal extremities, classically sparing the face (Fig. 2). This is the predominant presentation of adult-onset MIS, although children may also be affected. In adults, it is strongly associated with SM and requires surveillance and staging. The monomorphic pattern in pediatric patients has also been associated with more severe and persistent disease beyond adolescence [20, 33, 34]. The number of lesions ranges from less than a dozen to near-universal involvement [20]. This morphology is persistent over time and carries a risk of anaphylaxis [20, 35]. Notably, as this classification schema is largely intuitive and based on experience, limitations in distinguishing monomorphic and polymorphic disease exist. In one small study, interobserver variability in the classification of MPCM in pediatric patients was demonstrated even among experts [36].
Fig. 2.
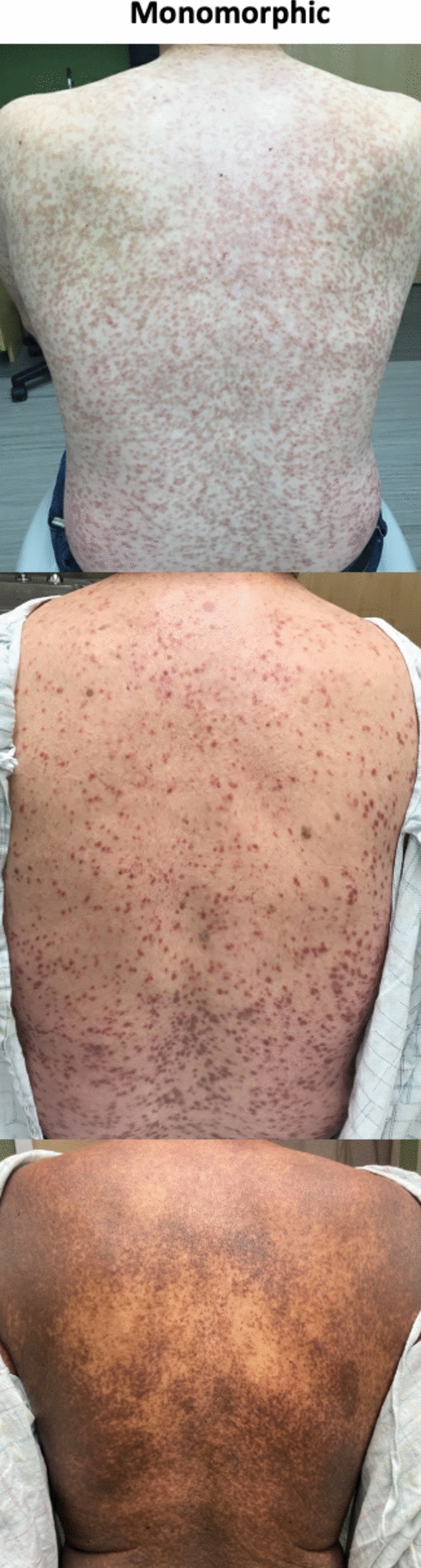
Monomorphic maculopapular cutaneous mastocytosis in adults of different skin types
Systemic Symptoms
Patients with SM frequently present with extracutaneous manifestations including abdominal pain, nausea/emesis, bowel pattern changes, musculoskeletal pain, vascular instability, and neuropsychiatric complaints leading to an impaired quality of life [37–39]. Fifty-six percent of adult patients with SM may also experience anaphylaxis, [40–42] and pathologic fractures, due to premature osteopenia or osteoporosis, are reported in 28–56% of patients with SM [43–45]. The prevalence of idiopathic anaphylaxis in patients with pediatric mastocytosis has also been reported to be higher than the general pediatric population; however, the prevalence of other triggers is lower [46]. Moreover, the prevalence of anaphylaxis in pediatric mastocytosis is lower than in adult patients with mastocytosis [42, 46]. A complete review of systems is imperative in patients presenting with MIS, as the presence of additional features may increase the suspicion for SM [47].
Histopathology
A skin biopsy should be considered in the initial diagnostic work-up if MIS is suspected and clinical manifestations are not conclusive. Pathologists should be notified of the clinical suspicion on the requisition as histopathological features can be subtle [48]. The duration of the lesion typically does not affect pathology; however, older lesions may not be as responsive when testing for Darier’s sign. Given that fibroblasts can be activated by MCs [49], the biopsy site may have an exaggerated fibrotic response. Therefore, the authors tend to biopsy areas covered by clothing such as the trunk.
Mastocytosis in the skin is characterized by the abnormal accumulation of MCs in the dermis of affected skin. In skin biopsies, MC distribution, number, and density, as well as the presence of additional “non-MC” findings, such as pigmentary change, dermal vascular proliferation and dilatation, eosinophilia, and dermal fibrosis, are all of importance. Mast cells can be highlighted by positive cluster of differentiation (CD) 117 (most sensitive) and tryptase (most specific) immunostaining [50]. When suspecting mastocytosis, additional antigens of aberrant expression in MCs, such as CD2, CD25, and more recently, CD30 [51], may be tested though have less utility in the skin than in other organs. The MC receptor Mas-related G protein-coupled receptor X2 (MRGPRX2) may also be analyzed in patients with MPCM [52]. The density of MCs in normal skin is variable and dependent on the location and the presence of other inflammatory dermatoses [53]; thus, an evaluation by an experienced dermatopathologist is important when considering the diagnosis, and a clinicopathologic correlation remains necessary. Discrimination of isolated CM from SM with cutaneous involvement is often not possible based on skin histopathology alone. As discussed previously, the term MIS is favored when describing an abnormal cutaneous proliferation of MC until an appropriate evaluation and staging have been performed [54]. Anecdotally, the authors have found that cases signed out as MIS in the adult population are more likely to prompt an additional evaluation compared with cases signed out as compatible with CM or urticaria pigmentosa.
The patterns of MC distribution were recently updated by Drabent et al. into “sheet-like,” “subepidermal,” and “perivascular” patterns [55]. The former two are strongly specific for mastocytosis. The presence of a dense sheet-like proliferation of MCs makes the histologic diagnosis of DCM and mastocytoma more straightforward than MPCM, which often presents with more subtle histologic findings. When not in a sheet-like or subepidermal pattern, evaluation of the MC component of the inflammatory infiltrate is important to discriminate mastocytosis from other inflammatory disorders associated with increased MCs (Fig. 3). A 40% cut-off for the percentage of MCs among inflammatory cells has been shown to have a sensitivity of >75% and a specificity approaching 100%. In cases wherein the MC component is <40%, additional features (including number of MCs per mm2) may improve sensitivity [55]. A retrospective histopathologic study by King et al. suggests that a MC density >66 per mm2 is a useful threshold to distinguish MPCM from chronic urticaria [56].
Fig. 3.

Histopathologic images showing mast cell infiltration in the skin from a maculopapular cutaneous mastocytosis presentation. (a) A brisk infiltrate of mast cells in the superficial and deep reticular dermis is present with a perivascular and interstitial distribution (hematoxylin and eosin, 20×). (b, c) The mast cells reveal an abnormal morphology characterized by a combination of epithelioid and spindle cell features (hematoxylin and eosin, 100× [b] and 200× [c]). (d) The mast cells are diffusely positive for cluster of differentiation 117 (20×)
A recently published scoring model has been proposed by experts in the field (Table 2), though the model does not account for presentations in skin of color [57]. This model has not yet been widely adopted in clinical practice, and further data may validate such tools for implementation in the future. Testing the skin biopsy for the presence of KIT D816V using a high-sensitivity assay enhances the robustness of this model but is not widely available (see Sect. 4 for more details).
Table 2.
Proposed scoring model to assist with the pathologic diagnosis of MPCM
| Variable | Score (points) |
|---|---|
| ≥27 subepidermal MC/HPF | 3 |
| ≥12 subepidermal MC/HPF | 2 |
| ≥7 subepidermal MC/HPF | 1 |
| Intermediate or strong basal pigmentationa | 1 |
| Interstitial distribution of MCs | 1 |
| ≥3 MC clusters | 1 |
| Interpretation | |
| MPCM highy likely (Sp = 97.2%, Sn = 90.6%) | 4–6 |
| MPCM unconfirmed (Sp = 38.9%, Sn = 100%) | 1–3 |
| MPCM excluded | 0 |
HPF high-power field, MC mast cell, MPCM maculopapular cutaneous mastocytosis, Sn sensitivity, Sp specificity
aApplies to Caucasian individuals only
Adapted from Gebhard et al. J Eur Acad Dermatol Venereol. 2022;36(8):1367–75 [57]
Laboratory Studies
As mentioned, the risk of SM is highest in patients with monomorphic MPCM but rarely occurs in those with DCM morphology [58, 59]. Laboratory evaluations are useful in diagnosis, risk stratification, and assessment of disease burden in patients with known or suspected SM (Fig. 4). Evaluation of serum tryptase levels is indicated in all variants of mastocytosis; further laboratory testing is generally not indicated for children with CM alone. Although an elevated basal serum tryptase (BST) level is a valuable clinical parameter, the oft-held view that adults with a serum tryptase level of < 20 ng/mL do not require further evaluation is not supported by the literature or clinical experience [60]. In a retrospective cohort from the Toulouse Excellence Center for Mastocytosis, 27% of adults with cutaneous involvement in the setting of SM were found to have tryptase levels below this metric, with 15% of patients demonstrating levels of < 11.3 ng/mL [61]. Tryptase genotyping is also recommended to interpret whether an elevated BST value is due to different tryptase genotypes (including hereditary alpha tryptasemia) or an MC abnormality [13, 62–64].
Fig. 4.

Evaluation of adult and pediatric patients with mastocytosis in the skin. ABD abdominal, DCM diffuse cutaneous mastocytosis, HαT hereditary alpha-tryptasemia, MC mast cell, MPCM maculopapular cutaneous mastocytosis, PB peripheral blood, SBT serum basal tryptase, yo years old
The KIT D816V somatic variant is the driving mutation in ~ 95% of patients with SM [65, 66]. High-sensitivity (≥ 0.01%) quantitative polymerase chain reaction is recommended over Sanger- or next-generation sequencing-based methods for KIT p.D816V detection [67–69]. Peripheral blood (PB) KIT D816V quantitative polymerase chain reaction and BST are both recommended in the preliminary diagnostic evaluation for SM using the World Health Organization diagnostic criteria [70]. Next-generation sequencing panels focused on myeloproliferative and myelodysplastic neoplasms are also recommended for patients with SM to evaluate for additional mutations suggestive of SM with an associated myeloid hematologic neoplasm.
Treatment and Management
Initial treatment and subsequent evaluation of clinically diagnosed or biopsy-proven MIS are largely based on the subform/variant. All patients should undergo a physical examination to assess for hepatomegaly, splenomegaly, and lymphadenopathy, and a complete review of systems to identify features suggestive of systemic disease. Patients and families should be counseled on the risk of anaphylaxis and be trained in the use of an epinephrine autoinjector. All patients should be offered treatment directed at their cutaneous and extracutaneous symptoms even in the absence of SM or if the SM evaluation is still ongoing. Skin hydration is paramount, and pruritus, wheals, and flushing may be treated with non-sedating second-generation H1 antihistamines (dosed at up to four times the standard dose based on data in patients with chronic spontaneous urticaria) and/or MC stabilizers such as cromolyn [71]. Phototherapy, including the use of narrowband ultraviolet B and 8-methoxypsoralen plus ultraviolet A light, has demonstrated a benefit in adult patients with CM and SM [72–74]; although emerging evidence suggesting susceptibility to a cutaneous malignancy in patients with SM has led to caution with use. Omalizumab, an anti-immunoglobulin E monoclonal antibody, may be considered in patients with urtication and significant mediator symptoms; however, two small prospective clinical trials were unable to prove a definitive advantage, despite a tendency toward symptom reduction [75–77].
Patients with MIS who should be referred for evaluation and consideration of a BM biopsy (preferably at a Center of Excellence [78] with expertise and access to high-complexity molecular testing) are (1) adult patients (onset at ≥18 years of age) with monomorphic MPCM; (2) patients with familial forms of DCM; (3) patients with DCM and features suggestive of systemic disease such as splenomegaly, pathologic fractures, and cytopenias/cytoses; and (4) pediatric patients with monomorphic MPCM wherein there is suspicion of SM such as positive PB KIT D816V or organomegaly (Fig. 4) [79]. As stated previously, BST values do not independently determine whether an adult patient with MIS is a candidate for a BM biopsy as BST values may be less than the < 20 ng/mL (minor criterion) cut-off in patients with SM [61, 67]. Further, the most common etiology of an elevated baseline serum tryptase is hereditary alpha tryptasemia [80]; when possible, genetic testing for hereditary alpha tryptasemia can be included in the work-up. Scoring criteria, such as the European Competence Network on Mastocytosis/Fuchs’ score, can be beneficial in risk stratification and to facilitate informed decision making as patients who lack positivity for KIT D816V mutations and have a low score may be considered for close clinical monitoring alone [81]. Children who are clinically stable with a polymorphic pattern (which is not typically seen in adult-onset disease) can also be followed with PB laboratories including serum tryptase and blood counts at 6- to 12-month intervals. [20, 79]. If there is a suspicion for systemic disease in a child based on an elevated serum tryptase and organomegaly, a PB KIT D816V mutation should be ordered along with an abdominal ultrasound to verify organomegaly [82]. Patients requiring BM studies or additional therapy beyond symptomatic support can be referred to a Center of Excellence or reference center with multidisciplinary expertise to include hematology [83]. A BM evaluation at an experienced center is important as errors in diagnosis and classification occur more frequently in centers with a low case volume [67, 84, 85]. There are currently 21 identified Center of Excellence facilities within the USA along with five additional reference centers [86].
Historically, treatment options were limited for patients with SM and often reserved for those with advanced disease. Within the past decade, there have been significant advancements with the introduction of tyrosine kinase inhibitors (TKIs), which have the potential to induce partial or complete remission and improve patient-reported outcomes. Treatment with TKIs is reserved for patients with SM based on the underlying KIT driving mutation, SM subtype, and symptom burden. If a patient with SM is not adequately staged (e.g., when an appropriate pediatric or adult patient is not referred for a BM biopsy), they may be excluded from meaningful treatment.
Imatinib was the first TKI to be assessed and was found to be effective in some patients with SM who have wild-type or harbor mutations outside of codon 816 in KIT [47, 87–92]. Midostaurin, a multikinase inhibitor capable of inhibiting KIT harboring 816 codon substitutions, is currently US Food and Drug Administration approved for advanced SM. Midostaurin reduced the BM MC burden, spleen volume, and mastocytosis-related organ damage in an open-label phase II study of patients with advanced SM [93, 94]. Toxicity related to midostaurin was frequent, included cytopenias and gastrointestinal problems, and often required a dose reduction. A small phase II, non-randomized, single-center, open-label study of midostaurin treatment for patients with non-advanced disease demonstrated symptom improvement [95]. Avapritinib is a TKI that is selective for KIT harboring 816 codon substitutions and has been approved by the Food and Drud Administration at 200 mg once daily for advanced SM and at 25 mg once daily for ISM. Avapritinib in advanced SM led to complete or partial remission in most patients and improved survival [96–100]. In ISM, avapritinib use was associated with meaningful improvements in patient-reported outcomes and disease biomarkers [101, 102]. Additional targeted TKIs are currently under development and available through clinical trial protocols at experienced centers [103].
Conclusions
Mastocytosis presents in the skin as part of a heterogeneous group of clonal conditions affecting both adults and children. Classification criteria and staging of SM, as well as the treatment landscape, have evolved significantly in recent years. Despite this, many patients who warrant additional evaluation go without an appropriate referral. By addressing knowledge gaps related to diagnosis, terminology, evaluation, and treatment options, dermatologists will be better positioned to identify patients who require additional scrutiny and may benefit from novel therapeutics.
Acknowledgments
Medical editorial assistance was provided by Marcelene Yumul, MD, of Healthcare Consultancy Group, funded by Blueprint Medicines Corporation, Cambridge, MA, USA according to current policies established by the International Committee of Medical Journal Editors and Good Publication Practice guidelines.
Declarations
Funding
Blueprint Medicines Corporation provided funding for the preparation of this article.
Conflicts of Interest
Lauren M. Madigan served on an international advisory board for Blueprint Medicines and participated as a speaker in a non-Continuing Medical Education (CME) session on recognizing and diagnosis mastocytosis; Section editor, JAMA Dermatology. Nathan A. Boggs participated in a Blueprint Medicines-sponsored patient advocacy summit; participated in developing a Blueprint-sponsored webinar for the College of American Pathologists. Anton V. Rets participated in developing a Blueprint Medicines-sponsored webinar for the College of American Pathologists. Alejandro A. Gru is an investigator for Innate Pharma, Stemline Therapeutics, and Dren Bio; a consultant for Blueprint Medicines; and a speaker for Kyowa Kirin. Tsewang Tashi is a member of the advisory board and a primary investigator in Blueprint Medicines clinical trials, Cogent Biosciences, and PharmaEssentia; a member of the study steering committee for Blueprint Medicines clinical trials. David. A Wada and Melody C. Carter have no conflicts of interest that are directly relevant to the content of this article. Scott R. Florell is a consultant for Orlucent Inc.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Patients or their representatives provided consent for the publication of any identifiable photographs.
Availability of Data and Material
Not applicable.
Code Availability
Not applicable.
Authors’ Contributions
All authors contributed to the concept, research, and writing of the manuscript. All authors read and approved the final manuscript.
Footnotes
This article was revised due to retrospective open access.
Change history
7/23/2025
A Correction to this paper has been published: 10.1007/s40257-025-00972-6
References
- 1.Kristensen T, Vestergaard H, Moller MB. Improved detection of the KIT D816V mutation in patients with systemic mastocytosis using a quantitative and highly sensitive real-time qPCR assay. J Mol Diagn. 2011;13(2):180–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen SS, Skovbo S, Vetergaard H, et al. Epidemiology of systemic mastocytosis in Denmark. Br J Haematol. 2014;166(4):521–8. [DOI] [PubMed] [Google Scholar]
- 3.Valent P, Sperr WR, Schwartz LB, et al. Diagnosis and classification of mast cell proliferative disorders: delineation from immunologic diseases and non-mast cell hematopoietic neoplasms. J Allergy Clin Immunol. 2004;114(1):3–11. [DOI] [PubMed] [Google Scholar]
- 4.Brockow K. Epidemiology, prognosis, and risk factors in mastocytosis. Immunol Allergy Clin North Am. 2014;34(2):283–95. [DOI] [PubMed] [Google Scholar]
- 5.Carter MC, Metcalfe DD. Mastocytosis. In: Young NS, Gerson SL, Hight KH, editors. Clinical hematology. Philadelphia (PA): Mosby, Inc.; 2006.
- 6.Chang A, Tung RC, Schlesinger T, et al. Familial cutaneous mastocytosis. Pediatr Dermatol. 2001;18(4):271–6. [DOI] [PubMed] [Google Scholar]
- 7.Ben-Amitai D, Metzker A, Cohen HA. Pediatric cutaneous mastocytosis: a review of 180 patients. Isr Med Assoc J. 2005;7(5):320–2. [PubMed] [Google Scholar]
- 8.Bodemer C, Hermine O, Palmerini F, et al. Pediatric mastocytosis is a clonal disease associated with D816V and other activating c-KIT mutations. J Invest Dermatol. 2010;130(3):804–15. [DOI] [PubMed] [Google Scholar]
- 9.Valent P, Akin C, Escribano L, et al. Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria. Eur J Clin Invest. 2007;37(6):435–53. [DOI] [PubMed] [Google Scholar]
- 10.Arber DA, Orazi A, Hasserjian RP, et al. International Consensus Classification of myeloid neoplasms and acute leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022;140(11):1200–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khoury JD, Solary E, Abla O, et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia. 2022;36(7):1703–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Valent P, Sotlar K, Horny HP, et al. World Health Organization classification and diagnosis of mastocytosis: update 2023 and future perspectives. Immunol Allergy Clin North Am. 2023;43(4):627–49. [DOI] [PubMed] [Google Scholar]
- 13.Valent P, Akin C, Hartmann K, et al. Updated diagnostic criteria and classification of mast cell disorders: a consensus proposal. Hemasphere. 2021;5(11): e646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.NCCN clinical practice guidelines in oncology (NCCN Guidelines®) for systemic mastocytosis V.3.2024. © National Comprehensive Cancer Network, Inc. 2024. Available from: https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1490. Accessed 12 Sep 2024.
- 15.Kluin-Nelemans HC, Oldhoff JM, Van Doormaal JJ, et al. Cladribine therapy for systemic mastocytosis. Blood. 2003;102(13):4270–6. [DOI] [PubMed] [Google Scholar]
- 16.Pardanani A, Lim, KH, Lasho TL, et al. WHO subvariants of indolent mastocytosis: clinical details and prognostic evaluation in 159 consecutive adults. Blood. 2010;115(1):150–1. [DOI] [PubMed] [Google Scholar]
- 17.Alvarez-Twose I, Jara-Acevedo M, Morgado JM, et al. Clinical, immunophenotypic, and molecular characteristics of well-differentiated systemic mastocytosis. J Allergy Clin Immunol. 2016;137(1):168–78. [DOI] [PubMed] [Google Scholar]
- 18.Caplan RM. Urticaria pigmentosa and systemic mastocytosis. JAMA. 1965;194(10):1077–80. [PubMed] [Google Scholar]
- 19.Sezary ALCG, Chauvillon P. Dermographisme et mastocytose. Bull Soc Fr Dermatol Syph. 1936;43:359–61. [Google Scholar]
- 20.Hartmann K, Escribano L, Grattan C, et al. Cutaneous manifestations in patients with mastocytosis: consensus report of the European Competence Network on Mastocytosis; the American Academy of Allergy, Asthma & Immunology; and the European Academy of Allergology and Clinical Immunology. J Allergy Clin Immunol. 2016;137(1):35–45. [DOI] [PubMed] [Google Scholar]
- 21.Herrero-Moyano M, Capusan TM, Perez-Plaza A, et al. Intertriginous maculopapular mastocytosis in a patient with acute myeloid leukemia. JAAD Case Rep. 2017;3(1):61–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Simmons BJ, LeBlanc RE, Glass JS. Intertriginous mastocytosis: a rare presentation of an uncommon disease. Int J Dermatol. 2019;58(7):852–3. [DOI] [PubMed] [Google Scholar]
- 23.Sammut J, Mercieca L, Boffa MM, et al. Palmoplantar maculopapular cutaneous mastocytosis. Int J Dermatol. 2019;58(4):E79-80. [DOI] [PubMed] [Google Scholar]
- 24.Soilleux EJ, Brown VL, Bowling J. Cutaneous mastocytosis localized to a radiotherapy field. Clin Exp Dermatol. 2009;34(1):111–2. [DOI] [PubMed] [Google Scholar]
- 25.Griffiths WA, Daneshbod K. Pseudoxanthomatous mastocytosis. Br J Dermatol. 1975;93(1):91–5. [DOI] [PubMed] [Google Scholar]
- 26.Nabavi NS, Nejad MH, Feli S, et al. Adult onset of xanthelasmoid mastocytosis: report of a rare entity. Indian J Dermatol. 2016;61(4):468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Revert A, Jorda E, Ramon D, et al. Xanthelasmoid mastocytosis. Pediatr Dermatol. 1991;8(2):152–4. [DOI] [PubMed] [Google Scholar]
- 28.Hoang MV, Van Dang P, Van Bui D, et al. Acquired cutis laxa associated with cutaneous mastocytosis. Dermatol Online J. 2015;21(7):13030. [PubMed] [Google Scholar]
- 29.Mutreja D, Purohit A, Singh PK, et al. A 60-year-old lady with leonine facies: a rare diagnosis. Indian J Pathol Microbiol. 2012;55(4):566–8. [DOI] [PubMed] [Google Scholar]
- 30.Leung AKC, Lam JM, Leong KF. Childhood solitary cutaneous mastocytoma: clinical manifestations, diagnosis, evaluation, and management. Curr Pediatr Rev. 2019;15(1):42–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lange M, Niedoszytko M, Nedoszytko B, et al. Diffuse cutaneous mastocytosis: analysis of 10 cases and a brief review of the literature. J Eur Acad Dermatol Venereol. 2012;26(12):1565–71. [DOI] [PubMed] [Google Scholar]
- 32.Uzzaman A, Maric I, Noel P, et al. Pediatric-onset mastocytosis: a long term clinical follow-up and correlation with bone marrow histopathology. Pediatr Blood Cancer. 2009;53(4):629–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wiechers T, Rabenhorst A, Schick T, et al. Large maculopapular cutaneous lesions are associated with favorable outcome in childhood-onset mastocytosis. J Allergy Clin Immunol. 2015;136(6):1581-90.e3. [DOI] [PubMed] [Google Scholar]
- 34.Matito A, Azana JM, Torrelo A, et al. Cutaneous mastocytosis in adults and children: new classification and prognostic factors. Immunol Allergy Clin North Am. 2018;38(3):351–63. [DOI] [PubMed] [Google Scholar]
- 35.Brockow K, Akin C, Huber M, et al. Assessment of the extent of cutaneous involvement in children and adults with mastocytosis: relationship to symptomatology, tryptase levels, and bone marrow pathology. J Am Acad Dermatol. 2003;48(4):508–16. [DOI] [PubMed] [Google Scholar]
- 36.Brockow K. Unsatisfactory agreement using current classification of maculopapular cutaneous mastocytosis. J Eur Acad Dermatol Venereol. 2021;35(10):1917–8. [DOI] [PubMed] [Google Scholar]
- 37.Jennings S, Russell N, Jennings B, et al. The Mastocytosis Society survey on mast cell disorders: patient experiences and perceptions. J Allergy Clin Immunol Pract. 2014;2(1):70–6. [DOI] [PubMed] [Google Scholar]
- 38.Pyatilova P, Akin C, Alvarez-Twose I, et al. Refined treatment response criteria for indolent systemic mastocytosis proposed by the ECNM-AIM Consortium. J Allergy Clin Immunol Pract. 2022;10(8):2015–24. [DOI] [PubMed] [Google Scholar]
- 39.van Anrooij B, Kluin-Nelemans JC, Safy M, et al. Patient-reported disease-specific quality-of-life and symptom severity in systemic mastocytosis. Allergy. 2016;71(11):1585–93. [DOI] [PubMed] [Google Scholar]
- 40.Alvarez-Twose I, de Olano DG, Sanchez-Munoz L, et al. Clinical, biological, and molecular characteristics of clonal mast cell disorders presenting with systemic mast cell activation symptoms. J Allergy Clin Immunol. 2010;125(6):1269-78.e2. [DOI] [PubMed] [Google Scholar]
- 41.Brockow K, Jofer C, Behrendt H, et al. Anaphylaxis in patients with mastocytosis: a study on history, clinical features and risk factors in 120 patients. Allergy. 2008;63(2):226–32. [DOI] [PubMed] [Google Scholar]
- 42.Brockow K, Plata-Nazar K, Lange M, et al. Mediator-related symptoms and anaphylaxis in children with mastocytosis. Int J Mol Sci. 2021;22(5):2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Degboe Y, Eischen M, Nigon D, et al. Prevalence and risk factors for fragility fracture in systemic mastocytosis. Bone. 2017;105:219–25. [DOI] [PubMed] [Google Scholar]
- 44.Makovoz A, Wang J, Oshegbo G, et al. Assessment of osteoporosis and fracture risk in mastocytosis within a North American cohort. J Allergy Clin Immunol Pract. 2021;9(12):4459-67.e10. [DOI] [PubMed] [Google Scholar]
- 45.van der Veer E, van der Goot W, de Monchy JGR, et al. High prevalence of fractures and osteoporosis in patients with indolent systemic mastocytosis. Allergy. 2012;67(3):431–8. [DOI] [PubMed] [Google Scholar]
- 46.Matito A, Carter M. Cutaneous and systemic mastocytosis in children: a risk factor for anaphylaxis? Curr Allergy Asthma Rep. 2015;15(5):22. [DOI] [PubMed] [Google Scholar]
- 47.Lim KH, Tefferi A, Lasho TL, et al. Systemic mastocytosis in 342 consecutive adults: survival studies and prognostic factors. Blood. 2009;113(23):5727–36. [DOI] [PubMed] [Google Scholar]
- 48.Castells M, Metcalfe DD, Escribano L. Diagnosis and treatment of cutaneous mastocytosis in children: practical recommendations. Am J Clin Dermatol. 2011;12(4):259–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoermann G, Cerny-Reiterer S, Perne A, et al. Identification of oncostatin M as a STAT5-dependent mediator of bone marrow remodeling in KIT D816V-positive systemic mastocytosis. Am J Pathol. 2011;178(5):2344–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Algermissen B, Bauer F, Schadendorf D, et al. Analysis of mast cell subpopulations (MCT, MCTC) in cutaneous inflammation using novel enzyme-histochemical staining techniques. Exp Dermatol. 1994;3(6):290–7. [DOI] [PubMed] [Google Scholar]
- 51.Mitteldorf C, Kulberg A, Tronnier M, et al. Subcellular expression of CD30 in cutaneous mastocytosis: an important factor for targeted treatment. J Cutan Pathol. 2024;51(11):881–92. [DOI] [PubMed] [Google Scholar]
- 52.Deepak V, Komarow HD, Alblaihess AA, et al. Expression of MRGPRX2 in skin mast cells of patients with maculopapular cutaneous mastocytosis. J Allergy Clin Immunol Pract. 2021;9(10):3841-3.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Janssens AS, Heide R, den Hollander JC, et al. Mast cell distribution in normal adult skin. J Clin Pathol. 2005;58(3):285–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hollmann TJ, Brenn T, Hornick JL. CD25 expression on cutaneous mast cells from adult patients presenting with urticaria pigmentosa is predictive of systemic mastocytosis. Am J Surg Pathol. 2008;32(1):139–45. [DOI] [PubMed] [Google Scholar]
- 55.Drabent P, Polivka L, Agopian J, et al. Establishing diagnostic criteria for mastocytosis in skin biopsies. Histopathology. 2022;80(3):501–14. [DOI] [PubMed] [Google Scholar]
- 56.King AL, Montagnon CM, Todd A, et al. Quantifying mast cell and eosinophil cellular density in skin biopsy tissue from adults with maculopapular cutaneous mastocytosis as compared with urticaria and normal skin: a retrospective histopathologic study. Am J Dermatopathol. 2025;47(2):105–9. [DOI] [PubMed] [Google Scholar]
- 57.Gebhard J, Horny HP, Kristensen T, et al. Validation of dermatopathological criteria to diagnose cutaneous lesions of mastocytosis: importance of KIT D816V mutation analysis. J Eur Acad Dermatol Venereol. 2022;36(8):1367–75. [DOI] [PubMed] [Google Scholar]
- 58.Berezowska S, Flaig MJ, Rueff F, et al. Adult-onset mastocytosis in the skin is highly suggestive of systemic mastocytosis. Mod Pathol. 2014;27(1):19–29. [DOI] [PubMed] [Google Scholar]
- 59.Carter MC, Metcalfe DD, Clark AS, et al. Abnormal bone marrow histopathology in paediatric mastocytosis. Br J Haematol. 2015;168(6):865–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heymann WR. Telangiectasia macularis eruptiva perstans revisited. Dermatology world insights and inquiries. American Academy of Dermatology, 2016. Available from: https://www.aad.org/dw/dw-insights-and-inquiries/medical-dermatology/telangiectasia-macularis-eruptiva-perstans-revisited. [Accessed 14 Feb 2024].
- 61.Jendoubi F, Shourick J, Negretto M, et al. Cutaneous mastocytosis in adults with a serum tryptase level < 20 ng mL(-1): why we should investigate further. Br J Dermatol. 2021;185(2):453–5. [DOI] [PubMed] [Google Scholar]
- 62.Chovanec J, Tunc I, Hughes J, et al. Genetically defined individual reference ranges for tryptase limit unnecessary procedures and unmask myeloid neoplasms. Blood Adv. 2023;7(9):1796–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lyons JJ, Yu X, Hughes JD, et al. Elevated basal serum tryptase identifies a multisystem disorder associated with increased TPSAB1 copy number. Nat Genet. 2016;48(12):1564–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Waters AM, Park HJ, Weskamp AL, et al. Elevated basal serum tryptase: disease distribution and variability in a eregional health system. J Allergy Clin Immunol Pract. 2022;10(9):2424-35.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Garcia-Montero AC, Jara-Acevedo M, Teodosio C, et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: a prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood. 2006;108(7):2366–72. [DOI] [PubMed] [Google Scholar]
- 66.Nagata H, Worobec AS, Oh CK, et al. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc Natl Acad Sci U S A. 1995;92(23):10560–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Boggs NA, Sun X, Lyons JJ, et al. Challenges in applying diagnostic criteria for systemic mastocytosis. Blood Adv. 2023;7(13):3150–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sotlar K, Horny HP, Simonitsch I, et al. CD25 indicates the neoplastic phenotype of mast cells: a novel immunohistochemical marker for the diagnosis of systemic mastocytosis (SM) in routinely processed bone marrow biopsy specimens. Am J Surg Pathol. 2004;28(10):1319–25. [DOI] [PubMed] [Google Scholar]
- 69.Greiner G, Gurbisz M, Ratzinger F, et al. Digital PCR: a sensitive and precise method for KIT D816V quantification in mastocytosis. Clin Chem. 2018;64(3):547–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.World Health Organization. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Vol. 2. Lyon: International Agency for Research on Cancer; 2017.
- 71.Guillen-Aguinaga S, Jauregui Presa I, Aguinaga-Ontoso E, et al. Updosing nonsedating antihistamines in patients with chronic spontaneous urticaria: a systematic review and meta-analysis. Br J Dermatol. 2016;175(6):1153–65. [DOI] [PubMed] [Google Scholar]
- 72.Brazzelli V, Grassi S, Merante S, et al. Narrow-band UVB phototherapy and psoralen-ultraviolet A photochemotherapy in the treatment of cutaneous mastocytosis: a study in 20 patients. Photodermatol Photoimmunol Photomed. 2016;32(5–6):238–46. [DOI] [PubMed] [Google Scholar]
- 73.Christophers E, Honigsmann H, Wolff K, et al. PUVA-treatment of urticaria pigmentosa. Br J Dermatol. 1978;98(6):701–2. [DOI] [PubMed] [Google Scholar]
- 74.Godt O, Procksch E, Streit V, et al. Short- and long-term effectiveness of oral and bath PUVA therapy in urticaria pigmentosa and systemic mastocytosis. Dermatology. 1997;195(1):35–9. [DOI] [PubMed] [Google Scholar]
- 75.Distler M, Maul JT, Steiner UC, et al. Efficacy of omalizumab in mastocytosis: allusive indication obtained from a prospective, double-blind, multicenter study (XOLMA study). Dermatology. 2020;236(6):529–39. [DOI] [PubMed] [Google Scholar]
- 76.Jendoubi F, Gaudenzio N, Gallini A, et al. Omalizumab in the treatment of adult patients with mastocytosis: a systematic review. Clin Exp Allergy. 2020;50(6):654–61. [DOI] [PubMed] [Google Scholar]
- 77.McComish JS, Slade CA, Buizen L, et al. Randomized controlled trial of omalizumab in treatment-resistant systemic and cutaneous mastocytosis (ROAM). J Allergy Clin Immunol Pract. 2023;11(7):2248-50.e3. [DOI] [PubMed] [Google Scholar]
- 78.American Initiative in Mast Cell Diseases, Centers of Excellence. 2024. Available from: https://aimcd.net/centers/. [Accessed 16 Sep 2024].
- 79.Carter MC, Clayton ST, Komarow HD, et al. Assessment of clinical findings, tryptase levels, and bone marrow histopathology in the management of pediatric mastocytosis. J Allergy Clin Immunol. 2015;136(6):1673-9.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lyons JJ. Inherited and acquired determinants of serum tryptase levels in humans. Ann Allergy Asthma Immunol. 2021;127(4):420–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fuchs D, Kilbertus A, Kofler K, et al. Scoring the risk of having systemic mastocytosis in adult patients with mastocytosis in the skin. J Allergy Clin Immunol Pract. 2021;9(4):1705-12.e4. [DOI] [PubMed] [Google Scholar]
- 82.Czarny J, Zuk M, Zawrocki A, et al. New approach to paediatric mastocytosis: implications of KIT D816V mutation detection in peripheral blood. Acta Derm Venereol. 2020;100(10):149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Akin C. How to evaluate the patient with a suspected mast cell disorder and how/when to manage symptoms. Hematol Am Soc Hematol Educ Progr. 2022;2022(1):55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jawhar M, Schwaab J, Horny HP, et al. Impact of centralized evaluation of bone marrow histology in systemic mastocytosis. Eur J Clin Invest. 2016;46(5):392–7. [DOI] [PubMed] [Google Scholar]
- 85.McMurray JC, Pachecho CS, Schornack BJ, et al. Standardized indolent systemic mastocytosis evaluations across a healthcare system: implications for screening accuracy. Blood. 2024;144(4):408–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.American Initiative in Mast Cell Diseases, AIM Network. 2024. Available from: https://aimcd.net/centers/. [Accessed 19 Mar 2024].
- 87.Akin C, Fumo G, Yavuz AS, et al. A novel form of mastocytosis associated with a transmembrane c-kit mutation and response to imatinib. Blood. 2004;103(8):3222–5. [DOI] [PubMed] [Google Scholar]
- 88.Akin C, Metcalfe DD. The biology of Kit in disease and the application of pharmacogenetics. J Allergy Clin Immunol. 2004;114(1):13–9. [DOI] [PubMed] [Google Scholar]
- 89.Alvarez-Twose I, Matito A, Morgado JM, et al. Imatinib in systemic mastocytosis: a phase IV clinical trial in patients lacking exon 17 KIT mutations and review of the literature. Oncotarget. 2017;8(40):68950–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.de Melo CP, Machado-Neto JA, Scopim-Ribeiro R, et al. Familial systemic mastocytosis with germline KIT K509I mutation is sensitive to treatment with imatinib, dasatinib and PKC412. Leuk Res. 2014;38(10):1245–51. [DOI] [PubMed] [Google Scholar]
- 91.Huang L, Wang SA, Konoplev S, et al. Well-differentiated systemic mastocytosis showed excellent clinical response to imatinib in the absence of known molecular genetic abnormalities: a case report. Medicine (Baltimore). 2016;95(41): e4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang LY, Smith ML, Schultheis B, et al. A novel K509I mutation of KIT identified in familial mastocytosis: in vitro and in vivo responsiveness to imatinib therapy. Leuk Res. 2006;30(4):373–8. [DOI] [PubMed] [Google Scholar]
- 93.DeAngelo DJ, George TI, Linder A, et al. Efficacy and safety of midostaurin in patients with advanced systemic mastocytosis: 10-year median follow-up of a phase II trial. Leukemia. 2018;32(2):470–8. [DOI] [PubMed] [Google Scholar]
- 94.Gotlib J, Kluin-Nelemans HC, George TI, et al. Efficacy and safety of midostaurin in advanced systemic mastocytosis. N Engl J Med. 2016;374(26):2530–41. [DOI] [PubMed] [Google Scholar]
- 95.van Anrooij B, Oude Elberink JNG, Span LFR, et al. Midostaurin in patients with indolent systemic mastocytosis: an open-label phase 2 trial. J Allergy Clin Immunol. 2018;142(3):1006-8.e7. [DOI] [PubMed] [Google Scholar]
- 96.DeAngelo DJ, Radia DH, George TI, et al. Safety and efficacy of avapritinib in advanced systemic mastocytosis: the phase 1 EXPLORER trial. Nat Med. 2021;27(12):2183–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gotlib J, Castells M, Elberink HO, et al. Avapritinib versus placebo in indolent systemic mastocytosis. NEJM Evid. 2023;2(6):2200339. [DOI] [PubMed] [Google Scholar]
- 98.Gotlib J, Reiter A, Radia DH, et al. Efficacy and safety of avapritinib in advanced systemic mastocytosis: interim analysis of the phase 2 PATHFINDER trial. Nat Med. 2021;27(12):2192–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Reiter A, Gotlib J, Alvarez-Twose I, et al. Efficacy of avapritinib versus best available therapy in the treatment of advanced systemic mastocytosis. Leukemia. 2022;36(8):2108–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Reiter A, Schwaab J, DeAngelo DJ, et al. Efficacy and safety of avapritinib in previously treated patients with advanced systemic mastocytosis. Blood Adv. 2022;6(21):5750–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gotlib J, Castells M, Elberink HO, et al. Reductions in indolent systemic mastocytosis biomarker burden with avapritinib in the registrational, double-blind placebo-controlled PIONEER trial. Presented at: European Hematology Association Annual Meeting; June 8-15, 2023; Frankfurt, and virtual. Poster 1017.
- 102.Maurer M, Siebenhaar F, Broesby-Olsen S, et al. Avapritinib improved skin findings in patients with indolent systemic mastocytosis (ISM) in the registrational, double-blind, placebo-controlled PIONEER study. Presented at: 2023 AAAAI Annual Meeting; February 24-27, 2023; San Antonio (TX). Abstract L69.
- 103.ClinicalTrials.gov. 2024. Available from: https://www.clinicaltrials.gov/. [Accessed 28 Oct 2024].