

Abstract
Axially chiral indoles have garnered significant attention due to their synthetic and biological importance. However, atroposelective halogenation of this scaffold has been rarely explored. This study presents a catalytic and enantioselective iodination of N-arylindoles, achieving precise control over the C−N stereogenic axis. The reaction is facilitated by a chiral phosphoric acid, which promotes iodination at the C-3 position of N-arylindole, followed by iodine migration and deprotonation. A hydrogen-bonding donor on the aromatic ring plays a key role in achieving high enantioselectivities. Under optimized reaction conditions, a wide range of substrates are well-tolerated, and subsequent reactions with various carbonyl electrophiles maintain enantioselectivity. Computational studies provide insights into the origin of enantioselectivity at the plausible enantiodetermining step.

Subject terms: Synthetic chemistry methodology, Asymmetric catalysis, Asymmetric synthesis
Axially chiral indoles are crucial in synthetic and biological contexts, yet atroposelective halogenation remains underexplored. Here, the authors achieve catalytic and enantioselective iodination of N-arylindoles, utilizing a chiral phosphoric acid to control the C−N stereogenic axis.
Introduction
Catalytic and enantioselective halogenation offers an efficient pathway to access chiral halogenated compounds, which can be easily transformed into more complex molecules1–10. These transformations have been developed to control not only point chirality but also axial chirality, showcasing the advancements in asymmetric catalysis11–22 Since Miller’s pioneering work11, significant attention has been directed toward the atroposelective synthesis of biaryls12–15, diarylamines16, and alkenes17 via enantiodetermining halogenations using chiral organocatalysts (Fig. 1A). To expand accessibility to complex and versatile molecules, the continuous development of atroposelective synthesis of halogenated scaffolds is crucial, especially as the synthesis of biologically relevant heterocyclic biaryls can open new avenues for the development of novel chemotherapeutics and chemical probes23–25.
Fig. 1. Organocatalytic enantioselective halogenation.

A Organocatalytic atroposelective halogenation via enantiodetermining halogenation. B Catalytic enantioselective halocyclization of indoles. C Catalytic and atroposelective C2-functionalization of indoles. D Proposed mechanism for chiral phosphoric acid-catalyzed atroposelective iodination of indoles.
As indoles are privileged motifs commonly found in nature26,27, pharmaceuticals28–32, and materials science33, catalytic and enantioselective halogenations have been extensively studied. For example, chiral catalysts mediate halogenation reactions preferentially at the C-3 position of indoles, where stereochemistry is controlled. This intermediate then undergoes further stereoselective nucleophilic addition, yielding chiral indolines (Fig. 1B)34–38. Various catalysts have been employed for enantioselective halocyclizations, leading to fused heterocycles. If the nucleophilic attack is absent or slow, the halogen may migrate to the C-2 position39,40, producing 2-halogenated indoles. In this regard, we hypothesized that the stereogenic axis in N-arylindoles41 could be controlled by enantioselective halogenation at the C-2 position using a chiral organocatalyst. Catalytic methods for axially chiral N-arylindoles have been developed by Ackermann, Zhou/Xu/Shi, Ackermann/Wencel-Delord, Neidig/Wencel-Delord/Ackermann, and our group, introducing several functional groups at the C-2 position (Fig. 1C)42–46, contributing to the expansion of axially chiral indole-based scaffolds47–52, which represent a structurally important framework. Our halogenation strategy could provide key intermediate that may offer an alternative route to accessing previously valuable compounds and expanding the scope of accessible axially chiral N-arylindoles.
To realize our hypothesis, halogen migration must occur faster than internal or external nucleophilic attacks on the iminium ion intermediate. Two relevant strategies for controlling atroposelectivity in this reaction53–61 are: (1) central-to-axial chirality conversion59, and (2) dynamic kinetic resolution60,61. In the first scenario, the C-3 stereogenic center is initially controlled in a highly enantioselective manner, which subsequently governs atroposelectivity through chirality transfer. Alternatively, based on dynamic kinetic resolution, the absolute configuration is established in the subsequent steps62, while stereoselectivity during the iodination step may be less critical. Herein, we present a chiral phosphoric acid-catalyzed63,64 highly atroposelective iodination of N-arylindoles (Fig. 1D). To the best of our knowledge, organocatalytic halogenation of arylindoles to control a stereogenic axis has not yet been explored. Our methodology accommodates a range of N-arylindole substrates, which can be further transformed into 2-substituted indoles. Both experimental and computational mechanistic investigations were conducted to support our mechanistic insights.
Results and discussion
Reaction optimizations
To demonstrate our hypothesis, we initially designed substrate 1a, which contains a methyl group at the C-3 position of the indole and a benzamide moiety on the opposing phenyl ring to form favorable secondary interactions with the catalyst (Table 1). Catalytic reactions of 1a were performed using N-bromosuccinimide (NBS) and N-iodosuccinimide (NIS) in the presence of a chiral phosphoric acid (P1) (Table 1, entries 1 and 2). Both reactions produced the desired halogenated products, however, the enantioselectivities varied significantly (2a, 41% yield, 50:50 er vs 3a, 71% yield, 97:3 er). Inspired by the excellent enantioselectivity observed in the iodination, we tested several iodine sources (entries 3−6). While iodine and iodine monochloride did not yield 3a, N-iodophthalimide (NIP) and 1,3-diiodo-5,5-dimethylhydantoin (DIH) gave the desired product, albeit with slightly reduced enantioselectivity. To further optimize the reaction, we screened a range of chiral phosphoric acids, as summarized in entries 7−15. Among the tested catalysts, the tricyclohexyl-substituted catalyst (P2) was identified as the optimal choice. While the catalysts containing 9-anthracenyl (P3) or 9-phenanthryl (P4) groups provided moderate enantioselectivity, catalysts bearing 3,5-substituted phenyl (P5 and P6) or 4-substituted phenyl group (P7−P9) showed low enantioselectivity. The results indicated that proximal bulkiness at the 3,3′ positions of the catalyst was highly correlated with improved enantioselectivity. Despite the 1,1′-spirobiindane-7,7′-diol (SPINOL)-derived catalyst (P10) displaying high enantioselectivity, it resulted in a low chemical yield. With the optimal catalyst P2, further reaction parameters were optimized (see Supplementary Table 1−4). Enantioselectivity was slightly enhanced when the reaction was conducted at −20 °C (entry 16, 87% yield, 98:2 er). To validate the configurational stability of 3a, racemization experiments were conducted. When 3a was dissolved in dimethylsulfoxide and exposed to 100 °C, gradual racemization was observed. Based on these observations, the rotational barrier was calculated to be 30.2 kcal/mol, indicating that 3a maintains its configuration stably at room temperature (Supplementary Fig. 4−6 and Supplementary Table 5−7). During the optimization process, we did not observe any halocyclization product resulting from a nucleophilic attack by the benzamide. However, we did detect the incorporation of succinimide into the intermediate when the reaction was performed in a chlorinated solvent (vide infra).
Table 1.
Optimization of reaction conditions
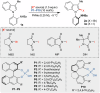 | ||||
|---|---|---|---|---|
| Entry | X+ | Catalyst | Yielda (%) | erb |
| 1 | NBS | P1 | 2a, 41 | 50:50 |
| 2 | NIS | P1 | 3a, 71 | 97:3 |
| 3 | I2 | P1 | <5 | - |
| 4 | I-Cl | P1 | <5 | - |
| 5 | NIP | P1 | 3a, 68 | 88:12 |
| 6 | DIH | P1 | 3a, 67 | 84:16 |
| 7 | NIS | P2 | 3a, 94 | 96:4 |
| 8 | NIS | P3 | 3a, 14 | 70:30 |
| 9 | NIS | P4 | 3a, 37 | 34:66 |
| 10 | NIS | P5 | 3a, 75 | 51:49 |
| 11 | NIS | P6 | 3a, 61 | 54:46 |
| 12 | NIS | P7 | 3a, 76 | 38:62 |
| 13 | NIS | P8 | 3a, 72 | 41:59 |
| 14 | NIS | P9 | 3a, 74 | 32:68 |
| 15 | NIS | P10 | 3a, 43 | 3:97 |
| 16c | NIS | P2 | 3a, 87 | 98:2 |
aIsolated yields.
bEnantiomeric ratios were determined by chiral-phase high-performance liquid chromatography analysis.
cThe reaction was performed at −20 °C.
Evaluation of the substrate scopes
Next, we explored the substrate scope, which demonstrated the high compatibility of our methodology (for detailed procedures, NMR spectra and HPLC chromatograms, see Supplementary Methods and Supplementary Data 3 and 4). Initially, various substituents were introduced to the lower aromatic ring while retaining the benzamide group (Fig. 2). Substrates (1b−1d), containing methyl and methoxy groups at the ortho, meta, or para positions relative to the benzamide, were highly tolerant, providing high yields and enantioselectivities. For substrates 1c and 1d, the reactions were conducted at room temperature to achieve high conversion. Notably, when a methoxy group was introduced at the ortho position to the stereogenic axis, a kinetic resolution effect was observed. To achieve high enantioselectivity, the reaction of 1e had to be stopped after 33 h at less than 50% conversion (34% yield and 83:17 er). Extending the reaction time to 72 h resulted in lower enantioselectivity (79% yield and 51:49 er). Subsequently, we replaced the benzamide group with various amines and amides to investigate the versatility and limitations of our methodology. The substrate bearing a 4-methoxybenzamide group reacted with N-iodosuccinimide at room temperature, yielding the desired product in 74% yield and 96:4 er.
Fig. 2. Substrate scope.

Unless otherwise noted, the reactions were conducted with 1 (0.050 mmol, 1.0 equiv), N-iodosuccinimide (0.075 mmol, 1.5 equiv), P2 (0.005 mmol, 10 mol%), PhMe (0.2 mL, 0.25 M). aThe reaction was performed at rt. bThe reaction was performed at −5 °C. cThe reaction was performed in CH2Cl2. dThe reaction was performed with 20 mol% of P2 at −5 °C.
In addition, substrates with benzylamino (1g) and methyl carbamate (1h) groups, instead of benzamide, proved compatible. In the presence of catalyst P2, these reactions proceeded smoothly, affording the desired products with excellent enantioselectivities (3g, 89% yield and 98:2 er at room temperature; 3h, 59% yield and 97:3 er). While reactions involving substrates bearing sulfonamide (1i) or phosphoramidate (1j) groups did not give the desired products under the optimized reaction conditions, slight modifications to the reaction parameters enabled product formation, albeit with moderate enantioselectivities (3i, 74% yield and 86:14 er in CH2Cl2; 3j, 72% yield, 81:19 er with 20 mol% of P2 at −5 °C). When the substrate lacked a hydrogen-bonding donor, the reaction yielded the desired products with low enantioselectivities (3k, 50:50 er; 3l, 70:30 er; 3m, 71:29 er; 3n, 66:34 er). These findings underscore the significance of secondary interactions with the chiral phosphoric acid catalyst.
We then explored the scope of indole derivatives, which demonstrate high tolerance at the C-5 position and steric dependency at the C-3 position (Fig. 3). When a methoxy group was introduced at the C-5 position of the indole, the catalytic reaction yielded the desired product (3o) with 52% yield and 93:7 er. Substitutions at the bottom aromatic ring were also well-tolerated in the presence of a C-5 methoxy group, as exemplified by 3p and 3q (3p, 53% yield and 93:7 er; 3q, 61% yield and 97:3 er). In the case of 1r, which bears a chloro group at the C-5 position, the reaction proceeded slowly at −20 °C. Increasing the temperature to −5 °C afforded the desired product in 55% yield and 97:3 er. When modifying the methyl group at the C-3 position, the enantioselectivity proved to be sensitive to the size of the substituent. While an ethyl group was highly tolerable, providing 3s in 74% yield and 98:2 er, the reaction of the propyl-substituted substrate (1t) exhibited slightly lower selectivity (64% yield and 87:13 er). Due to the steric bulk of the isopropyl group, the reaction with 1u proceeded slowly and required a higher temperature (26% yield and 89:11 er at room temperature). The catalytic iodination of 3-benzyl-N-arylindole (1v) allowed for some control over the sterogenic axis, yielding the product in 32% yield and 84:16 er at room temperature. However, the enantioselectivity was lower in the catalytic reaction of 3-phenyl-N-arylindole (3w), which afforded the product in 55% yield and 60:40 er at room temperature. The absolute stereochemistry was determined by the single crystal X-ray structure of 3s (see Supplementary Data 1, Supplementary Fig. 7, and Supplementary Tables 8−12) and further confirmed by comparing the experimental ECD spectrum with the calculated one of 3a (see Supplementary Figs. 9 and 10 in Supplementary Data 5).
Fig. 3. Substrate scope of substituted indoles.

Unless otherwise noted, the reactions were conducted with 1 (0.050 mmol, 1.0 equiv), N-iodosuccinimide (0.075 mmol, 1.5 equiv), P2 (0.005 mmol, 10 mol%), PhMe (0.2 mL, 0.25 M). aThe reaction was performed at −5 °C. bThe reaction was performed at rt.
Computational studies and mechanistic consideration
To investigate the reaction mechanism, we first calculated the rotational barriers of the substrate and halogenated compounds using Gaussian 16 software65 as shown in Fig. 4A (See Supplementary Tables 18−20 in Supplementary Data 5). Geometries were optimized using the B3LYP-D3/6-31 g(d) level of theory, and single-point energies of the optimized geometries were computed at the M06-2X/def2TZVP level of theory with the inclusion of solvation energy corrections using a self-consistent reaction field based on SMD implicit solvent model with dimethylsulfoxide as a solvent. The rotational barrier of 1a was calculated to be 16.4 kcal/mol, indicating rapid rotation at the reaction temperature. As expected, the rotational barriers for both 2a and 3a were significantly higher (31.0 kcal/mol and 31.8 kcal/mol, respectively). This implies that the low enantioselectivity of 2a was not due to the low rotational barrier of the product. We hypothesize that the size difference between bromine and iodine atoms alters the substrate’s pose in the enantiodeterming transition state, leading to variations in enantioselectivity. Regarding to the reaction mechanism, it is noteworthy that compound 4a was observed, where succinimide was incorporated at the C-2 position with low enantioselectivity (54:46 er) when the catalytic reaction was performed in chloroform (Fig. 4B). This observation suggests that an external nucleophile competes in the reaction. To further probe this we designed and synthesized the 3-hydroxyethyl substrate (1x), which was subjected to the optimized reaction conditions (Fig. 4C)34–38. In the initial reaction, a series of products were observed due to the instability of the halocyclization product. Remarkably, we isolated the major cyclized product (3x) by adding methanol after full substrate consumption, though the enantioselectivity was low (68:32 er). This result implies that 3-iodination initially occurs similarly to halocyclization and that the enantioselectivity of the iodination step may not be high. Next, we conducted a kinetic isotope effect (KIE) experiment (see Supplementary Fig. 1−3), which revealed no significant KIE (Fig. 4D). This finding suggests that neither hybridization change nor deprotonation is involved in the rate-determining step. Based on these experiments results and supporting literature, a plausible reaction mechanism is proposed in Fig. 4E. Following the initial catalytic iodination, the 3-iodo intermediate (Int I) is formed. Although succinimide can attack the C-2 position at this stage in a chlorinated solvent, iodine migration preferentially occurs in toluene, leading to the formation of the 2-iodo intermediate (Int II). Since the iodination step may not be highly selective and rotations around the stereogenic axis in Int I and Int II are likely rapid, the enantiodetermining step is proposed to be the deprotonation of Int II, proceeding via a dynamic kinetic process. Despite the proposed and evidenced reaction mechanism, the possibility that iodine migration and deprotonation occur simultaneously cannot be completely ruled out. To further validate this hypothesis, the rotational barriers of (2S)-Int II and (2R)-Int II were calculated to be 15.5 kcal/mol and 17.9 kcal/mol, respectively (See Supplementary Tables 21 and 22 in Supplementary Data 5). The low rational barrier is presumably due to the fact that the C-2 position is an sp3-hybridized carbon, which places the iodine atom out of the same plane, thereby reducing steric repulsion66. These results further support that the enantiodetermining step would be the deprotonation of Int II.
Fig. 4. Mechanistic studies.

A Calculated rotational barriers of 1a, 2a, and 3a. B Catalytic reaction of 1a in CHCl3. C Catalytic reaction of 1x. D Kinetic isotope experiment of 1a. E Plausible reaction mechanism. F Calculated transition state structures of 3a.
To further elucidate the origin of the observed enantioselectivity, computational studies were conducted on four possible transition states derived from (2S)-Int II and (2R)-Int II (See Supplementary Table 23 in Supplementary Data 5). Conformations for the transition state structures were generated using the MMFF force field and further optimized with the two-layer quantum mechanical (QM)/semiempirical (SE) ONIOM model67 due to the bulky substituents of the chiral phosphoric acid (P2). The QM layer was applied to the phosphoric acid group in P2 and the substrates, while the SE layer was applied to the remaining atoms of P2. The QM layer was treated with the B3LYP-D3/6-31 g(d) level of theory, while the SE layer was treated with PM6 for geometry optimization and frequency calculations. Single-point energies of these optimized structures were calculated using M06-2X/def2-TZVP for the QM layer and PM6 for the SE layer, with solvation energy corrections included (SMD = toluene). As shown in Fig. 4F, the four diastereomeric transition state structures were obtained for the formation of 3a (TS1-3a and TS3-3a) and ent-3a (TS2-ent-3a and TS4-ent-3a), which were consistent with our experimental results. The corrected Gibbs free energies of TS1-3a and TS3-3a were lower than those of TS2-ent-3a and TS4-ent-3a, respectively (2.65 kcal/mol between TS1-3a and TS2-ent-3a and 2.15 kcal/mol between TS3-3a and TS4-ent-3a).
To explain these results, we conducted a visual analysis of non-covalent interactions using Multiwfn software68, applying the independent gradient model method based on Hirshfeld partitioning of molecular density (IGMH)69–71. This analysis clearly revealed intermolecular interactions among the catalyst, succinimide, and substrate (See Supplementary Figs. 11−14 in Supplementary Data 5). In our studies, steric interactions between the opposite aromatic ring (including the benzamide) and the bulky substituents of the catalyst were commonly observed. However, the least steric interactions were found in TS1-3a, leading to the most stable transition state structure. In TS2-ent-3a, the indole ring was positioned slightly higher compared to TS1-3a, resulting in two additional steric interactions: one between the C7-position of the indole and the catalyst, and another between succinimide and the catalyst. While the steric interaction between succinimide and the catalyst persisted in TS3-3a, no steric interaction was observed between the carbocycle of indole and the catalyst, although weak interactions were found between the 3-position of indole and the catalyst. TS4-ent-3a exhibited the most steric interactions, similar to those in TS2-ent-3a. Consequently, these steric interactions contribute to the high enantioselectivities observed, despite the low rotational barriers in the intermediates.
Synthetic applications
To validate the practicality of our methodology, a 2-mmol scale reaction was performed as shown in Fig. 5A (for detailed procedures, NMR spectra and HPLC chromatograms, see Supplementary Methods and Supplementary Data 3 and 4). When toluene was used as the solvent, the reaction mixture became cloudy, resulting in a low yield of the desired product. This issue was resolved by using a 10:1 mixture of toluene and dichloromethane as the solvent, which afforded the desired product (3a) in 75% yield and 94:6 er. Next, the reaction product was further transformed, as shown in Fig. 5B. The benzamide (3a) and benzylamine (3g) products were easily methylated or benzylated, yielding the desired compounds 5a−5c. The absolute stereochemistry was additionally confirmed through the X-ray structure of 5c (see Supplementary Data 2, Supplementary Fig. 8, and Supplementary Tables 13−17). The methylated atropisomeric compounds were carefully lithiated and reacted with various aldehydes and ketones. While the reactions of the benzoylated compound (5a) with benzophenone, diethyl ketomalonate, and benzaldehyde provided the desired products (5aa and 5ab), albeit in low yields due to decomposition during the reaction, the benzylated compound (5b) reacted with acetone, 3-pentanone, and acetaldehyde in high yields with minimal erosion of enantioselectivities. Although reactions with aldehydes exhibited low diastereoselectivities (1.5:1−2:1 dr), the enantioselectivities were retained.
Fig. 5. Synthetic utility.

A 2-mmol scale reaction. B Further transformations.
Conclusions
In conclusion, we have demonstrated the atroposelective iodination of indoles catalyzed by a chiral phosphoric acid. By introducing a hydrogen-bonding donor to the opposite aromatic ring, a secondary non-covalent interaction is formed with the catalyst, leading to high enantioselectivities. We have proven the broad substrate scope of our methodology, its scalability to 2-mmol, and the potential for further modifications of atropisomeric compounds. Based on our observations, the initial 3-iodination step is not highly selective, and atroposelectivity would be determined by the final deprotonation step, as supported by our computational studies. Given the biological significance of indole compounds, we believe that our methodology could serve as a foundation for accessing a wide variety of atropisomeric and therapeutic compounds, and our effort to medicinal chemistry study of this scaffold is currently on the way.
Methods
General procedure for atroposelective iodination of indoles
To an oven dried 5 mL vial equipped with a magnetic stir bar, substrate 1 (0.05 mmol, 1.0 equiv), P2 (5.0 mg, 0.005 mmol, 0.1 equiv), NIS (16.9 mg, 0.075 mmol, 1.5 equiv), and PhMe (0.2 mL, 0.25 M) were added. The vial was sealed with a Teflon cap and further secured with Parafilm M®. The reaction mixture was stirred at –20 °C until complete consumption of 1. The crude material was purified by flash column chromatography using an eluent of EtOAc/Hx or CH2Cl2/Hx to afford the desired material 3. The enantioselectivity was determined by chiral-phase HPLC.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
This work is supported by a National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (2020R1C1C1006231, 2022R1A4A1018930, RS-2023-00259659, and RS-2024-00351238). This work is also supported by the POSCO Science Fellowship of POSCO TJ Park Foundation (Y.K.). The authors thank Dr H. Ju (Western Seoul Center of Korea Basic Science Institute) for technical support with single-crystal X-ray crystallography.
Author contributions
A.K. and H.-AC. contributed equally to this work. A.K., H.-AC., B.O., J.S., and Y.K. conceived and designed the experiments. A.K., H.-AC., and B.O. performed the chemical experiments. A.K., J.S., and Y.K. performed the computational studies. J.S. and Y.K. wrote the paper. All authors analyzed the results and commented on the manuscript.
Peer review
Peer review information
Communications Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work.
Data availability
Detailed experimental procedures and characterizations of new compounds are available in Supplementary Information. The X-ray crystallographic coordinates for structures reported in this Article have been provided as Supplementary Data 1 and 2 and have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers CCDC 2380810 (3s) and 2383264 (5c). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. 1H and 13C NMR spectra and HPLC chromatograms can be found in the Supplementary Data 3 and 4, respectively. Computational chemistry details are available in Supplementary Data 5. Reprints and permissions information is available online at www.nature.com/reprints. Correspondence and requests for materials should be addressed to Y.K.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Ahreum Kim, Hyun-A Cho.
Supplementary information
The online version contains supplementary material available at 10.1038/s42004-025-01584-1.
References
- 1.Zhu, Y. et al. Modern approaches for asymmetric construction of carbon–fluorine quaternary stereogenic centers: synthetic challenges and pharmaceutical needs. Chem. Rev.118, 3887–3964 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Landry, M. L. & Burns, N. Z. Catalytic enantioselective dihalogenation in total synthesis. Acc. Chem. Res.51, 1260–1271 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu, Y., Leng, H.-J., Li, Q.-Z. & Li, J.-L. Catalytic strategies for the asymmetric construction of cyclic frameworks with a halogenated tetrasubstituted stereocenter. Adv. Synth. Catal.362, 3926–3947 (2020). [Google Scholar]
- 4.China, H., Kumar, R., Kikushima, K. & Dohi, T. Halogen-induced controllable cyclizations as diverse heterocycle synthetic strategy. Molecules25, 6007 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wong, J. & Yeung, Y.-Y. Recent advances in C–Br bond formation. Synlett32, 1354–1364 (2021). [Google Scholar]
- 6.Maria Faisca Phillips, A. & Pombeiro, A. J. L. Recent developments in enantioselective organocatalytic cascade reactions for the construction of halogenated ring systems. Eur. J. Org. Chem.2021, 3938–3969 (2021). [Google Scholar]
- 7.Liu, S., Zhang, B.-Q., Xiao, W.-Y., Li, Y.-L. & Deng, J. Recent advances in catalytic asymmetric syntheses of functionalized heterocycles via halogenation/chalcogenation of carbon-carbon unsaturated bonds. Adv. Synth. Catal.364, 3974–4005 (2022). [Google Scholar]
- 8.Lee, S. & Chung, W.-j. Enantioselective halogenation via asymmetric phase-transfer catalysis. Bull. Korean Chem. Soc.43, 896–911 (2022). [Google Scholar]
- 9.Shibatomi, K. Organocatalytic synthesis of chiral halogenated compounds. Chem. Rec.23, e202300061 (2023). [DOI] [PubMed] [Google Scholar]
- 10.Nishiyori, R., Mori, T., Okuno, K. & Shirakawa, S. Chiral sulfide and selenide catalysts for asymmetric halocyclizations and related reactions. Org. Biomol. Chem.21, 3263–3275 (2023). [DOI] [PubMed] [Google Scholar]
- 11.Gustafson, J. L., Lim, D. & Miller, S. J. Dynamic kinetic resolution of biaryl atropisomers via peptide-catalyzed asymmetric bromination. Science328, 1251–1255 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diener, M. E., Metrano, A. J., Kusano, S. & Miller, S. J. Enantioselective synthesis of 3-Arylquinazolin-4(3H)-ones via peptide-catalyzed atroposelective bromination. J. Am. Chem. Soc.137, 12369–12377 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miyaji, R., Asano, K. & Matsubara, S. Bifunctional organocatalysts for the enantioselective synthesis of axially chiral Isoquinoline N-Oxides. J. Am. Chem. Soc.137, 6766–6769 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Snodgrass, H. M., Mondal, D. & Lewis, J. C. Directed evolution of flavin-dependent halogenases for site- and atroposelective halogenation of 3-Aryl-4(3H)-Quinazolinones via kinetic or dynamic kinetic resolution. J. Am. Chem. Soc.144, 16676–16682 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rose, B. T. et al. High-level data fusion enables the chemoinformatically guided discovery of chiral disulfonimide catalysts for atropselective iodination of 2-Amino-6-arylpyridines. J. Am. Chem. Soc.144, 22950–22964 (2022). [DOI] [PubMed] [Google Scholar]
- 16.Vaidya, S. D., Toenjes, S. T., Yamamoto, N., Maddox, S. M. & Gustafson, J. L. Catalytic atroposelective synthesis of N-Aryl Quinoid compounds. J. Am. Chem. Soc.142, 2198–2203 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu, S. et al. Urea group-directed organocatalytic asymmetric versatile dihalogenation of alkenes and alkynes. Nat. Catal.4, 692–702 (2021). [Google Scholar]
- 18.Zhao, K. et al. Enhanced reactivity by torsional strain of cyclic diaryliodonium in Cu-Catalyzed enantioselective ring-opening reaction. Chem4, 599–612 (2018). [Google Scholar]
- 19.Zheng, D.-S., Zhang, W.-W., Gu, Q. & You, S.-L. Rh(III)-Catalyzed Atroposelective C–H Iodination of 1-Aryl Isoquinolines. ACS Catal.13, 5127–5134 (2023). [Google Scholar]
- 20.Linde, S. T. et al. Atroposelective brominations to access chiral biaryl scaffolds using high-valent Pd-catalysis. Chem. Sci.14, 3676–3681 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yao, C.-Z. et al. Stereogenic-at-Cobalt(III) complex catalyzed halocyclization of alkynes: enantioselective access to axially chiral ortho-Halo-C2-indoles. J. Org. Chem.88, 6146–6158 (2023). [DOI] [PubMed] [Google Scholar]
- 22.Yao, L., Gashaw Woldegiorgis, A., Huang, S., Wang, Y. & Lin, X. Palladium-catalyzed directed atroposelective C−H iodination to synthesize axial chiral biaryl N-Oxides via enantioselective desymmetrization strategy. Chem. Eur. J.29, e202203051 (2023). [DOI] [PubMed] [Google Scholar]
- 23.Toenjes, S. T. & Gustafson, J. L. Atropisomerism in medicinal chemistry: challenges and opportunities. Future Med. Chem.10, 409–422 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perreault, S., Chandrasekhar, J. & Patel, L. Atropisomerism in drug discovery: a medicinal chemistry perspective inspired by atropisomeric class I PI3K Inhibitors. Acc. Chem. Res.55, 2581–2593 (2022). [DOI] [PubMed] [Google Scholar]
- 25.Glunz, P. W. Recent encounters with atropisomerism in drug discovery. Bioorg. Med. Chem. Lett.28, 53–60 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Kochanowska-Karamyan, A. J. & Hamann, M. T. Marine Indole alkaloids: potential new drug leads for the control of depression and anxiety. Chem. Rev.110, 4489–4497 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gribble G. W. Indole ring synthesis: From natural products to drug discovery (John Wiley & Sons, 2016).
- 28.Zhang, M.-Z., Chen, Q. & Yang, G.-F. A. review on recent developments of indole-containing antiviral agents. Eur. J. Med. Chem.89, 421–441 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sravanthi, T. V. & Manju, S. L. Indoles — A promising scaffold for drug development. Eur. J. Pharm. Sci.91, 1–10 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Patil, S. A., Patil, R. & Miller, D. D. Indole molecules as inhibitors of tubulin polymerization: potential new anticancer agents. Future Med. Chem.4, 2085–2115 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Kumari, A. & Singh, R. K. Medicinal chemistry of indole derivatives: current to future therapeutic prospectives. Bioorg. Chem.89, 103021 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Chadha, N. & Silakari, O. Indoles as therapeutics of interest in medicinal chemistry: Bird’s eye view. Eur. J. Med. Chem.134, 159–184 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Manickam, M., Iqbal, P., Belloni, M., Kumar, S. & Preece, J. A. A brief review of carbazole-based photorefractive liquid crystalline materials. Isr. J. Chem.52, 917–934 (2012). [Google Scholar]
- 34.Lozano, O. et al. Organocatalyzed enantioselective fluorocyclizations. Angew. Chem. Int. Ed.50, 8105–8109 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Xie, W. et al. Highly enantioselective bromocyclization of tryptamines and its application in the synthesis of (−)-Chimonanthine. Angew. Chem. Int. Ed.52, 12924–12927 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Liu, H. et al. Highly asymmetric bromocyclization of tryptophol: unexpected accelerating effect of DABCO-Derived Bromine Complex. Org. Lett.16, 1908–1911 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Cai, Q., Yin, Q. & You, S.-L. Chiral-amine-catalyzed asymmetric bromocyclization of tryptamine derivatives. Asian J. Org. Chem.3, 408–411 (2014). [Google Scholar]
- 38.Liang, X.-W., Liu, C., Zhang, W. & You, S.-L. Asymmetric fluorinative dearomatization of tryptamine derivatives. Chem. Commun.53, 5531–5534 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Budylin, V. A., Kost, A. N. & Matveeva, E. D. Indole chemistry. Chem. Heterocycl. Compds.8, 52–56 (1972). [Google Scholar]
- 40.Nakashima, K., Kudo, Y., Matsushima, Y., Hirashima, S.-i & Miura, T. Synthesis of 2-Substituted Indoles via Migration Reaction of 3-Substituted Indoles with Triflic Acid. Chem. Pharm. Bull.72, 336–339 (2024). [DOI] [PubMed] [Google Scholar]
- 41.Ototake, N. et al. Catalytic enantioselective synthesis of atropisomeric indoles with an N–C Chiral Axis. Chem. Eur. J.16, 6752–6755 (2010). [DOI] [PubMed] [Google Scholar]
- 42.Li, Y., Liou, Y.-C., Chen, X. & Ackermann, L. Thioether-enabled palladium-catalyzed atroposelective C–H olefination for N–C and C–C axial chirality. Chem. Sci.13, 4088–4094 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiang, A.-L. et al. Pd-Catalyzed Atroposelective C–H olefination: diverse synthesis of axially chiral Biaryl-2-carboxylic Acids. Org. Lett.26, 5670–5675 (2024). [DOI] [PubMed] [Google Scholar]
- 44.Luc, A. et al. Double cobalt-catalyzed atroposelective C–H activation: one-step synthesis of atropisomeric indoles bearing vicinal C–C and C–N diaxes. Chem Catal.3, 100765 (2023). [Google Scholar]
- 45.Zhang, Z.-J. et al. Iron-catalyzed stereoselective C–H alkylation for simultaneous construction of C–N axial and C-central chirality. Nat. Commun.15, 3503 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim, A., Lee, C., Song, J., Lee, S. K. & Kwon, Y. All-round catalytic and atroposelective strategy via dynamic kinetic resolution for N-/2-/3-arylindoles. Nat. Commun.14, 5502 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen, Z.-H. et al. Organocatalytic enantioselective synthesis of axially chiral N,N′-Bisindoles. Angew. Chem. Int. Ed.62, e202300419 (2023). [DOI] [PubMed] [Google Scholar]
- 48.Liu, Y.-W., Chen, Y.-H., Cheng, J. K., Xiang, S.-H. & Tan, B. Enantioselective synthesis of 3-arylindole atropisomers via organocatalytic indolization of iminoquinones. Chem. Synth.3, 11 (2023). [Google Scholar]
- 49.Wang, J.-Y. et al. Design and catalytic asymmetric synthesis of furan-indole compounds bearing both axial and central chirality. Angew. Chem. Int. Ed.63, e202316454 (2024). [DOI] [PubMed] [Google Scholar]
- 50.Yang, S. et al. Catalytic asymmetric diastereodivergent synthesis of 2-Alkenylindoles bearing both axial and central chirality. Precis. Chem.2, 208–220 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen, K.-W. et al. Organocatalytic atroposelective synthesis of N−N axially chiral indoles and pyrroles by De novo ring formation. Angew. Chem. Int. Ed.61, e202116829 (2022). [DOI] [PubMed] [Google Scholar]
- 52.Liu, S.-J. et al. Rational design of axially chiral styrene-based organocatalysts and their application in catalytic asymmetric (2+4) cyclizations. Angew. Chem. Int. Ed.61, e202112226 (2022). [DOI] [PubMed] [Google Scholar]
- 53.Cheng, J. K., Xiang, S.-H., Li, S., Ye, L. & Tan, B. Recent advances in catalytic asymmetric construction of atropisomers. Chem. Rev.121, 4805–4902 (2021). [DOI] [PubMed] [Google Scholar]
- 54.Feng, J., Lu, C.-J. & Liu, R.-R. Catalytic asymmetric synthesis of atropisomers featuring an aza axis. Acc. Chem. Res.56, 2537–2554 (2023). [DOI] [PubMed] [Google Scholar]
- 55.He, X.-L., Wang, C., Wen, Y.-W., Wang, Z. & Qian, S. Recent advances in catalytic atroposelective construction of pentatomic heterobiaryl scaffolds. ChemCatChem13, 3547–3564 (2021). [Google Scholar]
- 56.Sun, H.-R., Sharif, A., Chen, J. & Zhou, L. Atroposelective synthesis of heterobiaryls through ring formation. Chem. Eur. J.29, e202300183 (2023). [DOI] [PubMed] [Google Scholar]
- 57.Zhang, H.-H. & Shi, F. Organocatalytic atroposelective synthesis of indole derivatives bearing axial chirality: strategies and applications. Acc. Chem. Res.55, 2562–2580 (2022). [DOI] [PubMed] [Google Scholar]
- 58.Zhang, Y.-C., Jiang, F. & Shi, F. Organocatalytic asymmetric synthesis of indole-based chiral heterocycles: strategies, reactions, and outreach. Acc. Chem. Res.53, 425–446 (2020). [DOI] [PubMed] [Google Scholar]
- 59.Min, X.-L., Zhang, X.-L., Shen, R., Zhang, Q. & He, Y. Recent advances in the catalytic asymmetric construction of atropisomers by central-to-axial chirality transfer. Org. Chem. Front.9, 2280–2292 (2022). [Google Scholar]
- 60.Kim, A. & Kwon, Y. Catalytic atroposelective dynamic kinetic resolution of substituted indoles. Synlett33, 201–206 (2021). [Google Scholar]
- 61.Shi, Q., Fang, F. & Cheng, D.-J. Organocatalytic atroposelective dynamic kinetic resolution involving ring manipulations. Adv. Synth. Catal.366, 1269–1284 (2024). [Google Scholar]
- 62.Wu, Q.-H. et al. Organocatalytic olefin C–H functionalization for enantioselective synthesis of atropisomeric 1,3-dienes. Nat. Catal.7, 185–194 (2024). [Google Scholar]
- 63.Uraguchi, D. & Terada, M. Chiral Brønsted acid-catalyzed direct mannich reactions via electrophilic activation. J. Am. Chem. Soc.126, 5356–5357 (2004). [DOI] [PubMed] [Google Scholar]
- 64.Akiyama, T., Itoh, J., Yokota, K. & Fuchibe, K. Enantioselective mannich-type reaction catalyzed by a chiral Brønsted acid. Angew. Chem. Int. Ed.43, 1566–1568 (2004). [DOI] [PubMed] [Google Scholar]
- 65.Frisch, M. J. et al. Gaussian 16 Revision C.01. (Gaussian Inc. Wallingford CT, 2016).
- 66.Liu, Y. et al. Photoredox catalytic deracemization enabled enantioselective and modular access to axially chiral N-Arylquinazolinones. Angew. Chem. Int. Ed.63, e202411236 (2024). [DOI] [PubMed] [Google Scholar]
- 67.Dapprich, S., Komáromi, I., Byun, K. S., Morokuma, K. & Frisch, M. J. A new ONIOM implementation in Gaussian98. Part I. The calculation of energies, gradients, vibrational frequencies and electric field derivatives1Dedicated to Professor Keiji Morokuma in celebration of his 65th birthday.1. THEOCHEM461-462, 1–21 (1999). [Google Scholar]
- 68.Lu, T. A comprehensive electron wavefunction analysis toolbox for chemists. Multiwfn. J. Chem. Phys.161, 082503 (2024). [DOI] [PubMed] [Google Scholar]
- 69.Lu, T. & Chen, Q. Independent gradient model based on Hirshfeld partition: a new method for visual study of interactions in chemical systems. J. Comput. Chem.43, 539–555 (2022). [DOI] [PubMed] [Google Scholar]
- 70.Lefebvre, C. et al. Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density. Phys. Chem. Chem. Phys.19, 17928–17936 (2017). [DOI] [PubMed] [Google Scholar]
- 71.Johnson, E. R. et al. Revealing noncovalent interactions. J. Am. Chem. Soc.132, 6498–6506 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
Detailed experimental procedures and characterizations of new compounds are available in Supplementary Information. The X-ray crystallographic coordinates for structures reported in this Article have been provided as Supplementary Data 1 and 2 and have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers CCDC 2380810 (3s) and 2383264 (5c). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. 1H and 13C NMR spectra and HPLC chromatograms can be found in the Supplementary Data 3 and 4, respectively. Computational chemistry details are available in Supplementary Data 5. Reprints and permissions information is available online at www.nature.com/reprints. Correspondence and requests for materials should be addressed to Y.K.