

Abstract
Hepatocellular carcinoma (HCC) typically develops in the context of chronic liver disease, where prolonged hepatocyte exposure to inflammation drives the synergistic accumulation of genetic and epigenetic alterations. Epigenetic regulation encompasses multiple mechanisms that govern the transcription machinery accessibility to DNA. This process is regulated by the addition and removal of covalent marks on chromatin, which can either affect DNA-histone interactions or serve as scaffolds for other proteins, among other mechanisms. Recent research has revealed that epigenetic alterations can disrupt chromatin homeostasis, redirecting transcriptional regulation to favour cancer-promoting states. Consequently, these alterations play a pivotal role in the acquisition of cancer hallmarks and provide insights into several biological processes involved in hepatocarcinogenesis. This review highlights the key epigenetic mechanisms underlying the development, progression and dissemination of HCC, with a particular focus on DNA methylation and histone post-translational modifications. This knowledge is relevant for guiding the development of innovative therapeutic approaches based on epigenetic modulators.
Keywords: Gastrointestinal Cancer, Epigenetics, Hepatocellular Carcinoma, Hepatocarcinogenesis
Introduction
Hepatocellular carcinoma (HCC), the most common primary liver cancer, is a leading cause of cancer-related mortality worldwide. Despite the exponential growth in genomic and transcriptomic data, the complex nature of this disease continues to challenge the identification of effective therapeutic targets. The inherent heterogeneity of solid tumours is further amplified in HCC by the diverse underlying aetiologies, including chronic hepatitis B and C infections, long-lasting alcohol consumption, metabolic dysfunction-associated steatohepatitis (MASH) and exposure to aflatoxin B1.1 These factors, along with ethnic differences and disease stages, contribute to distinct genetic profiles in HCC, influencing specific mutations and pathway activations. For instance, chronic HBV-associated HCC exhibits a high prevalence of mutations in TP53, CCNA2 and CCNE1, with activation of classical proliferation pathways, whereas HCC linked to MASH, alcoholic steatohepatitis, or HCV infection is mainly characterised by mutations in the beta-catenin gene (CTNNB1) and activation of the Wnt/β-catenin pathway.1 2
Emerging evidence indicates that the aetiology of liver disease also influences the epigenetic landscape of HCC,3 extending the heterogeneity of HCC beyond genetic alterations. The term ‘epigenetic’ encompasses processes that lead to potentially heritable phenotypic changes without altering the underlying DNA sequence.4 These mechanisms regulate not only gene transcription but also other DNA-based processes, such as DNA repair, replication and recombination.5 6 Epigenetics enables cells with identical genomes to exhibit diverse phenotypes, adapt to environmental changes, maintain lineage-specific gene expression, support cell differentiation and stem cell renewal, ensure genome integrity and regulate proliferation.7 Epigenetic regulation involves the reversible and dynamic addition or removal of chemical groups on either DNA (via DNA methylation) or histones (through histone post-translational modifications, HPTMs) by enzymes known as ‘writers’ and ‘erasers’, respectively. Other major epigenetic mechanisms include non-covalent interactions, such as the binding of long non-coding RNAs (lncRNA), ATP-dependent nucleosome remodelling and the incorporation of histone variants.4 8 It is widely accepted that epigenetic processes primarily (1) modify chromatin structure and compaction3 and (2) control chromatin interactions with ‘readers’, protein mediators that recognise specific modifications and direct specific downstream outcomes.
Epigenetic mechanisms respond to various signals, including metabolic perturbations, cell cycle regulators, DNA damage sensors and signalling cascades triggered by growth factors, hormones, developmental cues or cellular stress. Collectively, these signals give rise to an exceptionally dynamic and complex epigenetic network that drives widespread changes in cell phenotype and behaviour.8 Hepatocytes are particularly reliant on epigenetic mechanisms to adapt to constant exposure to nutrients and metabolite fluctuations, xenobiotics and interactions with immune and microbial elements.9 Epigenetic mechanisms allow hepatocytes to rapidly adapt to stressors while maintaining cellular homeostasis without genetic alterations. Hence, epigenetic disruptions can undermine this balance, increasing the risk of HCC development.10 Indeed, epigenetic alterations in both hepatocytes and non-parenchymal liver cells can shift gene regulation towards a cancer-promoting state, contributing to several hallmarks of cancer, including proliferation, immune evasion and metastasis.11 12
In this review, we aim to provide a comprehensive analysis of the epigenetic mechanisms involved in HCC development and progression, focusing on how covalent chromatin modifications influence cellular homeostasis and tumourigenesis (table 1). Additionally, we will examine the interactions between DNA methylation and HPMTs and their implications for liver cancer progression.
Table 1. Epigenetic modulators in HCC: expression alterations, functional roles and prognostic implications.
| Epigenetic modulators | Expression in HCC patients | Primary epigenetic substrate | Effect on HCC | Prognosis associated with expression | Reference |
|---|---|---|---|---|---|
| Writers | |||||
| ASH1L | Up-regulated | H3K4, H3K36 | Promotes immunosuppression via M2-like macrophage recruitment and polarisation | Poor | 50 |
| Aurora A | Up-regulated | H3T118 | Drives radioresistance via NF-κB activation | Poor | 90 |
| Aurora B | Up-regulated | H3S10, H3S28, H2AXS121, H3.3S31 | Enhances the expression of growth-promoting genes | Poor | 85 |
| CBP/p300 | Up-regulated | H3K27, H3K18 | Contributes to malignant transformation, proliferation, apoptotic sensitivity and invasion | Poor | 32 |
| KMT1C | Up-regulated | H3K9 | Enhances proliferation and adaptation to hypoxia | Unknown | 47 |
| EZH2 | Up-regulated | H3K27 | Represses tumour suppressor miRNAs and Wnt antagonists while interacting with signalling pathways | Poor | 11 41 42 |
| HAT1 | Up-regulated | H4K5, H4K12, H2AK5 | Promotes cell growth and regulates glucose metabolism | Poor | 33 34 |
| PAD2 | Up-regulated | H3R2, H3R8, H3R17, H3R26 | Regulates proliferation and migration | Unknown | 78 |
| PAD4 | Up-regulated | H1R54, H3R2, H3R8, H3R17, H3R26, H4R3 | Promotes metastasis via E-cadherin suppression, drives NET formation for immune evasion and angiogenesis and activates HIF target genes under hypoxia | Unknown | 74 77 |
| PARP1 | Up-regulated | Glutamate, aspartate and serine histone residues | Contributes to impairment of DNA repair mechanisms | Unknown | 96 |
| PARP10 | Down-regulated | Glutamate, aspartate and serine histone residues | Hinders metastasis by impairing EMT, migration and invasion | Unknown | 98 |
| PARP14 | Up-regulated | Glutamate histone residues | Promotes the Warburg effect | Poor | 100 |
| Patt1 | Down-regulated | N-terminal residues of H4 and H2A | Enhances apoptosis | Unknown | 37 |
| PCAF | Down-regulated | H3K9 | Promotes cell autophagy and apoptosis | Unknown | 35 36 |
| PRMT1/2/4/9 | Up-regulated | Arginine histone residues | Promote proliferation, migration and invasion | Poor | 71 72 |
| PRMT5 | Up-regulated | H3R8, H2AR3, H4R3 | Enhances lipid biosynthesis, cell proliferation, metastatic potential and self-renewal | Poor | 71 72 |
| SETDB1 | Up-regulated | H3K9 | Enhances proliferation and migration | Poor | 45 |
| SMYD2 | Up-regulated | H3K36, H3K4 | Reprogrammes glutamine metabolism | Poor | 47 |
| SMYD3 | Up-regulated | H3K4, H4K5 | Enhances proliferation and invasiveness | Poor | 33 34 |
| Erasers | |||||
| HDAC1/2 | Up-regulated | N-terminal lysines of H2A, H2B, H3 and H4 | Repress tumour suppressor p21 expression | Poor | 26 |
| HDAC3 | Up-regulated | N-terminal lysines of H2A, H2B, H3 and H4 | Enhances proliferation and invasiveness | Unknown | 24 |
| HDAC5 | Up-regulated | N-terminal lysines of H2A, H2B, H3 and H4 | Enhances migration and invasion under hypoxia | Poor | 26 |
| HDAC6 | Down-regulated | N-terminal lysines of H2A, H2B, H3 and H4 | Suppresses angiogenesis and metastatic and antiphagocytic potential | Poor | 30 |
| HDAC8 | Up-regulated | N-terminal lysines of H2A, H2B, H3 and H4 | Promotes proliferation and inhibits apoptosis | Unknown | 25 |
| KDM1B | Up-regulated | H3K4 | Enhances proliferation | Poor | 52 |
| KDM4B | Up-regulated | H3K9 | Enhances proliferation | Poor | 56 |
| KDM1 | Up-regulated | H3K4, H3K9 | Maintains the shift from mitochondrial to glycolytic metabolism | Poor | 59 |
| KDM2A | Up-regulated | H3K36 | Promotes proliferation, migration and EMT | Poor | 53 |
| KDM3A | Up-regulated | H3K9 | Promotes proliferation by inducing expression of HIF1 target genes | Poor | 54 55 |
| KDM5B | Up-regulated | H3K4 | Represses expression of tumour suppressor molecules | Poor | 60 62 |
| KDM5C | Up-regulated | H3K4 | Promotes metastasis by repressing BMP7 transcription | Poor | 64 |
| KDM6B | Up-regulated | H3K27 | Modulates EMT by inducing SNAI2 transcription | Poor | 63 |
| KDM7B | Up-regulated | H3K9, H3K27, H4K20 | Promotes migration and invasion | Unknown | 64 |
| KDM8 | Down-regulated | H3K36 | Inhibits proliferation by activating CDKN1A expression | Poor | 11 66 |
| RIOX2 | Up-regulated | H3K9 | Unknown/Non-established | Poor | 11 |
| SIRT1 | Up-regulated | H3K9, H3K14, H4K16 | Disrupts bile acid homeostasis, sustains telomerase activity and activates PI3K/PTEN/AKT signalling | Poor | 26 |
| SIRT2 | Up-regulated | H4K16, H3K9 | Promotes EMT | Poor | 26 |
| SIRT7 | Up-regulated | H3K18, H3K36 | Suppresses p53 activity | Poor | 26 31 |
| Reader-domain containing proteins | |||||
| BRD4 | Up-regulated | Acetylated lysine histone residues | Promotes EMT and enhances oncogene expression | Unknown | 105 |
| BRD7 | Down-regulated | H3K9ac, H3K14ac, H4K8ac, H4K12ac and H4K16ac. | Activates p53 pathway | Poor | 109 |
| BRD9 | Up-regulated | Acetylated lysine histone residues | Activates the Wnt/β-catenin and TUFT1/AKT signalling pathways | Poor | 106 |
| BPTF | Up-regulated | H3K4me3, H4K12Ac, H4K16Ac, H4K20Ac | Promotes hTERT expression | Poor | 107 |
| BRPF1 | Up-regulated | H2AK5ac, H4K12ac, H4K8ac, H4K5ac, H3K14ac | Regulates oncogene expression | Poor | 108 |
| CHD1L | Up-regulated | poly-ADP-ribosylated serine residues | Promotes proliferation, migration, and metastasis by upregulating oncogene expression and enhancing autophagy | Poor (after surgical resection) | 110 112 |
| CHD5 | Down-regulated | H3K27me3, H3K4 | Impairs cell motility and invasion | Poor | 113 |
| MBD2 | Up-regulated | Methylated CpGs | Promotes proliferation, migration, and invasion | Poor | 115 |
| MeCP2 | Up-regulated | Methylated CpGs | Promotes proliferation by activating ERK1/2 and inhibiting p38 activity | Unknown | 117 118 |
| UHRF1 | Up-regulated | Hemimethylated DNA, H3K9me3, H3R2 | Modulates the stemness of tumour-initiating cells | Poor | 119 123 |
| UHRF2 | Up-regulated | Hemimethylated DNA, 5hmC, H3K9me2/3 | Reduces apoptosis and promotes malignant phenotype | Poor | 124 |
EMT, epithelial-mesenchymal transition; HCC, hepatocellular carcinoma; HIF, hypoxia inducible factor; hTERT, human telomerase reverse transcriptase; NETs, neutrophil extracellular traps; SNAI2, Snail family transcriptional repressor 2.
DNA methylation
DNA methylation refers to the covalent transfer of a methyl group from the metabolite S-adenosylmethionine (SAM) to the 5’ carbon of cytosine residues at CpG dinucleotides9 (figure 1). Methylation at CpG islands in transcription start sites and repetitive regions typically results in strong transcriptional repression and long-term gene silencing. On the other hand, methylation within gene bodies can promote transcription elongation and influence splicing.12 This reaction is catalysed by DNA methyltransferases (DNMTs) that are classified into two groups based on their functions. DNMT1 is primarily responsible for the maintenance of DNA methylation patterns through the methylation of hemi-methylated DNA, thereby ensuring the heritability of these marks after replication.9 On the other hand, DNMT3A and DNMT3B preferentially target unmethylated sites, being the main regulators of de novo methylation.4 Additionally, DNMT3L, a member of the DNMT3 family that lacks methyltransferase activity, acts as a positive regulator of de novo DNA methylation by interacting with DNMT3A and DNMT3B.13 However, this dichotomous classification of DNMT functions is overly simplistic. It has been reported that DMNT3 also plays a role in maintaining and dynamically remodelling DNA methylation patterns in differentiated cells.14 DNA methylation is also regulated by Ten-Eleven Translocation (TET) methylcytosine dioxygenases. These enzymes mediate the stepwise oxidation of 5-methylcytosine into 5-hydroxymethylcytosine, 5-formylcytosine and finally into 5-carboxylcytosine.15 Research suggests that intermediates of DNA demethylation may have regulatory functions of their own, influencing gene expression and chromatin structure independently of complete demethylation.15 Notably, 5-hydroxymethylcytosine levels are decreased in HCC and may serve as a prognostic marker.16 DNA demethylation is then completed either by the base excision repair machinery or through passive dilution of the epigenetic mark, as it is not recognised by DNMT1.15 This intricate balance between methylation and demethylation highlights the complexity and dynamism of this epigenetic process.
Figure 1. DNA methylation and demethylation dynamics. (A) DNA methylation is maintained by DNMT1 on hemimethylated DNA after replication, while DNMT3A/3B catalyse de novo methylation on unmethylated DNA. In mammals, this modification is primarily found at CpG dinucleotides. (B) DNMTs use SAM as a methyl donor to convert cytosine into 5mC, while TET enzymes oxidise 5mC to 5hmC, 5fC and 5caC, which can be passively diluted through replication or actively removed via BER. 5caC, 5-carboxylcytosine; 5fC, 5-formylcytosine; 5hmC, 5-hydroxymethylcytosine; 5mC, 5-methylcytosine; α-KG, α-ketoglutarate; BER, base excision repair; DNMTs, DNA methyltransferases; Me, methylation; SAH, S-adenosylhomocysteine; SAM, S-adenosylmethionine; Succ., succinate; TETs, Ten-Eleven Translocation enzymes.

Dysregulation of DNA methylation is a well-established factor in HCC pathogenesis. Even at preneoplastic stages, both genome-wide hypomethylation and specific gene promoter hypermethylation are evident.3 In general, hypomethylation contributes to chromosomal instability, and hypermethylation silences tumour suppressor genes, such as TFPI2, CDKN1A, CDKN2B, CDKN2A HHIP, SFRP2, APC, SOCS1, CDH1 and HAI-2.3 A study by Calvisi et al demonstrated the lack of correlation between DNA methylation patterns and different aetiologic factors, highlighting the ubiquity of these epigenetic alterations in HCC.17 These changes progressively accumulate from normal liver to cirrhosis, dysplasia and HCC at various stages.18 Additionally, methylation patterns in non-tumoural adjacent liver tissue show prognostic value in patients with HCC.18
Histone post-translational modifications
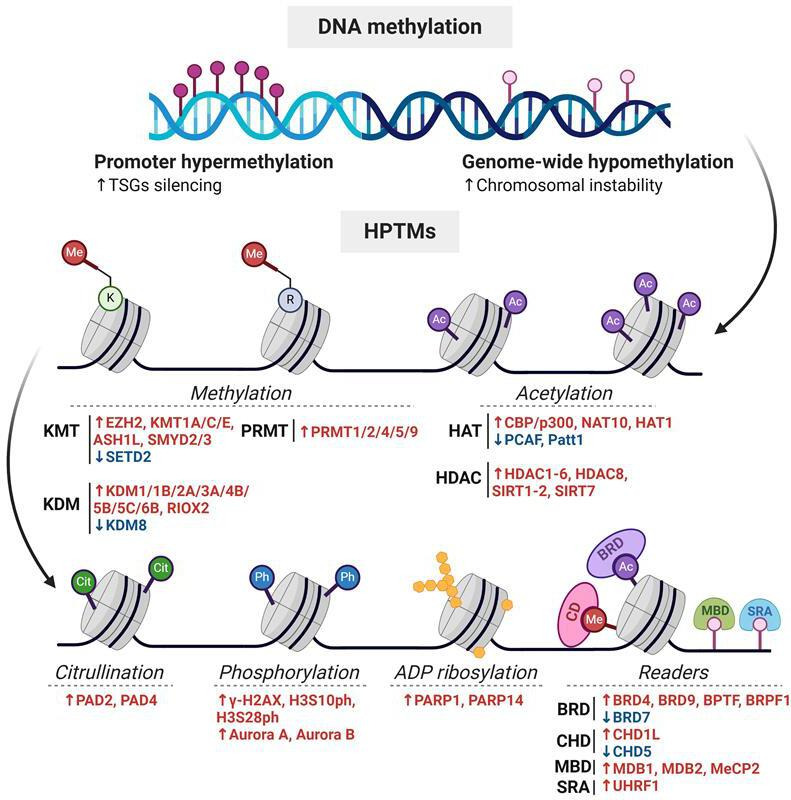
HPTMs encompass a wide range of epigenetic marks, including acetylation, methylation, citrullination, phosphorylation and ADP-ribosylation, among others3 19 (figure 2). HPTMs are mainly located on the N-terminal tails of histones, but they can be found on their globular domains as well.19 Interestingly, the transcriptional impact of HPTMs depends on their position, molecular nature and the surrounding chromatin context. Some HPTMs, such as acetylation and phosphorylation, promote chromatin condensation or relaxation through electrostatic and stereochemical interactions that directly affect nucleosome stability, while others act as molecular scaffolds for the recruitment of effector proteins.20 The dysregulation of the enzymatic machinery responsible for writing, reading and erasing HPTMs is frequently observed in human HCC, resulting in a histone code that disrupts the epigenetic balance required for the maintenance of normal gene expression.19
Figure 2. Molecular dynamics of the most studied histone post-translational modifications (HPTMs) in hepatocellular carcinoma (HCC). Key modifications involved in HCC include lysine methylation and acetylation, regulated by KMTs/KDMs and HATs/HDACs, respectively, which influence chromatin accessibility and transcriptional activity. Additional PTMs, such as arginine methylation by PRMTs, phosphorylation by kinases, citrullination by PADs and ADP-ribosylation by PARPs also contribute to epigenetic remodelling. Some of these modifications, such as acetylation, citrullination, phosphorylation and ADP-ribosylation, alter the charge of the targeted amino acid residues, thereby affecting histone-DNA interactions and chromatin compaction. In contrast, methylation does not change the residue’s charge but can still influence chromatin structure by serving as docking sites for regulatory proteins. Ac, acetylation; Ac-CoA, acetyl-coenzyme A; ADPr, ADP-ribose; α-KG, α-ketoglutarate; BER, base excision repair; Cit, citrullination; CoA, coenzyme A; HATs, histone acetyltransferases; HDACs, histone deacetylases; JmjC, Jumonji C domain-containing demethylases; KDMs, lysine demethylases; KMTs, lysine methyltransferases; MARylation, mono-ADP-ribosylation; Me, methylation; PADs, peptidyl arginine deiminases; PARPs, poly(ADP-ribose) polymerases; PARylation, poly-ADP-ribosylation; Ph, phosphorylation; PRMTs, protein arginine methyltransferases; SAH, S-adenosylhomocysteine; SAM, S-adenosylmethionine; Succ., succinate.

Histone acetylation
Histone acetylation involves the dynamic addition and removal of acetyl groups into the ε-amino group of lysine residues by histone acetyltransferases (HATs) and histone deacetylases (HDACs), respectively.9 20 Human HATs catalyse the transfer of an acetyl group from Acetyl-CoA to lysine residues. These enzymes are categorised into two major groups: GNAT (including HAT1, General Control Non-repressed protein 5 (GCN5), P300/CBP-associated factor (PCAF)) and MYST (comprising Tip60, MOF, MOZ, MORF, HBO1). A third group, called p300/CBP, is often referred to as an ‘orphan class’ because, despite having acetylase activity, it does not possess true consensus HAT domains.21 HDACs, on the other hand, are a family of enzymes classified into four classes based on their homology to yeast analogues: class I: Rpd3-like proteins (HDACs 1, 2, 3 and 8), located in the nucleus; class II: Hda1-like proteins (HDACs 4, 5, 6, 7, 9 and 10), with both nuclear and cytoplasmic locations; class III: Sir2-like proteins (SIRT 1–7), also termed sirtuins; class IV: HDAC11, which combines features of classes I and II.21
The addition of negatively charged acetyl groups neutralises the positive charge of lysines, weakening the interaction between nucleosomes and DNA, which results in an open chromatin structure conducive to transcriptional activity.7 Chromatin relaxation can also be facilitated by the binding of readers through bromodomains (BrDs), small modules that can recognise these acetylated modifications.20 However, emerging research suggests that the effects of acetylation extend beyond its electrostatic effect. For example, histone acetylation may act as a rheostat to regulate intracellular pH (pHi). Studies have shown that in certain tumours with low pHi, a reduction in histone acetylation is observed. Histone deacetylation appears to promote the release of acetate anions, which are coexported with protons, thereby contributing to the stabilisation of pHi.22
Altered expression of HATs and HDACs is a major cause of disrupted histone acetylation dynamics, with HDACs being the most extensively studied due to their therapeutic relevance. Numerous studies report the upregulation of different HDACs in HCC, such as HDAC1 and HDAC2, and their inhibition has demonstrated efficacy in suppressing proliferation and inducing tumour cell death in various HCC cell lines.23 The expression of these enzymes has also been correlated with a worse prognosis in different HCC cohorts.23 Additionally, HDAC3 serves as a biomarker for tumour recurrence following liver transplantation in HBV-associated HCC, playing an active role in regulating tumour cell proliferation and invasion.24 HDAC8 is also significantly upregulated in HCC tumour tissue, and its knockdown inhibits HCC cell proliferation and induces apoptosis.25 Moreover, multiple class II and III HDACs (HDAC4, HDAC5, SIRT1, SIRT2 and SIRT7) are overexpressed in HCC and correlate with poorer prognosis,26,29 while HDAC6 acts as a tumour suppressor in HCC.27 30 31
Similarly, an imbalance in the acetylation/deacetylation ratio can also result from altered HAT expression. Inagaki et al reported the upregulation of CBP/p300 in both HCC and extrahepatic metastatic HCC compared with non-malignant liver tissues, with p300 expression strongly correlating with HCC aggressiveness and serving as a potential predictor of patient survival.32 Other HATs upregulated in HCC and associated with poor prognosis include NAT10 and HAT1.33 34 Conversely, PCAF, an acetyltransferase capable of suppressing HCC growth as it promotes cell autophagy and accelerates apoptosis, is frequently downregulated in liver cancer tissues.35 36 Apart from correlating to tumour-node-metastasis (TNM) stages, PCAF expression is also associated with tumour metastasis.36 In this context, PCAF downregulation significantly enhances HCC cell migration and invasion through epithelial-mesenchymal transition (EMT).35 Similarly, decreased levels of Patt1, a GNAT family acetyltransferase whose expression is also lower in HCC tissues, result in the hypoacetylation of pro-apoptotic genes and the subsequent attenuation of apoptosis in hepatoma cells.37
Histone methylation
Unlike the clear link between acetylation and transcriptional activation, the effects of histone methylation are far more complex and unpredictable. This process begins with the transfer of a methyl group from S-adenosyl-L-methionine to a lysine or arginine residue on histones. Both amino acids can undergo mono-, di- or, in the case of lysine, tri-methylation, with each modification leading to a distinct transcriptional outcome.3 In all cases, the positive charge of the residues remains unchanged. Instead, histone methylation drives chromatin reconfiguration through two main mechanisms: it can either influence the addition of other histone marks—such as preventing acetylation of lysine residues that are already methylated—or recruit a highly specific array of proteins that recognise these modifications. Both mechanisms contribute to a complex functional landscape.10 Importantly, lysine and arginine methylation can interact, either synergising or counteracting each other’s effect on the recruitment of readers. For example, the binding affinity of the transcription factor SPIN1 to the trimethylated (me3) lysine (K) 4 on histone 3 (H3) is influenced by the methylation status of arginine 8 on the same histone.38
Histone lysine methylation
Lysine methylation is carried out by six main classes of histone lysine methyltransferases (KMTs). The catalytic activity of nearly all members of this large family is driven by a Su(var)3–9, enhancer of zeste, or a trithorax (SET) domain. The exception is the disruptor of telomeric silencing 1-like methyltransferase, which functions via a seven-beta-strand domain.39 Apart from being highly specific for lysine residues, most KMTs are incapable of catalysing all degrees of methylation.39 This creates a tightly intertwined relationship between these marks and their writers, characterised by a certain extent of enzymatic redundancy that allows context-dependent regulation of specific activities.39 Conversely, histone lysine demethylases (KDMs) counterbalance KMTs by removing methylation marks. KDMs have been classified into at least eight families based on structural and functional similarities. KDM1 family includes KDM1A and KDM1B, which use a flavin adenine dinucleotide-dependent amine oxidase domain to specifically remove mono and dimethyl marks, but not trimethyl marks.3 Meanwhile, the Jumonji C (JmjC) domain found in the remaining families (KDM2-8) enables the removal of all three degrees of methylation states through their α-ketoglutarate-dependent hydroxylase activity.40 Hence, the final methylation state results from the balance of the activities of all KMTs and KDMs, along with the availability of necessary cofactors like α-ketoglutarate and flavin adenine dinucleotide.
The transcriptional outcome of histone lysine methylation depends on both the lysine residue involved and the methylation state. For example, trimethylation at histone 3 (H3) lysine (K) 4 is linked to gene activation, whereas trimethylation at H3K9 or H3K27 generally correlates with silenced chromatin states.3 Additionally, certain lysine methylation marks interact closely with other histone modifications and DNA methylation to finely regulate gene expression. For instance, bivalent chromatin domains marked by both H3K4 and H3K27 methylation maintain genes in a poised state, allowing them to shift towards either activation or repression as needed.20
Over the past few years, the understanding of the impact of histone lysine methylation on HCC pathobiology has continuously expanded. Altered expression of KMTs and KDMs can result in abnormal transcriptional activation or repression, depending on the enzyme’s target and the cellular context. Interestingly, KMTs and KMDs have been reported to be more frequently mutated in HCC patients than HATs or BrDs.11 Among KMTs, EZH2 (also known as KMT6) has garnered significant attention due to growing evidence of its increased expression in HCC tissues, along with an enrichment in H3K27me3 repressive mark.11 41 42 This correlates with greater tumour aggressiveness and worse prognosis11 and even holds diagnostic power, with a sensitivity of 95.8% and a specificity of 97.8%.42 Mechanistically, the protumourigenic effects of EZH2 are linked to the epigenetic suppression of tumour suppressor microRNAs expression (eg, miR-200c),43 repression of Wnt antagonists3 and its interaction with components of multiple signalling pathways such as CDKN2A, FOXO3, E2F1 and NOTCH2.44 In addition, KMT1A/SUV39H1, the primary KMT responsible for establishing the repressive H3K9me3 mark, contributes to HCC development and metastasis. This also applies to KMT1E/SETDB1, identified as the most significantly upregulated epigenetic regulator in human HCC. Additionally, SETDB1 overexpression is associated with poor prognosis, tumour aggressiveness and disease progression, and its inhibition reduces HCC cell proliferation and migration.45 More recently, the deletion of SETD2, a tumour suppressor in various cancer types mutated in approximately 5% of HCC patients, was shown to be sufficient to induce spontaneous HCC development in mice, underscoring its critical role in regulating DNA damage and lipid metabolism in the liver.46 Other KMTs upregulated in HCC that have been shown to promote tumour development include ASH1L, KMT1C/G9a, SMYD2/KMT3C and SMYD3.1147,50 Among these, KMT1C role in HCC progression and metastasis has been linked to enhanced cell proliferation and adaptation to hypoxia.47 Finally, SMYD3’s H3K4 trimethyltransferase activity has been demonstrated to promote gene transcription in HCC cells, enhancing proliferation and invasiveness through the upregulation of sphingosine-1-phosphate receptor 1, cyclin-dependent kinase 2 and matrix metalloproteinase 2, among others.49 51
Of equivalent clinical relevance, the dysregulation of KDMs in HCC and its implications for disease progression are actively being investigated. Overexpression of KDM1/LSD1, KDM1B/AOF1, KDM2A/FBXL11, KDM3A/JMJD1A, KDM4B/JMJD2B, KDM5B/JARID1B, KDM5C/JARID1C, KDM6B/JMJD3 and RIOX2 in HCC tissue has been identified as a poor prognostic factor and is linked to increased tumour aggressiveness.1152,58 While the precise mechanisms driving the oncogenic effects of many of these enzymes remain unclear, specific roles have been elucidated for certain KDMs. For example, KDM1 has been shown to play a role in maintaining the shift from mitochondrial to glycolytic metabolism in human HCC cells, a hallmark of most cancers.59 Additionally, KDM2A promotes HCC cell proliferation, migration and EMT enhancement through unc-5 netrin receptor B antisense RNA 1 (UNC5B-AS1).53 In the case of KDM3A, there is evidence of an upregulation of the enzyme on hypoxic conditions and of its contribution to HCC cell proliferation, potentially through the removal of repressive H3K9 methylation on hypoxia inducible factor 1 target gene promoters, such as adrenomedullin.54 55 Notably, KDM3A expression is a significant predictor of HCC recurrence after hepatic resection,54 and its inhibition has been shown to reduce EMT and invasion under hypoxia, while maintaining cells in an undifferentiated state. On the other hand, KDM5B represses the transcription of several tumour suppressor molecules (eg, CDKN1B, CDKN2B, miR-448, PTEN) through the demethylation of H3K4me3 on their promoters.60,62 Therefore, when overexpressed in HCC tissue, this enzyme drives HCC cell proliferation, promotes the malignant phenotype and contributes to hepatocarcinogenesis,60 61 while enhancing the metastatic capacity in vivo and facilitating EMT, migration and invasion of HCC cells in vitro.62 Other demethylases with similar pro-invasive and pro-metastatic effects are KDM5C, KDM6B and KDM7B/PHF8. KDM5C exerts its effect by repressing bone morphogenetic protein-7 transcription through the removal of H3K4me3, while KDM6B removes H3K27me3 in Snail family transcriptional repressor 2 promoter to induce its transcription.63 64 Additionally, KDM7B/PHF8 promotes migration and invasion through the upregulation of SNAI1, vimentin and RB1-inducible coiled-coil 1 (FIP200).65
Conversely, KDM8/JMJD5 appears to play a tumour-suppressive role in HCC, as its expression is frequently downregulated and correlates with a better prognosis.11 Notably, Wu et al found that the decrease in KDM8 expression in HCC specimens is partly due to the dysregulation of other histone-modifying enzymes, specifically involving an enrichment of H3K27 and H3K9 trimethylation, along with a reduction of H3K9ac, H3K27ac and H3K4me2/3 on KDM8 promoter. Functionally, the authors demonstrated that KDM8 knockdown leads to a downregulation of the cell cycle-negative regulator CDKN1A, thereby accelerating G1/S transition.66
Histone arginine methylation
To date, three families of protein arginine (R) methyltransferases (PRMTs) have been identified.67 These enzymes methylate the guanidinium group of arginine residues on both histone and non-histone proteins, producing specific methylated arginine products.68 Although all three types of PRMTs can generate monomethylarginine, only type I PRMTs can produce symmetric dimethylarginine, while type II PRMTs are responsible for symmetric dimethylarginine.68 Arginine demethylation was long elusive, but it is now understood that methyl arginine can be directly removed by JmjC-containing demethylase or converted into citrulline by peptidyl-arginine deiminases (PADs).68 On methylation, the functional properties of the guanidinium group remain intact, whereas PAD-catalysed conversion results in the loss of the guanidinium group’s charge and functionality, preventing any possibility of remethylation.68 Additionally, the interplay between arginine and lysine methylation is crucial in fine-tuning gene expression, particularly in poised promoters. For example, symmetrical dimethylation of H3R2 enhances the trimethylation of H3K4, an active transcriptional mark, while its asymmetrical dimethylation antagonises this process, reducing H3K4me3 levels and thereby suppressing transcriptional activation.69 This illustrates how different methylation patterns at the same site can exert opposing effects on transcriptional regulation.
The strong association between PRMT dysregulation and carcinogenesis has sparked significant clinical interest in targeting arginine methylation as a therapeutic approach.42 However, due in part to its later discovery, research on arginine methylation in HCC remains limited, particularly when compared with lysine methylation.37 PRMT5 emerges as one of the first PRMTs identified as a potential therapeutic target in HCC, due to its marked upregulation in HCC tissues and its negative correlation with overall patient survival.43 Further studies have demonstrated that PRMT5 overexpression plays a critical role in HCC tumourigenesis by modulating key transcription factors, including sterol regulatory element binding transcription factor 1, B cell translocation gene 2, β-catenin and hepatocyte nuclear factor 4α. This results in the hyperactivation of lipid biosynthesis, increased cell proliferation and metastatic potential, and enhanced self-renewal of cancer cells.70 71 Other PRMTs with pro-tumourigenic functions in HCC include PRMT1, PRMT2, PRMT4 and PRMT9, which also associate with poor clinical prognosis.71 72 Like PRMT5, the overexpression of these PRMTs has shown to significantly promote HCC cell proliferation, migration and invasion in vitro. This occurs through the activation of multiple signalling pathways, including STAT3, AKT/mTOR and PI3K/Akt/GSK‐3β/Snail.71
Histone citrullination
In humans, five PAD enzymes convert a positively charged arginine residue (whether methylated or not) into neutral citrulline, preventing further methylation of that residue.73 Histone citrullination affects the transcriptional pattern of cells due to its influence on the methylation status of arginine and lysine residues. For example, citrullination at H3R2 by PAD4 can shift a promoter from a poised to a more repressive state by inhibiting the recruitment of the H3K4 by the MLL methyltransferase.67 73 Additionally, PAD4 cooperates with HDAC1 to repress transcription. In contrast, PAD2 and the methyltransferase polycomb repressor 2 (PRC2) engage in a competitive dynamic to regulate enhancer activity. The repressive H3K27me3 mark, established by PRC2, hinders PAD2’s ability to catalyse H3R26 citrullination, and vice versa.73 It should be noted that histone citrullination by PAD4 also plays a crucial role in NETosis, a form of cell death characterised by the release of chromatin into the extracellular space.73 This process has been associated with several cancer hallmarks, including tumour growth, metastasis and immune escape.
The role of citrullination in HCC has not been as thoroughly studied as other epigenetic modifications, such as methylation or acetylation. However, emerging evidence indicates that dysregulation of histone citrullination, particularly when mediated by PAD4, may contribute to HCC pathogenesis.74 For instance, elevated serum levels of citrullinated H3 have shown potential as both diagnostic and prognostic markers in patients with advanced HCC.75 A study by Lin et al further demonstrated that PAD4-mediated histone citrullination promotes HCC cell metastasis by regulating the expression of E-cadherin, a key molecule involved in cell adhesion and metastasis suppression.76 Furthermore, PAD4 activity is crucial for the formation of neutrophil extracellular traps (NETs), which contribute to HCC progression probably by promoting angiogenesis, immune evasion and metastasis.77 In this line, PAD4 is believed to enhance chromatin decondensation, facilitating the release of chromosomal DNA necessary for NET formation.77 Additionally, hypoxia-induced PAD4 expression in HCC cells enhances histone citrullination at hypoxia response elements, leading to greater DNA accessibility and activation of HIF target genes, which promote angiogenesis and tumour growth.74 Elevated citrullinated H3 may also contribute to chemoresistance in HBV-related HCC by upregulating the autophagy regulator Beclin1, thereby enhancing autophagy.75 78 While PAD4 shows a clear pro-tumourigenic role in HCC, the impact of PAD2 on tumourigenesis appears to be multifaceted and context-dependent. In fact, although PAD2 is upregulated in HCC, its inhibition paradoxically promotes HCC cells’ proliferation and migration.78
Histone phosphorylation
Significant efforts have been made to understand how histone phosphorylation influences chromatin behaviour, unveiling its critical roles in other chromatin-related processes, such as DNA damage response (DDR), nucleosome assembly, definition of chromatin regions and apoptosis.79 Phosphorylation of different histone serine, threonine or tyrosine residues is meticulously regulated by specific kinases and phosphatases. Remarkably, there is less knowledge about histone phosphatases than for kinases. It is accepted that, although phosphatases are fewer in number, they achieve the necessary specificity by interacting with a wide array of targeting subunits, which direct phosphatases to specific cellular compartments or protein substrates.80
Histone phosphorylation impacts chromatin accessibility by integrating effects seen with both histone acetylation and methylation. As acetylation, phosphorylation confers a negative charge to histone serine, threonine and tyrosine residues, leading to an alteration in overall histone charge that affects histone–histone or histone–DNA interactions. Additionally, phosphorylated residues, like methylated ones, can either facilitate or hinder the binding of specific readers, acting independently or alongside adjacent HPTMs. In line with this, there are KS (lysine-serine) domains, such as H3K9S10 and H3K27S28, where the lysine can be methylated, and the adjacent serine phosphorylated, creating a combinatorial code that determines chromatin dynamics.81 The final outcomes of histone phosphorylation events depend heavily on the cellular context and the specific residues targeted. For instance, phosphorylation of H3 in serines (S) 10 (H3S10) and 28 (H3S28) plays a critical role in chromatin condensation during mitosis, while the phosphorylation of H2AXS139 (commonly referred to as γ-H2AX) is pivotal in the DDR.79
The mechanisms by which histone phosphorylation contributes to the development and progression of HCC are complex and multifaceted. They involve not only the direct regulation of key cancer-related genes but also interactions with other epigenetic modifications and transcriptional factors. For example, γH2AX, a well-established marker of DNA double-strand breaks (DSBs), is crucial for recruiting DDR proteins to the site of damage, facilitating the repair response.82 Defects in DSB repair mechanisms can lead to genomic instability, a hallmark of cancer, yet paradoxically, may also be exploited to induce cancer cell death.82 In HCC, elevated levels of γ-H2AX in preneoplastic lesions suggest its potential as a predictive biomarker.83 Furthermore, its accumulation is associated with large tumour size, vascular invasion, advanced TNM stage and poorer patient outcomes after liver transplantation.84 Beyond its role as a marker of DNA damage, γ-H2AX may contribute to HCC progression by promoting angiogenesis, possibly through modulation of the EGFR/HIF-1α/VEGF signalling pathways.84
After γH2AX, H3S10ph ranks as the second most studied histone phosphorylation in oncology, though its role in HCC has been comparatively less explored. H3S10ph is mediated by Aurora B kinase, which facilitates chromatin compaction during cell cycle progression by interacting with other chromatin modifications, like methylation at H3K9. This phospho-methyl switch between H3K9 methylation and H3S10 phosphorylation influences chromatin accessibility and gene expression during cell cycle transitions.29 This modification is often upregulated in HCC, driving the overexpression of growth-promoting genes and contributing to tumour growth and aggressiveness.85 In diethyl nitrosamine-induced carcinogenesis, H3S10 phosphorylation is enhanced, particularly at the promoters of Brf1 and Pol III genes, both linked to HCC development in mice. The same study shows that inhibiting H3S10ph reduces the expression of these genes, leading to decreased cell proliferation and transformation.86 While H3S28ph has also been shown to regulate Pol III-mediated transcription, its direct role in HCC remains unclear.87 This gap in knowledge highlights the need for further research to elucidate how these epigenetic modifications influence the pathogenesis and progression of HCC.
On the other hand, it is known that H1.2 is phosphorylated at T146 by DNA-dependent protein kinase (DNA-PK) in response to DNA damage.88 In HCC, the role of metastasis-associated 1 (MTA1) has been shown to promote tumour growth and metastasis by interfering with this phosphorylation process. MTA1 disrupts DNA-PK-mediated H1.2T146 phosphorylation, preventing the binding of phosphorylated H1.2 to its target genes. This, in turn, facilitates the expression of genes involved in cancer proliferation, invasion and metastasis, such as MMP-9, MMP-7 and cyclin D1. Furthermore, the oncogenic effects of MTA1 are suppressed by the ectopic expression of H1.2T146ph in HCC cell lines, highlighting the importance of this phosphorylation in the molecular pathology of HCC.89
Perturbations in the balance of epigenetic modifications observed in cancer can arise from the dysregulation of their writers or erasers. Specifically, altered histone kinase expression or activity can result in aberrant histone phosphorylation, which in turn affects critical processes such as DNA repair, cell proliferation and chromatin remodelling. In HCC, kinases such as DNA-PK and Aurora kinases often exhibit altered activity levels, playing a critical role in tumour progression and therapy resistance. Research has consistently demonstrated the overexpression of Aurora A in HCC tissues and its contribution to radioresistance through the activation of the NF-κB signalling pathway, emphasising its potential as a therapeutic target.90 Aurora B is also frequently upregulated in HCC and is associated with mutations in p53 and β-catenin as well as with Aurora A overexpression. The latter indicates a significant interrelation between the two kinases, despite their distinct chromosomal locations. Beyond this genetic correlation, Aurora B overexpression is considered an independent risk factor for the poor prognosis of HCC patients.91 In contrast, the role of histone phosphatases in hepatocarcinogenesis remains poorly understood.
Histone ADP ribosylation
Histone poly ADP-ribosylation (PARylation) is a multiple-step process initiated by the transfer of ADP-ribose from nicotinamide adenine dinucleotide to specific residues in histones.92 This reaction begins with the transfer of a mono-ADP-ribose moiety, followed by the addition of further ADP-ribose units (elongation step) to generate linear or branched structures through the formation of glycosidic bonds between riboses.93 For the moment, at least 17 ADP-ribosyltransferases have been described. However, while most of these enzymes are mono(ADP-ribosyl) transferases, only PARP-1, PARP-2, PARP-5a and PARP-5b are able to synthesise polymers of ADP-ribose (PAR).92 93 Remarkably, PARP-1 and PARP-2 have redundant functions, as do PARP-5a and PARP-5b.94 In the nucleus, serine residues are the main target for auto-PARylation and histone-PARylation, with recent discoveries showing that histone glutamic acid and aspartic acid residues are susceptible to this modification as well.94 On the other hand, PAR chain removal is carried out by the PAR glycohydrolase PARG and by the PAR hydrolase ARH3.92 93
After histones, PARP-1 stands as the most abundant protein within the nucleus.93 Even more, it is well known that PARP-1/PARP-2 play critical roles as regulators of transcription, nucleosome remodelling and DDR by modifying not only histones and other DNA-associated proteins.95 Under physiological conditions, the activity of PARP-1/PARP-2 on nucleosomes promotes sustained transcription by facilitating chromatin relaxation.94 PARP-1/PARP-2 can extensively PARylate the core of histones to promote chromatin relaxation during a wide variety of DNA damage contexts and serve as scaffolds for DNA repair machinery.95 PARP-1 also acts as a DNA damage sensor, thanks to its N-terminal Zn finger domains and facilitates DNA repair.93 Meanwhile, PARP-2 is recruited to DNA damage sites thanks to its tryptophan, glycine and arginine (WGR) motif.92
Research on ADP ribosylation in cancer has historically centred on its role in DDRs, focusing on its importance in repairing and maintaining genomic integrity. An example of this is how the interaction of the hepatitis B virus X protein with PARP1 may impair DNA repair mechanisms and alter PARP1-induced epigenetic modifications, leading to abnormal gene expression during the early stages of hepatocarcinogenesis.96 However, recent studies have extended the scope of ADP ribosylation beyond direct histone ribosylation to include the modulation of numerous biological processes that are essential for cancer cell survival and metastasis. Recent findings on HCC have highlighted the critical role of PARP10 and PARP14 in disease progression and metabolic adaptation. While PARP10 can act as both a tumour suppressor and an oncogene,97 increasing evidence supports a tumour-suppressive role in HCC. For instance, it has been reported that PARP10 significantly hinders HCC metastasis through the mono-ADP-ribosylation (MARylation) of Aurora A kinase, leading to a reduction of its kinase activity.98 This has a negative impact on EMT and thereby reduces tumour cell migration and invasion capability. The relevance of PARP10 in halting cancer progression is further emphasised by its diminished expression in metastatic HCC compared with primary tumours and adjacent non-cancerous tissues.98 Additionally, the inactivation of PARP10 through a feedback mechanism involving PLK1 and NF-κB signalling has been linked to accelerated HCC progression.99 This is partly due to PARP10’s role in modulating the NF-κB pathway by interfering with the ubiquitin-mediated degradation of NF-κB essential modulator (NEMO), a key component of the IκB kinase (IKK) complex.99 As a result, the phosphorylation and subsequent inactivation of PARP10 by PLK1 lead to the activation of NF-κB signalling. In turn, NF-κB transcriptionally inhibits PARP10 expression, causing its downregulation and promoting HCC development. Conversely, the MARylation of PLK1 by PARP10 dampens PLK1’s kinase activity and oncogenic potential in HCC,99 illustrating the complex interplay between phosphorylation and MARylation.
Further research into the metabolic adaptations of HCC has identified PARP14 as a key regulator of the Warburg effect, a metabolic hallmark of cancer characterised by elevated aerobic glycolysis.100 PARP14 indirectly fosters this metabolic pathway by maintaining low PKM2 activity, which is critical for sustaining aerobic glycolysis and supporting tumour cell survival. In the absence of PARP14, PKM2 is phosphorylated and activated by the pro-apoptotic kinase c-Jun N-terminal Kinase 1 (JNK1), promoting the conversion of glucose to pyruvate and impairing the Warburg effect. Moreover, PARP14 is overexpressed in primary HCC tumours and is associated with poor prognosis.100
HPTMs and DNA readers
A fast-growing collection of reader domains links different sets of histone and DNA modifications to otherwise unexplainable transcriptional outcomes. Grouped into families based on unifying structural features, these domains are integral components of downstream effectors that are recruited to specific epigenetic marks to regulate gene expression.101 Some reader domain families recognise multiple HPTMs, while others display high specificity.102 For instance, chromodomains specifically bind to H3K9 or H3K27 dimethylation or trimethylation marks.101 Similarly, methylated residues can also be recognised by malignant brain tumour, Tudor and plant homeodomain domains.102 Meanwhile, 14-3-3 protein domains and macrodomains bind to phospho-ribosyl and ADP-ribosyl-histones, respectively. In addition, reader domains such as PWWP have bivalent engagement through the recognition of specific HPTMs and DNA sequences, allowing for increased selectivity.101 Noteworthy, most of these domains are present in dozens of proteins. For example, as many as sixty-one different (although highly conserved) BrDs are encoded in the human proteome, distributed across forty-six nuclear and cytoplasmic proteins, including histone methyltransferases, HATs and HAT-associated proteins, transcriptional co-activators and mediators, helicases, and ATP-dependent chromatin remodelling complexes. BrDs target chromatin-modifying enzymes to specific genomic sites, often acting as transcriptional co-activators. For instance, BRD4 recruits P-TEFb, which phosphorylates RNA polymerase II, thereby facilitating transcriptional elongation.103 Conversely, chromodomains are associated with gene silencing. In this context, the chromodomain-containing protein HP1 promotes chromatin compaction by recruiting multiple repressive factors, including HDACs and DNMTs, to methylated H3K9.102
DNA methylation, on the other hand, can recruit methyl-binding proteins (MBPs) to methylated sites, where they function as scaffolds to recruit members of chromatin remodelling complexes.104 MBPs are broadly categorised into three main families: methyl-CpG binding domain (MBD) proteins, which specifically recognise symmetrically methylated CpG dinucleotides; SET-associated and Really Interesting New Gene (RING)-associated domain-containing proteins, which display higher binding affinity towards hemi-methylated regions; and methyl-CpG binding zinc finger proteins, which can bind to both methylated and unmethylated DNA.104 Collectively, these MBP families translate DNA methylation marks into transcriptional activation or repression, depending on the cellular context.
Epigenetic readers are pivotal in interpreting the histone code, and their dysregulation can lead to abnormal gene expression, fuelling oncogenesis, tumour growth and metastasis. Among these, BRD4 has attracted considerable attention for its elevated expression in HCC tissues, where it promotes EMT. BRD4 binds acetylated histones, particularly H3K27ac, which are highly present in large enhancers known as super-enhancers, driving the transcription of oncogenes.105 Other BRD-containing proteins, including BRD9, Bromodomain Phd-Finger Transcription Factor (BPTF) and BRPF1, also contribute to HCC progression through distinct mechanisms that are only partially understood. BRD9’s oncogenic role is likely associated with the activation of the Wnt/β-catenin and TUFT1/AKT signalling pathways.106 Meanwhile, BPTF influences the transcriptional upregulation of human telomerase reverse transcriptase,107 and BRPF1 regulates various oncogenes, including E2F2 and EZH2.108 In contrast, BRD7 appears to activate the p53 pathway in HCC, indicating a potential tumour-suppressive function.109 While less studied, chromodomain proteins are increasingly recognised for their roles in hepatocarcinogenesis.
CHD1L, a chromatin-remodelling protein, frequently amplified in HCC, transcriptionally activates several oncogenes during liver carcinogenesis, promoting cell survival and spontaneous liver tumour formation in mouse models.110 Additionally, CHD1L promotes HCC cell migration and metastasis through ZKSCAN3-mediated autophagy,111 and its overexpression may drive HCC dedifferentiation by facilitating chromatin access for critical developmental transcription factors.112 On the other hand, CHD5 is emerging as a critical tumour suppressor in HCC, orchestrating an intricate tumour-suppressive network.113 Its inactivation, often due to promoter hypermethylation and PRC2-mediated H3K27me3, correlates with metastasis and poor prognosis.113
Regarding DNA methylation readers, the MBD protein family is the most extensively studied in HCC. Recent findings highlight MBD1 as a key driver of HCC progression and metastasis, operating in an axis that involves lncRNA SNHG20 and miR-5095. LncRNA SNHG20 stabilises MBD1 expression by acting as a sponge for miR-5095, which otherwise suppresses MBD1 expression.114 Similarly, a regulatory axis involving MBD2, lncRNA LOC105369748 and miR-5095 has been identified, paralleling the mechanism of MBD1.115 This suggests analogous pathways for gene expression regulation in liver cancer, based on intricate interactions among specific lncRNAs, miRNAs and MBD proteins. Additionally, MBD2 overexpression is associated with poor survival in HCC after hepatic resection.116 Methyl-CpG-binding protein 2 (MeCP2), another MBD protein, also displays copy number amplification in HCC, promoting cell proliferation by activating ERK1/2 and inhibiting p38 activity.117 118
Altered expression of SET-associated and RING-associated domain-containing methylated-cytosine readers has also been extensively documented in HCC. Through this domain, Ubiquitin Like With PHD And Ring Finger Domains 1 (UHRF1) recognises hemi-methylated DNA during replication,104 ensuring DNA methylation maintenance and supporting DNMT stability and functionality. Frequently overexpressed in HCC, UHRF1 has been associated with tumour proliferation, metastasis and poor prognosis.117 119 Interestingly, its silencing induces global hypomethylation and epigenetically reprogrammes cancer cells towards differentiation, effectively reducing cancer stem cell traits and tumour growth in HCC models.120 However, studies in zebrafish hepatocytes revealed that UHRF1 overexpression may also drive DNA hypomethylation by decreasing DNMT1 levels and its interaction with DNA.121 This process characterises an aggressive HCC phenotype marked by genomic instability, mutations in TP53 and the disruption of the TP53-induced senescence pathway. Emerging evidence further implicates circUHRF1 in immune modulation, specifically by impairing Natural Killer (NK) cell function and contributing to resistance to anti-PD1 immunotherapy.122 UHRF1 also boosts CSF1 expression through DNA hypomethylation at its promoter, thereby fostering tumour-associated macrophage accumulation.123 These examples illustrate how a complex interplay between epigenetic dysregulation and the immune environment dynamics can contribute to HCC progression. UHRF2, while less studied, has been suggested to exert a similar pro-tumoural role in HCC.124
Impact of metabolism on the regulation of epigenetic machinery
At this point, it is important to mention an additional regulatory layer arising from the intersection between epigenetic and metabolic processes. Since many epigenetic modifiers depend on cellular metabolites as cofactors or substrates, metabolite fluctuations can directly influence chromatin state and gene expression by modulating the epigenome.3 Additionally, the accumulation of certain cellular metabolites can suppress the activity of key epigenetic enzymes. For example, elevated levels of the ketone body β-hydroxybutyrate inhibit HDACs, while tricarboxylic acid (TCA) cycle intermediates like succinate and fumarate inhibit α-ketoglutarate-dependent enzymes, including TET demethylases and KDMs.3
Moreover, reactive metabolites can induce non-enzymatic covalent modifications (NECMs) to nucleotides and histones, such as glycation resulting from interactions with reducing sugars. These modifications directly influence chromatin structure and function, thereby affecting cell phenotype. Unlike enzyme-mediated post-translational modifications, NECMs depend primarily on the cellular microenvironment and tend to accumulate progressively over time. In turn, epigenetic mechanisms can regulate the expression of metabolic genes, shaping the cellular metabolic profile.125 Furthermore, certain metabolic enzymes, including those involved in the synthesis of SAM and acetyl-CoA, can translocate to the nucleus and directly regulate metabolite levels at specific genomic loci.19 With hepatocytes at the core of intermediary metabolism, this interplay between epigenetics and metabolism becomes particularly relevant in the liver. For instance, an imbalance in the homeostasis of the methyl donor SAM within hepatocytes affects both DNA and histone methylation patterns, potentially contributing to HCC development.19
Limitations, conclusions and future prospects
This review offers a comprehensive analysis of the epigenetic mechanisms that influence hepatocarcinogenesis. It covers well-documented alterations in DNA and histone methylation patterns as well as abnormalities in emerging HPTMs in HCC, such as ADP-ribosylation and citrullination (figure 3). Additionally, it provides an updated overview of the role of epigenetic readers in shaping the dysregulated transcriptional landscape characteristic of HCC. However, certain epigenetic regulators relevant to HCC development, such as non-coding RNAs, were not extensively discussed, as they fall beyond the scope of this review and have been described in detail elsewhere.126 127
Figure 3. Epigenetic aberrations in DNA methylation and HPTMs in hepatocellular carcinoma (HCC). (A) Global DNA hypomethylation contributes to genomic instability, while promoter hypermethylation leads to the silencing of tumour suppressor genes, promoting tumour progression. (B) HPTM writers, readers and erasers that are frequently overexpressed (red) or downregulated (blue) in HCC are depicted. Dysregulated expression of these epigenetic modulators disrupts histone modification patterns, leading to aberrant chromatin remodelling and altered accessibility. The resulting transcriptional reprogramming contributes to tumour growth, immune evasion, and therapy resistance. BRD, bromodomain-containing protein; CHD, chromodomain helicase DNA-binding protein; HAT, histone acetyltransferase; HDAC, histone deacetylase; HPTMs, histone post-translational modifications; KDM, lysine demethylase; KMT, lysine methyltransferase; MBD, methyl-CpG binding domain protein; Me, methylation; PRMT, protein arginine methyltransferase; SRA, SET and RING-associated domain-containing proteins; TSGs, tumour suppressor genes.
Elucidating the epigenetic dependencies of HCC is essential for the development of novel therapeutic strategies. Although numerous HPTMs have been identified over the past decade, their roles in HCC remain largely unexplored. Among these are lactylation, associated with glycolytic metabolism; succinylation, linked to the TCA cycle; and hydroxybutyrylation, derived from ketone bodies.128 Investigating these modifications systematically is especially relevant in a metabolically dynamic liver, where intermediary metabolites are closely tied to chromatin regulation. The reciprocal interplay between tumour metabolic reprogramming and epigenetic regulation has emerged as a promising area of research. Oncometabolites can influence the activity of epigenetic enzymes, while epigenetic alterations drive the deregulated expression of metabolic genes.128 However, the impact of this metabolic-epigenetic crosstalk on hepatocarcinogenesis, particularly in the context of metabolic dysfunction-associated steatotic liver disease, remains poorly characterised.
Epigenetic research of HCC has traditionally centred on hepatocytes due to their central role in liver function and hepatocarcinogenesis. However, the epigenetic regulation of the HCC tumour microenvironment (TME) has emerged as a critical area of study, given its influence on tumour initiation and progression. Increasing attention is now directed towards elucidating the epigenetic landscape of key non-parenchymal populations, such as hepatic stellate cells and tumour-associated macrophages. For instance, it is now well established that histone acetylation and DNA methylation regulate the immunosuppressive functions of myeloid-derived suppressor cells and modulate the expression of immune checkpoints, including PD-L1.129 130 Despite substantial knowledge gaps regarding the role of TME epigenetic reprogramming in HCC progression, these findings highlight the importance of viewing epigenetics as a multifaceted phenomenon encompassing multiple cell types rather than one confined to hepatocytes. Similarly, research is shifting from the study of individual epigenetic marks and modulators to integrative approaches that explore the interplay between distinct epigenetic mechanisms and their interactions with metabolic pathways, structural chromatin elements and other regulatory processes. This evolution reflects the growing recognition of the intricate and interconnected nature of epigenetic regulation in HCC.
To address this complexity, integrating artificial intelligence with advanced technologies such as single-cell multiomics, spatial transcriptomics and network pharmacology is becoming essential. These cutting-edge approaches enable high-throughput screening to uncover patterns and dependencies that would otherwise remain undetectable. This information could be used to predict potential toxicities, off-target effects and compensatory mechanisms, thus overcoming some of the current limitations of epigenetic therapies. For instance, epigenetic drugs (‘epidrugs’) with promising preliminary results, such as the DNMT inhibitor guadecitabine, have faced toxicity issues in clinical trials for HCC. Moreover, several epidrugs, including decitabine for DNA methylation and belinostat for histone modifications, have been approved for haematologic malignancies; however, their use in solid tumours, including HCC, remains limited. Beyond monotherapy, epidrugs are increasingly being explored in combination therapies to enhance treatment responses and overcome resistance mechanisms.3 Given the reversible nature of epigenetic alterations, these therapies hold promise for reprogramming malignant gene expression without the risks associated with direct modification of the DNA sequence.131 For example, guadecitabine has been shown to activate the expression of endogenous retroviruses that are epigenetically silenced in HCC cells.132 This leads to an innate immune response that could improve the efficacy of immune checkpoint inhibitors. A clinical trial is currently evaluating the combination of guadecitabine and the anti-PD-L1 antibody durvalumab in digestive tumours, including HCC.127 The growing body of experimental evidence reinforces the potential of epigenetic therapies, emphasising the importance of addressing the challenges associated with their development and clinical implementation.
Footnotes
Funding: This research was funded by grants from PICT-2021-I-A-00975 (GM, JB), PICT-2018-1036 (JB) and PICT-2021- CAT-II-0012 (GM, JB).
prepub: Prepublication history for this paper is available online. To view these files, please visit the journal online (https://doi.org/10.1136/egastro-2025-100186).
Patient consent for publication: Not applicable.
Ethics approval: Not applicable.
Provenance and peer review: Not commissioned; externally peer-reviewed.
Patient and public involvement: Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
References
- 1.Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7:6. doi: 10.1038/s41572-020-00240-3. [DOI] [PubMed] [Google Scholar]
- 2.Péneau C, Imbeaud S, La Bella T, et al. Hepatitis B virus integrations promote local and distant oncogenic driver alterations in hepatocellular carcinoma. Gut. 2022;71:616–26. doi: 10.1136/gutjnl-2020-323153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fernández-Barrena MG, Arechederra M, Colyn L, et al. Epigenetics in hepatocellular carcinoma development and therapy: The tip of the iceberg. JHEP Rep. 2020;2:100167. doi: 10.1016/j.jhepr.2020.100167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu X, Zhao H, Wang R, et al. Cancer epigenetics: from laboratory studies and clinical trials to precision medicine. Cell Death Discov. 2024;10:28. doi: 10.1038/s41420-024-01803-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fernandez A, O’Leary C, O’Byrne KJ, et al. Epigenetic Mechanisms in DNA Double Strand Break Repair: A Clinical Review. Front Mol Biosci. 2021;8:685440. doi: 10.3389/fmolb.2021.685440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sansam CG, Pietrzak K, Majchrzycka B, et al. A mechanism for epigenetic control of DNA replication. Genes Dev. 2018;32:224–9. doi: 10.1101/gad.306464.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arechederra M, Recalde M, Gárate-Rascón M, et al. Epigenetic Biomarkers for the Diagnosis and Treatment of Liver Disease. Cancers (Basel) 2021;13:1265. doi: 10.3390/cancers13061265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Strahl BD, Allis CD. The language of covalent histone modifications. Nature New Biol. 2000;403:41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 9.Toh TB, Lim JJ, Chow E-H. Epigenetics of hepatocellular carcinoma. Clin Transl Med. 2019;8:13. doi: 10.1186/s40169-019-0230-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hardy T, Mann DA. Epigenetics in liver disease: from biology to therapeutics. Gut. 2016;65:1895–905. doi: 10.1136/gutjnl-2015-311292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bayo J, Fiore EJ, Dominguez LM, et al. A comprehensive study of epigenetic alterations in hepatocellular carcinoma identifies potential therapeutic targets. J Hepatol. 2019;71:78–90. doi: 10.1016/j.jhep.2019.03.007. [DOI] [PubMed] [Google Scholar]
- 12.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–92. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 13.Veland N, Lu Y, Hardikar S, et al. DNMT3L facilitates DNA methylation partly by maintaining DNMT3A stability in mouse embryonic stem cells. Nucleic Acids Res. 2019;47:152–67. doi: 10.1093/nar/gky947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu Z, Shi J, Chen Q, et al. Regulation of de novo and maintenance DNA methylation by DNA methyltransferases in postimplantation embryos. J Biol Chem. 2025;301:107990. doi: 10.1016/j.jbc.2024.107990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu X, Zhang Y. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet. 2017;18:517–34. doi: 10.1038/nrg.2017.33. [DOI] [PubMed] [Google Scholar]
- 16.Liu C, Liu L, Chen X, et al. Decrease of 5-hydroxymethylcytosine is associated with progression of hepatocellular carcinoma through downregulation of TET1. PLoS ONE. 2013;8:e62828. doi: 10.1371/journal.pone.0062828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Calvisi DF, Ladu S, Gorden A, et al. Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma. J Clin Invest. 2007;117:2713–22. doi: 10.1172/JCI31457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hernandez-Meza G, von Felden J, Gonzalez-Kozlova EE, et al. DNA Methylation Profiling of Human Hepatocarcinogenesis. Hepatology. 2021;74:183–99. doi: 10.1002/hep.31659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dai Z, Ramesh V, Locasale JW. The evolving metabolic landscape of chromatin biology and epigenetics. Nat Rev Genet. 2020;21:737–53. doi: 10.1038/s41576-020-0270-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao Z, Shilatifard A. Epigenetic modifications of histones in cancer. Genome Biol. 2019;20:245. doi: 10.1186/s13059-019-1870-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eckschlager T, Plch J, Stiborova M, et al. Histone Deacetylase Inhibitors as Anticancer Drugs. Int J Mol Sci. 2017;18:1414. doi: 10.3390/ijms18071414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McBrian MA, Behbahan IS, Ferrari R, et al. Histone acetylation regulates intracellular pH. Mol Cell. 2013;49:310–21. doi: 10.1016/j.molcel.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ler SY, Leung CHW, Khin LW, et al. HDAC1 and HDAC2 independently predict mortality in hepatocellular carcinoma by a competing risk regression model in a Southeast Asian population. Oncol Rep. 2015;34:2238–50. doi: 10.3892/or.2015.4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu L-M, Yang Z, Zhou L, et al. Identification of histone deacetylase 3 as a biomarker for tumor recurrence following liver transplantation in HBV-associated hepatocellular carcinoma. PLoS ONE. 2010;5:e14460. doi: 10.1371/journal.pone.0014460. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Wu J, Du C, Lv Z, et al. The up-regulation of histone deacetylase 8 promotes proliferation and inhibits apoptosis in hepatocellular carcinoma. Dig Dis Sci. 2013;58:3545–53. doi: 10.1007/s10620-013-2867-7. [DOI] [PubMed] [Google Scholar]
- 26.Zhao J, Gray SG, Greene CM, et al. Unmasking the pathological and therapeutic potential of histone deacetylases for liver cancer. Expert Rev Gastroenterol Hepatol. 2019;13:247–56. doi: 10.1080/17474124.2019.1568870. [DOI] [PubMed] [Google Scholar]
- 27.Yang HD, Kim HS, Kim SY, et al. HDAC6 Suppresses Let‐7i‐5p to Elicit TSP1/CD47‐Mediated Anti‐Tumorigenesis and Phagocytosis of Hepatocellular Carcinoma. Hepatology. 2019;70:1262–79. doi: 10.1002/hep.30657. [DOI] [PubMed] [Google Scholar]
- 28.Tsai C-L, Liu W-L, Hsu F-M, et al. Targeting histone deacetylase 4/ubiquitin-conjugating enzyme 9 impairs DNA repair for radiosensitization of hepatocellular carcinoma cells in mice. Hepatology. 2018;67:586–99. doi: 10.1002/hep.29328. [DOI] [PubMed] [Google Scholar]
- 29.Jiang H, Zhang X, Tao Y, et al. Prognostic and clinicopathologic significance of SIRT1 expression in hepatocellular carcinoma. Oncotarget. 2017;8:52357–65. doi: 10.18632/oncotarget.14096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang HD, Kim HS, Kim SY, et al. HDAC6 Suppresses Let-7i-5p to Elicit TSP1/CD47-Mediated Anti-Tumorigenesis and Phagocytosis of Hepatocellular Carcinoma. Hepatology. 2019;70:1262–79. doi: 10.1002/hep.30657. [DOI] [PubMed] [Google Scholar]
- 31.Zhao J, Wozniak A, Adams A, et al. SIRT7 regulates hepatocellular carcinoma response to therapy by altering the p53-dependent cell death pathway. J Exp Clin Cancer Res. 2019;38:252. doi: 10.1186/s13046-019-1246-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Inagaki Y, Shiraki K, Sugimoto K, et al. Epigenetic regulation of proliferation and invasion in hepatocellular carcinoma cells by CBP/p300 histone acetyltransferase activity. Int J Oncol. 2016;48:533–40. doi: 10.3892/ijo.2015.3288. [DOI] [PubMed] [Google Scholar]
- 33.Li Q, Liu X, Jin K, et al. NAT10 is upregulated in hepatocellular carcinoma and enhances mutant p53 activity. BMC Cancer. 2017;17:605. doi: 10.1186/s12885-017-3570-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jin X, Tian S, Li P. Histone Acetyltransferase 1 Promotes Cell Proliferation and Induces Cisplatin Resistance in Hepatocellular Carcinoma. Oncol Res. 2017;25:939–46. doi: 10.3727/096504016X14809827856524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Q, Liu Z, Xu M, et al. PCAF inhibits hepatocellular carcinoma metastasis by inhibition of epithelial-mesenchymal transition by targeting Gli-1. Cancer Lett. 2016;375:190–8. doi: 10.1016/j.canlet.2016.02.053. [DOI] [PubMed] [Google Scholar]
- 36.Jia Y-L, Xu M, Dou C-W, et al. P300/CBP-associated factor (PCAF) inhibits the growth of hepatocellular carcinoma by promoting cell autophagy. Cell Death Dis. 2016;7:e2400. doi: 10.1038/cddis.2016.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Z, Liu Y, Wang H, et al. Patt1, a novel protein acetyltransferase that is highly expressed in liver and downregulated in hepatocellular carcinoma, enhances apoptosis of hepatoma cells. Int J Biochem Cell Biol. 2009;41:2528–37. doi: 10.1016/j.biocel.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 38.Luise C, Robaa D, Regenass P, et al. Structure-Based Design, Docking and Binding Free Energy Calculations of A366 Derivatives as Spindlin1 Inhibitors. Int J Mol Sci. 2021;22:11. doi: 10.3390/ijms22115910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Husmann D, Gozani O. Histone lysine methyltransferases in biology and disease. Nat Struct Mol Biol. 2019;26:880–9. doi: 10.1038/s41594-019-0298-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang J, Hu Y, Zhang B, et al. The JMJD Family Histone Demethylases in Crosstalk Between Inflammation and Cancer. Front Immunol. 2022;13:881396. doi: 10.3389/fimmu.2022.881396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Au SLK, Ng IOL, Wong CM. Epigenetic dysregulation in hepatocellular carcinoma: focus on polycomb group proteins. Front Med. 2013;7:231–41. doi: 10.1007/s11684-013-0253-7. [DOI] [PubMed] [Google Scholar]
- 42.Cai M-Y, Tong Z-T, Zheng F, et al. EZH2 protein: a promising immunomarker for the detection of hepatocellular carcinomas in liver needle biopsies. Gut. 2011;60:967–76. doi: 10.1136/gut.2010.231993. [DOI] [PubMed] [Google Scholar]
- 43.Xu L, Lin J, Deng W, et al. EZH2 facilitates BMI1-dependent hepatocarcinogenesis through epigenetically silencing microRNA-200c. Oncogenesis. 2020;9:101. doi: 10.1038/s41389-020-00284-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gao S-B, Xu B, Ding L-H, et al. The functional and mechanistic relatedness of EZH2 and menin in hepatocellular carcinoma. J Hepatol. 2014;61:832–9. doi: 10.1016/j.jhep.2014.05.015. [DOI] [PubMed] [Google Scholar]
- 45.Wong C-M, Wei L, Law C-T, et al. Up-regulation of histone methyltransferase SETDB1 by multiple mechanisms in hepatocellular carcinoma promotes cancer metastasis. Hepatology. 2016;63:474–87. doi: 10.1002/hep.28304. [DOI] [PubMed] [Google Scholar]
- 46.Li X, Li Q, Ju L, et al. Deficiency of Histone Methyltransferase SET Domain‐Containing 2 in Liver Leads to Abnormal Lipid Metabolism and HCC. Hepatology. 2021;73:1797–815. doi: 10.1002/hep.31594. [DOI] [PubMed] [Google Scholar]
- 47.Bárcena-Varela M, Caruso S, Llerena S, et al. Dual Targeting of Histone Methyltransferase G9a and DNA-Methyltransferase 1 for the Treatment of Experimental Hepatocellular Carcinoma. Hepatology. 2019;69:587–603. doi: 10.1002/hep.30168. [DOI] [PubMed] [Google Scholar]
- 48.Zuo S-R, Zuo X-C, He Y, et al. Positive Expression of SMYD2 is Associated with Poor Prognosis in Patients with Primary Hepatocellular Carcinoma. J Cancer. 2018;9:321–30. doi: 10.7150/jca.22218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang H, Zheng Z, Zhang R, et al. SMYD3 promotes hepatocellular carcinoma progression by methylating S1PR1 promoters. Cell Death Dis. 2021;12:731. doi: 10.1038/s41419-021-04009-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Du Y, Wu S, Xi S, et al. ASH1L in Hepatoma Cells and Hepatic Stellate Cells Promotes Fibrosis-Associated Hepatocellular Carcinoma by Modulating Tumor-Associated Macrophages. Adv Sci (Weinh) 2024;11:2404756. doi: 10.1002/advs.202404756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Y, Xie B, Lin W, et al. Amplification of SMYD3 promotes tumorigenicity and intrahepatic metastasis of hepatocellular carcinoma via upregulation of CDK2 and MMP2. Oncogene. 2019;38:4948–61. doi: 10.1038/s41388-019-0766-x. [DOI] [PubMed] [Google Scholar]
- 52.Zhao Z-K, Yu H-F, Wang D-R, et al. Overexpression of lysine specific demethylase 1 predicts worse prognosis in primary hepatocellular carcinoma patients. World J Gastroenterol. 2012;18:6651–6. doi: 10.3748/wjg.v18.i45.6651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang X, Pan J, Wang G, et al. UNC5B-AS1 promotes the proliferation, migration and EMT of hepatocellular carcinoma cells via regulating miR-4306/KDM2A axis. Cell Cycle. 2021;20:2114–24. doi: 10.1080/15384101.2021.1962632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yamada D, Kobayashi S, Yamamoto H, et al. Role of the hypoxia-related gene, JMJD1A, in hepatocellular carcinoma: clinical impact on recurrence after hepatic resection. Ann Surg Oncol. 2012;19 Suppl 3:S355–64. doi: 10.1245/s10434-011-1797-x. [DOI] [PubMed] [Google Scholar]
- 55.Park S-J, Kim J-G, Son TG, et al. The histone demethylase JMJD1A regulates adrenomedullin-mediated cell proliferation in hepatocellular carcinoma under hypoxia. Biochem Biophys Res Commun. 2013;434:722–7. doi: 10.1016/j.bbrc.2013.03.091. [DOI] [PubMed] [Google Scholar]
- 56.Lu J-W, Ho Y-J, Lin L-I, et al. JMJD2B as a potential diagnostic immunohistochemical marker for hepatocellular carcinoma: a tissue microarray-based study. Acta Histochem. 2015;117:14–9. doi: 10.1016/j.acthis.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 57.Shigekawa Y, Hayami S, Ueno M, et al. Overexpression of KDM5B/JARID1B is associated with poor prognosis in hepatocellular carcinoma. Oncotarget. 2018;9:34320–35. doi: 10.18632/oncotarget.26144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tang B, Qi G, Tang F, et al. Aberrant JMJD3 Expression Upregulates Slug to Promote Migration, Invasion, and Stem Cell-Like Behaviors in Hepatocellular Carcinoma. Cancer Res. 2016;76:6520–32. doi: 10.1158/0008-5472.CAN-15-3029. [DOI] [PubMed] [Google Scholar]
- 59.Sakamoto A, Hino S, Nagaoka K, et al. Lysine Demethylase LSD1 Coordinates Glycolytic and Mitochondrial Metabolism in Hepatocellular Carcinoma Cells. Cancer Res. 2015;75:1445–56. doi: 10.1158/0008-5472.CAN-14-1560. [DOI] [PubMed] [Google Scholar]
- 60.Wang D, Han S, Peng R, et al. Depletion of histone demethylase KDM5B inhibits cell proliferation of hepatocellular carcinoma by regulation of cell cycle checkpoint proteins p15 and p27. J Exp Clin Cancer Res. 2016;35:37. doi: 10.1186/s13046-016-0311-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guo J-C, Liu Z, Yang Y-J, et al. KDM5B promotes self-renewal of hepatocellular carcinoma cells through the microRNA-448-mediated YTHDF3/ITGA6 axis. J Cell Mol Med. 2021;25:5949–62. doi: 10.1111/jcmm.16342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tang B, Qi G, Tang F, et al. JARID1B promotes metastasis and epithelial-mesenchymal transition via PTEN/AKT signaling in hepatocellular carcinoma cells. Oncotarget. 2015;6:12723–39. doi: 10.18632/oncotarget.3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lachat C, Boyer-Guittaut M, Peixoto P, et al. Epigenetic Regulation of EMT (Epithelial to Mesenchymal Transition) and Tumor Aggressiveness: A View on Paradoxical Roles of KDM6B and EZH2. Epigenomes. 2018;3:1. doi: 10.3390/epigenomes3010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ji X, Jin S, Qu X, et al. Lysine-specific demethylase 5C promotes hepatocellular carcinoma cell invasion through inhibition BMP7 expression. BMC Cancer. 2015;15:801. doi: 10.1186/s12885-015-1798-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhou W, Gong L, Wu Q, et al. PHF8 upregulation contributes to autophagic degradation of E-cadherin, epithelial-mesenchymal transition and metastasis in hepatocellular carcinoma. J Exp Clin Cancer Res. 2018;37:215. doi: 10.1186/s13046-018-0890-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu B-H, Chen H, Cai C-M, et al. Epigenetic silencing of JMJD5 promotes the proliferation of hepatocellular carcinoma cells by down-regulating the transcription of CDKN1A 686. Oncotarget. 2016;7:6847–63. doi: 10.18632/oncotarget.6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fuhrmann J, Thompson PR. Protein Arginine Methylation and Citrullination in Epigenetic Regulation. ACS Chem Biol. 2016;11:654–68. doi: 10.1021/acschembio.5b00942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lorton BM, Shechter D. Cellular consequences of arginine methylation. Cell Mol Life Sci. 2019;76:2933–56. doi: 10.1007/s00018-019-03140-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Migliori V, Müller J, Phalke S, et al. Symmetric dimethylation of H3R2 is a newly identified histone mark that supports euchromatin maintenance. Nat Struct Mol Biol. 2012;19:136–44. doi: 10.1038/nsmb.2209. [DOI] [PubMed] [Google Scholar]
- 70.Zhang B, Dong S, Li Z, et al. Targeting protein arginine methyltransferase 5 inhibits human hepatocellular carcinoma growth via the downregulation of beta-catenin. J Transl Med. 2015;13:349. doi: 10.1186/s12967-015-0721-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lei Y, Han P, Tian D. Protein arginine methyltransferases and hepatocellular carcinoma: A review. Transl Oncol. 2021;14:101194. doi: 10.1016/j.tranon.2021.101194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Du P, Luo K, Li G, et al. PRMT4 promotes hepatocellular carcinoma progression by activating AKT/mTOR signaling and indicates poor prognosis. Int J Med Sci. 2021;18:3588–98. doi: 10.7150/ijms.62467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhu D, Zhang Y, Wang S. Histone citrullination: a new target for tumors. Mol Cancer. 2021;20:90. doi: 10.1186/s12943-021-01373-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang Y, Lyu Y, Tu K, et al. Histone citrullination by PADI4 is required for HIF-dependent transcriptional responses to hypoxia and tumor vascularization. Sci Adv. 2021;7:35. doi: 10.1126/sciadv.abe3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lu M, Zhang X, Xu Y, et al. Elevated histone H3 citrullination is associated with increased Beclin1 expression in HBV-related hepatocellular carcinoma. J Med Virol. 2020;92:1221–30. doi: 10.1002/jmv.25663. [DOI] [PubMed] [Google Scholar]
- 76.Zhu D, Lu Y, Wang Y, et al. PAD4 and Its Inhibitors in Cancer Progression and Prognosis. Pharmaceutics. 2022;14:2414. doi: 10.3390/pharmaceutics14112414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhu W, Fan C, Dong S, et al. Neutrophil extracellular traps regulating tumorimmunity in hepatocellular carcinoma. Front Immunol. 2023;14:1253964. doi: 10.3389/fimmu.2023.1253964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Guo W, Zheng Y, Xu B, et al. Investigating the expression, effect and tumorigenic pathway of PADI2 in tumors. Onco Targets Ther. 2017;10:1475–85. doi: 10.2147/OTT.S92389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rossetto D, Avvakumov N, Côté J. Histone phosphorylation: a chromatin modification involved in diverse nuclear events. Epigenetics. 2012;7:1098–108. doi: 10.4161/epi.21975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bauman AL, Scott JD. Kinase- and phosphatase-anchoring proteins: harnessing the dynamic duo. Nat Cell Biol. 2002;4:E203–6. doi: 10.1038/ncb0802-e203. [DOI] [PubMed] [Google Scholar]
- 81.Sabbattini P, Sjoberg M, Nikic S, et al. An H3K9/S10 methyl-phospho switch modulates Polycomb and Pol II binding at repressed genes during differentiation. Mol Biol Cell. 2014;25:904–15. doi: 10.1091/mbc.E13-10-0628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Georgoulis A, Vorgias CE, Chrousos GP, et al. Genome Instability and γH2AX. Int J Mol Sci. 2017;18:1979. doi: 10.3390/ijms18091979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Matsuda Y, Wakai T, Kubota M, et al. DNA damage sensor γ -H2AX is increased in preneoplastic lesions of hepatocellular carcinoma. ScientificWorldJournal. 2013;2013:597095. doi: 10.1155/2013/597095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xiao H, Tong R, Ding C, et al. γ-H2AX promotes hepatocellular carcinoma angiogenesis via EGFR/HIF-1α/VEGF pathways under hypoxic condition. Oncotarget. 2015;6:2180–92. doi: 10.18632/oncotarget.2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Komar D, Juszczynski P. Rebelled epigenome: histone H3S10 phosphorylation and H3S10 kinases in cancer biology and therapy. Clin Epigenetics. 2020;12:147. doi: 10.1186/s13148-020-00941-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhong Q, Shi G, Zhang Q, et al. Role of phosphorylated histone H3 serine 10 in DEN-induced deregulation of Pol III genes and cell proliferation and transformation. Carcinogenesis. 2013;34:2460–9. doi: 10.1093/carcin/bgt219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang Q, Zhong Q, Evans AG, et al. Phosphorylation of histone H3 serine 28 modulates RNA polymerase III-dependent transcription. Oncogene. 2011;30:3943–52. doi: 10.1038/onc.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Thorslund T, Ripplinger A, Hoffmann S, et al. Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature New Biol. 2015;527:389–93. doi: 10.1038/nature15401. [DOI] [PubMed] [Google Scholar]
- 89.Li Y-H, Zhong M, Zang H-L, et al. MTA1 Promotes Hepatocellular Carcinoma Progression by Downregulation of DNA-PK-Mediated H1.2T146 Phosphorylation. Front Oncol. 2020;10:567. doi: 10.3389/fonc.2020.00567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jeng Y-M, Peng S-Y, Lin C-Y, et al. Overexpression and amplification of Aurora-A in hepatocellular carcinoma. Clin Cancer Res. 2004;10:2065–71. doi: 10.1158/1078-0432.ccr-1057-03. [DOI] [PubMed] [Google Scholar]
- 91.Lin Z-Z, Jeng Y-M, Hu F-C, et al. Significance of Aurora B overexpression in hepatocellular carcinoma. Aurora B Overexpression in HCC. BMC Cancer. 2010;10:461. doi: 10.1186/1471-2407-10-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gupte R, Liu Z, Kraus WL. PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev. 2017;31:101–26. doi: 10.1101/gad.291518.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.van Beek L, McClay É, Patel S, et al. PARP Power: A Structural Perspective on PARP1, PARP2, and PARP3 in DNA Damage Repair and Nucleosome Remodelling. Int J Mol Sci. 2021;22:10. doi: 10.3390/ijms22105112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Krishnakumar R, Kraus WL. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell. 2010;39:8–24. doi: 10.1016/j.molcel.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Smith R, Lebeaupin T, Juhász S, et al. Poly(ADP-ribose)-dependent chromatin unfolding facilitates the association of DNA-binding proteins with DNA at sites of damage. Nucleic Acids Res. 2019;47:11250–67. doi: 10.1093/nar/gkz820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Na T-Y, Ka N-L, Rhee H, et al. Interaction of hepatitis B virus X protein with PARP1 results in inhibition of DNA repair in hepatocellular carcinoma. Oncogene. 2016;35:5435–45. doi: 10.1038/onc.2016.82. [DOI] [PubMed] [Google Scholar]
- 97.Zhou Z, Wei B, Liu Y, et al. Depletion of PARP10 inhibits the growth and metastatic potential of oral squamous cell carcinoma. Front Genet. 2022;13:1035638. doi: 10.3389/fgene.2022.1035638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhao Y, Hu X, Wei L, et al. PARP10 suppresses tumor metastasis through regulation of Aurora A activity. Oncogene. 2018;37:2921–35. doi: 10.1038/s41388-018-0168-5. [DOI] [PubMed] [Google Scholar]
- 99.Tian L, Yao K, Liu K, et al. PLK1/NF-κB feedforward circuit antagonizes the mono-ADP-ribosyltransferase activity of PARP10 and facilitates HCC progression. Oncogene. 2020;39:3145–62. doi: 10.1038/s41388-020-1205-8. [DOI] [PubMed] [Google Scholar]
- 100.Iansante V, Choy PM, Fung SW, et al. PARP14 promotes the Warburg effect in hepatocellular carcinoma by inhibiting JNK1-dependent PKM2 phosphorylation and activation. Nat Commun. 2015;6:7882. doi: 10.1038/ncomms8882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Weaver TM, Morrison EA, Musselman CA. Reading More than Histones: The Prevalence of Nucleic Acid Binding among Reader Domains. Molecules. 2018;23:2614. doi: 10.3390/molecules23102614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Park I-G, Jeon M, Kim H, et al. Coordinated methyl readers: Functional communications in cancer. Semin Cancer Biol. 2022;83:88–99. doi: 10.1016/j.semcancer.2021.03.015. [DOI] [PubMed] [Google Scholar]
- 103.Altendorfer E, Mochalova Y, Mayer A. BRD4: a general regulator of transcription elongation. Transcription. 2022;13:70–81. doi: 10.1080/21541264.2022.2108302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mahmood N, Rabbani SA. DNA Methylation Readers and Cancer: Mechanistic and Therapeutic Applications. Front Oncol. 2019;9:489. doi: 10.3389/fonc.2019.00489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tsang FH-C, Law C-T, Tang T-CC, et al. Aberrant Super-Enhancer Landscape in Human Hepatocellular Carcinoma. Hepatology. 2019;69:2502–17. doi: 10.1002/hep.30544. [DOI] [PubMed] [Google Scholar]
- 106.Fang D, Wang M-R, Guan J-L, et al. Bromodomain-containing protein 9 promotes hepatocellular carcinoma progression via activating the Wnt/β-catenin signaling pathway. Exp Cell Res. 2021;406:112727. doi: 10.1016/j.yexcr.2021.112727. [DOI] [PubMed] [Google Scholar]
- 107.Zhao X, Zheng F, Li Y, et al. BPTF promotes hepatocellular carcinoma growth by modulating hTERT signaling and cancer stem cell traits. Redox Biol. 2019;20:427–41. doi: 10.1016/j.redox.2018.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cheng CL-H, Tsang FH-C, Wei L, et al. Bromodomain-containing protein BRPF1 is a therapeutic target for liver cancer. Commun Biol. 2021;4:888. doi: 10.1038/s42003-021-02405-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chen C-L, Mo H-Q, Jiang Y-H, et al. BRD7 inhibits tumor progression by positively regulating the p53 pathway in hepatocellular carcinoma. J Cancer. 2021;12:1507–19. doi: 10.7150/jca.50293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li Y, Chen L, Chan THM, et al. SPOCK1 is regulated by CHD1L and blocks apoptosis and promotes HCC cell invasiveness and metastasis in mice. Gastroenterology. 2013;144:179–91. doi: 10.1053/j.gastro.2012.09.042. [DOI] [PubMed] [Google Scholar]
- 111.Zhang X, Bai Y, Huang L, et al. CHD1L augments autophagy-mediated migration of hepatocellular carcinoma through targeting ZKSCAN3. Cell Death Dis. 2021;12:950. doi: 10.1038/s41419-021-04254-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Liu M, Chen L, Ma N, et al. CHD1L promotes lineage reversion of hepatocellular carcinoma through opening chromatin for key developmental transcription factors. Hepatology. 2016;63:1544–59. doi: 10.1002/hep.28437. [DOI] [PubMed] [Google Scholar]
- 113.Xie C-R, Li Z, Sun H-G, et al. Mutual regulation between CHD5 and EZH2 in hepatocellular carcinoma. Oncotarget. 2015;6:40940–52. doi: 10.18632/oncotarget.5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Xu B, Li C, Yang B, et al. LncRNA SNHG20 silencing inhibits hepatocellular carcinoma progression by sponging miR-5095 from MBD1. Am J Transl Res. 2023;15:4314–31. [PMC free article] [PubMed] [Google Scholar]
- 115.E C, Li C, Li H, et al. Silencing of a novel lncRNA LOC105369748 suppresses the progression of hepatocellular carcinoma by sponging miR-5095 from MBD2. J Cell Physiol. 2019;234:18504–12. doi: 10.1002/jcp.28486. [DOI] [PubMed] [Google Scholar]
- 116.Liu W, Wang N, Lu M, et al. MBD2 as a novel marker associated with poor survival of patients with hepatocellular carcinoma after hepatic resection. Mol Med Rep. 2016;14:1617–23. doi: 10.3892/mmr.2016.5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shi R, Zhao H, Zhao S, et al. Molecular subtypes, prognostic and immunotherapeutic relevant gene signatures mediated by DNA methylation regulators in hepatocellular carcinoma. Aging (Albany NY) 2022;14:5271–91. doi: 10.18632/aging.204155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhao LY, Zhang J, Guo B, et al. MECP2 promotes cell proliferation by activating ERK1/2 and inhibiting p38 activity in human hepatocellular carcinoma HEPG2 cells. Cell Mol Biol (Noisy-Le-Grand) 2013;Suppl 59:OL1876–81. [PubMed] [Google Scholar]
- 119.Liu X, Ou H, Xiang L, et al. Elevated UHRF1 expression contributes to poor prognosis by promoting cell proliferation and metastasis in hepatocellular carcinoma. Oncotarget. 2017;8:10510–22. doi: 10.18632/oncotarget.14446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang Y, Hu P, Wang F, et al. UHRF1 inhibition epigenetically reprograms cancer stem cells to suppress the tumorigenic phenotype of hepatocellular carcinoma. Cell Death Dis. 2023;14:381. doi: 10.1038/s41419-023-05895-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mudbhary R, Hoshida Y, Chernyavskaya Y, et al. UHRF1 overexpression drives DNA hypomethylation and hepatocellular carcinoma. Cancer Cell. 2014;25:196–209. doi: 10.1016/j.ccr.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhang P-F, Gao C, Huang X-Y, et al. Cancer cell-derived exosomal circUHRF1 induces natural killer cell exhaustion and may cause resistance to anti-PD1 therapy in hepatocellular carcinoma. Mol Cancer. 2020;19:110. doi: 10.1186/s12943-020-01222-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhang J, Zhang H, Ding X, et al. Crosstalk between macrophage-derived PGE2 and tumor UHRF1 drives hepatocellular carcinoma progression. Theranostics. 2022;12:3776–93. doi: 10.7150/thno.69494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhang Y, Wu K, Liu Y, et al. UHRF2 promotes the malignancy of hepatocellular carcinoma by PARP1 mediated autophagy. Cell Signal. 2023;109:110782. doi: 10.1016/j.cellsig.2023.110782. [DOI] [PubMed] [Google Scholar]
- 125.Zhang T, Gong Y, Meng H, et al. Symphony of epigenetic and metabolic regulation-interaction between the histone methyltransferase EZH2 and metabolism of tumor. Clin Epigenetics. 2020;12:72. doi: 10.1186/s13148-020-00862-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Manna D, Sarkar D. Non-Coding RNAs: Regulating Disease Progression and Therapy Resistance in Hepatocellular Carcinoma. Cancers (Basel) 2020;12:1243. doi: 10.3390/cancers12051243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Braghini MR, Lo Re O, Romito I, et al. Epigenetic remodelling in human hepatocellular carcinoma. J Exp Clin Cancer Res. 2022;41:107. doi: 10.1186/s13046-022-02297-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sun L, Zhang H, Gao P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell. 2022;13:877–919. doi: 10.1007/s13238-021-00846-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Xu L, Zhou C, Liang Y, et al. Epigenetic modifications in the accumulation and function of myeloid-derived suppressor cells. Front Immunol. 2022;13:1016870. doi: 10.3389/fimmu.2022.1016870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ma X, Wu J, Wang B, et al. Epigenetic modifications: Critical participants of the PD‑L1 regulatory mechanism in solid tumors (Review) Int J Oncol. 2022;61:134. doi: 10.3892/ijo.2022.5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cramer SA, Adjei IM, Labhasetwar V. Advancements in the delivery of epigenetic drugs. Expert Opin Drug Deliv. 2015;12:1501–12. doi: 10.1517/17425247.2015.1021678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tao S, Liang S, Zeng T, et al. Epigenetic modification-related mechanisms of hepatocellular carcinoma resistance to immune checkpoint inhibition. Front Immunol. 2022;13:1043667. doi: 10.3389/fimmu.2022.1043667. [DOI] [PMC free article] [PubMed] [Google Scholar]