

Abstract
To define the bioequivalence (BE) of two orally administered film coated tablets containing 40 mg lurasidone hydrochloride. A single-dose, open-label, randomized two-way crossover trial was conducted under fed conditions, with a washout period of at least one week. Blood samples were drawn over a period of 72 h and plasma concentrations were analysed using a Liquid Chromatography tandem Mass Spectrometry (LC–MS/MS) method. The Pharmacokinetic (PK) parameters were calculated using Phoenix WinNonlin software, based on non-compartmental analysis. The two one-sided hypothesis at the α = 0.05 level of significance was tested by constructing the 90% confidence interval of two one-sided t-tests for the geometric mean ratios test versus reference preparation with analysis of variance (ANOVA). After oral administration of 40 mg lurasidone hydrochloride under fed conditions, the mean area under the curve (AUCt, AUC∞) and maximum plasma concentrations (Cmax) were 175 ng/mL*h; 184 ng/mL*h and 60 ng/mL, respectively for the test product and 185 ng/mL*h; 198 ng/mL*h and 59 ng/mL respectively for the reference product. 90% Confidence Intervals (CI) for all PK parameters were within the acceptance range of 80.00–125.00%. BE was demonstrated between the generic product and the innovator product in healthy Caucasian male subjects. No clinically meaningful differences in safety or tolerability were observed.
Keywords: Bioequivalence, Pharmacokinetics, Lurasidone, Variation
Subject terms: Pharmaceutics, Pharmacology
Introduction
Schizophrenia is a severe psychiatric disorder with a diverse clinical presentation, including both positive symptoms, such as hallucinations and delusions, and negative symptoms, such as depression and emotional flatness1. These symptom domains often overlap and contribute significantly to the overall functional impairment and diminished quality of life experienced by patients2. Depressive symptoms in patients with schizophrenia have traditionally been overlooked or even neglected because they are not considered as fundamental as positive and negative symptoms in the choice of medication. While first-generation antipsychotics are generally more effective in controlling positive symptoms, second-generation and third-generation (atypical) antipsychotics, including lurasidone, have shown efficacy in addressing negative symptoms3. The atypical antipsychotics including lurasidone, have recently been introduced as monotherapy or adjuncts for the treatment of depressive and negative symptoms in patients with schizophrenia. Lurasidone also has a favorable relapse prevention profile, and its tolerability is enhanced by minimal impact on metabolic parameters, weight, and prolactin levels4. Although all antipsychotics should be used during pregnancy only after a benefit / risk evaluation performed by the physician, nearly all antipsychotics drugs belong to pregnancy category C. Lurasidone is one of the modest antipsychotic agents of which the pregnancy category is B5 Animal studies in rats and rabbits have not shown an increased risk of birth defects. Furthermore, a case report of a woman taking lurasidone throughout her pregnancy found that the baby was born healthy, without any birth defects. A study involving 134 pregnant women who used lurasidone during pregnancy found no significant patterns of birth defects6.
When administered with food, 120 mg of lurasidone resulted in a two- to three-fold increase in both AUC and Cmax compared to when the drug was taken under fasting conditions7. Lurasidone is classified as a BCS Class 2 compound, characterized by high permeability (log P = 5.6) and low aqueous solubility (0.165 mg/mL). Due to its limited aqueous solubility, lurasidone exhibits low and variable bioavailability (9–19%), primarily due to dissolution rate-limited absorption following oral administration8. The impact of food on lurasidone absorption is comparable to other lipophilic atypical antipsychotics such as ziprasidone, which also exhibits a two- to threefold increase in bioavailability when administered with food7. The presence of dietary fat can modulate the absorption of certain drugs by enhancing the secretion of bile acids, which facilitate the solubilization of lipophilic compounds. Furthermore, dietary fat may increase the secretion of pancreatic enzymes, digestive hormones, and luminal fluid volume as well as lead to improved micelle formation, and delayed gastric emptying, resulting in greater systemic exposure. These effects are thought to enhance the solubility and absorption of lipophilic drugs. The impact of food on the pharmacokinetics of lurasidone is likely attributable to a combination of these physiological mechanisms, thereby contributing to the observed increase in systemic exposure when the drug is administered with food7. Given this pharmacokinetic behavior, regulatory agencies recommend that bioequivalence studies for lurasidone be conducted under fed conditions.
Therefore the summary of product characteristics (SmPC) of lurasidone recommends the intake of the medication with a meal. According to the Guideline on the Investigation of Bioequivalence of European Medicines Agency (EMA) it is recommended to perform the bioequivalence study under fed conditions, for products where the SmPC recommends intake of the reference medicinal product only in fed state.
Brand-name medications are typically 30–60% more expensive than their generic counterparts9,10. The test product used in this study is the first and currently the only generic version of lurasidone available in the Turkish market, meeting the market demand following its marketing authorization, even before reimbursement approval. Annual worldwide sales of oral solid forms containing lurasidone were $3058 million in the first six months of 2023, while the total for the first six months of 2024 was $375 million11. An important reason for this dramatic decline may be the economic advantage provided by generic products that have entered the market and become widely used. Because already in 2022 lurasidone generic products were on the top of the list of the most saving ANDA approved products12. In addition to the economic advantages, generic products also provide a significant treatment advantage in markets where original products do not enter, allowing more patients to access medicines.
This study aimed to evaluate the pharmacokinetics of lurasidone in healthy male Caucasian subjects and assess whether the test formulation is bioequivalent to the reference formulation. To be considered bioequivalent, the 90% CI of the ratio of the test to reference products for both AUC and Cmax should fall within the acceptance range of 80–125%.
Material and methods
Study design
This was a randomized, open-label, two-period, two-sequence crossover study conducted under fed conditions.
Sample size was determined based on the published nomogram table for a multiplicative model13. At a power value of 80%, a test/reference parameter ratio in the range between 0.90 and 1.10 and a probability level of α = 5%, a necessary sample size of 58 subjects is obtained from an assumed intra-subject coefficient of variation (ISCV) of 27.5%. This assumed ISCV coincided with internal data from previous investigations. The mathematical basis of power calculation is reflected by Owen’s algorithm used for sample size determination, with the following formula13:
P (t1 ≥ t (1 – α, n – 2) and t2 ≤ – t (1 – α, n – 2) / bioequivalence holds), where t1 and t2 can be expressed by:
t1 = (ln (μT/μR) – ln θ1)/σ
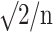
t2 = (ln (μT/μR) – ln θ2)/σ
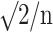 , where θ1, θ2; lower and upper acceptance limits; σ: square root of residual variance from ANOVA (indicative of intra-subject variability); n: number of subjects; μT, μR: means for test and reference target variables.
, where θ1, θ2; lower and upper acceptance limits; σ: square root of residual variance from ANOVA (indicative of intra-subject variability); n: number of subjects; μT, μR: means for test and reference target variables.
A total of 56 healthy male subjects completed the study, receiving both the test and reference formulations in two different sequences (TR or RT) with a washout period of at least one week between doses. The sequence of the treatment intake was determined randomly and was not stratified by BMI, age, or another factors as these factors were limited with the inclusion and exclusion criteria. Each subject stayed overnight at the clinical facility for each dosing period, from the evening before administration until approximately 12 h post-dose. Ambulatory blood samples were collected up to 72 h post-dose.
The study was un-blinded due to the low risk of bias in PK studies. Knowing the administered product was not expected to influence the PK assessments. However, the bioanalytical phase was blinded until completion of the analysis.
Screening procedure included an assessment of medical-surgical history as well as life style and habits, physical examination, registration of gender, birth date and ethnic group, measurement of body temperature, blood pressure and heart rate after 5 min supine rest, respiratory rate, registration of a 12-lead electrocardiogram (ECG) and determination of weight, height and body mass index (BMI). Additionally laboratory blood investigation tests (haemoglobin, haematocrit, leukocytes, erythrocytes, platelet count, sodium, potassium, calcium, chloride, aspartate transaminase, alanine transaminase, gamma glutamyl transferase, alkaline phosphatase, creatinine, total protein, total bilirubin, blood glucose, blood urea nitrogen, uric acid, serological analysis for HIV-Ab, HBsAg and HCV) and urine evaluation (pH, protein, glucose, ketones, blood (erythrocytes), leukocytes, bilirubin, urobilinogen, nitrites, specific gravity, sediment) inclusive of drug screen in urine (qualitative determination of amphetamines, cannabinoids, benzodiazepines, cocaine, opioids, barbiturates) were performed. Screening procedures were performed within 9 days prior to the subjects’ first dosing day. Administration of any prescribed systemic or topical medication within 2 weeks and OTC medication (including herbal remedies) within 1 week prior to the start of the study was determined as an exclusion criterion. Concomitant medication (including herbal remedies) was generally not allowed for the duration of the trial.
Male subjects aged between 18 and 55 years within a normal BMI range (18.5–30 kg/m2) and who had signed the informed consent, were enrolled. Subjects with known contraindications or hypersensitivities to lurasidone or similar products, with a history or diagnosis of any cardiovascular, respiratory, hepatic, renal, gastrointestinal, endocrine, neurological, immunologic, hematologic or psychiatric disorder and with positive test results for hepatitis B surface antigen, hepatitis C virus, and/or human immunodeficiency virus were excluded.
The study was conducted in accordance with the 1964 Declaration of Helsinki and its later amendments. The study protocol, the informed consent form (ICF), case report form (CRF) and related other documents was reviewed and approved by the Ethics Committee, Ethics Committee for Bioavailability-Bioequivalence Trials of Erciyes University and the Turkish Medicines and Medical Devices Agency of MoH. Written informed consent was obtained from every subject before any trial-related activities were started. The investigator retained one copy of the consent forms and subjects were given another copy of the forms.
Study products
Both investigational medicinal products used in this study contained 40 mg lurasidone hydrochloride equivalent to 37.2 mg lurasidone and were as following:
Test product: Lurasidone hydrochloride 40 mg Film Coated Tablets, Batch No: 160950803, exp. date: 03.2018 manufactured by Sanovel İlaç Sanayi ve Ticaret A.S., at their facility in Silivri – Istanbul, Türkiye under Good Manufacturing Practices.
18 months long-term (25 °C + % 60 RH) stability studies were completed with successfully results during December 2017. According to the Guideline on the requirements for the chemical and pharmaceutical quality documentation concerning investigational medicinal products in clinical trials—EMA/CHMP/QWP/545525/2017 “an extrapolation of + max 12 months to long-term stability data available (at least 6-months) may be acceptable for a shelf life of more than 12 months”. The re-test date of 03.2018 was thereby justified for the test product.
Reference product: LATUDA® 37 mg Film Coated Tablets, manufactured by Anderson Brecon (UK) Ltd.—UK and distributed by Sunovion Pharmaceuticals Europe Ltd.—UK. The reference product was purchased from the UK Market.
Treatment phase and blood sampling
Subjects were admitted to the clinical facility, Gaziantep University Farmagen IKU Merkezi, (Centre for GCP) (Gaziantep, Türkiye), the day before drug administration. After hospitalization, subjects received an evening snack (total caloric value of approx. 600 kcal) at about 7:30 pm and had to finish until 9:00 pm. After fasting for at least 10 h and consuming a high-fat breakfast 30 min before dosing, either test or reference formulation were administered orally together with 240 mL tap water.
For investigation of lurasidone pharmacokinetics, blood samples were collected prior to administration at 0 (pre-dose) and at 0.5, 1, 1.50, 2, 2.50, 3, 3.50, 4, 4.50, 5, 6, 8, 12, 24, 36, 48 and 72 hours post-dose. The date and time at which each sample was taken were recorded. Standard lunch and dinner were provided 4 h and 10 h after drug administration, respectively. Xanthine-containing food or beverages and fruit juices were not allowed during the stay in the clinical facility.
At pre-determined time points 7 mL of blood were taken from a cubital or forearm vein by either an indwelling catheter or individual venepuncture into tubes with K2EDTA as anticoagulating agent. After sampling, blood samples were immediately transferred to a water bath cooled with ice where they remained for not more than 20 min. The blood samples were centrifuged (3,000 rpm, 4–7 °C, 10 min) and the supernatant plasma from each sample was given within 20 min into 3.5 mL labelled polypropylene storage tubes (two tubes per sample, at least 1.5 mL per tube) and transferred to a deep freezer (< − 70 °C) where they were kept frozen until shipped to the analytical facility Analytisches Zentrum Biopharm GmbH Berlin, Germany.
Bioanalytical method
An HPLC–MS/MS method for quantifying lurasidone was validated in November 2016 and revalidated in April 2018. The validation demonstrated the method’s selectivity, intra-assay and inter-assay precision and accuracy, lower limit of quantification, dilution integrity, carry-over, recovery, matrix effect, linearity of the standard curve (ranging from 1.00 to 500.00 ng/mL in plasma), and stability under various conditions (bench-top, freeze–thaw, auto-sampler, extract stability, and long-term stability). The results confirmed that the method can reliably quantify lurasidone in plasma with sufficient accuracy, precision, specificity, and sensitivity. Additionally, 96.43% of ISR (incurred sample re-analyses) samples met the acceptance criteria during the measurement of study samples.
This validated HPLC–MS/MS method was then used to analyze lurasidone concentrations in plasma samples from the current study. Lurasidone and the internal standard (IS) (Lurasidone-d8 hydrochloride) were separated under isocratic conditions with a mobile phase composed of acetonitrile and 5 mM ammonium acetate in water (pH 5.0) using a C18 HPLC column. Ionization and detection were performed using a triple quadrupole mass spectrometer (ABSCIEX API 4000, Darmstadt, Germany) with electrospray ionization (ESI) in positive ion mode. Multiple reaction monitoring (MRM) mode was used for quantification, monitoring the parent-to-product ion transitions: 493.400 → 166.500 for lurasidone and 501.400 → 166.500 for lurasidone-d8 hydrochloride.
Chromatographic data were collected using Analyst software (version 1.5.2). The method and sequence file were prepared and downloaded before the injection of the first sample. A standard curve was generated for each assay day by plotting the peak area ratios of the analyte to the internal standard against their respective standard concentrations. Sample concentrations were determined by comparing the peak area ratios in the sample to the weighted (1/x) regression equation derived from the standard curve. The calibration function and sample concentration calculations were carried out using the validated software dbLabCal (version V3).
PK evaluation and statistics
Individual PK parameters including AUCt, AUC∞, Cmax, time to maximum plasma concentration (tmax) and half-life (t½) were calculated for each treatment. Target variables were defined as the intra-individual bioavailability ratios of the test to the reference preparation in the measures of AUCt and Cmax for lurasidone. The bioequivalence criteria were met if the 90% CI of the T/R ratio for AUCt and Cmax of lurasidone fell within 80.00%-125.00%. Further PK measures (AUC∞, tmax, t½) were also determined.
PK evaluation was performed with the program Phoenix WinNonlin, for non-compartmental assessment of data. For tmax, a non-parametric evaluation using two one-sided Wilcoxon tests comparing differences was applied.
Safety evaluation
All adverse events (AE) whether spontaneously reported by the participants or observed by the investigator, during regularly performed questionings prior to both study periods and at study time 1, 4, 6, 12, 24, 36, 48 and 72 h post-administration, were recorded. In addition, blood pressure (systolic, diastolic), pulse rate and body temperature (axillar) was measured at 0 h (pre-dose), 1, 3, 4, 6 and 12 h post-dose in each period and recorded into the CRF’s of each subject. One ECG was recorded for each subject during entry and final examinations.
Results
Ninety-nine Caucasian male subjects were screened and sixty-six (Intention-to-Treat population) of them were enrolled in the study. Fifty-six subjects (Per Protocol Population), between 18 and 53 years of age and with a BMI range of 18.7–29.8 kg/m2, completed the study regularly according to the Study Protocol so that plasma samples of fifty-six completed cases for each treatment were available for the analysis of lurasidone concentrations. For safety the intention-to-treat population was used whereas PK parameters were compared for the per protocol population of fifty-six subjects. Flowchart showing the number of participants and summary of the demographic data for the per protocol population can be seen in Fig. 1 and Table 1, respectively.
Fig. 1.

Flowchart showing the number of participants.
Table 1.
Summary of demographic data of the per protocol population.
| Study 1 | ||||
|---|---|---|---|---|
| Parameter | Age (y) | Weight (kg) | Height (cm) | BMI (kg/m2) |
| Mean value | 23.1 | 76.4 | 178.2 | 24.0 |
| Standard deviation | 5.8 | 10.4 | 5.9 | 2.8 |
| CV (%) | 25.3 | 13.7 | 3.3 | 11.6 |
| Minimum | 18 | 60 | 168 | 18.7 |
| Maximum | 53 | 102 | 192 | 29.8 |
| Number | 56 | 56 | 56 | 56 |
CV coefficient of variation, y year, kg kilogram, cm centimetre, BMI Body Mass Index, m2 square metre.
PK results
Lurasidone PK parameters AUCt, AUC∞ and Cmax, were similar between the test and reference products (Table 2). In order to achieve a better approximation to a normal distribution, Cmax and AUC are logarithmically transformed (base e) before analysis. Mean lurasidone plasma concentration—time profiles stratified by treatments are presented in Fig. 2 both as linear (± SEM) (a) and loglinear (b) plots.
Table 2.
PK parameters (arithmetic means ± s.d.) of 37 mg lurasidone under fed conditions; n = 56 subjects.
| Treatment | AUCt (ng/mL*h) | AUC∞ (ng/mL*h) | Cmax (ng/mL) | tmax (h) | t½ (h) |
|---|---|---|---|---|---|
| Test product | 174.50 ± 92.50 | 184.40 ± 97.92 | 59.85 ± 27.42 | 1.99 ± 1.13 | 4.77 ± 3.76 |
| Reference product | 184.94 ± 98.08 | 197.56 ± 102.94 | 59.48 ± 26.06 | 2.29 ± 1.19 | 4.53 ± 3.58 |
s.d. standard deviation, AUCt area under the curve until time t, AUC∞ area under the curve until infinity, Cmax maximum plasma concentration, tmax time to maximum plasma concentration, t½ half-life, ng nanogram, ml millilitre, h hour.
Fig. 2.

Mean concentrations of lurasidone in plasma after administration of different (T and R) 40 mg lurasidone hydrochloride containing preparations under fed conditions; linear (± SEM) (a) and loglinear (b) plot; n = 56 (prepared by Phoenix WinNonlin software).
For evaluation of BE between test preparation and reference preparation, the 90% CI (ANOVA; two one-sided t-tests) for the T/R ratios of geometric means of the PK target variables AUCt and Cmax were calculated based on the assumption of a lognormal distribution of individual data. Other PK parameters AUC∞ and tmax served as supportive data. AUC∞ was evaluated in the same way as AUCt, applying a multiplicative model; tmax was subjected to non-parametric analysis (90% CI of two one-sided Wilcoxon tests). The observed 90% CI including pertaining point estimators (ratios T/R of geometric means) are shown in Table 3. NOVA reveals no significant period effect (p = 0.4057) and no significant sequence effect (p = 0.2514). A slight formulation effect (p = 0.038) was found, because the test/reference ratio of 92.95% for geometric means of AUCt was below 100%.
Table 3.
Statistical assessment of bioequivalence comparing T and R containing 40 mg lurasidone hydrochloride, n = 56.
| Pharmacokinetic parameter: (ANOVA; two one-sided t-tests) | 90% CI | Point estimators (ratios T/R of geometric means) (%) | Intra-subject CV (%) |
|---|---|---|---|
| AUCt | 87.81–98.38% | 92.95 | 18.09 |
| Cmax | 93.46–108.11% | 100.52 | 23.30 |
| AUC∞ | 87.95–98.47% | 93.06 | 17.99 |
ANOVA analysis of variance, CI confidence interval, T test product, R reference product, CV coefficient of variation, AUCt area under the curve until time t, Cmax maximum plasma concentration, AUC∞ area under the curve until infinity.
According to the intra-subject CV, the necessary sample size of 58 subjects (58 subjects represented the a priori fixed per protocol population for pharmacokinetic evaluation; 56 subjects completed the study regularly), estimated prior to the study based on an intra-individual AUC and Cmax variability of 27.5%, a test/reference parameter ratio between 0.90 and 1.10 and a statistical power of 80%, was confirmed for parameter AUCt and Cmax by the ANOVA results.
Safety results
In the course of the study, 41 treatment emergent adverse events occurred in 29 out of 66 enrolled subjects. 30 treatment emergent adverse events (16 × sleepiness, 4 × nausea, 3 × dizziness, 3 × hot flush, 2 × headache, 1 × feeling of restlessness, 1 × vomiting, 1 × epistaxis) were of mild intensity, two treatment emergent adverse events (1 × nausea, 1 × feeling of restlessness) were of moderate intensity and eight treatment emergent adverse events (6 × feeling of restlessness, 1 × vomiting, 1 × pruritus) were of severe intensity.
Five further adverse events (1 × ALT high, 2 × leukocytes high, 1 × leukocytes low, 1 × glucose high) were attributable to clinical laboratory findings, when data of final examination were found to be significantly out of the normal range. ECG’s recorded at final examination showed sinus tachycardia for two subjects but these were judged to be not clinically relevant by the Principal Investigator. 20 treatment emergent adverse events were recorded after the test preparation, 21 treatment emergent adverse events were recorded after the reference preparation. Four subjects left the study prematurely due to adverse events (2 due to vomiting within the first 4 h post dose, 1 due to pruritus and the last 1 subject as he felt nausea).
All adverse events were already reported in the SmPC of the reference product, except epistaxis and high leukocytes in the blood. All treatment emergent adverse events recovered without sequelae. No serious adverse events were experienced during the study. Test and reference preparation were acceptably well tolerated. No difference between the preparations was observed. All adverse events classified according to their severity and grouped according to treatments can be seen in Table 4.
Table 4.
Classification of all adverse events seen with both study products.
| Drug relationship | Adverse event (number) | Adverse events by treatment at onset of AE | |
|---|---|---|---|
| Test | Reference | ||
| Possible | Sleepiness (15) | 9 | 6 |
| Possible | Feeling of restlessness (8) | 3 | 5 |
| Possible | Nausea (5) | – | 5 |
| Possible | Vomiting (2) | – | 2 |
| Possible | Dizziness (3) | 2 | 1 |
| Possible | Hot flush (3) | 2 | 2 |
| Possible | Headache (2) | 1 | 1 |
| Possible | Epistaxis (1) | 1 | – |
| Possible | Pruritus (1) | 1 | – |
| Possible | Clin. Lab.: ALT high last treatment: R | – | 1 (last treatment) |
| Possible | Clin. Lab.: leukocytes high last treatment: T | 1 (last treatment) | – |
| Possible | Clin. Lab.: leukocytes low last treatment: R | – | 1 (last treatment) |
| Unlikely | Clin.Lab.: glucose high last treatment: T | 1 (last treatment) | – |
| Unlikely | Clin. Lab.: leukocytes high last treatment: R | – | 1 (last treatment) |
Discussion
As food increases the bioavailability of the active substance it is recommended to take the lurasidone products after a meal. This study was therefore performed under fed conditions in order to reflect the real life pharmacokinetics. The aim of the present trial was to investigate the BE between the test and reference preparations containing lurasidone hydrochloride. This clinical trial was conducted with the aim to investigate whether any differences concerning the rate and extent of absorption exist between both preparations, which were targeted to be used interchangeable. The reference drug in the present study is already registered and commercially available in the UK. For the purpose of registration, the efficacy and safety of this drug has been proved in clinical trials. This drug therefore served as reference and a basis for comparison with the test product manufactured by Sanovel Ilaç Sanayi ve Ticaret A.S., Türkiye. Study results were well comparable with literature data. After administration of a single oral dose of 40 mg lurasidone hydrochloride under fed conditions AUCt, Cmax, and tmax values were seen to be about 163 ng/mL*h, 51 ng/mL and 2 h, respectively14. The same published PK parameters after administration of a single oral dose of 80 mg lurasidone hydrochloride under fed conditions were reported to be about 353 ng/mL*h, 113 ng/mL and 1.5 h respectively14. The results of our study are confirmed by the literature data and the comparison of the pharmacokinetic parameters in between doses indicate dose proportional pharmacokinetic properties of lurasidone.
The food effect reported for lurasidone7 was seen when comparing the Cmax data in the literature which was reported to be 113 ng/mL14 under fed conditions but only 71 ng/mL15 under fasting conditions. In the contrary a food effect could not be followed for AUC data from the literature (322 ng/mL*h vs 310 ng/mL*h) published by Hu et al.14 and Vargas et al.15 under fed and fasting conditions, respectively. It is known that pharmacokinetic parameters are variable between subjects and phase 1 studies of the originator products are performed on a very limited number of subjects. Therefore publishing new pharmacokinetic data after the originator studies is very valuable in order to understand the pharmacokinetics of active substances better.
Sleepiness was the most frequently observed adverse event with a number of 15 cases seen in 13 subjects of 66 subjects (appr. 20%), followed by feeling restlessness seen in 8 subjects (appr. 12%) in the study. These values are less than reported in the literature data which change between 34 and 100% for somnolence7,15 which defines similar adverse events. On the other hand also in similar or less percentages of patients, somnolence or sedation cases were observed in short-term registration studies performed in Schizophrenia patients. The commonly observed AEs (that occurred mostly and at greater incidence than in the placebo-treated patients) included somnolence (15% for lurasidone vs. 7% for placebo), nausea (14% vs. 3%), akathisia (9% vs. 2%), viral infections (10% vs. 6%), and in smaller percentages vomiting, diarrhea, dry mouth, rhinitis, oropharyngeal pain, tachycardia as well as dizziness16. In a 12-month, double blind safety and tolerability study, clinically stable adult outpatients with schizophrenia (n = 629) the three most frequent AEs in the lurasidone group were nausea (16.7%), insomnia (15.8%), and sedation (14.6%)17. Nausea frequency observed in our study results (7.5%) were seen to be under the literature and label data. Dizziness was observed in our study to a limited degree which is also similarly reported in the label information. Although one subject exhibited transient ALT elevations with values twice (106 U/L) the upper limit of the normal range (reference range 1.0–50.0 U/L), only 5 days later the values turned back to a slightly high level (80 U/L) and was graded by the principle investigator to be of no clinically significance. The frequency of elevated ALT values are given in the SmPC of the reference product as uncommon and compatibly it was seen in our study also only in 1 case.
In summary, 56 subjects completed the study under fed conditions. No significant difference was seen in the tmax value for the test product or reference product and the median difference for tmax between reference and test formulation was found to be 0.25 h according to Wlicoxon analysis carried out on the 90% probability level. After the 7-day washout period no lurasidone was detected in the pre-dose samples, which showed the absence of residual effect of the previous period.
A reference product is a medicinal product authorised and marketed on the basis of a full dossier, i.e. including chemical, biological, pharmaceutical, pharmacological-toxicological and clinical data. This information is not fully available in the public domain. Authorizations for generic products are therefore linked to the ‘original’ authorized medicinal product, which is legally allowed once the data protection time of the dossier of the reference product has expired. For this kind of marketing authorization application, it has to be demonstrated that the pharmacokinetic (PK) profile of the product is similar to the PK profile of the reference product. To this end the Marketing Authorization Holder performs a bioequivalence study in which the PK profile of the test product is compared with the PK profile of the reference product. A bioequivalence study is the widely accepted means of demonstrating that difference of use of different excipients and different methods of manufacture have no influence on efficacy and safety. Showing the bioequivalence is a globally accepted way to prove the interchangeability of the generic product with the reference product. Therefore the test product can be used interchangeably with the reference product.
Conclusion
Bioequivalence between test and reference products has been demonstrated in male Caucasian subjects after administration of single oral doses under fed conditions.
Acknowledgements
We would like to acknowledge the effort of Ali Türkyılmaz for his contribution to this work.
Author contributions
All authors contributed to the study conception and design. Gökçe Sözer was the coordinator of the sponsor. All study documentation was prepared, Ethical and Health Authority submissions was performed by Selma Alime Koru. The clinical part of the study was performed by Aydın Erenmemişoğlu, Muradiye Nacak and Erol Durucu. Analysis of the plasma samples was performed by Martin Reinsch. Statistical and Pharmacokinetic analysis as well as reporting was performed by Wolfgang Martin. The first draft of the manuscript was written by Selma Alime Koru and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Funding
This study and manuscript was funded by Sanovel İlac Sanayi ve Ticaret A.S. – Türkiye.
Data availability
The data that support the findings of this study are not openly available due to reasons of confidentiality but will be available from the corresponding author upon request.
Declarations
Competing interests
All study partners were paid by Sanovel İlac Sanayi ve Ticaret A.S. – Türkiye for their efforts on realising the study and the preparation of the manuscript, on a company basis. No author was personally paid. Gökçe Sözer, Muradiye Nacak, Erol Durucu, Aydın Erenmemişoğlu, Wolfgang Martin, Ewald Ottmann, Martin Reinsch, Selma Alime Koru declare that they have no more conflicts of interest.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Rognoni, C., Bertolani, A. & Jommi, C. Second-generation antipsychotic drugs for patients with schizophrenia: Systematic literature review and meta-analysis of metabolic and cardiovascular side effects. Clin. Drug Investig.41(4), 303–319 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kanchanatawan, B., Thika, S., Anderson, G., Galecki, P. & Maes, M. Affective symptoms in schizophrenia are strongly associated with neurocognitive deficits indicating disorders in executive functions, visual memory, attention and social cognition. Prog. Neuropsychopharmacol. Biol. Psychiatry80(Pt C), 168–176 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Fiorillo, A. et al. The role of lurasidone in managing depressive symptoms in people with schizophrenia: A review. Brain Sci.14(3), 225 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fiorillo, A. et al. Lurasidone in adolescents and adults with schizophrenia: From clinical trials to real-world clinical practice. Expert Opin. Pharmacother.23, 1801–1818 (2022). [DOI] [PubMed] [Google Scholar]
- 5.Cruz, M. P. Lurasidone HCl (latuda), an oral, once-daily atypical antipsychotic agent for the treatment of patients with schizophrenia. Pharm. Ther.36(8), 489–492 (2011). [PMC free article] [PubMed] [Google Scholar]
- 6.Organization of Teratology Information Specialists (OTIS). Mother to baby | fact sheets [internet]. brentwood (TN). Lurasidone (Latuda®). https://www.ncbi.nlm.nih.gov/books/NBK583258/ (2024).
- 7.Preskorn, S., Ereshefsky, L., Chiu, Y. Y., Poola, N. & Loebel, A. Effect of food on the pharmacokinetics of lurasidone: Results of two randomized, open-label, crossover studies. Hum. Psychopharmacol.28(5), 495–505 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Madan, J. R., Pawar, K. T. & Dua, K. Solubility enhancement studies on lurasidone hydrochloride using mixed hydrotropy. Int. J. Pharm. Investig.5(2), 114–120. 10.4103/2230-973X.153390 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zarowitz, B. J. The generic imperative. Geriatr. Nurs.29(4), 223–226 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Lewek, P. & Kardas, P. Generic drugs: The benefits and risks of making the switch. J. Fam. Pract.59(11), 634–640 (2010). [PubMed] [Google Scholar]
- 11.IMS data, November 2024.
- 12.Conrad, R., Nance, S., Tillman, Z. & Davis, K. Estimating cost savings from new generic drug approvals in. FDA U.S. FOOD & DRUG Report.https://www.fda.gov/media/182435/download (2022).
- 13.Diletti, E., Hauschke, D. & Steinijans, V. W. Sample size determination for bioequivalence assessment by means of confidence intervals. Int. J. Clin. Pharmacol. Ther. Toxicol.30(Suppl. 1), 51–58 (1992). [PubMed] [Google Scholar]
- 14.Hu, C. et al. Single- and multiple-dose pharmacokinetics, safety and tolerability of lurasidone in healthy Chinese subjects. Clin. Drug Investig.37(9), 861–871 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Vargas, M. & Villarraga, E. A. Bioequivalence study of two formulations containing lurasidone 80 mg tablets in healthy Colombian volunteers. J. Bioequiv. Availab.8(5), 220–223 (2016). [Google Scholar]
- 16.Sumitomo Pharma Co. Ltd Latuda: US Package Insert for Latuda (lurasidone HCl) tablets for oral use (2025). https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/200603s041lbl.pdf. Accessed 28 Apr 2025.
- 17.Citrome, L. et al. Long-term safety and tolerability of lurasidone in schizophrenia: A 12-month, double-blind, active-controlled study. Int. Clin. Psychopharmacol.27(3), 165–176 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are not openly available due to reasons of confidentiality but will be available from the corresponding author upon request.