

Abstract
Photoinduced intramolecular electron transfer (ET) is essential for understanding charge transport in biological and synthetic systems. This study examines ET in peptide His-Glu-Tyr-Gly (1) and the conjugate His-Gln(BP)-Tyr-Gly (2) with benzophenone (BP) as a photoactive electron acceptor and His or Tyr as donors. Time-resolved and field-dependent chemically induced dynamic nuclear polarization (CIDNP) techniques were employed to investigate ET mechanisms and kinetics. Peptide 1 with 3,3’,4,4’-tetracarboxy benzophenone as a photosensitizer initially forms two types of radical with radical center at either His or Tyr residue, the consequent intra- and intermolecular ET electron transfer from Tyr residue to the His radical takes place with rate constants ke(intra)=(1.5±0.5)×105 s− 1 and ke(inter)=(1.3±0.4)×107 M− 1s− 1 at pH 8.8. Conjugate 2 forms two types of biradicals under irradiation: with radical centers at Tyr and BP across the entire pH range, and with radical centers at His and BP at slightly basic pH. Field-dependent CIDNP revealed nonzero electronic exchange interaction (2Jex = − 8.78 mT) at acidic pH, indicating proximity between BP and Tyr radicals. Low-field CIDNP spectra showed strong emissive polarization patterns, with pH-dependent exchange interaction and biradical geometry. Notably, no electron transfer from tyrosine to histidine radicals was observed in the conjugate 2, distinguishing its behavior from peptide 1.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-025-04831-6.
Subject terms: Biophysical chemistry, NMR spectroscopy, Solution-state NMR
Introduction
The transfer of electrons (ET) over long distances in living cells is a crucial process in a vast array of biochemical activities, including respiration and photosynthesis1–4. Despite the extensive study of numerous model peptide systems5–10, it is evident that the kinetics of the process are influenced by the molecular dynamics of the system and a number of molecular structure parameters, including donor/acceptor distance, amino acid sequence, terminal amino group charge, and reactant protonation state. Further investigation is needed to define a clear mechanism and fully understand the role of these and other factors. Photoinduced electron transfer is a special case of electron transfer. It is a light-driven process that forms a charge-separated state consisting of a donor radical cation, D•+, and an acceptor radical anion, A•−. In an intramolecular photoinduced ET process, D and A are part of the same molecule. In an intermolecular photoinduced ET process, they belong to different molecules. Photoinduced ET occurs in many chemical and biological scenarios, including photovoltaic devices, molecular photo-switches, and natural and artificial photosynthesis11–16. The electrochemical and excited state properties of the acceptor and donor moieties modulate photoinduced ET. However, it is important to consider that charge separation or recombination (back electron transfer) can be influenced by weak interactions of electron spins with each other, with a magnetic field and spin of surrounding nuclei, which is often neglected because thermodynamically they are of minor importance. This interaction significantly changes the quantum yield of the photoreaction. The cation of the donor radical and the anion of the acceptor radical interact electrostatically and magnetically. The strength of this interaction is determined by three factors: the distance between the radicals, their relative orientation, and, in the case of intramolecular ET, the nature of the linker connecting the radicals. The recombination rate of the singlet and triplet charge separation states differs significantly. The back electron transfer reaction from the singlet state is much faster than from the triplet state. The transition between these states is forbidden due to the difference in the total spin quantum number. However, spin interactions of electrons with each other, with an external magnetic field, and with spins of magnetic nuclei remove this spin prohibition and induce intersystem crossing transitions between singlet and triplet states. This has an important effect on the lifetime of the charge separation state. Studying magnetic and spin effects in the reversible electron transfer reaction allows to exploit the controlling effect of very weak interactions of electron spins with external magnetic fields and surrounding nuclei17–21.
While researchers in various fields of science have shown great interest in intramolecular photoinduced ET processes, these processes have been less extensively studied compared to intermolecular processes. This is particularly true for processes occurring in living organisms and in various model systems that simulate them. Biomimetic models designed for this purpose include the Tyr/Trp and His/Tyr pairs, that serve as an illustrative example of a redox-active site found in natural enzymes6,7,22–27. It is established that Tyr residues facilitate long-range electron transfer processes in a number of enzymes11,28–32. Tyr exhibits a lower oxidation potential (0.9 V)33 than His (1.17 V)34. However, in the presence of chemical agents with high oxidizing ability (e.g., photo-excited benzophenone derivatives), both His and Tyr residues can undergo oxidation. When the initial His• radical is formed, the intramolecular electron transfer from Tyr to His• occurs35–37. Whereas the reaction with the participation of Tyr in ET processes are mostly studied by time-resolved methods of laser pulsed photolysis or EPR spectroscopy4,6,7,38, it is difficult or almost impossible to study directly the reactions involving the histidine radicals using these methods: histidine radicals are poor chromophores which precludes their optical detection, and they are too short-lived at ambient conditions which does not allow them to be detected by EPR. In investigating the structure and properties of transient histidine radicals in the presence of other amino acid the chemically induced dynamic nuclear polarization (CIDNP) method plays a pivotal role35,36,39,40. By employing UV irradiation directly within the NMR spectrometer probe, the CIDNP technique enables the characterization of paramagnetic species generated during irradiation36,41,42. CIDNP is manifested by NMR signals with non-equilibrium nuclear spin populations − either enhanced absorption or emission − arising in nuclei with spin density of an unpaired electron in intermediate paramagnetic particles, which serve as precursors to polarized diamagnetic compounds43,44. Analysis of CIDNP signal patterns provides valuable information on the signs of hyperfine coupling constants (HFCCs), differences in g-factors of paramagnetic species, and other parameters essential for elucidating radical reaction mechanisms42,45,46. Previously, a time-resolved version of CIDNP was employed to investigate the kinetics of electron transfer from tyrosine to short-lived histidine radicals in peptides of varying structures35,36. Furthermore, the complete reaction scheme of histidine radical reduction by tyrosine for free amino acids and their N-acetylated derivatives was established, and electron transfer was identified as the reaction mechanism in the pH range from 6 to 1240. The influence of the protonation state of the reactants on the reaction rate constants was studied in detail, and it was found that the reduction of the histidine radical by tyrosine is always irreversible.
The aim of this work was to reveal and characterize photoinduced intramolecular ET reactions in model objects, synthetic tetrapeptide 1 − His-Glu-Tyr-Gly, and newly developed tetrapeptide/photosensitizer conjugate 2 − His-Gln(BP)-Tyr-Gly (Fig. 1). In this conjugate we have introduced the photosensitizer 4-aminobenzophenone by conjugation with glutamate midway between the histidine (at the N-terminus) and tyrosine moieties. In such a system, intramolecular ET is initiated by UV light and occurs between the photoexcited triplet benzophenone and the amino acid residue with the low oxidation potential. The peptide 1 can only undergo intermolecular benzophenone-sensitized photoinduced oxidation. Both of these biomimetic chemical compounds can be used as model systems to provide detailed mechanistic investigation of the underlying elementary reaction steps that are of biological relevance.
Fig. 1.

Structures of the His-Glu-Tyr-Gly peptide 1, 3,3’,4,4’-tetracarboxy benzophenone TCBP, and His-Gln(BP)-Tyr-Gly conjugate 2, in neutral aqueous solution.
Two variations of the CIDNP method were employed: time-resolved and field dependent. Time-resolved CIDNP has been used to study the photoinduced oxidation of peptide 1 by triplet-excited 3,3’,4,4’-tetracarboxy benzophenone (TCBP, Fig. 1). A combination of time-resolved and field-dependent CIDNP techniques was applied to the study of photo-induced reactions of the peptide/photosensitizer conjugate 2. The details of these experimental methods are summarized in Materials and Methods Section. The CIDNP technique allows the sensitive detection not only of transient mono-radicals but also biradicals even if the biradicals cannot be observed by EPR methods due to their short lifetime. Furthermore, CIDNP is capable of detecting biradicals in liquids at ambient temperature. The reason for this is that CIDNP is formed on the nanosecond to microsecond time scale and persists during typical nuclear relaxation time in diamagnetic molecules, providing a “frozen signature“41 of the elusive radicals. Analyzing the CIDNP as a function of time and magnetic field allows to determine the reaction mechanism, the rate constants of the radical reactions and the magnetic resonance parameters of such radicals. At present, the study of the CIDNP field dependence of the products of biradical conversion is the most reliable indirect method for demonstrating the involvement of biradicals in chemical reactions. Previously, it was shown that CIDNP kinetics in biradicals can provide information about the competition between spin dynamics, the chemical reaction of decarboxylation, which transforms primary radicals into secondary radicals, and the molecular dynamics of a linker47–54.
In the present study we (1) analyzed CIDNP kinetic data obtained for the photoreaction of TCBP and peptide 1 with the aim to determine the rate constant of photoinduced intramolecular ET in peptide 1; (2) measured CIDNP kinetics in the photoreaction of conjugate 2 in order to determine the pathways of the primary photochemical reaction of triplet benzophenone quenching and to identify the contribution of the potential intramolecular ET from a tyrosine residue to a histidine radical; (3) measured CIDNP field dependences in photoreaction of conjugate 2 to reveal the biradicals formation and characterize their magnetic properties.
Results and discussion
Selection of experimental conditions
It is known from previous studies that the processes of photoinduced oxidation of histidine and tyrosine55, as well as the electron transfer from tyrosine to the histidine radical36,40, are determined by the protonated state of the reactants. In order to obtain the pKa values that determine the protonated states (SI, Figure S1), a chemical shift titration of the conjugate 2 was performed. It turned out that conjugate 2 with neutral imidazole and charged amino group is poorly soluble in water and precipitated during titration, therefore experiments were possible only at pH below pKa of imidazole (typical value ~ 6 for N-terminal histidine) or above pKa of amino group (typical value ~ 7.6). Thus, pH values of ~ 4 and ~ 8.7 (and higher) were chosen. Accordingly, to determine the rate constant of intramolecular electron transfer in oxidized peptide 1, experiments were carried out at pH 8.8. The only acidity constant that could be determined was that for phenol of tyrosine residue: pKa=10.8 (see Materials and Methods Section for details).
Electron transfer from Tyr residue to His radical in His-Glu-Tyr-Gly peptide
As in our previous studies, radicals were generated in the photoinduced reaction with the photosensitizer 3,3’,4,4’-tetracarboxy benzophenone, rather than 4-aminobenzophenone, which was employed in the synthesis of conjugate 2. This was due to the low solubility of 4-aminobenzophenone in water. CIDNP effects in the photochemical reaction of TCBP with His and Tyr and manifestation of the reduction reaction in the CIDNP kinetics are described in detail in our previous publications36,40,55 and are only briefly summarized below. In the photoreaction with triplet-excited TCBP, two types of peptide radicals can be formed, with a radical center at either the His residue or the Tyr residue. As demonstrated in our previous studies, histidine and tyrosine exhibit comparable reactivity towards triplet-excited TCBP at neutral and slightly basic pH40. The reaction mechanism for both amino acids at neutral and slightly basic pH is proton-coupled electron transfer (PCET)56. At pH 8.8, the reactions leading to the triplet radical pairs (RP) formation are as follows (here the superscript T denotes the triplet state of the radical pair under overbar):
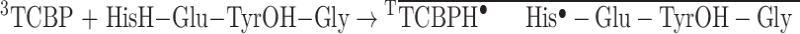 |
1 |
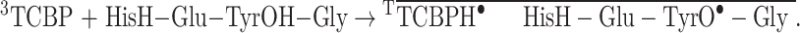 |
2 |
Here H in HisH refers to imidazolyl proton, and OH in TyrOH refers to phenyl moiety. The two types of radical pairs are formed (shown in Fig. 2), then eventually recombine back to the diamagnetic initial compounds. This process leads to characteristic polarized NMR signal patterns of His, Tyr and TCBP. By recording the polarized NMR spectra at a certain delay after the laser flash that initiates the reaction, the kinetics of CIDNP can be obtained at the time-scale from one microsecond to about 100 microseconds (this is referred to as the ‘kinetic window’ of the method), applying the technique known as time-resolved CIDNP41,57–61. It is an elegant method to study intra- and intermolecular ET reactions in peptides36,62. This technique allows the identification of transfer pathways and precise determination of electron transfer rates, which is beneficial for understanding the mechanisms of peptide radical formation.
Fig. 2.

Two types of peptide 1 radicals formed in the photoinduced reactions of 3,3′,4,4′-tetracarboxy benzophenone and His-Glu-Tyr-Gly (HisH-Glu-TyrOH-Gly) in slightly basic solution. Intramolecular (as well as intermolecular) electron transfer from Tyr to His• occurs, whereas TyrO• radical is not reduced by histidine.
Figure 3 shows the CIDNP spectra obtained for the photoreaction of TCBP with HisH-Glu-TyrOH-Gly at pH 8.8. The upper spectrum, obtained without delay following the laser flash, contains CIDNP signals from His and Tyr residues, as well as from TCBP as reversible reaction products formed at the geminate reaction stage. The CIDNP signals are in accordance with Kaptein’s rule for the net CIDNP effect63. According to this rule, the CIDNP sign for the product of geminate recombination is determined by the sign of the HFCC of the nucleus under consideration in the radical, by the difference between the g-factor of this radical and of the partner radical, and by the precursor multiplicity (triplet in our case). TCBP radical64 has a g-factor that is greater than that of the histidine radical65, and less than that of the tyrosine radical66. Thus, enhanced absorption occurs for protons with positive HFCCs in Tyr radical, and negative HFCCs in His radical: H2, H4 protons of His, H2,6, and β protons of Tyr67. Emission occurs for protons with positive HFCCs in His radical, and negative HFCCs in Tyr radical: β protons of His, H3,5 of Tyr67. The polarization of TCBP formed in radical pairs with a His radical is of an opposite sign to that formed in radical pairs with a Tyr radical. The resulting CIDNP of TCBP protons is determined by (i) the proportion of peptide radicals of each type (with radical center at histidine or tyrosine residue) formed in the quenching reaction, and (ii) the ratio of the CIDNP enhancement factors for TCBP protons in the two types of radical pairs. The initial positive polarization of H2,2’ and H6,6’ and negative polarization of H5,5’ of TCBP reflects predominance of CIDNP formed in a radical pair with Tyr radical, the same as in the photoreaction of a dye with peptides of different structures containing His and Tyr residues56.
Fig. 3.

1H CIDNP spectra obtained for the photoreaction of 2 mM TCBP with 40 mM His-Glu-Tyr-Gly at pH = 8.8 at 0 µs (top) and 100 µs (bottom) after the laser pulse. The aromatic protons of histidine are represented by signals of two stereoisomers.
Our preceding study36 provided a solid background for determining intramolecular ET rate constant in the oxidized peptide 1. In that study, the pH-dependences of the observed ET rate constant were analyzed for peptides of different structures at concentrations 20 and 40 mM. Intramolecular ET equally contributes at both concentrations, while intermolecular ET contribution is concentration-dependent. At the pH = 8.8 chosen for this study, Tyr is mainly present in solution with neutral phenol (TyrOH, pKa=10.8), the proportion of tyrosine in the form of tyrosinate anion is ~ 1%. However, the rate constant of intermolecular ET from tyrosinate was found to be about two orders of magnitude higher, than that from neutral tyrosine, and contributes to the total intermolecular ET at pH 8.8. No intramolecular ET from tyrosinate anion was revealed. In the case of neutral tyrosine, both intramolecular and intermolecular electron transfer reactions were found to occur.
The histidine radical is present in neutral and basic aqueous solutions in its neutral form with an unpaired electron localized on the imidazole ring65,68. The reduction of the His radical leads to the formation of the His anion, which protonates rapidly in the reaction with water (pKa of HisH > 14)68. Cation radical of Tyr formed after ET quickly deprotonates69. Thus, the net intramolecular ET reaction involves neutral phenol and imidazole (as product), and is as follows:
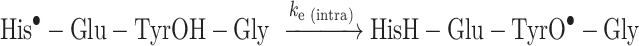 |
3 |
The corresponding net intermolecular reactions at pH 8.8 are:
 |
4 |
 |
5 |
The CIDNP kinetic data obtained in this case for H2 of His residue are shown in Fig. 4. Measurements were performed at two peptide 1 concentrations, 20 mM and 40 mM, to reveal the contributions of intra- and intermolecular ET from Tyr residues to His radicals in the peptide 1 like in the previous study36. CIDNP effects in the photochemical reaction of TCBP with His and Tyr and the manifestation of the reduction reaction in the CIDNP kinetics are described in detail in our previous publications36,39,40. They are only briefly summarized here. In the case of the peptide 1 under study, the fast decay of the His H2 signal is a clear indication of the ET reaction from the Tyr residue to the His radical. Indeed, as a result of the nuclear spin selectivity of T0⟷S transitions, the nuclear spin polarization of His residue in the His•–Glu–TyrOH–Gly radicals which escaped the initial in-cage recombination is opposite to that in the product of the recombination reaction of the geminate RP. However, ET from Tyr residue restores the diamagnetic state of His residue in these escaped radicals, leading to cancelation of CIDNP signals of His protons which manifests in a fast CIDNP decay. Tyr radicals formed via ET carry no nuclear polarization. However, these radicals participate in F-pair processes with TCBP radicals, in which additional CIDNP is formed. An increase is actually observed in the CIDNP kinetics of Tyr protons relative to the case when there is no ET. However, this increase is less pronounced than the decay of the histidine CIDNP, which is affected by ET directly. Similarly, for TCBP radicals, in the reaction with Tyr radicals, an additional CIDNP is formed, but its sign is opposite to the polarization formed in radical reactions with His. This dependence of the CIDNP sign on the partner of the radical pair makes the CIDNP kinetics of TCBP more sensitive to ET compared to CIDNP kinetics of Tyr, and the CIDNP growth for TCBP is expressed much more strongly than for Tyr. To determine the observed rate constants of ET, CIDNP kinetics for the protons of all reaction participants were simulated (SI, Figure S3). The simulation procedure was developed and applied earlier in the study of intra- and intermolecular electron transfer from tyrosine to the histidine radical in various systems, which is described in detail in the corresponding publications36,40.
Fig. 4.

CIDNP kinetics for proton H2 of the His residue, obtained during the photoreaction between 2 mM TCBP and 20 mM (open symbols) or 40 mM (solid symbols) of the peptide His-Glu-Tyr-Gly at pH 8.8. The normalization coefficient is selected so that the first point in the calculated kinetics has an amplitude equal to 1. Dashed line – calculations with the observed rate constants of ET is equal to 0.
The ET rate constants obtained from the simulation of the CIDNP kinetics are 4.0 × 105 s− 1 and 6.5 × 105 s− 1 at 20 and 40 mM of His–Glu–Tyr–Gly, respectively. These observed rate constants are contributed by the rate constants of the intra- and intermolecular ET, the latter is the product of the second-order reaction rate constant and the peptide 1 concentration. Solving simple algebraic equations gives (1.5±0.5)×105 s−1 for the rate constant of the intramolecular ET, and (1.3±0.4)×107 M−1s−1 for the intermolecular ET at pH 8.8. As mentioned above, the intramolecular ET rate constant determined in the present study for peptide 1 was assigned to the reaction involving neutral tyrosine (Eq. 3), while the rate constant of intermolecular ET refers to both reaction Eqs. (4) and (5). To determine the individual rate constants of reactions (4) and (5), pH-dependence analysis is required; however, firstly, this was not the aim of the present study, and secondly, it is impossible due to the low solubility of peptide 1 in a neutral aqueous solution. The value 1.5 × 105 s−1 is comparable to that obtained for His-Tyr peptide, 2.6 × 105 s−1, 36 and is the reference for conjugate 2.
Electron transfer in His-Gln(BP)-Tyr-Gly conjugate
Based on previously obtained data on quenching rate constants55 and rate constants for the reduction of histidine radicals by tyrosine40, we assume that photoirradiation of such biomimetic conjugate leads to the formation of a primary spin-correlated biradical consisting of the benzophenone radical and the radical of one of the amino acids residues. The main feature of biradicals is the non-zero electronic exchange interaction 2Jex between the radical centers throughout the lifetime of the biradical70. The electronic exchange interaction splits the singlet and triplet states of the biradical at zero magnetic field, where the energy gap is equal to 2|Jex|. The magnitude of the exchange interaction depends on the overlap of the unpaired electron orbitals of the radicals. A shorter distance between the radicals results in a larger 2|Jex| that suppresses the T0⟷S transitions at high magnetic field, while a longer distance results in a smaller electron coupling making the T0⟷S transitions more efficient. At high magnetic field, the Zeeman splitting becomes much larger than the average singlet-triplet splitting, B>>2|Jex|. Under these conditions, the transitions T–⟷S (or T+⟷S) are not efficient and CIDNP effect is formed in T0⟷S transitions according to radical-pair mechanism71–73. Due to the impossibility of the radical centers separation, the biradicals can only recombine generating geminate CIDNP. The magnetic interactions of the electrons and nuclei in the biradicals, namely the difference in their g-factors and the hyperfine interaction between the unpaired electron and the nuclei, drive the transition from the T0 to the S state. The rates of these transitions are not the same for the different configurations of the nuclear spins. In this way, all the biradicals produced are divided into sub-ensembles: those with nuclear spin states for which the rate of transition from T0 to S is higher, and those with nuclear spin states with lower rate. Thus, the CIDNP kinetics show an initial increase, followed by a maximum and then a decay to zero (upper dashed line in Figure 5)74. According to the radical-pair T0⟷S (or S⟷T0, which is fully equivalent) mechanism, CIDNP spectra of biradicals recorded at high magnetic field show the NMR signals of emission and enhanced absorption, by contrast to low field, where emission effects observed in the case of triplet-born biradicals with negative Jex due to T–⟷S transitions.
Fig. 5.

Schematic representation of CIDNP kinetics formed by S-T0 (upper dashed line) and S-T− (lower dashed line) mechanisms in biradicals for spin 1/2 nuclei with a negative HFCC, A = − 0.5 mT, at a magnetic field B satisfying the inequality |A| < B < 2|Jex|. Solid line shows the resulting curve.
TR CIDNP in His-Gln(BP)-Tyr-Gly conjugate
Representative CIDNP spectra obtained after photoirradiation of the His-Gln(BP)-Tyr-Gly conjugate at pH 4.0, 8.8 and 11.9 are shown in Fig. 6. The spectra were obtained without delay after the laser pulse using detecting RF-pulse of 5 µs and contain signals of products formed as a result of the recombination reaction. CIDNP for tyrosine protons was observed at all pH values, and for histidine protons – in the pH range from 8.7 to 10.8. These observations correspond to changes in the relative reactivity of amino acid residues: histidine with protonated imidazole (pH 4) is less reactive than tyrosine; histidine with neutral imidazole has reactivity comparable to that of tyrosine (neutral to slightly basic pH); tyrosinate anion residue is more reactive than histidine one (pH 11.9). Thus, when the photoreaction is carried out at pH 8.8, two types of biradicals are formed: the first − consisting of benzophenone and tyrosine radicals, the second − consisting of benzophenone and histidine radicals (Fig. 7). At pH 4.0 and 11.9 only one type of biradical is formed − with radical centers on benzophenone and tyrosine moieties. The signs of the CIDNP signals are in accordance with Kaptein’s rule for triplet precursor multiplicity63, with the known signs of the HFCCs for polarized protons, and a g-factor of the benzophenone radical greater64 than that of the histidine radical65 and less than that of the tyrosine radical66.
Fig. 6.

CIDNP spectra of 0.3 mM His-Gln(BP)-Tyr-Gly at pH 8.8, 4.0, 11.9 obtained just after the laser pulse. The number of scans was equal to 64. The detecting radiofrequency pulse (RF-pulse) was 5 µs.
Fig. 7.

Two types of His-Gln(BP)-Tyr-Gly biradicals formed under photooxidation at slightly basic solution. There was no intra- or intermolecular electron transfer from Tyr to His• detected. Exchange interaction in two types of His-Gln(BP)-Tyr-Gly biradicals is designated: JHis and JTyr.
In order to answer the question of whether intramolecular electron transfer from tyrosine residue to histidine radical takes place, CIDNP kinetic data for H2 of His residue in the pH range from 8.7 to 10.8 were analyzed. It turned out that the kinetics are pH-independent in this range, the data corresponding to the utmost values are shown in Fig. 8 (A).
Fig. 8.

CIDNP kinetics obtained during irradiation of 0.3 mM His-Gln(BP)-Tyr-Gly for (A) H2 of His residue at pH 8.7 (circles) and 10.8 (triangles); (B) H3,5 of Tyr residue at pH 4.0 (circles) and 11.8 (squares). Detecting RF-pulse was 0.5 µs. Number of scans 128.
Typically, the geminate evolution of the biradical spin states takes hundreds of nanoseconds75. In contrast to peptide 1, all reaction products of conjugate 2 photoirradiation are in-cage, as there is no diffusion separation of the biradicals. In general, at high magnetic field, S⟷T0 mixing is the dominant pathway of triplet-to-singlet interconversion75. The triplet biradicals resulting from the photochemical reaction can be divided into sub-ensembles with respect to nuclear spin orientation. These sub-ensembles have different rates of singlet-triplet conversion: the faster S⟷T0 transitions are followed by slower ones, and eventually all sub-ensembles of the T0 term acquire singlet character and yield diamagnetic products. Accordingly, the CIDNP passes through a maximum and goes to zero with time (Fig. 5)75. However, we were not able to use detection RF pulses shorter than 0.5 µs, so our time resolution did not allow to detect the initial CIDNP growth. The finite RF pulse duration is taken into account by defining the points on the time scale that correspond to the time between the laser pulse and the center of RF-pulse, which previous studies have shown to be a good approximation. Therefore, the time in Fig. 8 is shifted relative to the delay τ between the laser pulse and the RF pulse by half the length of the latter: in Fig. 8τ = 0 µs corresponds to the time 0.25 µs, τ = 0.25 µs − to the time 0.5 µs, etc.
At high magnetic field, S⟷T0 transitions caused by g-factor difference of the radical centers occur on a nanosecond time scale (~ 15 ns at B = 4.7 T and Δg ~ 10− 3). On the same time scale, the formation and decay of CIDNP in spin-correlated radical pairs occurs. The limiting stage that determines the lifetime of triplet biradicals is the time of electron dipole-dipole relaxation from the T± states to the T0 state, depleted as a result of interconversion to S state and recombination. Spin evolution of repopulated T0 state leads to CIDNP formation according to S⟷T0 mechanism. Thus, the decay of CIDNP kinetics in Fig. 8 (A) is determined by dipole-dipole relaxation, and the characteristic decay rate constant kd=1.6 × 106 s− 1 can be roughly attributed to the inverse relaxation time. Since the characteristic decay time is an order of magnitude greater than the assumed intramolecular ET rate constant, the latter has no chance of manifesting itself during the lifetime of the biradical.
The coincidence of kinetics at pH 8.8 and 10.8 indicates that there is no acceleration of CIDNP decay upon deprotonation of tyrosine with increasing pH. This means that during the biradical lifetime, there is no intermolecular electron transfer from the tyrosinate anion at the conjugate concentration of 0.3 mM, while the absence of intramolecular ET from tyrosinate anion is in accordance with the previous investigation36.
CIDNP kinetics for H3,5 protons of the Tyr residue were obtained during irradiation of 0.3 mM His-Gln(BP)-Tyr-Gly in the acidic (pH 4.0) and basic (pH 11.9) conditions (see Fig. 8, B). The decay was much faster than that of His protons and is essentially represented by only the first point in the CIDNP kinetics. The decay rate constant kd is estimated at 7.1 × 106 s− 1. In contrast to the case of His, a non-zero stationary CIDNP is observed. In the case of CIDNP formed in the reaction of free radicals, non-zero stationary CIDNP is determined by nuclear paramagnetic relaxation in the radicals caused by the anisotropy of hyperfine interaction, and the latter determines the characteristic relaxation time of 10–1000 µs. In the present case, non-zero stationary CIDNP may be caused by nuclear relaxation in biradical. So, it was revealed that two types of His-Gln(BP)-Tyr-Gly biradicals formed under photooxidation differ in their characteristics, in particular in the dipole-dipole relaxation time.
Thus, time-resolved CIDNP experiments on the synthetic peptide His-Glu-Tyr-Gly and the conjugate His-Gln(BP)-Tyr-Gly identified intramolecular and intermolecular electron transfer. However, to characterize the magnetic interactions influencing ET processes, field-dependent CIDNP studies are necessary. Therefore, in the next part of the article we describe the results of the experiments with the conjugate His-Gln(BP)-Tyr-Gly that determined a range of magnetic fields where the exchange interaction matches the electronic Zeeman interaction. The obtained CIDNP field dependences allowed to prove the biradical nature of the short-lived RP intermediate of the photochemical reaction, and to characterize the biradical geometry in acidic and basic conditions. In addition, the numerical modelling of CIDNP field dependences using the DFT calculated HFCCs allowed to extract the values the exchange interaction. This integrated approach demonstrates the effectiveness of combining time-resolved and field-dependent CIDNP methods to probe fast ET processes in biomimetic systems.
Field-dependent CIDNP in His-Gln(BP)-Tyr-Gly conjugate
The efficiency of triplet-singlet transition, and therefore photo-CIDNP formation in biradicals is highly dependent on magnetic field strength. When an external magnetic field is applied, the triplet state is split by the Zeeman interaction into T+, T0 and T− sublevels. The singlet and one of the triplet sublevels cross at the magnetic field B = 2|Jex|. Two regimes (cases) of magnetic field influence can be distinguished: (1) when the external magnetic field is comparable to 2|Jex|, we will call it the low-field case, and (2) when the external magnetic field is much larger than 2|Jex|, we will call it the high-field case. If we look at biradicals with a long carbon chain (e.g. eight methylene groups and more connecting the radical centers), such as the conjugate His-Gln(BP)-Tyr-Gly studied in this work, then comparatively small negative values of the electronic exchange interaction are characteristic for them: |Jex|<0.1 T, Jex <0. When Jex is negative, at zero magnetic field the singlet electronic state S is below the triplet manifold, and at magnetic field strength equal to 2|Jex| there is an S-T– electron energy level crossing (Fig. 9). In the presence of isotropic hyperfine interaction (HFI) with magnetic nuclei, the level crossing (LC) turns into a level anti-crossing (LAC) for the SβN and T–αN levels of the biradical (subscript “N” denotes the nuclear spin state); the SαN and T–βN levels are not mixed by the hyperfine interaction. The HFI in the vicinity of LAC induces coherent electron-nuclear spin transitions |T–αN>⟷|SβN > with the conservation of the z-projection of total electron and nuclear spin. Therefore, the spin mixing at the LAC leads to a predominance of the βN nuclear spin state in the recombination product formed from singlet electronic state and thus to the formation of emissive CIDNP in diamagnetic product. This is shown in the bottom plot as the Lorentz-peak shaped line in Figure 9.
Fig. 9.
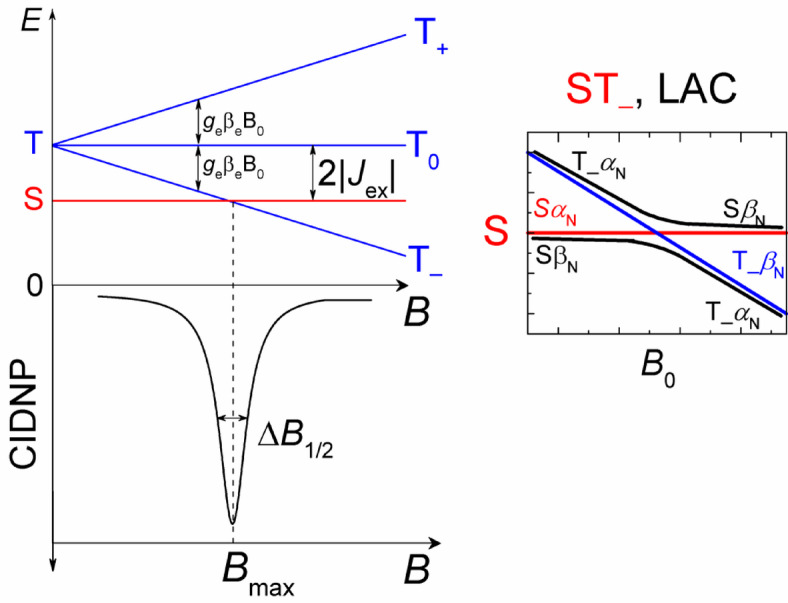
Energy level diagram of a biradical with negative exchange interaction at external magnetic field B. The region of S and T− level crossing (LC) is magnified in the insert. It shows that LC is turned to the level anti-crossing (LAC) by hyperfine interaction at Bmax where the coherent electron and nuclear spin flip-flop process |T–αN> ⟷|SβN > takes place, explaining the dependence of CIDNP on the magnetic field.
Magnetic field dependence of CIDNP allows us to reveal exchange interaction in short-lived biradicals formed in photoinduced intramolecular ET reaction in conjugate His-Gln(BP)-Tyr-Gly. The magnitude of CIDNP effect formed via J-resonance mechanism at low magnetic fields is significantly higher than that formed via S⟷T0 mechanism at high fields. Typical NMR spectra obtained before irradiation and CIDNP spectra of the His-Gln(BP)-Tyr-Gly conjugate at magnetic field 1 mT and 7 mT at pH 3.9 and 8.7 of the aqueous solution are shown in Fig. 10 (CIDNP spectrum at pH 11.8 is given in Figure S4 in Supporting information). In the CIDNP spectra obtained at 7 mT, the same protons are polarized as at high magnetic field, but all the signals are emissive. This is in agreement with the J-resonance mechanism of CIDNP generation at low field:70 the CIDNP sign does not depend on the sign of the HFCC, but depends on the product of the sign of the exchange interaction Jex and the multiplicity γ of the precursor (γ > 0 for triplet precursor and γ < 0 for singlet precursor). The emission sign of the signals corresponds to the T–⟷S mechanism of CIDNP formation. However, stationary CIDNP spectra provide only qualitative information about the distribution of the HFCCs in the products, since the absolute and relative line intensities depend strongly on the experimental conditions and the relationship between irradiation time, sample transfer time, and the relaxation time of the different nuclei.
Fig. 10.

(A, B) (top) 400 MHz 1H NMR spectrum (32 scans), obtained for the 0.12 mM solution of His-Gln(BP)-Tyr-Gly in D2O. (middle and bottom) 400 MHz 1H CIDNP spectra (16 scans), taken after the irradiation of the 0.12 mM solution of His-Gln(BP)-Tyr-Gly in D2O at Bpol = 7 mT (middle) or Bpol = 1 mT (bottom). The optical density was 1.4 cm− 1 at 265 nm. The irradiation time was 3 s per scan. The temperature was 25 °C. The pH values of aqueous solution were 3.9 (A), and 8.7 (B).
At magnetic field comparable to the typical value of proton HFCC (about 1 mT and below), no net polarization is expected because there is no preferred direction in space with respect to hyperfine interaction. However, in the case of two or more spins, a strong spin order is formed at zero field because the nuclear spin states are selected with respect to their total momentum76,77. This results in an unusual spectral pattern observed after the system is brought to a high detection field (see CIDNP spectra obtained at 1 mT in Fig. 10). Moreover, in this situation the singlet spin order of methylene protons is formed and hyperpolarization in diamagnetic product can be long-lived, relaxing much slower than the longitudinal magnetization78. The reason is that dipolar relaxation (the dominant relaxation mechanism for protons) cannot drive singlet-triplet transitions; consequently, singlet-triplet relaxation is caused by less efficient mechanisms and slows down. For the β protons of amino acids, the strong spin order is formed at 1 T and below. Therefore, for the β protons at the magnetic field of 7 mT, absorption/emission NMR signals are observed in addition to the net emission signal.
The field dependence of CIDNP for His-Gln(BP)-Tyr-Gly protons at pH 3.9 in aqueous solution is shown in Fig. 11, two other cases for pH 8.7 and 11.8 are shown in Figure S5 in SI. At pH 3.9 only polarized signals from tyrosine and benzophenone residue protons are observed, since histidine with protonated imidazole is less reactive than tyrosine. At pH 8.7, histidine with deprotonated imidazole exhibits a reactivity comparable to tyrosine, and the observed histidine residue signals overlap with the comparable in size signals of tyrosine and benzophenone residues. At pH 11.7, mainly the signals of tyrosine and benzophenone are observed, which indicates the increased reactivity of tyrosinate anion towards benzophenone triplets.
Fig. 11.

Experimental 1H CIDNP field dependencies for BP and Tyr protons obtained in photoreactions of 0.12 mM of His-Gln(BP)-Tyr-Gly in D2O at pH 3.9. The zooming factor of the vertical axis in (B) is multiplied by 7.5. Lines show the result of CIDNP calculation with parameters listed in Table S1 in Supporting information.
The observed CIDNP magnetic field dependencies have the same shape for all polarized protons at each pH studied, with almost coinciding maxima (Bmax) but slightly different width at half height (w1/2). The presence and coincidence of emission maxima for all protons can readily be accounted for T–⟷S mechanism of CIDNP formation. The T–⟷S intersystem crossing involves simultaneous flip of electron and one of the nuclear spins from α to β spin states (|T–αN > states transfer to |SβN > irrespective of the sign of HFI). The field dependence of CIDNP is determined by the magneto-resonance parameters (exchange and hyperfine interactions) of intermediate biradicals47,70,79.
To get more detailed information about the exchange interaction in the biradical of conjugate His-Gln(BP)-Tyr-Gly, we simulated the CIDNP field dependence obtained at pH 3.9 using the numerical approach published by some of us recently80. The simulation is shown by a solid line in the Fig. 11. Simulation parameters are listed in Table S1 in SI. Further data supporting the use of the simulation parameters values are found in Tables S2-S3 and Figure S6 in SI.
From fitting the CIDNP data we were able to reveal a non-zero exchange coupling 2Jex = −8.78 mT in biradical of conjugate His-Gln(BP)-Tyr-Gly at acidic pH. It is smaller and opposite in sign to values 2Jex = 17–30 mT obtained using the same procedure for a series of donor-bridge-acceptor compounds D-X-A studied by some of us earlier53,79. The TPSS/def2-SVP81,82 method accounting for the solvent (water) via the SMD83 model was used to optimize the geometries of biradical in its triplet state formed from His-Gln(BP)-Tyr-Gly conjugate (Fig. 12), see Materials and Methods section for details. The distance 9.1 Å between the Tyr and BP residues is much shorter than about 20 Å in biradical of the donor-bridge-acceptor compounds D-X-A79. However, the exchange interaction is smaller, which could be explained by two reasons. The first one is that positive Jex of D-X-A biradical indicates the involvement of the mediating solvent molecular orbital into the electron-electron coupling, extending its spatial range. The second one stems from the results of geometry optimization where the aromatic rings of BP and Tyr lie nearly in the same plane, thus suppressing the p-orbital overlap.
Fig. 12.

Optimized geometry of biradical in its triplet state formed from His-Gln(BP)-Tyr-Gly conjugate with neutral amino group of His. Electron density is located on BP• and Tyr• radical moieties, no spin density found on His residue. The calculation was done using TPSS/def2-SVP method accounting for the solvent (water) via the SMD model. Distances between centers are: His and Tyr residues – 10.6 Å; BP and His – 12.4 Å; BP and Tyr – 9.1 Å.
The value of 2Jex is a sensitive probe of the conformation of a short-lived biradical, reflecting the conformational ensemble of the precursor diamagnetic conjugate His-Gln(BP)-Tyr-Gly in solution. A comparison of the field dependences obtained at pH values of 3.9, 8.7 and 11.9 is shown in Fig. 13. Simulation of the field dependences measured at pH values 8.7 and 11.9 is underway and will be published elsewhere. It is seen that the half-width of the J-resonance curve increases with the increasing pH: Bmax is ~ 7 mT at pH 3.9, ~ 10 mT at pH 8.7, and ~ 14 mT at pH 11.8. It is known that the greater the exchange interaction is, the larger is the overlap of the orbitals occupied by the unpaired electrons. This could be either due to the closer proximity or more favorable mutual orientation of the radicals. In basic solution, His-Gln(BP)-Tyr-Gly contains tyrosine in the form of its anion, in acidic and neutral solution – in neutral form. However, at all pH values, the oxidized form of tyrosine is its neutral radical TyrO•, since its conjugated acid – cation radical TyrOH•+ with pKa< (−1) quickly deprotonates. Thus, the increase in exchange interaction in basic solution is apparently associated with a change in the structure of the benzophenone radical - from ketyl to anion-radical (pKa of ketyl radical of benzophenone is 9.4)84.
Fig. 13.

1H CIDNP field dependences for Tyr H3,5 (pH 3.9 and 11.7) and sum of Tyr H3,5 and His H4 (pH 8.7) protons obtained in the photoreactions of 0.12 mM of His-Gln(BP)-Tyr-Gly in D2O. The lines are guides for the eye.
Conclusion
This study provides a comparative analysis of photoinduced electron transfer in two model systems, the tetra-peptide His-Glu-Tyr-Gly and the conjugate His-Gln(BP)-Tyr-Gly (containing benzophenone BP), using chemically induced dynamic nuclear polarization. The findings reveal distinct differences in the mechanisms and kinetics of electron transfer processes in these systems, with an emphasis on the effects of molecular structure and pH on electron transfer dynamics. His-Glu-Tyr-Gly was studied by time-resolved CIDNP method in photosensitized reactions with 3,3’,4,4’-tetracarboxy benzophenone. At neutral and slightly basic pH, two types of radical pairs were detected due to the similar reactivity of histidine and tyrosine with triplet-excited TCBP under these conditions. Additionally, electron transfer from tyrosine to histidine radicals was observed. Analysis of the CIDNP kinetics demonstrated clear contributions from both intra- and intermolecular electron transfer pathways, and its rate constants were determined.
For the conjugate His-Gln(BP)-Tyr-Gly the kinetics and magnetic field dependence of 1H CIDNP were measured and analyzed at variable pH to reveal the influence of protonation states of reactants on photoinduced electron transfer. His-Gln(BP)-Tyr-Gly showed, like the peptide His-Glu-Tyr-Gly a complex pH-dependent behavior with respect to the electron transfer reaction. At acidic pH, electron transfer predominantly involved tyrosine, resulting in biradicals formed by benzophenone and tyrosine radicals. At neutral and slightly basic pH, both histidine and tyrosine participated in electron transfer, forming biradicals with distinct geometries but similar exchange interactions. At strongly basic pH, tyrosine in its deprotonated form (tyrosinate) dominated the electron transfer process. Magnetic field dependent CIDNP data revealed nonzero exchange interactions, indicating close spatial proximity of the radical centers of 4-amino benzophenone and Tyr residues. No electron transfer from tyrosine to histidine radicals was observed in the oxidized conjugate His-Gln(BP)-Tyr-Gly under any condition because of its short lifetime, contrasting with peptide His-Glu-Tyr-Gly. This study demonstrates how pH modulates electron transfer pathways and biradical characteristics in the conjugate His-Gln(BP)-Tyr-Gly, providing information into the interplay between protonation states and electron transfer dynamics. These findings underscore the utility of CIDNP in investigating complex processes including the formation of transient paramagnetic species in model systems and contribute to understanding photoinduced ET in biologically relevant contexts.
The methodological advancements in this study, based on the combination of the time-resolved and the magnetic field dependent CIDNP techniques provide a robust framework for electron transfer studies with atomic resolution. The replacement of high-power lasers with LEDs enhanced accessibility, portability, and safety, allowing rapid and stable experimentation. Usually, biochemists work with the low sample concentration (≤ 1µM) that typically require photosensitizers with long triplet-state lifetimes to ensure radical pair formation within the triplet lifetime. However, when the photosensitizer and quencher are linked and in close proximity, the triplet lifetime can be shorter, as exemplified by benzophenone (1.5 µs)85. This highlights the potential for designing compact and efficient electron transfer systems where close spatial arrangement compensates for shorter triplet-state lifetimes.
Materials and methods
Chemicals
The synthesis of the His-Glu-Tyr-Gly peptide and His-Gln(BP)-Tyr-Gly conjugate (Fig. 1) is described in Supporting information. The molecular structures as well as HPLC, ESI-MS and proton NMR spectra of the intermediate and final products of the synthesis are shown in Figures S7-S15 in the supporting information. Additional information is presented in Tables S4-S5. All chemicals for the synthesis were purchased from Sigma-Aldrich, Carl Roth GmbH, Iris, Acros, VWR and ABCR and if not explicitly mentioned used without any purification. The following chemicals were obtained from Sigma-Aldrich: DCl (35 wt% solution in D2O, 99 + atom % D), NaOD (40 wt% solution in D2O, 99 + atom % D), and D2O (99.9%).
Acidity of solutions and acidity constants of reactants in D2O
The 1H CIDNP measurements were performed in D2O. The acidity of the NMR samples was adjusted by the addition of small quantities of DCl or NaOD. The acidity of the D2O solutions was measured using a pH meter calibrated in H2O. The pH readings and pKa values obtained in this way correspond to what are known as pH* and pKa*, respectively, in accordance with the following formula: pKa=0.929pKa*+0.4286. Determined in this way was pKa* of phenol of tyrosine residue in conjugate 2 was 10.8 (see Figure S2 in SI). This gives pKa=10.4. However, for convenience, we show the exchangeable protons rather than deuterium atoms, and use the terms pH and pKa. Therefore, pH 10.8 here gives the conjugate with half neutral tyrosine and half tyrosinate anion.
Time-resolved CIDNP
Our setup for TR-CIDNP measurements has already been described in detail41. The samples purged with pure nitrogen gas and sealed in a standard NMR Pyrex ampule were irradiated in the probe of a Bruker DPX-200 NMR spectrometer (magnetic field 4.7 Т, resonance frequency of protons 200 MHz) by laser pulses from a COMPEX Lambda Physik XeCl excimer laser (308 nm, 15 ns pulse duration, pulse energy ~ 70 mJ). Light to the sample was guided using an optical system with a prism and a light-guide quartz rod (diameter 5 mm). The TR-CIDNP spectra were obtained in the following way: (1) saturation broadband radio frequency (RF) pulses, (2) a 15 ns laser pulse triggered by the spectrometer and (3) a detecting RF pulse with duration of 1 µs (under TR–CIDNP study of His-Glu-Tyr-Gly peptide) or 5 µs (to register CIDNP spectra for His-Gln(BP)-Tyr-Gly conjugate, shown in Fig. 6), or 0.5 µs (to study the CIDNP kinetics for His-Gln(BP)-Tyr-Gly conjugate, shown in Fig. 8) followed by FID acquisition. The laser pulse was synchronized with the front edge of the RF pulse. As the background signals from Boltzmann polarization were suppressed by saturation pulses, in the CIDNP spectra only the NMR signals from the polarized products of the cyclic photochemical reaction appear.
Field-cycling NMR and CIDNP
The experimental setup allowing fast field-cycling NMR experiments over a wide range of magnetic fields is described in detail in ref87. The setup allows the detection of high resolution NMR spectra at B0 = 9.4 T and light irradiation of the NMR sample53,54 at any desired magnetic field strength Bpol between 1 mT and 9.4 T by the precise positioning of the sample in the stray field of the spectrometer cryomagnet. The field resolution is determined by the field gradient across the illuminated sample volume. A 1H CIDNP experiment with mechanical field cycling is composed of five consecutive steps: (1) thermal polarization suppression at high magnetic field by application of a series 90° RF-pulses followed by a pulsed Bz-field gradient; (2) transfer of the sample to field Bpol; (3) CIDNP generation by light irradiation at the desired field Bpol; (4) transfer of the polarized reaction products to the observation field B0 = 9.4 T of the NMR spectrometer; (5) measurement of the NMR signal at B0 = 9.4 T. During step 3, the sample is irradiated for 3 s by a 265 nm 1 W LED light source via a quartz light guide sealed to it.
Simulation of magnetic field dependence of CIDNP
The theoretical model used for CIDNP simulation was as following. The Hamiltonian Ĥ comprises the Zeeman interaction of the electron spins, the isotropic hyperfine interaction, and the exchange interaction. Fixed average value exchange interaction 2Jex was used, and the effects of Jex modulation due to the relative mobility of the radicals was modeled by the amplitude of RJex parametrizing the dephasing rate of T⟷S coherences Wdeph~1/2∙(RJex)2∙τJex, τJex = 10−12 s. Longitudinal relaxation of an electron spin was described using simple Bloch model by a field independent relaxation time T1e. Values of 2Jex, RJex, and T1e were optimized to give the best fit of the experimental Tyr H2,6 and H3,5 CIDNP field dependences. In total, at least 15 protons and one nitrogen-14 in photoinduced biradical state of conjugate His-Gln(BP)-Tyr-Gly have non-zero HFCCs. This spin system is too large to be simulated within reasonable time using even an advanced personal computer. To reduce the computation time, some hyperfine interaction terms in Hamiltonian were approximated by an effective Schulten-Wolynes local field experienced only by the coupled electron88,89. Using this approximation, CIDNP field dependences of all protons were calculated separately, while including HFCCs of the majority of other magnetic nuclei in the amplitude of the Schulten-Wolynes local field. For example, CIDNP of six Tyr protons H2,6, H3,5, Hβ1 and Hβ2 was calculated using exact form of HFI, while the HFI with the magnetic nuclei of BP was approximated by Schulten-Wolynes local field 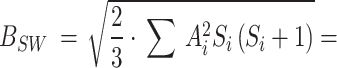 0.54 mT, where
0.54 mT, where 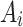 and
and 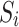 are the HFCC and spin of the i-th nucleus of BP.
are the HFCC and spin of the i-th nucleus of BP.
DFT calculations
Quantum chemical calculations were performed with the ORCA program package (version 5.0.2)90,91. The TPSSh/def2-TZVPP81,82 method was used to optimize the geometries of anion and ketyl radicals of 4-amino benzophenone. The 7 water molecules forming hydrogen bonds with C = O and -NH2 groups (3 near the C = O group and 4 near the NH2 group) are taken into account explicitly with the def2-TZVP basis82. To calculate their HFCCs and g-factors of the anion and the ketyl radical of 4-aminobenzophenone, the TPSSh/ma-def2-QZVPP method was used81,92 with def2-TZVPP basis for water molecules. The HFCCs and g-factor of 4-amino benzophenone radical were calculated to compare them with the literature; also these data were compared with HFCCs calculated for His-Gln(BP)-Tyr-Gly biradical to ensure the correctness of the method choice.
The geometry of the biradical in its triplet state formed from His-Gln(BP)-Tyr-Gly conjugate was optimized by the TPSS/def2-SVP method, 11 water molecules were taken into account (primarily near the oxygen atoms of Tyr, BP, and CO2-) explicitly. The TPSSh functional was used to calculate the HFCCs, the ma-def2-QZVPP basis was used for the tyrosine -CH2-C6H4-O and benzophenone -CO-NH-C6H4-CO-C6H5 fragments, the def2-SVP basis was used for the rest of the peptide and water molecules. SMD solvation model83 with water as a solvent was used in all cases. Results of DFT calculation are shown in Supporting Information.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
This work was supported by the Russian Science Foundation (project #23-73-01101) and the German Research foundation under contract Bu-911/29-1. We thank the Ministry of Science and Higher Education of the Russian Federation for granting access to the NMR equipment.
Author contributions
N.F.: Data curation (Field-dependent CIDNP experiments); Writing – original draft. K.H.: Resources; Synthesis. Writing – review & editing. O.M.: Data curation (time-resolved CIDNP experiments), Formal analysis (CIDNP kinetics simulation), Writing – original draft.I.Z.: Formal analysis (Field-dependent CIDNP simulation); Software; Validation; Writing – original draft. M.G.: Quantum chemical calculations. M.B.: Resources; Synthesis conceptualization. Writing – review & editing. T.W.: Resources; Synthesis. Writing – review & editing. G.B. Supervision; Conceptualization; Project administration; Writing – review & editing. A.Y. Supervision; Conceptualization; Writing – original draft; Writing – review & editing.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information files. Should any raw data files be needed in another format they are available from the corresponding author upon reasonable request. Source data are provided with this paper.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Natalya N. Fishman and Kevin Herr contributed equally to this work.
Contributor Information
Gerd Buntkowsky, Email: gerd.buntkowsky@chemie.tu-darmstadt.de.
Alexandra V. Yurkovskaya, Email: yurk@tomo.nsc.ru
References
- 1.Migliore, A., Polizzi, N. F., Therien, M. J. & Beratan, D. N. Biochemistry and theory of Proton-Coupled Electron transfer. Chem. Rev.114, 3381–3465. 10.1021/cr4006654 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hammes-Schiffer, S. Proton-Coupled Electron transfer: moving together and charging forward. J. Am. Chem. Soc.137, 8860–8871. 10.1021/jacs.5b04087 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stubbe, J., Nocera, D. G., Yee, C. S. & Chang, M. C. Y. Radical initiation in the class I ribonucleotide reductase: Long-range proton-coupled electron transfer? Chem. Rev.103, 2167–2201. 10.1021/Cr020421u (2003). [DOI] [PubMed] [Google Scholar]
- 4.Giese, B. et al. Electron relay race in peptides. J. Org. Chem.74, 3621–3625. 10.1021/jo900375f (2009). [DOI] [PubMed] [Google Scholar]
- 5.Pagba, C. V., McCaslin, T. G., Chi, S. H., Perry, J. W. & Barry, B. A. Proton-Coupled Electron transfer and a Tyrosine-Histidine pair in a photosystem II-Inspired beta-Hairpin maquette: kinetics on the picosecond time scale. J. Phys. Chem. B. 120, 1259–1272. 10.1021/acs.jpcb.6b00560 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Sibert, R. et al. Proton-coupled electron transfer in a biomimetic peptide as a model of enzyme regulatory mechanisms. J. Am. Chem. Soc.129, 4393–4400. 10.1021/ja068805f (2007). [DOI] [PubMed] [Google Scholar]
- 7.Barry, B. A. & Einarsdottir, O. Insights into the structure and function of redox-active tyrosines from model compounds. J. Phys. Chem. B. 109, 6972–6981. 10.1021/jp044749y (2005). [DOI] [PubMed] [Google Scholar]
- 8.Gao, J. A. et al. Electron transfer in peptides: the influence of charged amino acids. Angew Chem. Int. Ed.50, 1926–1930. 10.1002/anie.201003389 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Wang, M., Gao, J., Müller, P. & Giese, B. Electron transfer in peptides with cysteine and methionine as relay amino acids. Angew Chem. Int. Ed.48, 4232–4234. 10.1002/anie.200900827 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Giese, B. Electron transfer through DNA and peptides. Bioorg. Med. Chem.14, 6139–6143. 10.1016/j.bmc.2006.05.067 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Glover, S. D. et al. Photochemical tyrosine oxidation in the structurally Well-Defined alpha Y-3 protein: Proton-Coupled Electron transfer and a Long-Lived tyrosine radical. J. Am. Chem. Soc.136, 14039–14051. 10.1021/ja503348d (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whang, D. R. & Apaydin, D. H. Artificial photosynthesis: learning from nature. Chemphotochem2, 148–160. 10.1002/cptc.201700163 (2018). [Google Scholar]
- 13.Colvin, M. T. et al. Magnetic Field-Induced switching of the Radical-Pair intersystem crossing mechanism in a Donor – Bridge – Acceptor molecule for artificial photosynthesis. J. Am. Chem. Soc.133, 1240–1243. 10.1021/ja1094815 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Lübitz, W., Lendzian, F., Bittl, R. & Radicals Radical pairs and triplet States in photosynthesis. Acc. Chem. Res.35, 313–320. 10.1021/ar000084g (2002). [DOI] [PubMed] [Google Scholar]
- 15.Wasielewski, M. R. Photoinduced electron transfer in supramolecular systems for artificial photosynthesis. Chem. Rev.92, 435–461. 10.1021/cr00011a005 (1992). [Google Scholar]
- 16.Barry, B. A. & Babcock, G. T. Tyrosine radicals are involved in the photosynthetic oxygen-evolving system. Proc. Natl. Acad. Sci. U S A. 84, 7099–7103. 10.1073/pnas.84.20.7099 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halder, M., Parui, P. P., Gopidas, K. R., Nath, D. N. & Chowdhury, M. Magnetic field control of the back-electron-transfer process following photoinduced electron transfer (PIET) in biphenyl/phenylpyrylium salts in SDS micellar medium. J. Phys. Chem. A. 106, 2200–2206. 10.1021/jp012561y (2002). [Google Scholar]
- 18.Sun, X. N. et al. A molecular spin-photovoltaic device. Science357, 677–680. 10.1126/science.aan5348 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Wang, J. P., Chepelianskii, A., Gao, F. & Greenham, N. C. Control of exciton spin statistics through spin polarization in organic optoelectronic devices. Nat. Commun.3, 1191. 10.1038/ncomms2194 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Devir-Wolfman, A. H. et al. Short-lived charge-transfer excitons in organic photovoltaic cells studied by high-field magneto-photocurrent. Nat. Commun.5, 4529. 10.1038/ncomms5529 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Zhang, Y. et al. Spin-enhanced organic bulk heterojunction photovoltaic solar cells. Nat. Commun.3, 1043. 10.1038/ncomms2057 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Stuart-Audette, M. et al. Re-evaluation of intramolecular long-range electron transfer between tyrosine and Tryptophan in lysozymes - Evidence for the participation of other residues. Eur. J. Biochem.270, 3565–3571. 10.1046/j.1432-1033.2003.03741.x (2003). [DOI] [PubMed] [Google Scholar]
- 23.Weinstein, M., Alfassi, Z. B., Defelippis, M. R., Klapper, M. H. & Faraggi, M. Long-Range Electron-Transfer between tyrosine and Tryptophan in Hen Egg-White lysozyme. Biochim. Biophys. Acta. 1076, 173–178. 10.1016/0167-4838(91)90262-X (1991). [DOI] [PubMed] [Google Scholar]
- 24.Chen, X. et al. Proton-Regulated Electron transfers from tyrosine to Tryptophan in proteins: Through-Bond mechanism versus Long-Range hopping mechanism. J. Phys. Chem. B. 113, 16681–16688. 10.1021/jp9077689 (2009). [DOI] [PubMed] [Google Scholar]
- 25.DeFelippis, M. R., Faraggi, M. & Klapper, M. H. Evidence for through-bond long-range electron transfer in peptides. J. Am. Chem. Soc.112, 5640–5642. 10.1021/ja00170a039 (1990). [Google Scholar]
- 26.Odella, E., Moore, T. A. & Moore, A. L. Tuning the redox potential of tyrosine-histidine bioinspired assemblies. Photosynth Res.10.1007/s11120-020-00815-x (2021). [DOI] [PubMed] [Google Scholar]
- 27.Sheth, S. et al. Proton domino reactions at an imidazole relay control the oxidation of a TyrZ-His190 artificial mimic of photosystem II. Chem. : Eur. J.30, e202400862. 10.1002/chem.202400862 (2024). [DOI] [PubMed] [Google Scholar]
- 28.McCaslin, T. G. et al. Tyrosine, cysteine, and proton coupled electron transfer in a ribonucleotide reductase-inspired beta hairpin Maquette. Chem. Commun.55, 9399–9402. 10.1039/C9CC04067F (2019). [DOI] [PubMed] [Google Scholar]
- 29.Perez-Boada, M. et al. Versatile peroxidase oxidation of high redox potential aromatic compounds: Site-directed mutagenesis, spectroscopic and crystallographic investigation of three Long-range Electron transfer pathways. J. Mol. Biol.354, 385–402. 10.1016/j.jmb.2005.09.047 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Blomberg, M. R. A. The Redox-Active tyrosine is essential for proton pumping in cytochrome c oxidase. Front. Chem.910.3389/fchem.2021.640155 (2021). [DOI] [PMC free article] [PubMed]
- 31.Smith, W. L., Eling, T. E., Kulmacz, R. J., Marnett, L. J. & Tsai, A. L. Tyrosyl radicals and their role in Hydroperoxide-Dependent activation and inactivation of prostaglandin endoperoxide synthase. Biochemistry31, 3–7. 10.1021/bi00116a001 (1992). [DOI] [PubMed] [Google Scholar]
- 32.Reece, S. Y., Seyedsayamdost, M. R., Stubbe, J. & Nocera, D. G. Photoactive peptides for Light-Initiated Tyrosyl radical generation and transport into ribonucleotide reductase. J. Am. Chem. Soc.129, 8500–8509. 10.1021/ja0704434 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Harriman, A. Further comments on the redox potentials of Tryptophan and tyrosine. J. Phys. Chem.91, 6102–6104. 10.1021/j100308a011 (1987). [Google Scholar]
- 34.Navaratnam, S. & Parsons, B. J. Reduction potential of histidine free radicals: a pulse radiolysis study. J. Chem. Soc. Faraday Trans.94, 2577–2581. 10.1039/A803477J (1998). [Google Scholar]
- 35.Morozova, O. B. & Yurkovskaya, A. V. Intramolecular Electron transfer in the photooxidized peptides Tyrosine–Histidine and Histidine–Tyrosine: A Time-Resolved CIDNP study. Angew Chem. Int. Ed.49, 7996–7999. 10.1002/ange.201003780 (2010). [DOI] [PubMed] [Google Scholar]
- 36.Morozova, O. B., Stass, D. V. & Yurkovskaya, A. V. Kinetic evidence for the transiently shifted acidity constant of histidine linked to paramagnetic tyrosine probed by intramolecular electron transfer in oxidized peptides. Phys. Chem. Chem. Phys.23, 16698–16706. 10.1039/d1cp02408f (2021). [DOI] [PubMed] [Google Scholar]
- 37.Tanner, C., Navaratnam, S. & Parsons, B. J. Intramolecular electron transfer in the dipeptide, histidyltyrosine: a pulse radiolysis study. Free Radical Biol. Med.24, 671–678. 10.1016/S0891-5849(97)00340-7 (1998). [DOI] [PubMed] [Google Scholar]
- 38.Faller, P., Goussias, C., Rutherford, A. W. & Un, S. Resolving intermediates in biological proton-coupled electron transfer: A Tyrosyl radical prior to proton movement. Proc. Natl. Acad. Sci. U S A. 100, 8732–8735. 10.1073/pnas.1530926100 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morozova, O. B. & Yurkovskaya, A. V. Reduction of transient histidine radicals by tryptophan: influence of the amino group charge. Phys. Chem. Chem. Phys.23, 5919–5926. 10.1039/D0CP06366E (2021). [DOI] [PubMed] [Google Scholar]
- 40.Morozova, O. B. Reduction of Transient Histidine Radicals by Tyrosine: Influence of the Protonation State of Reactants. ChemPhysChem 21, 43–50, (2020). 10.1002/cphc.201901020 [DOI] [PubMed]
- 41.Morozova, O. B. & Ivanov, K. L. Time-resolved CIDNP of biologically important molecules. ChemPhysChem20, 197–215. 10.1002/cphc.201800566 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Goez, M. Photo-CIDNP spectroscopy. Annu. Rep. NMR Spectrosc.66, 77–147. 10.1016/S0066-4103(08)00403-1 (2009). [Google Scholar]
- 43.Bargon, J., Fischer, H. & Johnsen, U. Nuclear magnetic resonance emission lines during fast radical reactions. I. Recording methods and examples. Z. Naturforsch A: Phys. Sci.22, 1551–1555 (1967). [Google Scholar]
- 44.Bargon, J. & Fischer, H. Nuclear magnetic resonance emission lines during fast radical reactions. II. Chemically induced dynamic nuclear polarization. Z. Naturforsch A: Phys. Sci.22, 1556–1562 (1967). [Google Scholar]
- 45.Goez, M. An introduction to chemically induced dynamic nuclear polarization. Concepts Magn. Reson.7, 69–86. 10.1002/cmr.1820070105 (1995). [Google Scholar]
- 46.Hore, P. J. & Broadhurst, R. W. Photo-CIDNP of biopolymers. Prog Nucl. Magn. Reson. Spectrosc.25, 345–402. 10.1016/0079-6565(93)80002-B (1993). [Google Scholar]
- 47.Morozova, O. B., Yurkovskaya, A. V., Tsentalovich, Y. P. & Vieth, H. M. 1H and 13 C nuclear polarization in consecutive biradicals during the photolysis of 2,2,12,12-Tetramethylcyclododecanone. J. Phys. Chem. A. 101, 399–406. 10.1021/jp961456z (1997). [Google Scholar]
- 48.Morozova, O. B. et al. Study of consecutive biradicals from 2-Hydroxy-2,12-dimethylcyclododecanone by TR-CIDNP, TREPR, and laser flash photolysis. J. Phys. Chem. A. 101, 8803–8808. 10.1021/jp9711196 (1997). [Google Scholar]
- 49.Morozova, O. B., Tsentalovich, Y. P., Yurkovskaya, A. V. & Sagdeev, R. Z. Consecutive biradicals during the photolysis of 2,12-dihydroxy-2,12-dimethylcyclododecanone: low- and high-field chemically induced dynamic nuclear polarizations (CIDNP) study. J. Phys. Chem. A. 102, 3492–3497. 10.1021/jp980271k (1998). [Google Scholar]
- 50.Tsentalovich, Y. P. et al. Influence of molecular structure on the rate of intersystem crossing in flexible biradicals. J. Phys. Chem. A. 101, 8809–8816. 10.1021/jp972717n (1997). [Google Scholar]
- 51.Tsentalovich, Y. P. et al. Spin and molecular dynamics in Acyl-Containing biradicals: Time-Resolved Electron paramagnetic resonance and laser flash photolysis study. J. Phys. Chem. A. 106, 7121–7129. 10.1021/jp020098z (2002). [Google Scholar]
- 52.Yurkovskaya, A. V. et al. The influence of scavenging on CIDNP field dependences in biradicals during the photolysis of large-ring cycloalkanones. Chem. Phys.197, 157–166. 10.1016/0301-0104(95)00125-8 (1995). [Google Scholar]
- 53.Zhukov, I. et al. Mapping 13C hyperfine couplings and exchange interactions in short-lived charge separated States of rigid donor-bridge-acceptor dyads. J. Chem. Phys.155, 224201. 10.1063/5.0073193 (2021). [DOI] [PubMed] [Google Scholar]
- 54.Zhukov, I. V. et al. Exchange interaction in short-lived flavine adenine dinucleotide biradical in aqueous solution revisited by CIDNP (chemically induced dynamic nuclear polarization) and nuclear magnetic relaxation dispersion. Magn. Reson.2, 139–148. 10.5194/mr-2-139-2021 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Saprygina, N. N., Morozova, O. B., Grampp, G. & Yurkovskaya, A. V. Effect of amino group charge on the photooxidation kinetics of aromatic amino acids. J. Phys. Chem. A. 118, 339–349. 10.1021/Jp4097919 (2014). [DOI] [PubMed] [Google Scholar]
- 56.Morozova, O. B., Panov, M. S., Fishman, N. N. & Yurkovskaya, A. V. Electron transfer vs proton-coupled electron transfer as the mechanism of reaction between amino acids and triplet-excited benzophenones revealed by time-resolved CIDNP. Phys. Chem. Chem. Phys.20, 21127–21135. 10.1039/C8CP03591A (2018). [DOI] [PubMed] [Google Scholar]
- 57.Hore, P. J. & Kaptein, R. in NMR Spectroscopy: New Methods and Applications, ACS Symposium Series (ed G. C. Levy) 285–318 American Chemical Society (1982).
- 58.Miller, R. J. & Closs, G. L. Application of fourier transform-NMR spectroscopy to submicrosecond time-resolved detection in laser flash photolysis experiments. Rev. Sci. Instrum.52, 1876–1885 https://doi.org/10.1063/1.1136531 (1981). [Google Scholar]
- 59.Vollenweider, J. K., Fischer, H., Hennig, J. & Leuschner, R. Time-resolved CIDNP in laser flash photolysis of aliphatic ketones. A quantitative analysis. Chem. Phys.97, 217–234. 10.1016/0301-0104(85)87033-6 (1985). [Google Scholar]
- 60.Vollenweider, J. K. & Fischer, H. Time-resolved CIDNP in laser flash photolysis of di-tert-butyl ketone. Multiplet versus net effects. Chem. Phys.108, 365–372. 10.1016/0301-0104(86)80104-5 (1986). [Google Scholar]
- 61.Vollenweider, J. K. & Fischer, H. Absolute chemically induced nuclear polarizations and yields from geminate radical-pair reactions. A test of high-field radical-pair theories. Chem. Phys.124, 333–345. 10.1016/0301-0104(88)87058-7 (1988). [Google Scholar]
- 62.Morozova, O. B. & Yurkovskaya, A. V. Modulation of the rate of reversible Electron transfer in oxidized Tryptophan and tyrosine containing peptides in acidic aqueous solution. J. Phys. Chem. B. 119, 140–149. 10.1021/Jp511068n (2015). [DOI] [PubMed] [Google Scholar]
- 63.Kaptein, R. Simple rules for chemically induced dynamic nuclear polarization. J. Chem. Soc. Chem. Commun. 732–733. 10.1039/C29710000732 (1971).
- 64.Säuberlich, J., Brede, O. & Beckert, D. Photoionization of benzophenone carboxylic acids in aqueous solution. A FT EPR and optical spectroscopy study of radical cation decay. J. Phys. Chem.100, 18101–18107. 10.1021/jp960804u (1996). [Google Scholar]
- 65.Panov, M. S. et al. Photooxidation of histidine by 3,3’,4,4’-Benzophenone Tetracarboxylic acid in aqueous solution: Time-Resolved and Field-Dependent CIDNP study. Appl. Magn. Reson.45, 1019–1033. 10.1007/s00723-014-0577-8 (2014). [Google Scholar]
- 66.Tomkiewicz, M., McAlpine, R. D. & Cocivera, M. Photooxidation and decarboxylation of tyrosine studied by EPR and CIDNP [chemically-induced dynamic nuclear polarization] techniques. Can. J. Chem.50, 3849–3856. 10.1139/v72-606 (1972). [Google Scholar]
- 67.Morozova, O. B. et al. Time-resolved CIDNP: an NMR way to determine the EPR parameters of elusive radicals. Phys. Chem. Chem. Phys.13, 6619–6627. 10.1039/C0CP02449J (2011). [DOI] [PubMed] [Google Scholar]
- 68.Tsentalovich, Y. P., Morozova, O. B., Yurkovskaya, A. V., Hore, P. J. & Sagdeev, R. Z. Time-Resolved CIDNP and laser flash photolysis study of the photoreactions of N-Acetyl histidine with 2,2’-Dipyridyl in aqueous solution. J. Phys. Chem. A. 104, 6912–6916. 10.1021/jp000019o (2000). [Google Scholar]
- 69.Bansal, K. M. & Fessenden, R. W. Pulse radiolysis studies of the oxidation of phenols by SO4- and Br2- in aqueous solutions. Radiat. Res.67, 1–8. 10.2307/3574490 (1976). [PubMed] [Google Scholar]
- 70.Closs, G. L. & Doubleday, C. E. Determination of the average singlet-triplet splitting in biradicals by measurement of the magnetic field dependence of CIDNP [chemically induced dynamic nuclear polarization]. J. Am. Chem. Soc.95, 2735–2736. 10.1021/ja00789a082 (1973). [Google Scholar]
- 71.Kaptein, R. & Oosterhoff, L. J. Chemically induced dynamic nuclear polarization. III. Anomalous multiplets of radical coupling and disproportionation products. Chem. Phys. Lett.4, 214–216. 10.1016/0009-2614(69)80105-3 (1969). [Google Scholar]
- 72.Closs, G. L. & Closs, L. E. Induced dynamic nuclear spin polarization in photoreductions of benzophenone by toluene and ethylbenzene. J. Am. Chem. Soc.91, 4550–4552. 10.1021/ja01044a042 (1969). [Google Scholar]
- 73.Closs, G. L. Mechanism explaining nuclear spin polarizations in radical combination reactions. J. Am. Chem. Soc.91, 4552–4554. 10.1021/ja01044a043 (1969). [Google Scholar]
- 74.Tsentalovich, Y. P. et al. Kinetics of Nuclear-Polarization in the geminate recombination of biradicals. Chem. Phys.139, 307–315. 10.1016/0301-0104(89)80143-0 (1989). [Google Scholar]
- 75.Salikhov, K. M., Molin, Y. N., Sagdeev, R. Z. & Buchachenko, A. L. Spin Polarization and Magnetic Effects in Chemical Reactions (Elsevier, 1984).
- 76.Grosse, S., Yurkovskaya, A. V., Lopez, J. & Vieth, H. M. Field dependence of chemically induced dynamic nuclear polarization (CIDNP) in the photoreaction of N-Acetyl histidine with 2,2’-Dipyridyl in aqueous solution. J. Phys. Chem. A. 105, 6311–6319. 10.1021/jp004582i (2001). [Google Scholar]
- 77.Ivanov, K. L., Vieth, H. M., Miesel, K., Yurkovskaya, A. V. & Sagdeev, R. Z. Investigation of the magnetic field dependence of CIDNP in multi-nuclear radical pairs.part II. Photoreaction of tyrosine and comparison of model calculation with experimental data. Phys. Chem. Chem. Phys.5, 3470–3480. 10.1039/b304086k (2003). [Google Scholar]
- 78.Pravdivtsev, A. N., Yurkovskaya, A. V., Zimmermann, H., Vieth, H. M. & Ivanov, K. L. Magnetic field dependent long-lived spin States in amino acids and dipeptides. Phys. Chem. Chem. Phys.16, 7584–7594. 10.1039/C3cp55197k (2014). [DOI] [PubMed] [Google Scholar]
- 79.Zhukov, I. et al. Positive electronic exchange interaction and predominance of minor triplet channel in CIDNP formation in short lived charge separated States of D-X-A dyads. J. Chem. Phys.152, 014203. 10.1063/1.5131817 (2020). [DOI] [PubMed] [Google Scholar]
- 80.Zhukov, I. et al. Simulation of electron and nuclear spin dynamics in many-spin charge-separated States. J. Chem. Phys.16210.1063/5.0244106 (2025). [DOI] [PubMed]
- 81.Staroverov, V. N., Scuseria, G. E., Tao, J. & Perdew, J. P. Comparative assessment of a new nonempirical density functional: molecules and hydrogen-bonded complexes. J. Chem. Phys.119, 12129–12137. 10.1063/1.1626543 (2003). [Google Scholar]
- 82.Weigend, F. & Ahlrichs, R. Balanced basis sets of split Valence, triple zeta Valence and quadruple zeta Valence quality for H to rn: design and assessment of accuracy. Phys. Chem. Chem. Phys.7, 3297–3305. 10.1039/B508541A (2005). [DOI] [PubMed] [Google Scholar]
- 83.Marenich, A. V., Cramer, C. J. & Truhlar, D. G. Universal solvation model based on solute Electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B. 113, 6378–6396. 10.1021/jp810292n (2009). [DOI] [PubMed] [Google Scholar]
- 84.Okutsu, T., Muramatsu, H., Horiuchi, H. & Hiratsuka, H. Explanation to the difference in the ketyl radical formation yields of benzophenone and benzil. Chem. Phys. Lett.404, 300–303. 10.1016/j.cplett.2005.01.102 (2005). [Google Scholar]
- 85.Turro, N. J. Modern Molecular Photochemistry of Organic Molecules (University Science Books, 1991).
- 86.Krezel, A. & Bal, W. A formula for correlating pKa values determined in D2O and H2O. J. Inorg. Biochem.98, 161–166. 10.1016/j.jinorgbio.2003.10.001 (2004). [DOI] [PubMed] [Google Scholar]
- 87.Zhukov, I. V. et al. Field-cycling NMR experiments in ultra-wide magnetic field range: relaxation and coherent polarization transfer. Phys. Chem. Chem. Phys.20, 12396–12405. 10.1039/C7CP08529J (2018). [DOI] [PubMed] [Google Scholar]
- 88.Schulten, K. & Wolynes, P. G. Semiclassical description of electron spin motion in radicals including the effect of electron hopping. J. Chem. Phys.68, 3292–3297. 10.1063/1.436135 (1978). [Google Scholar]
- 89.Osintsev, A. M., Purtov, P. A. & Salikhov, K. M. Calculation of SNP effects in weak magnetic fields. Chem. Phys.174, 237–245. 10.1016/0301-0104(93)87008-B (1993). [Google Scholar]
- 90.Neese, F. The ORCA program system. Wiley Interdiscip Rev. Comput. Mol. Sci.2, 73–78. 10.1002/Wcms.81 (2012). [Google Scholar]
- 91.Neese, F. Software update: the ORCA program system—Version 5.0. WIREs Comput. Mol. Sci.12, e1606. 10.1002/wcms.1606 (2022). [Google Scholar]
- 92.Zheng, J., Xu, X. & Truhlar, D. G. Minimally augmented Karlsruhe basis sets. Theor. Chem. Acc.128, 295–305. 10.1007/s00214-010-0846-z (2011). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information files. Should any raw data files be needed in another format they are available from the corresponding author upon reasonable request. Source data are provided with this paper.