

Abstract
Diesel exhaust particles (DEPs), a major component of air pollution, are well-known to induce inflammation and vascular dysfunction. However, the molecular mechanisms linking DEP exposure to the disruption of the blood–retina barrier (BRB) remain poorly understood. Toll-like receptors (TLRs), particularly TLR2 and TLR4, play critical roles in inflammatory signaling and may contribute to DEP-induced retinal endothelial dysfunction. This study investigates the involvement of TLR2 and TLR4 in mediating DEP-induced disruption of the BRB and evaluates the protective effects of TLR inhibition using both in vitro and in vivo experiments. U937 human macrophages were exposed to DEPs of ultrafine size (<0.2 μm), and the mRNA expression of TNF-α and IL-1β was quantified. Conditioned media from DEP-exposed U937 cultures were then used to treat human retinal endothelial cells (HRECs). DEP exposure significantly increased TNF-α and IL-1β mRNA expression in U937 macrophages. Conditioned media from DEP-exposed U937 macrophages reduced claudin-5 and ZO-1 expression in HRECs, resulting in increased BRB permeability. Inhibition of TLR2 and TLR4 using C29 and TAK242, respectively, significantly attenuated TNF-α and IL-1β mRNA expression in DEP-exposed U937 macrophages and preserved BRB integrity by maintaining claudin-5 and ZO-1 expression in HRECs. In the mouse model, DEP exposure caused a marked reduction in claudin-5 and ZO-1 levels in retinal vessels, whereas treatment with C29 and TAK242 mitigated the loss of these tight junction proteins. This study demonstrates that DEPs induce inflammation and BRB dysfunction through TLR2 and TLR4 activation, leading to increased vascular permeability and potential retinal damage. Furthermore, TLR2 and TLR4 inhibition may be a promising therapeutic strategy to protect retinal health from air pollution–induced damage.
Keywords: Endothelial cell, Macrophage, Particulate matter, Retina, Toll-like receptor
INTRODUCTION
Particulate matter (PM), especially diesel exhaust particles (DEPs), is a critical environmental pollutant with substantial effects on human health. DEPs, a major component of urban air pollution, are primarily emitted from diesel engines and consist of a complex mixture of carbonaceous cores and adsorbed organic compounds, including polycyclic aromatic hydrocarbons and heavy metals (1, 2). Inhalation of DEPs can activate inflammatory pathways via stimulation of macrophages located in the respiratory tract and lung, leading to the release of pro-inflammatory cytokines such as tumor necrosis factor (TNF)-α and interleukin (IL)-1β (3-5). The upregulation of TNF-α and IL-1β underscores their importance as key mediators in the pathophysiological processes induced by particulate pollutants (6, 7).
The blood–retina barrier (BRB) plays an important role in retinal homeostasis by tightly regulating the exchange of nutrients, ions, and waste products between the bloodstream and the retinal tissue, thereby preserving the immune-privileged environment of the retina (8, 9). The BRB is formed by endothelial cells interconnected through tight junctions, with key proteins such as claudin-5 and ZO-1 being vital for its structural integrity (10, 11). These tight junction proteins create a selective barrier that prevents the influx of harmful substances and inflammatory cells while allowing necessary molecules to pass through (12, 13). Disruption of these proteins increases retinal vascular permeability and compromises the BRB, potentially leading to retinal edema and damage. Retinal endothelial dysfunction is a hallmark of vision-threatening conditions, including diabetic retinopathy and age-related macular degeneration (9, 10, 14). Therefore, understanding the mechanisms underlying BRB integrity and finding strategies to preserve it are critical for preventing complications associated with increased vascular permeability.
Toll-like receptors (TLRs) are pivotal to the innate immune system, functioning as pattern-recognition receptors that detect pathogen-associated molecular patterns and initiate inflammatory responses (15). Among these, TLR2 and TLR4 are particularly important. TLR2 recognizes a broad range of microbial components, including lipoproteins and peptidoglycans, whereas TLR4 primarily detects lipopolysaccharides from Gram-negative bacteria (16). Upon activation, these receptors initiate signaling cascades that stimulate the production of pro-inflammatory cytokines (17). Exposure to PM can activate TLR2 and TLR4 on macrophages, leading to the secretion of TNF-α and IL-1β (18, 19). The upregulation of these pro-inflammatory cytokines in response to PM exposure is believed to play a role in increased vascular permeability and endothelial dysfunction; however, the precise molecular mechanisms underlying these effects remain poorly understood. In this study, we evaluated the effects of ultrafine size (<0.2 μm) DEPs on TNF-α and IL-1β mRNA expression in U937 human macrophages, as well as on the expression of tight junction proteins in human retinal endothelial cells (HRECs). Additionally, we investigated the role of TLR2 and TLR4 in modulating DEP-induced inflammation and BRB permeability. To confirm in vitro findings, we examined the effects of DEP exposure and TLR inhibition in a mouse model, focusing on inflammatory responses and retinal vascular integrity in vivo.
RESULTS
Increased expression of TNF-α and IL-1β mRNA in U937 macrophages following DEP exposure
The mRNA expression levels of TNF-α and IL-1β in U937 human macrophages were assessed at 6, 24, and 48 h after DEP exposure (Fig. 1A). Quantitative reverse transcription PCR (RT-qPCR) analysis revealed a significant, time-dependent increase in TNF-α and IL-1β mRNA levels in U937 macrophages following DEP exposure (Fig. 1B, C). Both cytokines were markedly elevated at 6, 24, and 48 h compared with that in the control, indicating a sustained DEP-induced inflammatory response over time.
Fig. 1.

TNF-α and IL-1β mRNA expression in U937 human macrophages at 6, 24, and 48 h after DEP exposure followed by washing. (A) Schematic representation of the experimental procedure. (B, C) TNF-α and IL-1β mRNA expression. TNF-α (B) and IL-1β (C) mRNA levels were significantly elevated at each time point following DEP exposure compared with those in the control. *P < 0.05 vs. control.
Increased BRB permeability in HRECs treated with DEP-exposed U937–conditioned media
BRB integrity and permeability were evaluated in HRECs treated with DEP-exposed U937–conditioned media (Fig. 2A). Immunocytochemical staining demonstrated a marked reduction in claudin-5 and ZO-1 expression in HRECs treated with DEP-exposed U937–conditioned media (DEP 6 h, DEP 24 h, and DEP 48 h) compared with the control (Fig. 2B). Western blotting further confirmed a significant reduction in the protein levels of claudin-5 and ZO-1 in HRECs treated with DEP-exposed U937–conditioned media in a time-dependent manner (Fig. 2C, D). Consistent with these findings, transepithelial electrical resistance (TEER) permeability assays showed a significant time-dependent increase in BRB permeability of HRECs treated with DEP-exposed U937–conditioned media (Fig. 2E).
Fig. 2.

Claudin-5 and ZO-1 expression and permeability in human retinal endothelial cells (HRECs) treated with conditioned media from U937 macrophages exposed to DEP. (A) Schematic representation of the experimental procedure. (B) Immunocytochemical staining. Claudin-5 and ZO-1 expression in HRECs was markedly reduced after treatment with conditioned media from DEP-exposed U937 macrophages. (C, D) Western blot analysis. Claudin-5 (C) and ZO-1 (D) protein levels in HRECs were significantly decreased following treatment with DEP-exposed U937-conditioned media compared with the control. (E) TEER permeability assay. Permeability in HRECs was significantly increased after treatment with DEP-exposed U937-conditioned media compared with the control. Scale bar = 30 μm, *P < 0.05 vs. control.
TLR inhibition reduces TNF-α and IL-1β mRNA expression in U937 macrophages following DEP exposure
Expression levels of TLR2, TLR4, and TNF-α and IL-1β mRNA in U937 macrophages were analyzed at 24 h after DEP exposure alone and in the presence of TLR2 (C29) or TLR4 (TAK242) inhibitors (Fig. 3A). While TLR2 and TLR4 levels were significantly elevated after DEP exposure, they were restored following treatment with C29 and TAK242 before DEP exposure, respectively. These findings indicated that C29 and TAK242 appropriately inhibit DEP-induced overexpression of TLR2 and TLR4 (Fig. 3B, C). U937 macrophages treated with C29 or TAK242 prior to DEP exposure showed significantly lower expression of TNF-α and IL-1β mRNA than those that received DEP exposure alone (Fig. 3D, E). These results indicate that TLR2 and TLR4 are key mediators of DEP-induced pro-inflammatory responses in macrophages.
Fig. 3.

Expression levels of TLR2/TLR4 and TNF-α/IL-1β mRNA in U937 macrophages exposed to DEPs with TLR2 (C29) or TLR4 (TAK242) inhibitors. (A) Schematic representation of the experimental procedure. (B, C) TLR2 and TLR4 expression levels. Compared with those in the control, TLR2 (B) and TLR4 (C) levels in U937 macrophages were significantly elevated following DEP exposure. TLR2 and TLR4 expression levels were significantly restored following treatment with C29 and TAK242 before DEP exposure, respectively. (D, E) TNF-α and IL-1β mRNA expression. TNF-α (D) and IL-1β (E) mRNA levels were significantly lower in U937 macrophages treated with C29 or TAK242 before DEP exposure than in cells that received DEP exposure alone without TLR inhibitors. *P < 0.05 vs. control, †P < 0.05 vs. DEP.
TLR inhibition preserves BRB permeability of HRECs treated with DEP-exposed U937–conditioned media
The effect of TLR inhibition on BRB integrity and permeability was examined in HRECs that were treated with C29 or TAK242 prior to treatment with DEP-exposed U937–conditioned media (Fig. 4A). Immunocytochemistry revealed that claudin-5 and ZO-1 expression was markedly preserved in HRECs treated with C29 or TAK242 prior to treatment with DEP-exposed U937–conditioned media compared with those that did not receive C29 or TAK242 prior to treatment with DEP-exposed U937–conditioned media (Fig. 4B). Western blotting confirmed that treatment with C29 or TAK242 significantly mitigated the reduction of claudin-5 and ZO-1 protein levels (Fig. 4C, D). Furthermore, TEER permeability assays showed that treatment with C29 or TAK242 significantly attenuated the DEP-induced increase in BRB permeability, retaining permeability at near-normal levels (Fig. 4E).
Fig. 4.

Claudin-5 and ZO-1 expression and permeability in HRECs treated with conditioned media from U937 macrophages treated with C29 or TAK242 before exposure to DEPs. (A) Schematic representation of the experimental procedure. (B) Immunocytochemical staining. Claudin-5 and ZO-1 expression was markedly preserved in HRECs cultured in conditioned media from U937 macrophages treated with C29 or TAK242 before DEP exposure, than in HRECs that received DEP exposure alone without pretreatment with TLR inhibitors. (C, D) Western blot analysis. Claudin-5 (C) and ZO-1 (D) protein levels in HRECs were significantly restored following treatment with conditioned media from U937 macrophages treated with C29 or TAK242 before DEP exposure. (E) TEER permeability assay. Permeability was significantly lower in HRECs treated with conditioned media from U937 macrophages treated with C29 or TAK242 before DEP exposure, than in HRECs that received DEP exposure alone without pretreatment with TLR inhibitors. Scale bar = 30 μm, *P < 0.05 vs. control, †P < 0.05 vs. DEP.
TLR inhibition protects retinal vascular integrity in a mouse model
In a mouse model of DEP exposure, immunohistochemical staining revealed a significant reduction in claudin-5 and ZO-1 expression in CD31+ retinal vessels of mice exposed to DEP (Fig. 5A, B). Treatment with C29 or TAK242 effectively preserved the expression of claudin-5 and ZO-1 in mice exposed to DEP (Fig. 5B). Western blot analysis confirmed that C29 or TAK242 significantly prevented the DEP-induced reduction in claudin-5 and ZO-1 protein levels in retinal vessels, consistent with the results of immunohistochemical staining (Fig. 5C, D).
Fig. 5.
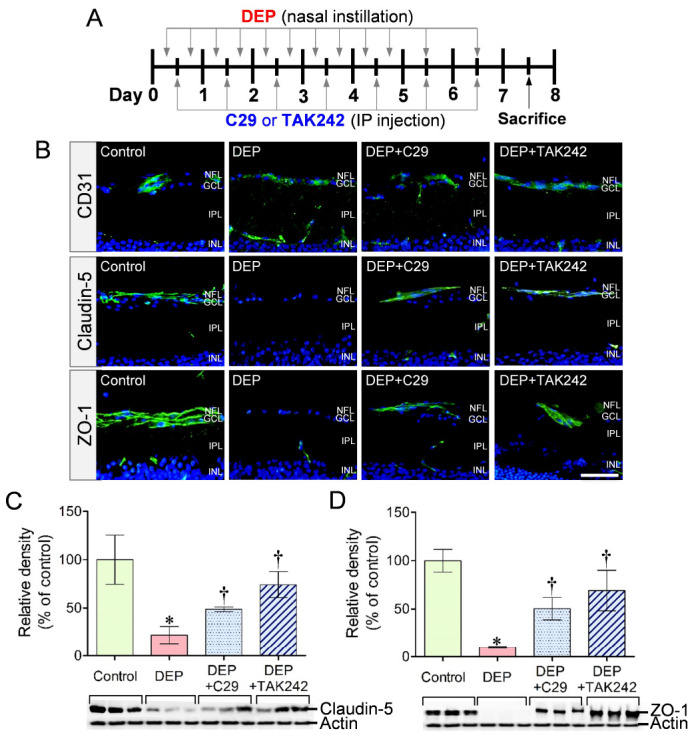
Claudin-5 and ZO-1 expression in retinal vessels of mice exposed to DEP and treated with C29 or TAK242. (A) Schematic representation of the experimental procedure. (B) Immunohistochemical staining. Claudin-5 and ZO-1 expression was markedly preserved in CD31+ retinal vessels of mice exposed to DEP with C29 or TAK242 than in those of mice exposed to DEP alone without TLR inhibitors. (C, D) Western blot analysis. Claudin-5 (B) and ZO-1 (C) protein levels in retinal lysates were significantly higher in mice that received DEP exposure with C29 or TAK242 than in those that received DEP exposure alone without TLR inhibitors. Scale bar = 50 μm, *P < 0.05 vs. control, †P < 0.05 vs. DEP (n = 3 per group).
DISCUSSION
To our knowledge, this is the first study to analyze the relationship between DEP-induced pro-inflammatory cytokines secretion by macrophages and BRB integrity using both a human cell system and a mouse model. Our findings demonstrate that DEP-induced activation of TLR2 and TLR4 in macrophages promotes the secretion of pro-inflammatory cytokines, particularly TNF-α and IL-1β, which compromise BRB integrity by increasing vascular permeability. Inhibition of TLR2 and TLR4 effectively reduced cytokine secretion and preserved BRB function, underscoring the therapeutic potential of targeting these receptors.
Previous studies have shown that DEP-induced inflammation is mediated primarily by TLR2 and TLR4 transmembrane receptors involved in innate immune signaling (19). Upon DEP exposure, TLR2 and TLR4 recognize specific components in DEP and activate downstream signaling pathways, including the MyD88–IRAK4 cascade and NF-κB transcription factor (17, 20). Although the exact DEP components recognized by TLR2 and TLR4 are not fully understood, endotoxins present in DEP are believed to play a critical role; earlier studies have reported that the level of endotoxins in PM influences the inflammatory response (18). Activation of these signaling pathways leads to the upregulation of TNF-α and IL-1β, which amplify immune responses and compromise BRB integrity by disrupting vascular endothelial tight junctions (6, 17). BRB damage associated with PM exposure may also be mediated through TLR-independent mechanisms, particularly those involving reactive oxygen species (ROS) and the NF-κB signaling pathway. Studies have indicated that PM exposure induces oxidative stress through the generation of ROS, which in turn activate inflammatory signaling and contribute to cellular injury (21, 22). ROS also activate NF-κB, a key transcription factor in inflammation, leading to the upregulation of pro-inflammatory cytokines and amplifying oxidative damage (23). In addition to oxidative mechanisms, nonoxidant-mediated mechanisms such as direct interactions between PM components and cellular structures can also trigger inflammatory responses, underscoring the multifaceted nature of PM-induced toxicity (24). Therefore, ROS and NF-κB signaling are crucial TLR-independent contributors to BRB disruption following PM exposure.
Tight junction proteins, particularly claudin-5 and ZO-1, are crucial for maintaining the structural and functional integrity of the BRB (10). Claudin-5, a transmembrane protein, regulates paracellular ion permeability and is a major contributor to TEER, which is an indicator of tight junction integrity (25). ZO-1, a linker protein, binds to the C-terminal of claudin-5 and stabilizes the tight junction complex (10, 26). Together, these proteins form a selective barrier that regulates molecular and cellular transport between the bloodstream and the retina. Our findings clearly show that DEP exposure disrupts claudin-5 and ZO-1 expression in retinal vascular endothelial cells via TLR2 and TLR4 activation in macrophages, resulting in increased BRB permeability. This disruption allows harmful substances to infiltrate retinal tissue, causing inflammation, edema, and potential retinal damage. BRB dysfunction may contribute to the onset or progression of retinal diseases such as glaucoma, age-related macular degeneration, and diabetic retinopathy (10, 27). Importantly, our results highlight the protective role of TLR2 and TLR4 inhibition in preserving BRB integrity and provide new insights into potential therapeutic strategies for retinal health.
A key aspect of this study is the validation of DEP-induced retinal damage mechanisms under two conditions: an in vitro human cell–based model and an in vivo mouse model. While in vitro human cell–based models are valuable for elucidating molecular mechanisms, they may not fully replicate the complexity of biological responses in living organisms. To address this limitation, we performed studies in an in vivo mouse model, which confirmed the effects of DEP exposure on retinal vascular integrity. However, this study has certain limitations. The TLR family consists of 13 receptors, with 10 identified in humans (excluding TLR11–TLR13) and 12 in mice (excluding TLR10) (28). This study focused solely on TLR2 and TLR4, using specific inhibitors (C29 and TAK242), which may not fully capture the complexity of DEP-induced inflammatory responses. Future research should investigate additional inflammatory pathways and other TLRs that may be involved in DEP-induced retinal and vascular damage. Moreover, because TLR inhibitors interfere with the production of inflammatory mediators, they may inadvertently weaken immune responses and increase susceptibility to infections (29). Therefore, while TLR inhibition holds therapeutic potential for PM-induced retinal damage, its long-term use must be carefully evaluated with regard to possible adverse effects. Additionally, this study utilized a short-term, 1-week DEP exposure model in mice. Recent findings support the potential impact of long-term fine PM exposure on BRB-related retinal damage. Mice exposed to fine PM for 3 weeks developed early retinal degenerative changes associated with age-related macular degeneration (11). An epidemiological study using optical coherence tomography imaging conducted every 2 years from 2009 to 2020 also reported that long-term fine PM exposure was associated with rapid thinning of the retinal nerve fiber layer, a hallmark of glaucoma (30). Considering the chronic nature of air pollution exposure, further studies should employ long-term exposure models to better understand BRB-related retinal damage and disease progression.
In summary, this study highlights the significant effects of DEP exposure on inflammation and BRB integrity. DEP-induced upregulation of TNF-α and IL-1β and disruption of tight junction proteins (claudin-5 and ZO-1) impair BRB function, leading to increased vascular permeability and potential retinal damage. Notably, inhibition of TLR2 and TLR4 effectively reduces inflammation and preserves BRB integrity, highlighting the therapeutic potential of targeting these receptors. These findings advance our understanding of the mechanisms underlying DEP-induced retinal damage and provide a foundation for developing strategies to mitigate the adverse effects of air pollution on retinal health.
MATERIALS AND METHODS
Cell culture
U937 human macrophages (Korean Cell Line Bank, Seoul, Korea) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Gibco, MA, USA) for 3 days at 37°C in a 5% CO2 incubator. HRECs (Innoprot, Bizkaia, Spain) were seeded onto fibronectin-coated (2 μg/cm2) culture dishes and cultured in endothelial cell medium containing 5% FBS, 1% endothelial cell growth supplement and 1% penicillin/streptomycin solution (Innoprot, Bizkaia, Spain) for 7 days at 37°C in a 5% CO2 incubator. Experiments involving cells were performed in at least 3 independent sets, with each set conducted in duplicate or triplicate cultures.
Preparation of DEP solution
Working suspensions of DEPs were prepared according to a previous method (31). Briefly, 2 mg of DEPs (NIST2975; Sigma-Aldrich, St. Louis, MO, USA) was suspended in 10 ml of phosphate-buffered saline (PBS) and vortexed, sonicated, and filtered through a syringe filter (0.2 μm; Sartorius, Göttingen, Germany).
Exposure of U937 macrophages to DEP
For DEP exposure, U937 macrophages were exposed to DEP solution (200 μg/ml) for 24 h, followed by washing with Dulbecco’s phosphate-buffered saline (DPBS), in accordance with a previous study (32). The cells were then resuspended in fresh DMEM. The cells and conditioned media were collected at 6, 24 and 48 h of culture, and the cells were washed (Fig. 1A). The collected cells were used for RT-qPCR experiments, and conditioned media (DEP 6 h, DEP 24 h and DEP 48 h media) were used for experiments involving HRECs.
TLR inhibition of U937 macrophages before DEP exposure
For TLR inhibition, U937 macrophages were treated with C29 (50 μM/ml, MedChemExpress, NJ, USA) or TAK242 (1 μM/ml, MedChemExpress, NJ, USA) for 1 h prior to DEP exposure. The cells were washed with DPBS and then exposed to DEP solution (200 μg/ml) for 24 h at 37°C in a 5% CO2 incubator. After DEP exposure, the cells were washed with DPBS and resuspended with fresh DMEM. The cells and conditioned media were collected at 24 h after DEP exposure and washing (Fig. 3A). The collected cells were used for RT-qPCR experiments, and conditioned media (DEP, DEP+C29 and DEP+TAK242 media) were used for experiments involving HRECs.
Treatment of HRECs with U937-conditioned media
To examine the effect of pro-inflammatory cytokines in U937-conditioned media on the expression of tight junction proteins, HRECs were initially cultured in endothelial cell medium, which was then replaced with DEP 6 h, DEP 24 h, or DEP 48 h media, and cultured for 24 h at 37°C in a 5% CO2 incubator. The HRECs were then collected and analyzed for claudin-5 and ZO-1 expression (Fig. 2A). To examine the effect of TLR2 and TLR4 inhibitors, the culture media for HRECs were replaced with DEP, DEP+C29 or DEP+TAK242 media for 24 h at 37°C in a 5% CO2 incubator, followed by analysis (Fig. 4A). For the control group, HRECs were initially cultured in endothelial cell medium, which was then replaced with U937-conditioned media not exposed to DEPs or TLR inhibitors.
TEER measurement
To examine the permeability of HRECs, TEER was measured using an endothelial volt/ohm meter with a Millicell-ERS-2 electro set (Millipore, MA, USA). HRECs were seeded on a six-well culture plate with polycarbonate membrane inserts (SPLInsert Standing, 3.0 μm pore size, SPL, Korea) at a density of 150,000 cells/cm2 and cultured for 7 days. The cells were treated with the U937-conditioned medium for 24 h at 37°C in a 5% CO2 incubator. The relative TEER value was calculated as a percentage of the control, which was set at 100%.
RT-qPCR
U937 macrophages were collected and lysed using Trizol reagent (Invitrogen, MA, USA) for isolation of total RNA. Reverse transcription reaction was performed for cDNA synthesis on a MyCycler (Bio-Rad, CA, USA) using AccuPower CycleScript RT Premix (Bioneer, Daejeon, Korea). Quantitative PCR was performed on a CFX Opus 96 Real-Time PCR System (Bio-Rad, CA, USA) using AccuPower 2x GreenStar qPCR Master Mix (Bioneer, Daejeon, Korea). PCR amplification was performed using primers specific for human TNF-α and IL-1β mRNA (Table 1) with the following thermal cycle: 50 cycles of denaturation at 95°C for 15 s, annealing at 58°C for 30 s, and extension at 72°C for 30 s. Target gene expression was determined by normalization to the endogenous Gapdh gene.
Table 1.
qPCR primers used in this study
| Gene | Direction | Sequence (5’→3’) |
|---|---|---|
| Human TNF-α | Forward | CTCTTCTGCCTGCTGCACTTTG |
| Reverse | ATGGGCTACAGGCTTGTCACTC | |
| Human IL-1β | Forward | CCACAGACCTTCCAGGAGAATG |
| Reverse | GTGCAGTTCAGTGATCGTACAGG | |
| Human GAPDH | Forward | GTCTCCTCTGACTTCAACAGCG |
| Reverse | ACCACCCTGTTGCTGTAGCCAA |
Experimental animals
Male Balb/c mice (7-weeks old, 20-22 g, n = 24) were obtained from a commercial source (DBL, Eumseong, Korea). Mice were housed under standard laboratory conditions under a 12-h light/dark cycle at 24-26°C and were provided ad libitum access to a commercial diet and water. Mice were divided into four groups: control (n = 6), DEP exposure group (DEP; n = 6), DEP exposure with C29 treatment group (DEP+C29; n = 6), and DEP exposure with TAK242 treatment group (DEP+TAK242; n = 6). Mice in each group were used for tissue preparation (n = 12; n = 3 per group) and Western blotting (n = 12; n = 3 per group). The study using experimental animals was approved by the Institutional Animal Care and Use Committee at Chungbuk National University (Approval No. CBNUA-24-0054-02). All procedures involving animals and animal care were conducted in accordance with the guidelines issued by the same committee.
Exposure of mice to DEP and TLR inhibition
Mice were lightly anesthetized with isoflurane, and 20 μl of DEP solution (0.4 mg/ml in saline) was gently instilled into the nasal cavities using a micropipette in the supine position. Mice were exposed to DEP twice a day at 12-h intervals for 5 days, followed by once-daily exposure for the next 2 days (Fig. 5A). For TLR inhibitor treatment, mice were intraperitoneally injected with C29 solution (30 mg/kg in saline, MedChemExpress) or TAK242 solution (30 mg/kg in saline, MedChemExpress) once a day for 7 days (Fig. 5A). The control group was treated with equal amounts of saline instead of DEP, C29, or TAK242 solution.
Tissue preparation
Mice were deeply anesthetized with a mixture of ketamine hydrochloride (100 mg/kg; Yuhan Co., Seoul, Korea) and xylazine hydrochloride (10 mg/kg; Bayer Korea, Seoul, Korea). After anesthetization, mice were transcardially perfused with precooled saline and 4% paraformaldehyde (PFA). The eyecup without the cornea, lens, and vitreous body was post-fixed in 4% paraformaldehyde (PFA) for 3 h. The eyecup was then cryoprotected in 30% sucrose in PBS overnight. The tissue was embedded in an optimal cutting temperature compound (Leica Biosystems, Richmond, IL, USA), rapidly frozen in 2-methyl butane (Junsei, Tokyo, Japan), adjusted to its freezing point using liquid nitrogen, and sectioned to 12-μm thickness using a cryostat (CM3050S; Leica Biosystems, Nussloch, Germany).
Immunofluorescence
HRECs were washed with PBS and fixed in 4% PFA in PBS for 3 h on ice. The cells and retinal tissues were incubated in PBS containing 10% normal horse or goat serum for 30 min to block nonspecific binding. The cells and tissues were incubated overnight at 4°C with anti-mouse claudin-5 (1:200, Invitrogen, MA, USA), anti-rabbit ZO-1 (1:100, Invitrogen, MA, USA), and anti-rat CD31 (1:50, BD bioscience, NJ, USA), respectively. Subsequently, cells and tissues were incubated for 2 h with Cy2-labeled horse anti-mouse IgG (1:500, Jackson ImmunoResearch Laboratories, PA, USA) for anti-claudin-5, Cy2-labeled goat anti-rabbit IgG (1:200, Jackson ImmunoResearch Laboratories, PA, USA) for anti-ZO-1, and Cy2-labeled goat anti-rat IgG (1:500, Jackson ImmunoResearch Laboratories, PA, USA) for anti-CD31 antibody. Between each step, cells and tissues were washed three times with PBS for 10 min. Following this, the stained cells and tissues were observed using a multipurpose microscope with an epifluorescence attachment (BX53, Olympus, Tokyo, Japan).
Western blotting
HREC and retina tissues were collected and homogenized in RIPA buffer (Elpis Biotech, Daejeon, Korea) and the lysates were centrifuged at 14,000 rpm and 4 °C for 10 min. The lysates were boiled for 5 min to denature and were then separated by electrophoresis on a 6-10% Tris-HCl gel. The separated proteins were transferred to PVDF membranes. The membranes were blocked with 5% skim milk in PBS containing 0.1% Tween 20 (PBSTw) for 1 h and were then incubated with the following primary antibodies: anti-mouse TLR2 (1:100, Santa Cruz Biotechnology, TX, USA), anti-mouse TLR4 (1:100, Santa Cruz Biotechnology), anti-mouse claudin-5 (1:500, Invitrogen), anti-rabbit ZO-1 (1:1000, Invitrogen) and anti-mouse actin (JLA20, 1:10, DSHB, IA, USA). After washing with PBSTw, membranes were incubated with secondary antibodies—peroxidase-conjugated anti-mouse IgG (1:1000, Vector Laboratories, CA, USA) and peroxidase-conjugated anti-rabbit IgG (1:1000, Vector Laboratories, CA, USA). The immunoreactive bands were reacted with chemiluminescence detection system (WestGlow PICO PLUS, Biomax, Korea) and were detected using a ChemiDoc imaging system (Bio-Rad, CA, USA).
Statistical analysis
Data were expressed as mean ± standard error of the mean (SEM). Statistical differences were analyzed using analysis of variance (ANOVA) followed by the Bonferroni’s method for post-hoc analysis using Prism 5 software (GraphPad, USA). P values less than 0.05 were defined as statistically significant.
ACKNOWLEDGEMENTS
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education (2021R1I1A3043435 and 2020R1I1A3073717).
Footnotes
CONFLICTS OF INTEREST
The authors have no conflicting interests.
References
- 1.Engels SM, Kamat P, Pafilis GS, et al. Particulate matter composition drives differential molecular and morphological responses in lung epithelial cells. PNAS Nexus. 2023;3:pgad415. doi: 10.1093/pnasnexus/pgad415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andrysík Z, Vondráček J, Marvanová S, et al. Activation of the aryl hydrocarbon receptor is the major toxic mode of action of an organic extract of a reference urban dust particulate matter mixture: the role of polycyclic aromatic hydrocarbons. Mutat Res. 2011;714:53–62. doi: 10.1016/j.mrfmmm.2011.06.011. [DOI] [PubMed] [Google Scholar]
- 3.Soler-Segovia D, De Homdedeu M, Sánchez-Díez S, et al. Immunological effects of diesel particles in a murine model of healthy mice. Toxics. 2024;12:530. doi: 10.3390/toxics12080530.4d0e2a0813f24a4b8c97754a57ebcf29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coillard A, Segura E. In vivo differentiation of human monocytes. Front Immunol. 2019;10:1907. doi: 10.3389/fimmu.2019.01907.73694f182a404160b3f73c01346c6db9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang S, Chen Y, Hong W, Li B, Zhou Y, Ran P. Chronic exposure to biomass ambient particulate matter triggers alveolar macrophage polarization and activation in the rat lung. J Cell Mol Med. 2022;26:1156–1168. doi: 10.1111/jcmm.17169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barros Ferreira L, Ashander LM, Ma Y, et al. Effects of tumor necrosis factor-α and interleukin-1β on human retinal endothelial cells. Cytokine. 2024;173:156407. doi: 10.1016/j.cyto.2023.156407. [DOI] [PubMed] [Google Scholar]
- 7.Versele R, Sevin E, Gosselet F, Fenart L, Candela P. TNF-α and IL-1β modulate blood-brain barrier permeability and decrease amyloid-β peptide efflux in a human blood-brain barrier model. Int J Mol Sci. 2022;23:10235. doi: 10.3390/ijms231810235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sterling JK, Rajesh A, Droho S, et al. Retinal perivascular macrophages regulate immune cell infiltration during neuroinflammation in mouse models of ocular disease. J Clin Invest. 2024;134:e180904. doi: 10.1172/JCI180904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ivanova E, Alam NM, Prusky GT, Sagdullaev BT. Blood-retina barrier failure and vision loss in neuron-specific degeneration. JCI Insight. 2019;5:e126747. doi: 10.1172/jci.insight.126747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O'Leary F, Campbell M. The blood-retina barrier in health and disease. FEBS J. 2023;290:878–891. doi: 10.1111/febs.16330. [DOI] [PubMed] [Google Scholar]
- 11.Gu Y, Sheng F, Gao M, et al. Acute and continuous exposure of airborne fine particulate matter (PM2.5): diverse outer blood-retinal barrier damages and disease susceptibilities. Part Fibre Toxicol. 2023;20:50. doi: 10.1186/s12989-023-00558-2.99b0839401884236a0ffb8849877896f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Campbell M, Humphries P. The blood-retina barrier: tight junctions and barrier modulation. Adv Exp Med Biol. 2012;763:70–84. doi: 10.1007/978-1-4614-4711-5_3. [DOI] [PubMed] [Google Scholar]
- 13.González-Mariscal L, Betanzos A, Nava P, Jaramillo BE. Tight junction proteins. Prog Biophys Mol Biol. 2003;81:1–44. doi: 10.1016/S0079-6107(02)00037-8. [DOI] [PubMed] [Google Scholar]
- 14.Soto I, Krebs MP, Reagan AM, Howell GR. Vascular inflammation risk factors in retinal disease. Annu Rev Vis Sci. 2019;5:99–122. doi: 10.1146/annurev-vision-091517-034416. [DOI] [PubMed] [Google Scholar]
- 15.Fitzgerald KA, Kagan JC. Toll-like receptors and the control of immunity. Cell. 2020;180:1044–1066. doi: 10.1016/j.cell.2020.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vijay K. Toll-like receptors in immunity and inflammatory diseases: past, present, and future. Int Immunopharmacol. 2018;59:391–412. doi: 10.1016/j.intimp.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moresco EMY, LaVine D, Beutler B. Toll-like receptors. Curr Biol. 2011;21:R488–R493. doi: 10.1016/j.cub.2011.05.039. [DOI] [PubMed] [Google Scholar]
- 18.Miyata R, van Eeden SF. The innate and adaptive immune response induced by alveolar macrophages exposed to ambient particulate matter. Toxicol Appl Pharmacol. 2011;257:209–226. doi: 10.1016/j.taap.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 19.Shoenfelt J, Mitkus RJ, Zeisler R, et al. Involvement of TLR2 and TLR4 in inflammatory immune responses induced by fine and coarse ambient air particulate matter. J Leukoc Biol. 2009;86:303–312. doi: 10.1189/jlb.1008587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ji J, Upadhyay S, Xiong X, et al. Multi-cellular human bronchial models exposed to diesel exhaust particles: assessment of inflammation, oxidative stress and macrophage polarization. Part Fibre Toxicol. 2018;15:19. doi: 10.1186/s12989-018-0256-2.1d02d3400e9b4c60a9f3ca11479ad8e0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valacchi G, Magnani N, Woodby B, Ferreira SM, Evelson P. Particulate matter induces tissue OxInflammation: from mechanism to damage. Antioxid Redox Signal. 2020;33:308–326. doi: 10.1089/ars.2019.8015. [DOI] [PubMed] [Google Scholar]
- 22.Lim EY, Kim GD. Particulate matter-induced emerging health effects associated with oxidative stress and inflammation. Antioxidants. 2024;13:1256. doi: 10.3390/antiox13101256.6d7af9124fe14550b98d7dcb3445e6de [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zeng Y, Zhu G, Zhu M, et al. Edaravone attenuated particulate matter-induced lung inflammation by inhibiting ROS-NF-κB signaling pathway. Oxid Med Cell Longev. 2022;2022:1–11. doi: 10.1155/2022/6908884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Øvrevik J, Refsnes M, Låg M, Holme J, Schwarze P. Activation of proinflammatory responses in cells of the airway mucosa by particulate matter: oxidant- and non-oxidant-mediated triggering mechanisms. Biomolecules. 2015;5:1399–1440. doi: 10.3390/biom5031399.5c9e52098d9f43c6a9443156583d3e58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Colegio OR, Itallie CV, Rahner C, Anderson JM. Claudin extracellular domains determine paracellular charge selectivity and resistance but not tight junction fibril architecture. Am J Physiol Cell Physiol. 2003;284:C1346–C1354. doi: 10.1152/ajpcell.00547.2002. [DOI] [PubMed] [Google Scholar]
- 26.Shin K, Fogg VC, Margolis B. Tight junctions and cell polarity. Annu Rev Cell Dev Biol. 2006;22:207–235. doi: 10.1146/annurev.cellbio.22.010305.104219. [DOI] [PubMed] [Google Scholar]
- 27.Han JH, Amri C, Lee H, Hur J. Pathological mechanisms of particulate matter-mediated ocular disorders: a review. Int J Mol Sci. 2024;25:12107. doi: 10.3390/ijms252212107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 29.Matsunaga N, Tsuchimori N, Matsumoto T, Ii M. TAK-242 (resatorvid), a small-molecule inhibitor of Toll-like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol Pharmacol. 2011;79:34–41. doi: 10.1124/mol.110.068064. [DOI] [PubMed] [Google Scholar]
- 30.Gayraud L, Mortamais M, Schweitzer C, et al. Association of long-term exposure to ambient air pollution with retinal neurodegeneration: the prospective Alienor study. Environ Res. 2023;232:116364. doi: 10.1016/j.envres.2023.116364. [DOI] [PubMed] [Google Scholar]
- 31.Block M, Wu X, Pei Z, et al. Nanometer size diesel exhaust particles are selectively toxic to dopaminergic neurons: the role of microglia, phagocytosis, and NADPH oxidase. FASEB J. 2004;18:1618–1620. doi: 10.1096/fj.04-1945fje. [DOI] [PubMed] [Google Scholar]
- 32.Roque PJ, Dao K, Costa LG. Microglia mediate diesel exhaust particle-induced cerebellar neuronal toxicity through neuroinflammatory mechanisms. Neurotoxicology. 2016;56:204–214. doi: 10.1016/j.neuro.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]