

Abstract
Pitt-Hopkins Syndrome (PTHS) is a rare neurodevelopmental disorder caused by mutations in the TCF4 gene (18q21.2), encoding the transcription factor 4 (TCF4). This protein is critical for central nervous system development and neuronal maturation. Mutations in TCF4, which range from point mutations to large deletions, result in varying clinical severity, including intellectual disability (ID), motor impairments, and autistic features. Despite its rarity, PTHS has gained increasing attention due to advances in understanding the genetic and molecular mechanisms underlying TCF4 function. Recent research has enhanced diagnostic approaches, including genetic testing techniques like genome sequencing, enabling more accurate identification of the disorder. Despite the evident enhancement in the PTHS management from a medical standpoint, the molecular underpinnings of the disorder progression remain puzzling. This is particularly the case where the disease is caused by point mutations. This review summarizes the latest findings on TCF4 function in PTHS, discusses the variability in mutation effects, highlights current diagnostic and therapeutic advancements, and attempts to explain the molecular bases of mutated TCF4 malfunctionality.
Keywords: Pitt-Hopkins syndrome (PTHS), Transcription factor 4 (TCF4), Neurodevelopment, Genetic disorder, Rare disease, Protein aggregation
Introduction to Pitt-Hopkins syndrome
The first description of the Pitt-Hopkins Syndrome (PTHS, OMIM 610954) appeared in the medical literature in 1978 [1]. PTHS is a rare genetic disorder resulting in abnormal development of the nervous system. The exact prevalence of PTHS is not yet known but, so far, few hundreds of cases have been reported, and the disease is classified as rare. However, the exact onset of the disease is difficult to establish since the disease is manifested with a wide range of symptoms.
Individuals with PTHS have distinctive facial features that are an important element in the diagnosis of the disease. They may be less prominent in infancy but become more defined with age. Patients with PTHS have thick facial features with deep-set eyes, raised eyelid crevices, thin eyebrows, wide lips with a protruding Cupid’s bow-shaped upper lip and a curved lower lip. Facial features are also characterized by a protruding lower part, a wide bridge of the nose, a wide nasal base with enlarged nostrils (the tip of the nose is lowered and can also be pointed). Ears are usually thick, and teeth are widely spaced. Many patients have slender or small hands and feet, broad fingertips and flat feet [2]. Myopia, strabismus and astigmatism have been described in more than half of the patients [3, 4]. Many suffer from scoliosis, postnatal growth retardation and microcephaly [5–8]. Sweetser et al. [2] have reported that people with PTHS often have high pain tolerance, lack of tears, and impaired sweating and dysregulation of body temperature. In all cases there is intellectual retardation and delayed psychomotor development. Developmental delay ranges from moderate to severe. Most patients with PTHS are nonverbal. Some can develop a few words, even fewer can string words together into sentences. Some affected individuals do not acquire the ability to walk unassisted at all, while others can walk with assistance. Those who walk unassisted often have a wide, unsteady gait [2, 9].
About half of the patients have breathing problems, occurring mainly between the ages of three and seven [6, 8–12]. These respiratory problems are characterized by hyperventilation (periods of rapid breathing) followed by periods of slowed or stopped breathing (apnea), and are often associated with strong emotions - anxiety or excitement [10, 11]. However, the diagnosis of PTHS should not be excluded due to the absence of respiratory problems, as they may start after 10 years, or may not start at all [7, 13]. Slightly less than half of PTHS patients have epileptic seizures, the type and severity of which vary from person to person [6, 14]. Seizures are represented by generalized tonic-clonic, atonic or focal seizures. In most PTHS cases, the onset of seizures occurs in the first decade of life [15], but some of them can develop epilepsy later in life [14]. Gastrointestinal disorders, primarily constipation, are also a characteristic symptom [9, 16]. In most patients, this problem begins in infancy. Constipation can be severe, requiring treatment [9]. Also, gastroesophageal reflux may occur [6].
Brain MRIs of PTHS patients have revealed that the most common brain abnormalities are hypoplasia or agenesis of the corpus callosum, reduction of the hippocampus, hypoplasia of the frontal lobes, and hyperintensity of the white matter in the temporal poles [9, 13, 17]. Patients with PTHS often show symptoms of autism spectrum disorders, characterized by impaired communication and social skills [2]. They also often have stereotypic head and hand movements [9, 13, 18]. They may experience behavioral problems, outbursts of aggression and screaming attacks [3, 14, 19]. Sleep disturbances are also common, including difficulty falling asleep and frequent awakenings during the night [6, 9]. However, many people with PTHS are described as sociable and cheerful with possible unprovoked laughter [2, 7, 13]. People with PTHS have a strong attachment to music and like to play with water [2].
PTHS diagnostics
Considering that the phenotypes of PTHS largely overlap with similar disorders on the autism spectrum and beyond, there is an urgent need to obtain a differential diagnosis. In addition to PTHS, two diseases not related to TCF4, but with a similar clinical presentation, have been described. They are characterized by similar to PTHS features, such as significant developmental delay, mental retardation, epilepsy and phenotypic features [20, 21]. The diseases are called Pitt-Hopkins-like syndrome-1 (PTHSL1; OMIM 610042) and Pitt-Hopkins-like syndrome-2 (PTHSL2; OMIM 614325) and are caused by mutations in CNTNAP2 (contactin-associated protein-like 2) and NRXN1 (neurexin 1) encoding genes [22–24], respectively. Both Pitt-Hopkins-like syndromes are inherited in an autosomal recessive manner, in contrast to TCF4 mutations associated with PTHS, which usually occur de novo or can be segregated in autosomal dominant manner. Differential diagnosis should be also made with Angelman syndrome (AS; OMIM 105830), Mowat-Wilson syndrome (MOWS; OMIM 235730), Rett syndrome (RTT; OMIM 312750), Joubert syndrome (JS), and Intellectual disability-hypotonic facies syndrome, X-linked, 1 (MRXHF1; OMIM 309580).
There are three diagnostic scales for scoring traits to assess the likelihood of a patient having PTHS and decide whether to perform genetic testing for the disease. Whalen et al. [9] proposed a scoring system with a maximum score of 20. If children over 3 years old have a score of 15 or more, then testing for PTHS is necessary, but if the child is younger than 3 years old, the total score must be at least 10 to perform genetic screening. In other cases, there is no indication for further testing for PTHS. In contrast, Marangi et al. [25] proposed a scale in which the maximum total score is 16, and a score ≥ 10 recommends genetic testing for PTHS, regardless of age. Unfortunately, these two sets of criteria were not accurate enough, so Zollino et al. [26] proposed a new clinical diagnostic criteria for PTHS, based on signs and symptoms. These are divided into cardinal and supportive, and each is rated accordingly. Cardinal symptoms include facial characteristics (narrow forehead; thin lateral eyebrows; wide nasal bridge/ridge/tip; flared nasal alae; full cheeks/prominent midface; wide mouth/full lips/cupid bow upper lip; thickened/overfolded helices) scored 4 points, if there are at least three of seven present. Cardinal symptoms also include severe ID with absent or limited speech (scored 2 points) and breathing regulation anomalies (scored 2 points). Supportive criteria include myopia, constipation, unstable gait and hands with slender fingers and/or abnormal palmar creases (scored 1 point for each). According to this scoring system, the clinical diagnosis of PTHS can be confirmed if a score is ≥ 9. If the score is 6–8 and characteristic facial features are present, further confirmation of the diagnosis by molecular testing is necessary. If the score is < 6, further studies for other etiologies than PTHS are indicated [26].
Molecular genetic testing
To diagnose patients suspected of having PTHS, molecular testing is recommended. The molecular bases of PTHS are characterised by significant heterogeneity. These may include point mutations, as well as insertion/deletion types, both small and large. These alterations can be either inherited in an autosomal dominant manner or arise de novo. It is important to note that PTHS can quite easily be mistaken for other rare symptoms that result in a similar phenotype. The utilisation of molecular testing enables the precise identification of mutations which are PTHS-related, and explicit them from similar pathologies. It is imperative to exercise caution when establishing a diagnosis in individuals with mutations in the 5’-terminal exons, as these exons are also associated with other syndromes. Molecular testing is offered for individuals with the characteristic facial features of PTHS and severe ID.
DNA sequencing of the TCF4 gene can serve as a basic tool for PTHS diagnosis. DNA sequencing is particularly important for findings of the nucleotide sequence changes that may cause the production of proteins with changed primary sequence. Microarray analysis may be useful for detection of large deletions in the TCF4 gene. Testing for methylation of specific loci can distinguish PTHS from Angelman syndrome [27]. Furthermore, translocations within the TCF4 gene can be analysed by means of karyotyping. Tested may not only be those who are suspected for PTHS but also their parents. It can be instrumental in verifying the de novo occurrence genetic of the variants. Consequently, the reporting of newly discovered gene aberrations to established databases is highly recommended [26].
TCF4 protein
PTHS has been linked to molecular changes in the TCF4 gene that lead to haploinsufficiency, or loss of function of transcription factor 4 (TCF4) [17, 28] also referred to as immunoglobulin transcription factor 2 (ITF2; e.g [29]), SL3-3 enhancer factor 2 (SEF2; e.g [30]), or E2-2 (e.g [31]). Importantly, transcription factor 4 (i.e., TCF4) should not be confused with TCF7L2 (transcription factor 7-like 2), which is also referred to as T cell factor 4. Therefore, in the older literature the acronym TCF4 was often used for TCF7L2. TCF4 is a member of the class I basic helix–loop–helix (bHLH) family of transcription factors, also named E-proteins, that plays an important role in a number of developmental processes [32, 33]. The TCF4 gene is 437 kb in length, located on chromosome 18q21.2 [34]. It consists of 41 exons, 20 of which are alternative 5′-exons, next 20 are internal protein-coding exons, and one is a 3′-non-coding exon [34]. As a result TCF4 exists in many isoforms, with tissue-specific pattern of expression [34] and different research teams studied different TCF4 isoforms, so in the references a reader may encounter different numbering of amino acids that were affected by missense mutations. In this work, we have paid special attention to sorting out the information on the location of each mutation in the TCF4 protein, and all the information refers to the canonical isoform of human TCF4, the B− isoform (UniProt ID P15884).
The canonical TCF4-B− isoform consists of several domains (for the details please refer to the Fig. 1). The C-terminal bHLH domain of TCF4 is able to bind Ephrussi box (E-Box) DNA element (5’-CANNTG-3’) [35–37] in the promoters and enhancers of certain genes [38–41]. In addition to the ability to bind a specific DNA sequence, bHLH is also a platform for homo- or heterodimerization with other bHLH family members [35, 36, 42]. This enables TCF4 functioning as a transcriptional activator [43–45] or repressor [14, 40, 46]. TCF4 regulates the neurogenesis and differentiation of cells by forming heterodimers with proneuronal activators belonging to the bHLH family, such as achaete-scute homolog 1 (ASCL1), protein atonal homolog 1 (ATOH1/MATH1) and neurogenic differentiation factor 1 (NEUROD1) [33, 47–51]. Importantly, reduced interaction between TCF4 and ASCL1 has been linked to respiratory symptoms of PTHS patients [17, 28, 52, 53], as ASCL1 has been shown to be part of pathway controlling the development of respiratory-related noradrenergic neurons [52, 54].
Fig. 1.

Schematic representation of TCF4 canonical isoform (B –) and its mutations (point substitutions) in PTHS. AD1 (1–99) and AD2 (325–432) are activation domain 1 [55, 56] and 2 [31, 55], respectively; CE (171–185) is conserved element [65] (marked in yellow); Rep (511–540) is repression domain [66] (marked in blue), bHLH (564–617) is basic helix-loop-helix domain [108] (b marked in red and HLH marked in green); NLS1 (156–178) and NLS2 (564–602) are nuclear localization signal 1 [34] and 2 [68], respectively (black dashed areas); NES1 (585–594) and NES2 (600–614) are nuclear export signals [68]. PTHS-related substitutions in TCF4 appear mostly in the basic, DNA-binding region of bHLH. Reported cases include: G358V [7, 46]; D535G [14, 46]; R565W [9]; R572G [14]; R572Q [9]; R574H [7]; R574P [7]; R574C [25]; R576Q [17]; R576W [17, 28]; R578P [18]; N581D [25]; A583P [9]; T602A [109]; K603E [109]; A610V [14]; V613I [109]
In addition to the conserved C-terminal bHLH domain responsible for dimerization and DNA binding, TCF4 has N-terminal functional domains, which in E-proteins are known to be responsible for transcriptional regulation [31, 55–59]. There is a variety of TCF4 isoforms that are expressed at varying levels in different organs [34]. Highest TCF4 expression levels were found in the developing human brain, shortly after birth [60]. The isoforms variable, besides their location in the body, is mainly the length of the N-terminal domain [34]. The canonical TCF4-B – isoform possesses two activation domains (AD1, AD2) (Fig. 1) that are known to modulate transcriptional activity [34, 55], whereas shorter isoforms have no or only partial AD1 [34]. AD1 and AD2 can interact with transcriptional activator p300 [57, 61] and AD1 can also interact with a repressor of transcription, runt-related transcription factor 1 (RUNX1T1) [62]. The demonstration that TCF4 can act as an activator [14, 40, 46] or a repressor of transcription [43–45] has revealed TCF4 as transcriptional switch. Recently, Sozańska et al. [63] showed that beside bHLH domain, TCF4-I – is completely disordered and the DNA binding has no effect on the conformation of N-terminus of the protein. It seems that TCF4 act as a hub protein mediating formation of the transcription initiation complex, where its bHLH serves as an anchor positioning the protein on an appropriate DNA sequence, whereas pliable N-terminal part of protein use its regulatory domains to interact with multiple partners [63, 64]. Besides ADs, TCF4 also contains two repression domains (Fig. 1). The first, conserved element (CE), is located between AD1 and AD2 and is capable to repress AD1 activity [65]. The second, called the repression domain (Rep), is located between AD2 and bHLH and has been shown to repress the activity of both AD1 and AD2 [66] but the exact molecular mechanism remains unrevealed. Another possible mode of intramolecular regulation appears to occur in TCF4 through a 4-residue sequence, RSRS, located between the Rep and bHLH domains in the “+” isoforms. Its presence has been shown to decrease transcriptional activity in some cell types [34, 67]. Transcriptional activity of transcription factors including TCF4 can be also regulated by its distribution in the cell. TCF4 was shown to contain two nuclear localization signals (NLS) and two nuclear export signals (NES) (Fig. 1), which suggests complexity of subcellular localization control. NLS-1, with the putative activity of nucleolar localization signal (NoLS), overlaps with the CE domain at the N-terminal region while NLS-2 overlaps NES-1 and NES-2 located in the bHLH domain at the C-terminal region of protein. The existence of overlapping signals indicate their putative masking/unmasking, dependent on the TCF4 interaction with partners [34, 68].
The function of TCF4 protein
In the nervous system, TCF4 has been shown to play a role in the differentiation of progenitor cells. TCF4 mutation leads to autonomous reduction of mature oligodendrocytes and concomitant reduction of myelination [69]. Myelination is an important process that involves the formation of a bilipid myelin layer around the axon, which ensures the rapid transmission of information necessary for cognitive, behavioural and emotional functions. Using mouse models with heterozygous Tcf4 mutations, Phan et al. [70] discovered the regulation of genes associated with oligodendrocytes and myelination in newborn and adults. As a result, they observed a higher number of progenitor cells than mature oligodendrocytes [70]. Also, they observed a reduced proportion of myelinated axons in the corpus callosum, and an increased proportion of unmyelinated axons in Tcf4+/− mice [70]. In contrast, Wedel et al. [69] found that the lack of TCF4 interaction with OLIG2 leads to impaired terminal oligodendrocyte differentiation. On the other hand, Papes et al. [71] performed studies on the relationship between loss of TCF4 function, neural progenitor cells (NPCs) proliferation and cortical neuron content using NPCs and neurons derived from induced pluripotent stem cells (iPSCs) of PTHS patients. Their results indicate that there are many defects in the development of neural tissue in PTHS patients. They observed abnormal morphology, structure, and size of brain organoids with proportionally more progenitor cells and fewer neurons, confirming defects in the proliferation and differentiation of NPCs [71]. This study supports the role of TCF4 in progenitor cells differentiation. Papes et al. [71] also observed that lack of TCF4 function decreases WNT signalling activity, induces lower expression of SOX transcription factors (SOX3 and SOX4), which in turn leads to decreased proliferation, increased cell aging and reduced neuronal differentiation.
The bHLH proteins play an important role during the development of the nervous system [51, 72]. Studies in Drosophila melanogaster and vertebrate models have played a key role in the discovery of complex networks of transcription factors that regulate the process of neuronal differentiation [51, 72]. Proneural transcription factors belonging to the bHLH family, such as ATOH1 and neurogenin-2 (NEUROG2), are expressed in neuronal precursor cells and coordinate neuronal differentiation programs. Many proneural factors form heterodimers with TCF4 to regulate gene expression during neuronal development. Homozygous Tcf4 knockout (Tcf4−/−) mice die within 24 h after birth, indicating that TCF4 is a key transcription factor required for proper development [73, 74]. These mice do not have any major anatomical defects, but detailed analysis of their brainstem has shown that there is impaired development of the pontine nucleus [74]. Deletion of Tcf4 in mice results in a reduction in the number of neurons forming the pontine nucleus and an accumulation of ectopic neurons outside this region. Importantly, these deficits are highly specific for Tcf4, as the development of this part of the brainstem in mice with knockout genes encoding other E proteins (Tcf3−/− and Tcf12−/−) is normal [74]. The development of the pontine nuclei is also dependent on the expression of the Atoh1 gene, whose product can heterodimerize with TCF4. Interestingly, Atoh1+/−/Tcf4+/− mutant mice also show defects in the development of pontine nuclei, while Tcf4+/− mice do not show these abnormalities [74]. The ability of TCF4 to form specific heterodimers may therefore be important for the development of specific neuronal subpopulations in the brain, and the studies described a role of TCF4 and TCF4:ATOH1 heterodimers in brain development. In addition, TCF4 has been shown to regulate the expression of several critical neurodevelopmental genes, including the promoters of CNTNAP2 and NRXN1 in vitro [46]. These genes encode related neuronal adhesion receptors, and their mutations are associated with a number of neurodevelopmental disorders [23, 48, 75, 76].
Sepp et al. [77] investigated the function of TCF4 isoforms in neurons. They hypothesized that TCF4 activity in primary neurons of the cerebral cortex depends on neuronal activity and protein kinase A (PKA). The results led to conclusion that in primary rat neurons, TCF4-controlled transcription is initiated by membrane depolarization depending on the cAMP–PKA pathway. It has been also suggested that Ca2+ influx through voltage-gated channels, in response to synaptic activity, is involved in triggering TCF4-driven transcription. This occurs because PKA phosphorylates the serine residue of TCF4 and this process is essential for the transcriptional activity of TCF4. Sepp et al. [77] also demonstrated that depolarization in primary neurons contributes to the activation of TCF4-ASCL1 heterodimers, which are involved in the induction of Gadd45g expression. Furthermore, when Tcf4 is silenced, the expression of the KCNQ1 and SCN10a genes increases in neurons of the rat medial prefrontal cortex, leading to reduced neuronal excitability [78]. Both genes encode ion channels, present mainly in the peripheral nervous system [78], and the binding properties of E-proteins to DNA were shown to be regulated by Ca2+ levels [17, 79, 80]. Ca2+-dependent proteins, such as calmodulin (CaM), interact directly with the bHLH domain [81], which is involved in the interaction of E-proteins with target DNA. In the presence of Ca2+, CaM primarily inhibits the binding of E-protein homodimers to DNA, whereas E-protein heterodimers are less sensitive [79]. This shows that alteration of the levels of CaM or Ca2+ in cells has a direct effect on E protein-mediated transcription [82, 83].
There have been many animal experiments confirming the role of TCF4 in neurodevelopment. In 2007, Brockschmidt et al. [84] analysed the embryonic expression of TCF4 in the striped Danio rerio. On the first day after fertilization, Tcf4 expression was evident in the telencephalon. On the second day, weak Tcf4 expression appeared in the hindbrain and increased in many regions of the midbrain. On the following and subsequent days, Tcf4 was also expressed in the retina, which may correlate with visual defects in individuals with PTHS. These results make D. rerio an important model for understanding PTHS [84]. The role of TCF4 in the development of organisms has also been studied in mouse models, in which Tcf4 mutations resulted in a PTHS-like phenotype. There are results indicating that Tcf4−/− mice die in less than 24 h after birth, confirming the necessity of TCF4 for postnatal survival [73, 85]. Also, using mouse models, Schoof et al. [86] classified TCF4 as an important factor for a proper brain architecture, as well as neuronal morphology and differentiation.
In addition to the neuronal system, TCF4 has been shown to play an important role in the development of the immune system [73, 87, 88]. It participates in regulation of the development of pDCs [87, 89], which play a key role in the response to most viruses due to their ability to produce massive amounts of type I interferon. TCF4 activity can be regulated negatively by interaction with repressor proteins - inhibitors of differentiation 2 (ID2), resulting in the formation of inactive heterodimers with TCF4. The resulting complex cannot bind to DNA or other transcription activators, leaving cells undifferentiated [90]. That originates from the fact that ID2 has no b motif, which is essential for DNA binding. TCF4 and its partners, with which it forms specific dimers, are involved in the control of lymphoid tissue development in the immune system, as studies using transgenic mice have shown that TCF4 can influence the development of both B and T cells [87, 88, 91].
TCF as transcription factor regulating gene expression
Identification of genes regulated by TCF4 is crucial for understanding the etiology of PTHS and finding new potential therapeutic targets. TCF4 binds the aforementioned DNA sequence known as the E-box (5’-CANNTG-3’), which is relatively short and thus common, making the identification of TCF4 target genes challenging [92, 93]. In 2013, Forrest et al. [92], used small interfering RNA (siRNA) to induce TCF4 silencing in the SH-SY5Y cell line to determine the effect of TCF4 silencing on the expression of other genes. They identified 1204 differentially expressed genes, of which 494 were up-regulated and the rest down-regulated. Analyses have shown that TCF4 is involved in processes such as apoptosis and NF-kB signaling. The latter is crucial for immune response, cell survival, and proliferation, so dysregulation of this pathway can lead to inflammation, autoimmune diseases, and cancer development. Forrest et al. [92] observed that in cells with silenced TCF4, the activity of components of the NF-kB pathway and caspase 3/7 were increased, leading to cell death. Using the MetaCore analysis tool, they were able to identify the effect of TCF4 deactivation on the expression of genes involved in the epithelial-mesenchymal transition (EMT), neurogenesis, and TGF-β controlling proliferation, differentiation and other functions of the cell. The results of this analysis showed a strong down-regulation of ligands from the TGF-β family, also the expression of the BMPR1A receptor and SMAD proteins which are involved in the TGF-β signaling pathway [92]. The EMT is the process by which an epithelial cell becomes a mesenchymal cell and occurs during embryonic development, and the formation of tumor metastases [94, 95]. In cells with silenced TCF4, SNAI2 and DEC1 that are regulators of EMT, were differentially expressed [92]. While the expression of SNAI2 was downregulated, the expression of DEC1 increased. Also, Forrest et al. [92]. suggested, that TCF4 regulation of IGF2 expression may be a determinant of cognitive dysfunction, as deactivation of TCF4 in cells results in changes in IGF signaling, as IGF2 plays a role in learning and memory and patients with PTHS have profound ID [96]. Also, NEUROG2 and ASCL1 genes, whose products directly interact with TCF4, appeared among the genes with the highest TCF4 knockdown downregulation [92]. Later, chromatin immunoprecipitation sequencing (Chip-seq) allowed the detection of more than 10 000 TCF4 binding sites throughout the genome, significantly enriched for the E-box binding motif [47, 93]. By comparison of the Chip-seq resulting list of TCF4 target genes [47] with genes presenting altered expression in previous TCF4 silencing experiment [92], Forrest et al. [47] determined that only 922 genes contained TCF4 binding sites, indicating that only a small group of genes functionally bind TCF4. In addition, the use of enrichment analysis to group biological processes with TCF4 target genes enabled to show that TCF4 plays an important role in neurogenesis, brain development, ion transport, and signal transduction.
TCF4 mutations in the development of PTHS
PTHS patients have been identified with a wide range of deletions of the TCF4 gene, ranging from about 1.2 up to 12 Mb [3, 4, 8, 9, 15, 17, 19, 28, 97, 98]. Sometimes larger and terminal deletions with a size up to 25 Mb occur and include also regions of 18q flanking TCF4. These result in severe ID and less typical, PTHS-like phenotypes as other genes deletion may contribute to the phenotype [8, 99]. Zollino et al. [26] proposed that these large deletions, including mosaic deletions [3–5] and mosaicism as a result from a ring chromosome [100], should rather be termed “18q deletion syndrome” to which the exact breakpoints should be added. Also, balanced translocations disrupting TCF4 have been reported, resulting in diverse, individual-specific phenotypes [13, 101–104].
Splice site, frameshift, and nonsense unique variants of TCF4, mostly located between exons 7 and 19 of TCF4, are the most common genetic abnormality reported in PTHS. Intragenic deletions causing a reading frame shift [9, 15, 84, 105, 106], as well as duplication of several exons [8] have been reported. It has been observed that structural variants of exons 1–4 lead to nonspecific mild ID, whereas these of exons 7–8 results in moderate to severe ID and optionally some of the phenotypic features of PTHS [105]. PTHS patients with mutations in exons 9–19 usually have all typical characteristics of PTHS, as mutations in this region affect all known functional protein isoforms [105]. Structural variants of upstream exons do not necessarily result in changes in all isoforms of TCF4, which may explain the milder symptoms in individuals with exons 1–4 or 7–8 TCF4 variants [105].
Variants in TCF4 underlying typical PTHS usually arise de novo. Therefore, an empiric recurrence risk is up to 2% [26]. What is important, there have been no reports of individuals with typical PTHS reproducing [2]. Several TCF4 variants with milder phenotypes, not fulfilling the diagnostic criteria of PTHS, have been segregating in an autosomal dominant pattern [104, 107], with recurrence risk of 50% [26].
Pathogenic missense mutations were identified in approximately 20% of PTHS individuals and are mostly gathering in the bHLH coding exon 18 [46, 110, 111], leading to point mutations in TCF4 bHLH (Fig. 1). This region of TCF4 is critical for dimer formation and DNA binding [35, 36, 42], and even the subtlest variations in this region may have its critical consequences. Amino acid residues that have been shown as substituted in PTHS and directly engaged in interaction with DNA, based on crystal structures of TCF4 bHLH in complex with E-box (6OD3, 6OD4 [112]), include R565 [9], R572 [9, 14], R574 [7, 25], R576 [17, 28], N581 [25] and K603 [109] (Fig. 2a). Missense mutations of R572, R574 and R576 were shown to affect DNA binding [110] and transcription activation [14, 46, 110]. Also, using molecular dynamics simulations, K603 substitution was predicted to affect DNA binding [109]. In the case of R565 and N581 substitutions, they negatively affected DNA binding, but effect on transcription activation was dependent on dimerization partner and cells used for test [113].
Fig. 2.

Mutations associated with PTHS affect the aggregation rate of TCF4. (A) Amino acid sequence of TCF4 bHLH with PTHS-related mutations sites marked red, with numbering corresponding to B – isoform. Arrows correspond to residues observed to interact with DNA (black – both 6OD3 and 6OD4, gray – only 6OD3 [112]). (B) PTHS-related variants and correlation of their ability to form nuclear puncta with propensity to aggregation. + indicates that nuclear puncta were observed, - that nuclear puncta were not observed (localization of TCF4 variant was the same as wild type), ND indicates no data available. AggreRATE-Pred Change in Aggregation Rate is  , where
, where 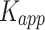 is apparent aggregation rate constant, in units of hour − 1, and
is apparent aggregation rate constant, in units of hour − 1, and 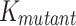 and
and 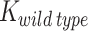 are aggregation rates of pathogenic variant and wild type protein, respectively.
are aggregation rates of pathogenic variant and wild type protein, respectively. 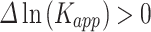 indicates that mutation leads to a higher propensity for aggregation (↑), whereas
indicates that mutation leads to a higher propensity for aggregation (↑), whereas  indicates that pathogenic variant has lower propensity for aggregation than wild type protein (↓)
indicates that pathogenic variant has lower propensity for aggregation than wild type protein (↓)
Interestingly, amino acid residues G358 [7, 46] located in AD2 and D535 [9, 14, 46] located in Rep domain, as well as R578 [18], A583 [9], T602 [109], A610 [14], V613 [109] located in bHLH have also been observed to be substituted in PTHS patients, though directly not interacting with E-box [112] (Fig. 2a). Substitution of G358 and D535, as expected, did not affect DNA binding [110], but were observed to slightly modulate transcription activation, that generally was up-regulated by TCF4 homodimers, and down-regulated by heterodimers [46, 110]. Surprisingly, substitutions of R578 or A583 significantly down-regulated both DNA binding and transcription activation [110, 113]. This suggested that these residues, located in helix 1, may be critical for bHLH in terms of structural stability (as they do not directly interact with E-box DNA). Potentially, R578P as well as A583P substitutions lead to conformational changes in helix 1, as proline is defined as a potent α-helical breaker [114, 115]. As result, this may lead to prevention of DNA binding and have its consequences in inhibition of TCF4-dependent transcription activation. Substitution of T602 (located in loop) was proposed to negatively affect DNA binding, but it has not been experimentally confirmed [109], A610 (located in helix 2) substitution did not affect DNA binding [110], but it down-regulated transcription activation [46, 110], and V613I mutation (helix 2) has been proposed to induce subtle conformational changes, without influence on DNA binding [109].
Based on the effects of PTHS-associated mutations on DNA binding and transcription activation by TCF4 described above, we were wondering how minor changes as a one-residue substitution could trigger such an impact. After all, many amino acid residues are involved in the binding of DNA by TCF4, and the substitution of one residue, sometimes not even directly involved in the interaction with DNA, is fatal to the function of TCF4. What is more, the surprising properties of some TCF4 pathogenic variants related to their cellular localization have been reported. Forrest et al. [46] observed that R574P, R576W and A610V TCF4 variants formed nuclear puncta, in contrast to homogenous intranuclear localization of wild type TCF4. In turn, Sirp et al. [113] observed nuclear puncta formed by R565W and N581D variants in primary neurons. It has been proposed that these mutations may affect the folding stability of TCF4, or may lead to dysfunction of NLS-2 [68], resulting in an aberrant nuclear pattern [113]. It is worth mentioning that forming of nuclear puncta by PTHS-related TCF4 pathogenic variants seems not to be they universal feature. R565W substitution was observed to cause TCF4 localization in nuclear puncta in primary neurons, but not in cell lines [113]. Three different reports indicate that some of PTHS-related TCF4 pathogenic variants may localize in nuclear puncta, but the data is not always in agreement [46, 110, 113]. This process seems to depend on the cell type and methodology used, and possibly these puncta have transient, impermanent nature depending on subtle differences in environmental conditions. However, to our knowledge, the mechanism of forming these puncta has not been experimentally confirmed so far.
What does mutated TCF4 do when it is not doing its job
As indicated above different types of TCF4 mutations cause PTHS. In case of point mutations, the protein is expressed but DNA binding and transcription of its responsive genes may be impaired, even if mutations affect residues which are not directly involved in DNA binding, as described above. A question arises why? Is there another factor that declines TCF4 from its natural functionality?
Seeking an answer this question, based on specific punctate pattern of PTHS linked variants, we decided to analyze the possibility that mutated TCF4 tends to aggregation. Up today, only a few reports relate to TCF4 aggregation and involve an elongation S649Lfs∗57 and G358V variant. Sepp et al. [110] observed that the TCF4 elongation triggered destabilization and aggregation of TCF4, and according to control it was an effect of gain of 56, and not loss of 19 native C-terminal amino acids. Zweier et al. [7], based on bioinformatic prediction, suggested that slightly reduced transcriptional activity of G358V variant may be due to its aggregation, although immunofluorescence data did not reveal visible TCF4 aggregates in cells transfected with the pathogenic variant protein.
Therefore, using bioinformatic tools, we performed analysis to determine whether TCF4 mutations associated with PTHS affect the aggregation propensity of the protein. For this purpose we used AggreRATE-Pred, a mathematical model for the prediction of change in aggregation rate upon point mutation [116]. This model uses experimental aggregation kinetics data [117] to quantitatively predict the change in aggregation rate upon mutation. The prediction is based on known 3D structures for the wild-type proteins in PDB [116].
Our analysis revealed that almost all the PTHS-related TCF4 variants with mutations in the bHLH have higher aggregation rate in comparison to the wild type, with exception of A583P presenting the opposite effect - the aggregation rate of the pathogenic variant is decreased compared to the wild type (Fig. 2b). The calculated effect of the mutations to increase the aggregation rate correlates quite well with the presence of nuclear puncta – which in most cases were observed for those mutations for which the change in aggregation rate is close to 1 or greater (R565W, R574P, R576W, N581D). The exception is A610V, for which Forrest et al. [46] observed the presence of nuclear punctate pattern, but the change in aggregation rate is about 0.5. For mutations not connected to the punctate pattern, we observed little effect on the aggregation rate, with a 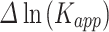 score of less than 1. Currently, the material properties of nuclear puncta formed by TCF4 pathogenic variants are not known. They might be aggregates of crosslinked proteins or they could have liquid nature. Interestingly, based on in silico analysis wild-type TCF4 was proposed to undergo liquid-liquid phase separation (LLPS) [118], but this was not experimentally confirmed to date. Further systematic molecular analyses on the material properties of nuclear puncta formed by TCF4 pathogenic variants are required.
score of less than 1. Currently, the material properties of nuclear puncta formed by TCF4 pathogenic variants are not known. They might be aggregates of crosslinked proteins or they could have liquid nature. Interestingly, based on in silico analysis wild-type TCF4 was proposed to undergo liquid-liquid phase separation (LLPS) [118], but this was not experimentally confirmed to date. Further systematic molecular analyses on the material properties of nuclear puncta formed by TCF4 pathogenic variants are required.
PTHS treatment
To date, there is no golden standard for treating PTHS, and treatment is mainly based on relief of symptoms. Patients with motor discoordination and lack of speech require physiotherapy and work with a speech therapist. Patients with PTHS also suffer from epilepsy and hyperventilation with breathlessness. Sodium valproate is often used as an antiepileptic drug, which exhibits sedative properties, reduces feelings of anxiety, and improves the mental state and mood of patients. In addition, high doses of valproate may have a mitigating effect on the clinical symptoms of respiratory distress associated with PTHS [12]. Acetazolamide was also mentioned to treat respiratory disorders in two patients with PTHS [119, 120]. To be able to treat PTHS, it is necessary to target the cause of the disease. In general, understanding the effects of TCF4 loss is essential for the development of therapeutic strategies leading to the activation of the damaged TCF4 gene. In recent years, many papers have presented potential treatment methods for PTHS. These methods are based on the use of negative regulators of histone deacetylases (HDACs) [121–123], regulation of the WNT signaling pathway [123], regulation of ion channels [124] and the use of viral gene therapy [71, 125].
In 2013, Sweatt [121] suggested that mutations in TCF4 underlying PTHS disrupt the mechanisms that regulate chromatin structure and gene transcription in areas of the brain important for memory and learning. He also formulated a hypothesis that increasing histone acetylation by inhibiting HDAC could rescue any memory impairment observed in PTHS. In 2016, Kennedy et al. [122] decided to test this hypothesis. As a potential model for PTHS they used Tcf4+/− mice with a deletion of the exons that code for the bHLH domain. They exposed them to a stress factor and demonstrated that those had shorter memory for stress factor than controls. In the next stage of experiments, they treated mice with silenced Tcf4 with inhibitor of histone deacetylases (HDACs), suberoylanilide hydroxamic acid (SAHA), and observed that memory of Tcf4+/− mice improved to the level of control. To confirm that selective HDAC inhibition is sufficient to restore memory, Kennedy et al. [122] performed ASO-mediated inhibition of HDAC. The results of this experiment showed improvements in spatial memory, object location memory and threat recognition in Tcf4+/− mice. The results of HDAC inhibition with SAHA treatment, as well as the use of ASO, provide evidence that some cognitive impairments in PTHS can be improved even during postnatal development. The effect of HDACs inhibition on TCF4 was also studied by Hennig et al. [123]. They used positive regulators of the WNT pathway (WNT3A, CHIR-99021), and inhibitors of HDACs (CI-994 or SAHA) to study the expression levels of TCF4 in NPCs derived from human iPSCs and human dermal fibroblasts from patients with PTHS. The results of this experiment showed a significant increase in the expression of TCF4 transcripts, indicating that TCF4 haploinsufficiency in PTHS patients can be treated with positive regulators of the WNT signaling pathway and negative regulators of HDAC [123].
Blocking ion channels has been proposed as another potential treatment for PTHS. It has been shown that silencing Tcf4 in rat medial prefrontal cortex neurons results in an increase in the expression of KCNQ1 and SCN10a, which encodes Nav1.8, a sodium channel subunit, leading to a decrease in neuronal excitability [78]. Blocking their expression is a potential therapeutic target for the treatment of PTHS. Ekins et al. [124] successfully used the Nav1.8 inhibitor, nicardipine, to rescue various behavioral deficits in a mouse model of PTHS.
Results of two papers have recently been published showing the potential application of gene therapy as a treatment for the root cause of PTHS. Papes et al. [71] in their study addressed the correction of PTHS phenotypes by manipulating the TCF4 gene using the CRISPR-Cas9 system. As expected, application of the CRISPR-Cas9 system resulted in amplification of both alleles, which resulted in an increase in TCF4 mRNA levels. The increased level of TCF4 transcripts also corrected the expression of target genes, and the number of mature neurons returned to normal. However, the addition of two lentiviral vectors disrupted the cellular aggregation of organoids [71]. To prevent ectopic TCF4 expression, a second procedure was tested in which an extra-copy of the TCF4 gene was overexpressed under the control of TCF4 binding motifs (µE5 boxes). An increase in TCF4 mRNA levels, correction of target gene expression and normalization of the number of mature neurons were observed, demonstrating an opportunity to correct abnormalities associated with TCF4 haploinsufficiency even during the development of the nervous system [71].
Concurrently, corresponding research by Kim et al. [125] related to embryonic and postnatal reactivation of Tcf4 expression was conducted. They applied viral gene therapy using the LoxP-Cre system to achieve embryonic cell-wide restoration of Tcf4 using 2 mouse models. The first mouse model of PTHS (Tcf4STOP/+) had the insertion of a loxP-P2A-GFP-STOP-loxP cassette upstream of the bHLH DNA binding domain in exon 18 of Tcf4 resulting in brain protein levels halved compared to control. The second mouse model contained the gene encoding Cre recombinase under the control of the Actb promoter. After crossing the two mouse models, sites flanked by loxP sequences were excised by the resulting Cre recombinase during embryonic development and TCF4 levels returned to normal [125]. Also analyzing the results of behavioral tests, Kim et al. [125] confirmed that embryonic activation of Tcf4 expression can completely prevent the occurrence of physiological and behavioral disorders associated with Tcf4 haploinsufficiency. Given the role of TCF4 in neuronal differentiation and maturation [86], Kim et al. [125] tested the effectiveness of correcting physiological and behavioral disorders in Tcf4 knockout mice by selectively restoring Tcf4 expression in glutamatergic and GABAergic neurons. Unfortunately, the results of behavioral tests showed that such selective activation of Tcf4 expression during embryonic development is not sufficient. The second part of the study was focused on postnatal Tcf4 repair, conducting viral gene therapy experiments using a mouse model of PTHS. First, the Cre gene, which is under the control of the human synapsin (hSyn; selective expression in neurons) promoter, was packed into a recombinant PHP.eB vector derived from adeno-associated virus 9 (AAV9). 17 days after injection of the vector to the brain ventricles of neonatal mice Cre became clearly expressed in the hippocampus, and after 60 days the expression of this protein was widespread throughout the brain. The results of behavioral tests revealed that after injection, mice had improved long-term memory, sociability, behavior, and regained control of limb strength, pointing to the possibility of using viral gene therapy to postnatally normalize Tcf4 expression in neural cells.
Conclusions
PTHS is a rare disorder characterized by significant motor delay, mental retardation, autistic characteristics, episodes of hyperventilation followed by apnea, epilepsy, and certain external characteristics that are important aspects for diagnosis [9, 25, 26]. PTHS is a disorder of nervous system development caused by mutations in the TCF4 gene, located on the human chromosome 18q21.2, encoding the TCF4 protein. TCF4 is involved in a variety of processes taking place in the human body, but it plays a major role in the developing and mature central nervous system and is critical for early brain development. It is involved in the development and differentiation of neurons, oligodendrocytes, plasmacytoid dendritic cells, lymphoid progenitors development and signal transduction. Recently, great strides have been made in understanding the role of TCF4 in the human body; however continuously publishing new reports on TCF4 indicates that it still remains incompletely understood. The effects of TCF4 mutations are still unpredictable. It has been shown that pathogenic TCF4 variants are found in ~ 0.7% of tested individuals with ID without a previous clinical suspicion of PTHS [126]. Missense mutations, nonsense mutations, splicing mutations, large deletions of the entire TCF4 gene, small deletions and insertions resulting in reading frame shifts, translocations, duplications and inversions have been identified in patients with PTHS. PTHS-related TCF4 point mutations may result in reduction of the DNA binding and transcription activation capacity of TCF4, as well as homo- or heterodimer formation [14, 46, 110, 113].
Here, we propose that aggregation of PTHS-linked point mutants may be responsible for disease progression, preventing TCF4 from performing its function. We were inspired to conduct the aggregation analysis by the fact that DNA binding in point mutants is affected, even though only one of the many amino acid residues responsible for DNA binding has been substituted, and even for these mutations within the bHLH domain that do not involve the residues responsible for DNA binding (R578 [110], A583 [113], and A610 [46, 110]). It seems that even the slightest disruption of such an evolutionarily well-preserved domain as bHLH can impair the function of the entire protein. The aggregation kinetics is sensitive not only to changes in amino acid sequence, but also changes in environmental conditions [127]. Though the results we obtained are the outcome of in silico analyses, we believe that our results can serve as base and pointing of direction for future experimental confirmation. Although the effects of PTHS-associated mutations on TCF4 function have been intensively studied by many teams, and some have been observed to cause TCF4 pathogenic variants to localize to nuclear puncta, these studies have not targeted the aggregation of TCF4 mutants [46, 110, 113]. We hope that our modest analyses presented in this work will in time become part of something larger, shedding a different light on the role of TCF4 pathogenic variants in the PTHS.
Acknowledgements
We are grateful to prof. Piotr Dobryszycki (WUST) for inspiring us to write this paper.
Abbreviations
- AD
Activation domain
- ASCL1
Achaete-scute homolog 1
- ATOH1
Protein atonal homolog 1
- bHLH
Basic helix-loop-helix
- CaM
Calmodulin
- CE
Conserved element
- CNTNAP2
Contactin associated protein-like 2
- E-box
Ephrussi box
- EMT
Epithelial-mesenchymal transition
- HDACs
Histone deacetylases
- ID
Intelectual disability
- ID2
Inhibitors of differentiation 2
- iPSCs
Induced pluripotent stem cells
- NES
Nuclear export signal
- NEUROG2
Neurogenin-2
- NLS
Nuclear localization signal
- NoLS
Nucleolar localization signal
- NPCs
Neural progenitor cells
- NRXN1
Neurexin 1
- PKA
Protein kinase A
- PTHS
Pitt-Hopkins Syndrome
- Rep
Repression domain
- SAHA
Suberoylanilide hydroxamic acid
- siRNA
Small interfering RNA
- TCF4
Transcription factor 4
Author contributions
Conceptualization: A.T., Investigation: N.S., Visualization: N.S. Writing – Original Draft Preparation: N.S., V.K., A.T., Writing – Review & Editing: N.S., V.K., B.G.M., A.O., A.T.
Funding
This work was supported by a subsidy from The Polish Ministry of Science and High Education for the Faculty of Chemistry of Wroclaw University of Science and Technology.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Pitt D, Hopkins IA. Syndrome of mental retardation, wide mouth and intermittent overbreathing. Aust Paediatr J. 1978. 10.1111/jpc.1978.14.3.182. [DOI] [PubMed] [Google Scholar]
- 2.Sweetser DA, Elsharkawi I, Yonker L, Steeves M, Parkin K, Thibert R, Pitt-Hopkins S, Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya. A., Eds.; 2018.
- 3.Giurgea I, Missirian C, Cacciagli P, Whalen S, Fredriksen T, Gaillon T, Rankin J, Mathieu-Dramard M, Morin G, Martin-Coignard D, et al. TCF4 deletions in Pitt-Hopkins syndrome. Hum Mutat. 2008;29:242–51. 10.1002/humu.20859. [DOI] [PubMed] [Google Scholar]
- 4.Stavropoulos DJ, MacGregor DL, Yoon G. Mosaic microdeletion 18q21 as a cause of mental retardation. Eur J Med Genet. 2010;53:396–9. 10.1016/j.ejmg.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 5.Rossi M, Labalme A, Cordier MP, Till M, Blanchard G, Dubois R, Guibaud L, Heissat S, Javouhey E, Lachaux A, et al. Mosaic 18q21.2 deletions including the TCF4 gene: A clinical report. Am J Med Genet Part A. 2012;158 A:3174–81. 10.1002/ajmg.a.35588. [DOI] [PubMed] [Google Scholar]
- 6.De Winter CF, Baas M, Bijlsma EK, Van Heukelingen J, Routledge S, Hennekam RCM. Phenotype and natural history in 101 individuals with Pitt-Hopkins syndrome through an internet questionnaire system. Orphanet J Rare Dis. 2016. 10.1186/s13023-016-0422-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zweier C, Sticht H, Bijlsma EK, Clayton-Smith J, Boonen SE, Fryer A, Greally MT, Hoffmann L, den Hollander NS, Jongmans M, et al. Further delineation of Pitt-Hopkins syndrome: phenotypic and genotypic description of 16 novel patients. J Med Genet. 2008;45:738–44. 10.1136/jmg.2008.060129. [DOI] [PubMed] [Google Scholar]
- 8.Goodspeed K, Newsom C, Morris MA, Powell C, Evans P, Golla S. Pitt-Hopkins syndrome: A review of current literature, clinical approach, and 23-Patient case series. J Child Neurol. 2018;33:233–44. 10.1177/0883073817750490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whalen S, Héron D, Gaillon T, Moldovan O, Rossi M, Devillard F, Giuliano F, Soares G, Mathieu-Dramard M, Afenjar A, et al. Novel comprehensive diagnostic strategy in Pitt-Hopkins syndrome: clinical score and further delineation of the TCF4 mutational spectrum. Hum Mutat. 2012;33:64–72. 10.1002/humu.21639. [DOI] [PubMed] [Google Scholar]
- 10.Giurgea I, Missirian C, Cacciagli P, Whalen S, Fredriksen T, Gaillon T, Rankin J, Mathieu-Dramard M, Morin G, Martin-Coignard D, et al. TCF4 deletions in Pitt-Hopkins syndrome. Hum Mutat. 2008. 10.1002/humu.20859. [DOI] [PubMed] [Google Scholar]
- 11.Steinbusch C, van Roozendaal K, Tserpelis D, Smeets E, Kranenburg-de Koning T, de Waal K, Zweier C, Rauch A, Hennekam R, Blok M, et al. Somatic mosaicism in a mother of two children with Pitt-Hopkins syndrome. Clin Genet. 2013;83:73–7. 10.1111/j.1399-0004.2012.01857.x. [DOI] [PubMed] [Google Scholar]
- 12.Maini I, Cantalupo G, Turco EC, Paolis F, De; Magnani C, Parrino L, Terzano MG, Pisani F. Clinical and polygraphic improvement of breathing abnormalities after valproate in a case of Pitt-Hopkins syndrome. J Child Neurol. 2012;27:1585–8. 10.1177/0883073811435917. [DOI] [PubMed] [Google Scholar]
- 13.Marangi G, Ricciardi S, Orteschi D, Lattante S, Murdolo M, Dallapiccola B, Biscione C, Lecce R, Chiurazzi P, Romano C, et al. The Pitt-Hopkins syndrome: report of 16 new patients and clinical diagnostic criteria. Am J Med Genet Part A. 2011;155:1536–45. 10.1002/ajmg.a.34070. [DOI] [PubMed] [Google Scholar]
- 14.De Pontual L, Mathieu Y, Golzio C, Rio M, Malan V, Boddaert N, Soufflet C, Picard C, Durandy A, Dobbie A, et al. Mutational, functional, and expression studies of the TCF4 gene in Pitt-Hopkins syndrome. Hum Mutat. 2009;30:669–76. 10.1002/humu.20935. [DOI] [PubMed] [Google Scholar]
- 15.Rosenfeld JA, Leppig K, Ballif BC, Thiese H, Erdie-Lalena C, Bawle E, Sastry S, Spence JE, Bandholz A, Surti U, et al. Genotype-Phenotype analysis of TCF4 mutations causing Pitt-Hopkins syndrome shows increased seizure activity with missense mutations. Genet Med. 2009. 10.1097/GIM.0b013e3181bd38a9. [DOI] [PubMed] [Google Scholar]
- 16.Comisi F, Esposito E, Marras M, Soddu C, Savasta S. Unusual inconsolable crying: an insight, case report, and review of the literature on the Pitt-Hopkins Gastrointestinal phenotype. Cureus. 2023;15:1–6. 10.7759/cureus.43781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Amiel J, Rio M, Pontual L. Mutations in TCF4, encoding a class I basic Helix-Loop-Helix transcription factor, are responsible for Pitt-Hopkins syndrome, a severe epileptic encephalopathy associated with autonomic dysfunction. Am J Hum Genet. 2007;80:988–93. 10.1086/515582. RedonR.Malan, V.; Boddaert, N.; Plouin, P.; Carter, N.P.; Lyonnet, S.; Munnich, A.; et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takano K, Lyons M, Moyes C, Jones J, Schwartz CE. 2% of patients suspected of having Angelman syndrome have TCF4 mutations. Clin Genet. 2010;78:282–8. 10.1111/j.1399-0004.2010.01380.x. [DOI] [PubMed] [Google Scholar]
- 19.Andrieux J, Lepretre F, Cuisset JM, Goldenberg A, Delobel B, Manouvrier-Hanu S, Holder-Espinasse M. Deletion 18q21.2q21.32 involving TCF4 in a Boy diagnosed by CGH-Array. Eur J Med Genet. 2008;51:172–7. 10.1016/j.ejmg.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 20.Orrico A, Galli L, Zappella M, Lam C-W, Bonifacio S, Torricelli F, Hayek G. Possible case of Pitt-Hopkins syndrome in Sibs. Am J Med Genet. 2001;103:157–9. 10.1002/ajmg.1523. [DOI] [PubMed] [Google Scholar]
- 21.Peippo MM, Simola KOJ, Valanne LK, Larsen AT, Kähkönen M, Auranen MP, Ignatius J. Pitt–Hopkins syndrome in two patients and further definition of the phenotype. Clin Dysmorphol. 2006;15:47–54. 10.1097/01.mcd.0000184973.14775.32. [DOI] [PubMed] [Google Scholar]
- 22.Strauss KA, Puffenberger EG, Huentelman MJ, Gottlieb S, Dobrin SE, Parod JM, Stephan DA, Morton DH. Recessive symptomatic focal epilepsy and mutant Contactin-Associated Protein-like 2. N Engl J Med. 2006;354:1370–7. 10.1056/NEJMoa052773. [DOI] [PubMed] [Google Scholar]
- 23.Zweier C, de Jong EK, Zweier M, Orrico A, Ousager LB, Collins AL, Bijlsma EK, Oortveld MAW, Ekici AB, Reis A, et al. CNTNAP2 and NRXN1 are mutated in Autosomal-Recessive Pitt-Hopkins-like mental retardation and determine the level of a common synaptic protein in Drosophila. Am J Hum Genet. 2009;85:655–66. 10.1016/j.ajhg.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smogavec M, Cleall A, Hoyer J, Lederer D, Nassogne M-C, Palmer EE, Deprez M, Benoit V, Maystadt I, Noakes C, et al. Eight further individuals with intellectual disability and epilepsy carrying Bi-Allelic CNTNAP2 aberrations allow delineation of the mutational and phenotypic spectrum. J Med Genet. 2016;53:820–7. 10.1136/jmedgenet-2016-103880. [DOI] [PubMed] [Google Scholar]
- 25.Marangi G, Ricciardi S, Orteschi D, Tenconi R, Monica M, Della; Scarano G, Battaglia D, Lettori D, Vasco G, Zollino M. Proposal of a Clinical Score for the Molecular Test for Pitt-Hopkins Syndrome. Am. J. Med. Genet. Part A. 2012;158(A):1604–1611. 10.1002/ajmg.a.35419 [DOI] [PubMed]
- 26.Zollino M, Zweier C, Van Balkom ID, Sweetser DA, Alaimo J, Bijlsma EK, Cody J, Elsea SH, Giurgea I, Macchiaiolo M et al. Diagnosis and management in Pitt-Hopkins syndrome: first international consensus statement. Clin Genet. 2019. [DOI] [PubMed]
- 27.Williams CA, Beaudet AL, Clayton-Smith J, Knoll JH, Kyllerman M, Laan LA, Magenis RE, Moncla A, Schinzel AA, Summers JA, et al. Angelman syndrome 2005: updated consensus for diagnostic criteria. Am J Med Genet Part A. 2006;140A:413–8. 10.1002/ajmg.a.31074. [DOI] [PubMed] [Google Scholar]
- 28.Zweier C, Peippo MM, Hoyer J, Sousa S, Bottani A, Clayton-Smith J, Reardon W, Saraiva J, Cabral A, Göhring I, et al. Haploinsufficiency of TCF4 causes syndromal mental retardation with intermittent hyperventilation (Pitt-Hopkins Syndrome). Am J Hum Genet. 2007;80:994–1001. 10.1086/515583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brandl L, Horst D, de Toni E, Kirchner T, Herbst A, Kolligs FT. ITF-2B protein levels are correlated with favorable prognosis in patients with colorectal carcinomas. Am J Cancer Res. 2015;5:2241–8. [PMC free article] [PubMed] [Google Scholar]
- 30.Larsson G, Schleucher J, Onions J, Hermann S, Grundström T, Wijmenga SS. A novel target recognition revealed by calmodulin in complex with the basic Helix–Loop–Helix transcription factor SEF2-1/E2‐2. Protein Sci. 2001;10:169–86. 10.1110/ps.28401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quong MW, Massari ME, Zwart R, Murre CA. New Transcriptional-Activation motif restricted to a class of Helix-Loop-Helix proteins is functionally conserved in both yeast and mammalian cells. Mol Cell Biol. 1993;13:792–800. 10.1128/mcb.13.2.792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Massari ME, Murre C, Helix-Loop-Helix P. Regulators of transcription in eucaryotic. Mol Cell Biol. 2000;20:429–40. 10.1128/mcb.20.2.429-440.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Forrest MP, Hill MJ, Quantock AJ, Martin-Rendon E, Blake DJ. The emerging roles of TCF4 in disease and development. Trends Mol Med. 2014;20:322–31. 10.1016/j.molmed.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 34.Sepp M, Kannike K, Eesmaa A, Urb M. Functional diversity of human basic Helix-Loop-Helix transcription factor TCF4 isoforms generated by alternative 5 9 exon usage and splicing. PLoS ONE. 2011;6. 10.1371/journal.pone.0022138. [DOI] [PMC free article] [PubMed]
- 35.Ephrussi A, Church GM, Tonegawa S, Gilbert W. B Lineage—Specific Interactions of an Immunoglobulin Enhancer with Cellular Factors in Vivo. Science (80-.). 1985;227:134–140. 10.1126/science.3917574 [DOI] [PubMed]
- 36.Ellenberger T, Arnaud M, Harrison SC. Crystal structure of transcription factor E47: E-Box recognition by a basic region Helix-Loop-Helix dimer. Genes Dev. 1994;8:970–80. 10.1101/gad.8.8.970. [DOI] [PubMed] [Google Scholar]
- 37.Murre C, McCaw PS, Vaessin H, Caudy M, Jan LY, Jan YN, Cabrera CV, Buskin JN, Hauschka SD, Lassar AB, et al. Interactions between heterologous Helix-Loop-Helix proteins generate complexes that bind specifically to a common DNA sequence. Cell. 1989;58:537–44. 10.1016/0092-8674(89)90434-0. [DOI] [PubMed] [Google Scholar]
- 38.Henthorn P, Kiledjian M, Kadesch T. Two distinct transcription factors that bind the Immunoglobulin enhancer ΜE5/ΚE2 motif. Sci (80-). 1990;247:467–70. 10.1126/science.2105528. [DOI] [PubMed] [Google Scholar]
- 39.Corneliussen B, Thornell A, Hallberg B, Grundström T. Helix-Loop-Helix transcriptional activators bind to a sequence in glucocorticoid response elements of retrovirus enhancers. J Virol. 1991;65:6084–93. 10.1128/jvi.65.11.6084-6093.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pscherer A, Dörflinger U, Kirfel J, Gawlas K, Rüschoff J, Buettner R, Schüle R. The Helix-Loop-Helix transcription factor SEF-2 regulates the activity of a novel initiator element in the promoter of the human somatostatin receptor II gene. EMBO J. 1996;15:6680–90. 10.1002/j.1460-2075.1996.tb01058.x. [PMC free article] [PubMed] [Google Scholar]
- 41.Yoon SO, Chikaraishi DM. Isolation of two E-Box binding factors that interact with the rat tyrosine hydroxylase enhancer. J Biol Chem. 1994;269:18453–62. 10.1016/S0021-9258(17)32330-X. [PubMed] [Google Scholar]
- 42.Voronova A, Baltimore D, Mutations That Disrupt DNA. Binding and Dimer Formation in the E47 Helix-Loop-Helix Protein Map to Distinct Domains. Proc. Natl. Acad. Sci. U. S. A. 1990;87:4722–4726. 10.1073/pnas.87.12.4722 [DOI] [PMC free article] [PubMed]
- 43.Petropoulos H, Skerjanc IS. Analysis of the Inhibition of myod activity by ITF-2B and Full-Length E12/E47. J Biol Chem. 2000;275:25095–101. 10.1074/jbc.M004251200. [DOI] [PubMed] [Google Scholar]
- 44.Lu Y, Sheng D-Q, Mo Z-C, Li H-F, Wu N-H, Shen Y-F. A negative regulatory Element-Dependent inhibitory role of ITF2B on IL-2 receptor α gene. Biochem Biophys Res Commun. 2005;336:142–9. 10.1016/j.bbrc.2005.08.050. [DOI] [PubMed] [Google Scholar]
- 45.Furumura M, Potterf SB, Toyofuku K, Matsunaga J, Muller J, Hearing VJ. Involvement of ITF2 in the transcriptional regulation of melanogenic genes. J Biol Chem. 2001;276:28147–54. 10.1074/jbc.M101626200. [DOI] [PubMed] [Google Scholar]
- 46.Forrest M, Chapman RM, Doyle AM, Tinsley CL, Waite A, Blake DJ. Functional analysis of TCF4 missense mutations that cause Pitt-Hopkins syndrome. Hum Mutat. 2012;33:1676–86. 10.1002/humu.22160. [DOI] [PubMed] [Google Scholar]
- 47.Forrest MP, Hill MJ, Kavanagh DH, Tansey KE, Waite AJ, Blake DJ. The psychiatric risk gene transcription factor 4 (TCF4) regulates neurodevelopmental pathways associated with schizophrenia, autism, and intellectual disability. Schizophr Bull. 2018. 10.1093/schbul/sbx164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blake DJ, Forrest M, Chapman RM, Tinsley CL, O’Donovan MC, Owen MJ. TCF4, schizophrenia, and Pitt-Hopkins syndrome. Schizophr Bull. 2010;36:443–7. 10.1093/schbul/sbq035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Flora A, Garcia JJ, Thaller C, Zoghbi HY. The E-Protein Tcf4 interacts with Math1 to regulate differentiation of a specific subset of neuronal progenitors. Proc Natl Acad Sci. 2007;104:15382–7. 10.1073/pnas.0707456104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Persson P, Jögi A, Grynfeld A, Påhlman S, Axelson H. HASH-1 and E2-2 are expressed in human neuroblastoma cells and form a functional complex. Biochem Biophys Res Commun. 2000;274:22–31. 10.1006/bbrc.2000.3090. [DOI] [PubMed] [Google Scholar]
- 51.Bertrand N, Castro DS, Guillemot F. Proneural genes and the specification of neural cell types. Nat Rev Neurosci. 2002;3:517–30. 10.1038/nrn874. [DOI] [PubMed] [Google Scholar]
- 52.de Pontual L. Noradrenergic neuronal development is impaired by mutation of the proneural HASH-1 gene in congenital central hypoventilation syndrome (Ondine’s Curse). Hum Mol Genet. 2003;12:3173–80. 10.1093/hmg/ddg339. [DOI] [PubMed] [Google Scholar]
- 53.Guillemot F, Lo L-C, Johnson JE, Auerbach A, Anderson DJ, Joyner AL. Mammalian Achaete-Scute homolog 1 is required for the early development of olfactory and autonomic neurons. Cell 1993, 75, 463–76, 10.1016/0092-8674(93)90381-Y [DOI] [PubMed]
- 54.Pattyn A, Goridis C, Brunet J-F. Specification of the central noradrenergic phenotype by the homeobox gene Phox2b. Mol Cell Neurosci. 2000;15:235–43. 10.1006/mcne.1999.0826. [DOI] [PubMed] [Google Scholar]
- 55.Aronheim A, Shiran R, Rosen A, Walker MD. The E2A gene product contains two separable and functionally distinct transcription activation domains. Proc Natl Acad Sci U S A. 1993. 10.1073/pnas.90.17.8063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Massari ME, Jennings PA, Murre C. The AD1 transactivation domain of E2A contains a highly conserved Helix which is required for its activity in both Saccharomyces Cerevisiae and mammalian cells. Mol Cell Biol. 1996;16:121–9. 10.1128/mcb.16.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bayly R, Chuen L, Currie RA, Hyndman BD, Casselman R, Blobel GA, LeBrun DP. E2A-PBX1 interacts directly with the KIX domain of CBP/P300 in the induction of proliferation in primary hematopoietic cells. J Biol Chem. 2004. 10.1074/jbc.M408654200. [DOI] [PubMed] [Google Scholar]
- 58.Denis CM, Chitayat S, Plevin MJ, Wang F, Thompson P, Liu S, Spencer HL, Ikura M, LeBrun DP, Smith SP. Structural basis of CBP/P300 recruitment in leukemia induction by E2A-PBX1. Blood. 2012. 10.1182/blood-2012-02-411397. [DOI] [PubMed] [Google Scholar]
- 59.Denis CM, Langelaan DN, Kirlin AC, Chitayat S, Munro K, Spencer HL, Lebrun DP, Smith SP. Functional redundancy between the transcriptional activation domains of E2A is mediated by binding to the KIX domain of CBP/P300. Nucleic Acids Res. 2014. 10.1093/nar/gku206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sirp A, Shubina A, Tuvikene J, Tamberg L, Kiir CS, Kranich L, Timmusk T. Expression of alternative transcription factor 4 MRNAs and protein isoforms in the developing and adult rodent and human tissues. Front Mol Neurosci. 2022;15. 10.3389/fnmol.2022.1033224. [DOI] [PMC free article] [PubMed]
- 61.Massari ME, Grant PA, Pray-Grant MG, Berger SL, Workman JL, Murre CA. Conserved motif present in a class of Helix-Loop-Helix proteins activates transcription by direct recruitment of the SAGA complex. Mol Cell. 1999. 10.1016/S1097-2765(00)80188-4. [DOI] [PubMed] [Google Scholar]
- 62.Zhang J, Kalkum M, Yamamura S, Chait BT, Roeder RG. E protein Silencing by the leukemogenic AML1-ETO fusion protein. Sci (80-). 2004. 10.1126/science.1097937. [DOI] [PubMed] [Google Scholar]
- 63.Sozańska N, Klepka BP, Niedzwiecka A, Zhukova L, Dadlez M, Greb-Markiewicz B, Ożyhar A, Tarczewska A. The molecular properties of the BHLH TCF4 protein as an intrinsically disordered hub transcription factor. Cell Commun Signal. 2025;23:154. 10.1186/s12964-025-02154-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Quednow BB, Brzózka MM, Rossner MJ. Transcription factor 4 (TCF4) and schizophrenia: integrating the animal and the human perspective. Cell Mol Life Sci. 2014;71:2815–35. 10.1007/s00018-013-1553-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Herbst A, Kolligs FT. A conserved domain in the transcription factor ITF-2B attenuates its activity. Biochem Biophys Res Commun. 2008. 10.1016/j.bbrc.2008.03.081. [DOI] [PubMed] [Google Scholar]
- 66.Markus M, Du Z, Benezra R. Enhancer-Specific modulation of E protein activity. J Biol Chem. 2002. 10.1074/jbc.M110659200. [DOI] [PubMed] [Google Scholar]
- 67.Liu Y, Ray SK, Yang XQ, Luntz-Leybman V, Chiu IM. A splice variant of E2-2 basic Helix-Loop-Helix protein represses the Brain-Specific fibroblast growth factor 1 promoter through the binding to an imperfect E-Box. J Biol Chem. 1998. 10.1074/jbc.273.30.19269. [DOI] [PubMed] [Google Scholar]
- 68.Greb-Markiewicz B, Kazana W, Zarębski M, Ożyhar A. The subcellular localization of BHLH transcription factor TCF4 is mediated by multiple nuclear localization and nuclear export signals. Sci Rep. 2019;9:15629. 10.1038/s41598-019-52239-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wedel M, Fröb F, Elsesser O, Wittmann MT, Lie DC, Reis A, Wegner M. Transcription factor Tcf4 is the preferred heterodimerization partner for Olig2 in oligodendrocytes and required for differentiation. Nucleic Acids Res. 2020;48:4839–57. 10.1093/nar/gkaa218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Phan BN, Bohlen JF, Davis BA, Ye Z, Chen H-Y, Mayfield B, Sripathy SR, Cerceo Page S, Campbell MN, Smith HL, et al. A Myelin-Related transcriptomic profile is shared by Pitt–Hopkins syndrome models and human autism spectrum disorder. Nat Neurosci. 2020;23:375–85. 10.1038/s41593-019-0578-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Papes F, Camargo AP, de Souza JS, Carvalho VMA, Szeto RA, LaMontagne E, Teixeira JR, Avansini SH, Sánchez-Sánchez SM, Nakahara TS, et al. Transcription factor 4 Loss-of-Function is associated with deficits in progenitor proliferation and cortical neuron content. Nat Commun. 2022;13:2387. 10.1038/s41467-022-29942-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Powell LM, Jarman AP. Context dependence of proneural BHLH proteins. Curr Opin Genet Dev. 2008;18:411–7. 10.1016/j.gde.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhuang Y, Cheng P, Weintraub HB. -Lymphocyte development is regulated by the combined dosage of three basic Helix-Loop-Helix genes, E2A, E2-2, and HEB†. Mol Cell Biol. 1996;16:2898–905. 10.1128/MCB.16.6.2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Flora A, Garcia JJ, Thaller C, Zoghbi HY. The E-Protein Tcf4 interacts with Math1 to regulate differentiation of a specific subset of neuronal progenitors. Proc Natl Acad Sci U S A. 2007;104:15382–7. 10.1073/pnas.0707456104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Alarcón M, Abrahams BS, Stone JL, Duvall JA, Perederiy JV, Bomar JM, Sebat J, Wigler M, Martin CL, Ledbetter DH, et al. Linkage, association, and gene-Expression analyses identify CNTNAP2 as an Autism-Susceptibility gene. Am J Hum Genet. 2008;82:150–9. 10.1016/j.ajhg.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kirov G, Rujescu D, Ingason A, Collier DA, O’Donovan MC, Owen MJ. Neurexin 1 (NRXN1) deletions in schizophrenia. Schizophr Bull. 2009;35:851–4. 10.1093/schbul/sbp079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sepp M, Vihma H, Nurm K, Urb M, Page SC, Roots K, Hark A, Maher BJ, Pruunsild P, Timmusk T. The intellectual disability and schizophrenia associated transcription factor TCF4 is regulated by neuronal activity and protein kinase A. J Neurosci. 2017;37:10516–27. 10.1523/JNEUROSCI.1151-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rannals MD, Hamersky GR, Page SC, Campbell MN, Briley A, Gallo RA, Phan BN, Hyde TM, Kleinman JE, Shin JH, et al. Psychiatric risk gene transcription factor 4 regulates intrinsic excitability of prefrontal neurons via repression of SCN10a and KCNQ1. Neuron. 2016;90:43–55. 10.1016/j.neuron.2016.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Saarikettu J, Sveshnikova N, Grundström T. Calcium/Calmodulin Inhibition of transcriptional activity of E-Proteins by prevention of their binding to DNA. J Biol Chem. 2004;279:41004–11. 10.1074/jbc.M408120200. [DOI] [PubMed] [Google Scholar]
- 80.Corneliussen B, Holm M, Waltersson Y, Onions J, Hallberg B, Thornell A, Grundström T. Transcription Factor Domains Nat. 1994;368:760–4. 10.1038/368760a0. Calcium/Calmodulin Inhibition of Basic-Helix-Loop-Helix. [DOI] [PubMed] [Google Scholar]
- 81.Onions J, Hermann S, Grundström T. Basic Helix-Loop-Helix protein sequences determining differential Inhibition by calmodulin and S-100 proteins. J Biol Chem. 1997;272:23930–7. 10.1074/jbc.272.38.23930. [DOI] [PubMed] [Google Scholar]
- 82.Hauser J, Saarikettu J, Grundström T. Calcium regulation of myogenesis by differential calmodulin Inhibition of basic Helix-Loop-Helix transcription factors. Mol Biol Cell. 2008;19:2509–19. 10.1091/mbc.e07-09-0886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hauser J, Sveshnikova N, Wallenius A, Baradaran S, Saarikettu J, Grundström T. B-Cell receptor activation inhibits AID expression through calmodulin Inhibition of E-Proteins. Proc Natl Acad Sci. 2008;105:1267–72. 10.1073/pnas.0708220105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Brockschmidt A, Todt U, Ryu S, Hoischen A, Landwehr C, Birnbaum S, Frenck W, Radlwimmer B, Lichter P, Engels H, et al. Severe mental retardation with breathing abnormalities (Pitt - Hopkins Syndrome) is caused by haploinsufficiency of the neuronal BHLH transcription factor TCF4. Hum Mol Genet. 2007;16:1488–94. 10.1093/hmg/ddm099. [DOI] [PubMed] [Google Scholar]
- 85.Thaxton C, Kloth AD, Clark EP, Moy SS, Chitwood RA, Philpot BD. Common pathophysiology in multiple mouse models of Pitt–Hopkins syndrome. J Neurosci. 2018;38:918–36. 10.1523/JNEUROSCI.1305-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schoof M, Hellwig M, Harrison L, Holdhof D, Lauffer MC, Niesen J, Virdi S, Indenbirken D, Schüller U. The basic Helix-loop‐helix transcription factor TCF4 impacts brain architecture as well as neuronal morphology and differentiation. Eur J Neurosci. 2020;51:2219–35. 10.1111/ejn.14674. [DOI] [PubMed] [Google Scholar]
- 87.Cisse B, Caton ML, Lehner M, Maeda T, Scheu S, Locksley R, Holmberg D, Zweier C, den Hollander NS, Kant SG, et al. Transcription factor E2-2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell. 2008;135:37–48. 10.1016/j.cell.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nagasawa M, Schmidlin H, Hazekamp MG, Schotte R, Blom B. Development of human plasmacytoid dendritic cells depends on the combined action of the basic Helix-loop‐helix factor E2‐2 and the Ets factor Spi‐B. Eur J Immunol. 2008;38:2389–400. 10.1002/eji.200838470. [DOI] [PubMed] [Google Scholar]
- 89.Ghosh HS, Cisse B, Bunin A, Lewis KL, Reizis B. Continuous expression of the transcription factor E2-2 maintains the cell fate of mature plasmacytoid dendritic cells. Immunity. 2010;33:905–16. 10.1016/j.immuni.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kee BL, E, Proteins Branch ID, Out. Nat Rev Immunol. 2009;9:175–84. 10.1038/nri2507. [DOI] [PubMed] [Google Scholar]
- 91.Bergqvist I, Eriksson M, Saarikettu J, Eriksson B, Corneliussen B, Grundström T, Holmberg D. The basic Helix-Loop-Helix transcription factor E2-2 is involved in T lymphocyte development. Eur J Immunol. 2000;30:2857–63. 10.1002/1521-4141(200010)30:10 <2857::AID-IMMU2857>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 92.Forrest MP, Waite AJ, Martin-Rendon E, Blake DJ. Knockdown of human TCF4 affects multiple signaling pathways involved in cell survival, epithelial to mesenchymal transition and neuronal differentiation. PLoS ONE. 2013;8:e73169. 10.1371/journal.pone.0073169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xia H, Jahr FM, Kim N-K, Xie L, Shabalin AA, Bryois J, Sweet DH, Kronfol MM, Palasuberniam P, McRae M, et al. Building a schizophrenia genetic network: transcription factor 4 regulates genes involved in neuronal development and schizophrenia risk. Hum Mol Genet. 2018;27:3246–56. 10.1093/hmg/ddy222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA. Epithelial-Mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest. 2009;119:1438–49. 10.1172/JCI38019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kalluri R, Weinberg RA. The basics of Epithelial-Mesenchymal transition. J Clin Invest. 2009;119:1420–8. 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Van Balkom IDC, Vuijk PJ, Franssens M, Hoek HW, Hennekam RCM, Development. Cognition, and behaviour in Pitt-Hopkins syndrome. Dev Med Child Neurol. 2012;54:925–31. 10.1111/j.1469-8749.2012.04339.x. [DOI] [PubMed] [Google Scholar]
- 97.Marangi G, Ricciardi S, Orteschi D, Lattante S, Murdolo M, Dallapiccola B, Biscione C, Lecce R, Chiurazzi P, Romano C, et al. The Pitt-Hopkins syndrome: report of 16 new patients and clinical diagnostic criteria. Am J Med Genet Part A. 2011. 10.1002/ajmg.a.34070. [DOI] [PubMed] [Google Scholar]
- 98.Kato Z, Morimoto W, Kimura T, Matsushima A, Kondo N. Interstitial deletion of 18q: comparative genomic hybridization array analysis of 46, XX,Del(18)(Q21.2.Q21.33). Birth defects res. Part A clin. Mol Teratol. 2010;88:132–5. 10.1002/bdra.20633. [DOI] [PubMed] [Google Scholar]
- 99.Hasi M, Soileau B, Sebold C, Hill A, Hale DE, O’Donnell L, Cody JD. The role of the TCF4 gene in the phenotype of individuals with 18q segmental deletions. Hum Genet. 2011;130:777–87. 10.1007/s00439-011-1020-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Takenouchi T, Yagihashi T, Tsuchiya H, Torii C, Hayashi K, Kosaki R, Saitoh S, Takahashi T, Kosaki K. Tissue-limited ring chromosome 18 mosaicism as a cause of Pitt–Hopkins syndrome. Am J Med Genet Part A. 2012;158A:2621–3. 10.1002/ajmg.a.35230. [DOI] [PubMed] [Google Scholar]
- 101.Kalscheuer VM, Feenstra I, Van Ravenswaaij-Arts CMA, Smeets DFCM, Menzel C, Ullmann R, Musante L, Ropers H-H. Disruption of the TCF4 gene in a Girl with mental retardation but without the classical Pitt-Hopkins syndrome. Am J Med Genet A. 2008;146A:2053–9. 10.1002/ajmg.a.32419. [DOI] [PubMed] [Google Scholar]
- 102.Talkowski ME, Rosenfeld JA, Blumenthal I, Pillalamarri V, Chiang C, Heilbut A, Ernst C, Hanscom C, Rossin E, Lindgren AM, et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell. 2012;149:525–37. 10.1016/j.cell.2012.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schluth-Bolard C, Labalme A, Cordier MP, Till M, Nadeau G, Tevissen H, Lesca G, Boutry-Kryza N, Rossignol S, Rocas D, et al. Breakpoint mapping by next generation sequencing reveals causative gene disruption in patients carrying apparently balanced chromosome rearrangements with intellectual deficiency and/or congenital malformations. J Med Genet. 2013;50:144–50. 10.1136/jmedgenet-2012-101351. [DOI] [PubMed] [Google Scholar]
- 104.Maduro V, Pusey BN, Cherukuri PF, Atkins P, Du Souich C, Rupps R, Limbos M, Adams DR, Bhatt SS, Eydoux P, et al. Complex translocation disrupting TCF4 and altering TCF4 isoform expression segregates as mild autosomal dominant intellectual disability. Orphanet J Rare Dis. 2016;11:1–15. 10.1186/s13023-016-0439-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bedeschi MF, Marangi G, Calvello MR, Ricciardi S, Leone FPC, Baccarin M, Guerneri S, Orteschi D, Murdolo M, Lattante S, et al. Impairment of different protein domains causes variable clinical presentation within Pitt-Hopkins syndrome and suggests intragenic molecular syndromology of TCF4. Eur J Med Genet. 2017. 10.1016/j.ejmg.2017.08.004. [DOI] [PubMed] [Google Scholar]
- 106.Lehalle D, Williams C, Siu VM, Clayton-Smith J. Fetal pads as a clue to the diagnosis of Pitt–Hopkins syndrome. Am J Med Genet Part A. 2011;155:1685–9. 10.1002/ajmg.a.34055. [DOI] [PubMed] [Google Scholar]
- 107.Kharbanda M, Kannike K, Lampe A, Berg J, Timmusk T, Sepp M. Partial deletion of TCF4 in three generation family with Non-Syndromic intellectual disability, without features of Pitt-Hopkins syndrome. Eur J Med Genet. 2016;59:310–4. 10.1016/j.ejmg.2016.04.003. [DOI] [PubMed] [Google Scholar]
- 108.Murre C, McCaw PS, A Baltimore D. New DNA binding and dimerization motif in Immunoglobulin enhancer binding, daughterless, MyoD, and Myc proteins. Cell. 1989;56:777–83. 10.1016/0092-8674(89)90682-X. [DOI] [PubMed] [Google Scholar]
- 109.Zhao T, Genchev GZ, Wu S, Yu G, Lu H, Feng J. Pitt–Hopkins syndrome: phenotypic and genotypic description of four unrelated patients and structural analysis of corresponding missense mutations. Neurogenetics. 2021;22:161–9. 10.1007/s10048-021-00651-8. [DOI] [PubMed] [Google Scholar]
- 110.Sepp M, Pruunsild P, Timmusk T. Pitt–Hopkins Syndrome-Associated mutations in TCF4 lead to variable impairment of the transcription factor function ranging from hypomorphic to Dominant-Negative effects. Hum Mol Genet. 2012;21:2873–88. 10.1093/hmg/dds112. [DOI] [PubMed] [Google Scholar]
- 111.Tamberg L, Sepp M, Timmusk T, Palgi M. Introducing Pitt-Hopkins Syndrome-Associated mutations of TCF4 to Drosophila daughterless. Biol Open. 2015;4:1762–71. 10.1242/bio.014696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yang J, Horton JR, Li J, Huang Y, Zhang X, Blumenthal RM, Cheng X. Structural basis for Preferential binding of human TCF4 to DNA containing 5-Carboxylcytosine. Nucleic Acids Res. 2019;47:8375–87. 10.1093/nar/gkz381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sirp A, Roots K, Nurm K, Tuvikene J, Sepp M, Timmusk T. Functional consequences of TCF4 missense substitutions associated with Pitt-Hopkins syndrome, mild intellectual disability, and schizophrenia. J Biol Chem. 2021;297:101381. 10.1016/j.jbc.2021.101381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Li SC, Goto NK, Williams KA, Deber CM. Alpha-Helical, but Not Beta-Sheet, Propensity of Proline Is Determined by Peptide Environment. Proc. Natl. Acad. Sci. U. S. A. 1996;93:6676–6681. 10.1073/pnas.93.13.6676 [DOI] [PMC free article] [PubMed]
- 115.Clark DP, Pazdernik NJ, McGehee MR, Molecular Biology. third.; Elsevier. 2019;ISBN 9780128132883.
- 116.Rawat P, Prabakaran R, Kumar S, Gromiha MM, AggreRATE-Pred. A mathematical model for the prediction of change in aggregation rate upon point mutation. Bioinformatics. 2020;36:1439–44. 10.1093/bioinformatics/btz764. [DOI] [PubMed] [Google Scholar]
- 117.Rawat P, Kumar S, Michael Gromiha M. An In-Silico method for identifying aggregation rate enhancer and mitigator mutations in proteins. Int J Biol Macromol. 2018;118:1157–67. 10.1016/j.ijbiomac.2018.06.102. [DOI] [PubMed] [Google Scholar]
- 118.Tsang B, Pritišanac I, Scherer SW, Moses AM, Forman-Kay JD. Phase separation as a missing mechanism for interpretation of disease mutations. Cell. 2020;183:1742–56. 10.1016/j.cell.2020.11.050. [DOI] [PubMed] [Google Scholar]
- 119.Verhulst SL, De Dooy J, Ramet J, Bockaert N, Van Coster R, Ceulemans B, De Backer W. Acetazolamide for Severe Apnea in Pitt-Hopkins Syndrome. Am. J. Med. Genet. Part A. 2012;158(A):932–934. 10.1002/ajmg.a.35247 [DOI] [PubMed]
- 120.Gaffney C, McNally P. Successful use of Acetazolamide for central apnea in a child with Pitt–Hopkins syndrome. Am J Med Genet Part A. 2015;167:1423–1423. 10.1002/ajmg.a.37034. [DOI] [PubMed] [Google Scholar]
- 121.Sweatt JD. Pitt-Hopkins syndrome: intellectual disability due to loss of TCF4-Regulated gene transcription. Exp Mol Med. 2013. [DOI] [PMC free article] [PubMed]
- 122.Kennedy AJ, Rahn EJ, Paulukaitis BS, Savell KE, Kordasiewicz HB, Wang J, Lewis JW, Posey J, Strange SK, Guzman-Karlsson MC, et al. Tcf4 regulates synaptic plasticity, DNA methylation, and memory function HHS public access graphical abstract INTRODUCTION. Cell Rep. 2016;16:2666–85. 10.1016/j.celrep.2016.08.004.Tcf4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hennig KM, Fass DM, Zhao W-N, Sheridan SD, Fu T, Erdin S, Stortchevoi A, Lucente D, Cody JD, Sweetser D, et al. WNT/β-Catenin pathway and epigenetic mechanisms regulate the Pitt-Hopkins syndrome and schizophrenia risk gene TCF4. Complex Psychiatry. 2017;3:53–71. 10.1159/000475666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ekins S, Puhl AC, Davidow A. Repurposing the dihydropyridine calcium channel inhibitor Nicardipine as a Nav1.8 inhibitor in vivo for Pitt Hopkins syndrome. Pharm Res. 2020;37:127. 10.1007/s11095-020-02853-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kim H, Gao EB, Draper A, Berens NC, Vihma H, Zhang X, Higashi-Howard A, Ritola KD, Simon JM, Kennedy AJ, et al. Rescue of behavioral and electrophysiological phenotypes in a Pitt-Hopkins syndrome mouse model by genetic restoration of Tcf4 expression. Elife. 2022;11:1–24. 10.7554/eLife.72290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mary L, Piton A, Schaefer E, Mattioli F, Nourisson E, Feger C, Redin C, Barth M, El Chehadeh S, Colin E, et al. Disease-Causing variants in TCF4 are a frequent cause of intellectual disability: lessons from Large-Scale sequencing approaches in diagnosis. Eur J Hum Genet. 2018;26:996–1006. 10.1038/s41431-018-0096-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Morel B, Varela L, Azuaga AI, Conejero-Lara F. Environmental conditions affect the kinetics of nucleation of amyloid fibrils and determine their morphology. Biophys J. 2010;99:3801–10. 10.1016/j.bpj.2010.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.