

Abstract
TP53-Y220C is a recurrent hotspot mutation in cancers and leukemias. It is observed predominantly in acute myeloid leukemia (AML)/myelodysplastic syndromes among hematological malignancies and is associated with poor outcome. The mutation creates a structural pocket in the p53 protein. PC14586 (rezatapopt) is a small molecule designed to bind to this pocket and thus restore a p53-wild type (p53-WT) conformation. We demonstrate that PC14586 converts p53-Y220C into a p53-WT conformation and activates p53 transcriptional targets, but surprisingly induces limited/no apoptosis in TP53-Y220C AML. Mechanistically, MDM2 induced by PC14586-activated conformational p53-WT and the nuclear exporter XPO1 reduce the transcriptional activities of p53, which are fully restored by inhibition of MDM2 and/or XPO1. Importantly, p53-WT protein can bind to BCL-2, competing with BAX in the BH3 binding pocket of BCL-2 and also binds to BCL-xL and MCL-1. However, such binding by PC14586-activated conformational p53-WT is not detected. Pharmacological inhibition of the BCL-2/BAX interaction with venetoclax fully compensates for this deficiency, induces massive cell death in AML cells and stem/progenitor cells in vitro and prolongs survival of TP53-Y220C AML xenografts in vivo. Collectively, we identified transcription-dependent and -independent mechanisms that limit the apoptogenic activities of reactivated conformational p53-WT and suggest approaches to optimize apoptosis induction in TP53-mutant leukemia. A clinical trial of PC14586 in TP53-Y220C AML/myelodysplastic syndromes has recently been initiated (NCT06616636).
Keywords: TP53, TP53-Y220C, MDM2, XPO1, BCL-2, AML stem/progenitor cells
Graphical Abstract

Introduction
Patients with TP53-mutant acute myeloid leukemia (AML) survive only several months1–4. They lack durable responses to chemotherapies3, stem cell transplantation, and targeted therapies such as combinations of the BCL-2 inhibitor venetoclax (VEN) with hypomethylating agents5,6. TP53 mutations emerge rapidly in 1/3 AML patients following exposure to chemotherapy and BCL-2 or MDM2 inhibition7,8 in initially TP53-wild type (WT) patients.
Mutations in the tumor suppressor TP53 are cancer-specific, thus constituting ideal therapeutic targets. However, attempts to targeting mutant p53 have not been successful. Current approaches focus on alternative mechanisms9, including targeting vulnerabilities exposed by p53 deficiencies10–13, destabilizing/depleting mutant p5314,15, bypassing p53, and functionally restoring p53 signaling pathways16–20. Agents reported to restore/stabilize the p53-WT conformation from mutant p53, such as APR-246, which failed in recent clinical trials21,22, or arsenic trioxide23, act through multiple p53-independent mechanisms. Until recently, no agents had been identified that selectively target p53-mutants to restore their functions.
p53-Y220C protein contains a well-defined pocket that impairs DNA binding and reduces p53-Y220C stability and melting temperature, leading to its rapid denaturation and aggregation at physiological temperatures24. This structural property makes p53-Y220C a druggable target suitable for structure-based drug design to restore functional p53. Several such drugs were recently identified25–29.
TP53-Y220C is a recurrent hotspot mutation in solid tumors30. In leukemias, the mutation is predominantly present in AML and myelodysplastic syndromes (MDS) and associated with poor outcomes31. It is the most frequent mutation in MDS patients, and the third frequent in AML patients among TP53 hotspot mutations at MD Anderson Cancer Center (MDACC)31. Novel therapies are urgently needed for these patients. PC14586 (rezatapopt, PMV Pharmaceuticals) was designed to fit into the structural pocket of p53-Y220C and restore p53-WT protein conformation and function29,32. Although PC14586 has documented activity against solid malignancies33,34, it has not been investigated in TP53-Y220C mutant AML/MDS.
p53 induces apoptosis by transcriptionally upregulating pro-apoptotic BCL-2 proteins35. Less investigated is its transcription-independent function of directly antagonizing anti-apoptotic BCL-2 proteins. We and others have shown that the transcription-independent function of p53 is sufficient to induce apoptosis36,37. We reported that p53 activation by blocking p53 interactions with its negative regulator MDM2 has activity in AML preclinically and in a clinical trial38,39. We demonstrated high synergy40 and synthetical lethality of combined BCL-2 inhibition and p53 activation by MDM2 inhibition in TP53-WT AML41, which achieved encouraging response rates in patients with relapsed/refractory AML7. Furthermore, we reported that in AML, apoptosis induced by inhibition of XPO1 (exportin 1), which mediates nuclear export of tumor suppressors, including p53, depends critically on p53 and that combined inhibition of XPO1 and MDM2 drastically increases nuclear p53 levels and apoptosis induction42 and strikingly enhances the transcriptional activity of activated p53. Co-inhibition of MDM2, XPO1, and BCL-2 greatly prolonged the survival of AML xenograft and patient-derived xenograft (PDX) models43.
In this study, we investigated mechanisms of action and efficacy of PC14586, identified transcription-dependent and -independent mechanisms that restrain the apoptogenic activities of reactivated conformational p53-WT, developed mechanism-based drug combinations to optimize apoptosis induction, and provide rationale for the clinical development of therapeutic strategies targeting TP53-Y220C in AML.
Methods
Cells, cell cultures, and treatments:
Isogeneic TP53-WT, TP53-knockout (KO), and TP53-mutant Molm13 and K562 cells were generated as described previously44. TP53-Y220C OCI-AML2 cells were generated by exposing TP53-WT OCI-AML2 cells to increasing concentrations (up to 5 μM) of RG7388 (idasanutlin) and were established by single-cell subcloning. The presence of TP53-Y220C and absence of other TP53 mutations were confirmed by Sanger-sequencing. AML patient samples with TP53-Y220C mutation (Table S1) and bone marrows from healthy donors (NBM) were obtained after written informed consent following the Institutional Review Board (IRB) approved protocols, in accordance with the Declaration of Helsinki. PDX#1 and PDX#5 (Table S1) was generated from AML patients with TP53-Y220C. Mutations in the PDX cells were confirmed by NGS-sequencing (MDACC molecular diagnostic laboratory) or droplet digital PCR (ddPCR). NBM-derived mesenchymal stromal cells (MSCs) were obtained as described previously45 under an IRB-approved protocol with informed consent. Cell lines, PDX cells, or mononuclear cells from primary samples were cultured as described previously46 and treated with PC14586 (PMV Pharmaceuticals), nutlin-3a, nutlin-3b, KPT-8602, VEN, or combinations.
Cell viability:
Cell viability was determined as described previously46,47. In cells from primary samples or PDX co-cultured with MSCs, annexin V (AnnV)/7-aminoactinomycin D (7AAD) positivity was determined in leukemia blasts (CD45+) and stem/progenitor cells (CD34+/CD34+CD38−). BAX activation and Cytochrome C levels were determined concomitantly using antibodies against activated BAX (clone 6A7, #556467, BD Biosciences, San Diego, CA) or Cytochrome C (#612310, BioLegend, San Diego, CA) by flow cytometry as described previously48. Clonogenic assay was conducted as described (www.stemcell.com/methocult-h4434-classic.html).
Immunoprecipitation, co-immunoprecipitation, and Western blotting:
For p53 immunoprecipitation (IP) and co-IP experiments, p53 protein was pulled down with antibodies against p53-WT (PAb1620, #102201, Caprico Bioscience) or p53-mutant (PAb240, NB200–103, Novus Biologicals, Centennial, CO) conformation. For BCL-2 co-IP experiments, BCL-2 was pulled down with a murine monoclonal antibody to human BCL-2 (Cell Signaling Technology, Danvers, MA) using the Dynabeads™ Protein G Immunoprecipitation Kit (ThermoFisher Scientific) following manufacturer’s instructions. IgG was used as control. Cytosolic and nuclear proteins were fractionated as described previously49. Protein levels were determined by Western blotting as described previously46,50 and briefly in Supplemental information.
p53 protein quantitation by flow cytometry:
Cells were stained with Ghost Dye™ Violet 510 (Tonbo Biosciences; San Diego, CA), washed and fixed with 4% paraformaldehyde in PBS, permeabilized with 100% methanol, and stained with the aforementioned antibodies selective for p53-WT or p53-mutant conformation and then with GAM-Alx647. The stained cells were evaluated (Cytoflex flow cytometer, Beckman Coulter Life Sciences, Indianapolis, IN) and analyzed (FlowJo analytic platform, BD Biosciences). p53 levels were expressed as the difference in the geometric mean fluorescence intensity (MFI) between cells stained with anti-p53 antibody and with IgG.
RNA-seq:
RNA was isolated from triplicate experiments and subjected to RNA-seq in the MDACC Advanced Technology Genomics Core (ATGC). Data analysis is shown in Supplemental Methods.
In vivo:
Mouse experiments were conducted following MDACC-IACUC approved protocols. Mice (male, NSG) were obtained from the Jackson Laboratory (Bar Harbor, ME). Cells were injected into mouse tail vein. All agents were given orally. Molm13 TP53-Y220C cells (0.5×106/mouse) were injected (6 to 10-week-old mice). Mice (5/group) were treated daily with vehicle, PC14586 (100 mg/kg), VEN (50 mg/kg), or both. PDX#1 cells derived from an AML patient sample with TP53-Y220C/TP53-P151A mutations (1.6×106/mouse) (Table S1) were injected (8-week-old mice). Once circulating human CD45+ (huCD45+) cells reached ≥1% as determined by flow cytometry, mice (10/group) were treated with vehicle, PC14586 (100 mg/kg daily), the MDM2 inhibitor RG7388 (50% active) (40 mg/kg, initially 8-day on, then 2-day off/5-day on), or both for three months. Disease progression and treatment responses were monitored by flow cytometry of huCD45+ cells. Spleens and BM cells (n=3 mice/group) were collected after 4 weeks of treatments. BM cells were subjected to CyTOF analysis (antibodies: Table S2) as described previously46,50. Survival was followed.
DNA-sequencing:
Single-cell DNA-sequencing (scDNA-seq) was conducted using the MissionBio Tapestri sc-sequencing platform. 7812 PDX cells from the BM of moribund mice treated with vehicle (59%; 99.9% huCD45+) or PC14586 (41%; 99.5% huCD45+) was sequenced. The HDF5 files were analyzed by the MissionBio python-based analysis package Mosaic. DdPCR using primers selective for the TP53-Y220C, -R175H, -R273H, -R248Q, and -R248W mutants (Bio-Rad; Hercules, CA) was conducted by ATGC, MDACC.
Statistical Analyses:
In vitro cell line experiments were performed in triplicate; results were expressed as means±standard errors of the means. CalcuSyn software was used to determine the mean combination index (CI) values51 for the ED50, ED75 and ED90. A CI<1 indicated a synergistic, CI=1 an additive, and CI>1 an antagonistic effect. The Student t-test was used to assess differences between groups; P values ≤0.05 were considered statistically significant. Mouse survival was estimated using the Kaplan–Meier method and data were analyzed using the log-rank test.
Results
PC14586 stabilizes p53-WT conformation, induces p53 signaling proteins, and suppresses growth in TP53-Y220C AML cells
PC14586 (Figure 1A) is a compound designed to specifically bind to the pocket in the p53 protein created by the TP53-Y220C mutation29. To determine its mechanism of action, we treated TP53-Y220C (Y220C/−)44 Molm13 and the isogenic TP53-WT control cells with PC14586 and the MDM2 inhibitor nutlin-3a, pulled down p53 using antibodies selective for p53-WT- or p53-mutant conformation, and determined p53 levels. PC14586 increased p53 protein with the WT and correspondingly decreased p53 with mutant conformation in TP53-Y220C cells, but not in TP53-WT cells. Conversely, nutlin-3a increased p53-WT in TP53-WT, but not in TP53-Y220C cells (Figure 1B). The same results were obtained in TP53-Y220C K562 and isogenic TP53-WT control cells44 treated with PC14586 and nutlin-3a (Figure S1A). Flow cytometry with the same antibodies selective for p53-WT and p53-mutant conformations confirmed the results in TP53-Y220C Molm13 cells (Figure 1C), indicating that PC14586 stabilizes p53-WT conformation in TP53-Y220C cells. PC14586 markedly induced the p53-WT targets MDM2 and p21 in TP53-Y220C Molm13 cells (Figure 1D), implying activation of p53 signaling.
Figure 1. PC14586 stabilizes the p53-WT conformation, induces the activation of p53 signaling, and has antileukemia activity in TP53-Y220C AML cells.
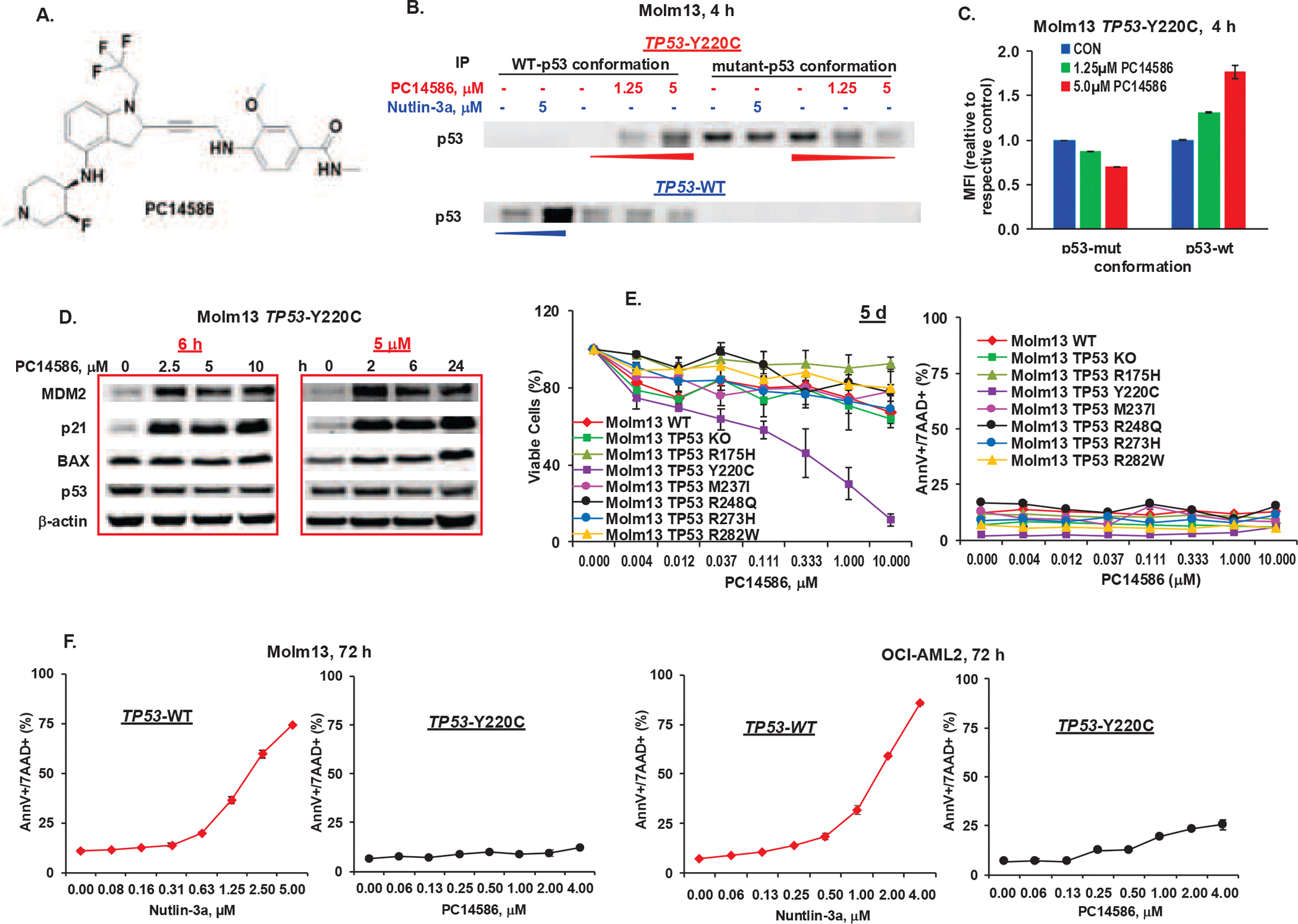
A. Structure of PC14586. B. TP53-Y220 and TP53-WT Molm13 cells were treated with the indicated concentrations of PC14586 or nutlin-3a for 4 h. The p53-WT and p53-mutant conformations were determined by IP with conformation-selective antibodies followed by Western blotting. C. TP53-Y220 Molm13 cells were treated with indicated concentrations of PC14586 for 4 h. The p53-WT and p53-mutant conformations were determined by flow cytometry with conformation-selective antibodies. D. TP53-Y220 Molm13 cells were treated with the indicated concentrations of PC14586 for 6 h or with 5 μM PC14586 for the indicated times. Protein levels were determined by Western blotting. E. Isogenic Molm13 cells with TP53-WT, KO, various mutations were treated with the indicated concentrations of PC14586 for 5 d. Viable cells and cell death were determined by flow cytometry. F. TP53-WT and TP53-Y220C Molm13 or OCI-AML2 cells were treated with the indicated concentrations of nutlin-3a or PC14586, respectively, for 72 h. Cell death was determined by flow cytometry. Mut, mutant; d, day; h, hour; MFI, mean fluorescence intensity; AnnV, annexin V; 7AAD, 7-aminoactinomycin D. Cell death and viability experiments were performed in triplicate; results were expressed as means±standard errors of the means.
To examine the biological consequences, we treated TP53-WT, TP53-KO, TP53-Y220C, and other TP53-mutant Molm13 and K562 cells44 with PC14586. PC14586 selectively suppressed the growth of TP53-Y220C cells (Figure 1E, Figure S1B), supporting the specificity of PC14586 for p53-Y220C. Surprisingly, we observed lack of apoptogenic activities in TP53-Y220C Molm13 and K562 cells (Figure 1E, Figure S1C). We next treated TP53-WT Molm13 or OCI-AML2 cells with nutlin-3a and isogenic TP53-Y220C cells with PC14586. Nutlin-3a induced pronounced apoptosis in TP53-WT cells, but PC14586 did not in TP53-Y220C Molm13 cells and induced only low levels of apoptosis in TP53-Y220C OCI-AML2 cells (~25% vs 85% maximal apoptosis induction in WT) (Figure 1F). Higher concentrations of PC14586 did not induce higher levels of p53, presumably owing to highly induced MDM2 levels (Figure 1D). We hypothesized that newly generated MDM2 could degrade the PC14586-reactivated p53.
MDM2 inhibition enhances p53 transcriptional activity and synergizes with PC14586 in apoptosis induction
To test the hypothesis that MDM2 inhibition antagonizes PC14586-induced MDM2 and enhances p53 transcriptional activity and function, we treated TP53-Y220C and TP53-WT Molm13 cells with PC14586, nutlin-3a, or both and performed RNA-seq. Principal component (Figure 2A), differential gene expression (DGE) (Figure 2B), and gene set enrichment analyses (GSEA) (Figure 2C) revealed that PC14586 had minimal effects on TP53-WT cells and nutlin-3a on TP53-Y220C cells. In TP53-WT cells, nutlin-3a greatly activated p53 signaling and adding PC14586 had minimal additional activity. However, in TP53-Y220C cells, PC14586 greatly changed the transcriptome and activated p53 signaling, which were markedly enhanced by nutlin-3a (Figure 2A–2C). Interestingly, like nutlin-3a and nutlin-3a/PC14586 in TP53-WT cells, PC14586 and PC14586/nutlin-3a significantly decreased Myc targets in TP53-Y220C cells (Figure 2C, Figure S2A) as previously reported43. Many PC14586-induced p53 targets35 were further upregulated in TP53-Y220C Molm13 cells treated with PC14586 plus nutlin-3a (Figure S2B). Western blotting validated the upregulation of several targets and the reduction of c-Myc (Figure 2D), confirming that MDM2 inhibition enhances p53 transcriptional activity by antagonizing the interaction of MDM2 with reactivated-p53. Although PC14586 or nutlin-3a alone lacked apoptosis induction in TP53-Y220C Molm13 cells, their combination synergistically induced cell death (CI<0.0001) and resulted in an even greater corresponding decrease in viable cells (Figure 2E). This synergism was also observed in TP53-Y220C OCI-AML2 cells (Figure S2C). Importantly, nutlin-3b, an inactive isomer of nutlin-3a had no effect on apoptosis induction in parental or TP53-Y220C AML cells and did not enhance apoptosis when combined with PC14586 (Figure S2D), supporting that PC14586-induced MDM2 hampers p53 transcriptional activity and apoptosis induction, which can be overcome by MDM2 inhibition.
Figure 2. PC14586 combined with MDM2 or XPO1 inhibition further upregulates p53 transcriptional activities and target proteins, synergistically induces cell death, and suppresses cell growth in TP53-Y220C cells.

TP53-WT or TP53-Y220C Molm13 cells were treated with PC14586 (4 μM), nutlin-3a (5 μM), or both for 4 h and subjected to RNA-seq (n=3) (A-C) and Western blotting (D). A. PCA of the treated cells. B. Heatmap of the DGE analysis (adjusted P ≤0.05, log2FC≥ 1, and baseMean ≥1000). C. Integrated Hallmark GSEA pathways (Adj P ≤0.001) with the x-axis representing different comparisons, y-axis representing various pathways, dot color representing −log10 P adj) x direction (i.e., red color indicates upregulation direction, while blue downregulation direction), and dot size representing absolute value of normalized enrichment scores. D. Validation of p53 target proteins by Western blotting. E. TP53-Y220C Molm13 cells were treated with PC14586, nutlin-3a or both for 72 h. Cell death and viable cells were determined by flow cytometry. F. TP53-Y220C Molm13 cells were treated with PC14586 (4 μM), KPT-8602 (200 nM), or both for 24 h. p53 localization and its target proteins were determined by Western blotting. G. TP53-Y220C Molm13 cells were treated with PC14586, KPT-8602, or both for 72 h. H-I. TP53-Y220C (H) or -WT (I) Molm13 cells were treated with PC14586, nutlin-3a, KPT-8602, or various combinations for 72 h. Cell death and viable cells were determined by flow cytometry. DEGs, differentially expressed genes; con, control; PC, PC14586; N3a, nutlin-3a; MWM, molecular weight marker; M/ml, million/ml; AnnV, annexin V; 7AAD, 7-aminoactinomycin D. Cell death and viability experiments were performed in triplicate; results were expressed as means±standard errors of the means.
XPO1 inhibition enhances p53 transcriptional activity and synergizes with PC14586 in apoptosis induction
We next determined if XPO1 inhibition increases the nuclear localization of PC14586-activated p53, hence enhancing its transcriptional activity. PC14586 and KPT-8602 combination indeed markedly increased nuclear p53, total MDM2, p21, and BAX levels (Figure 2F) and synergistically induced cell death in TP53-Y220C Molm13 cells (CI=0.045±0.038, Figure 2G). The combination was also highly synergistic in TP53-Y220C OCI-AML2 cells (Figure S2E). The inhibition of both MDM2 and XPO1 further enhanced the apoptogenic activity of PC14586 in TP53-Y220C Molm13 cells (Figure 2H).
In TP53-WT Molm13 cells treated with PC14586, nutlin-3a, KPT-8602, and combinations, nutlin-3a effectively induced apoptosis as expected, which was slightly more pronounced when combined with PC14586; KPT-8602 was more active in TP53-WT than TP53-Y220C cells, while the combination with PC14586 greatly induced apoptosis only at the highest dose; the three drug combination was most effective, largely due to the effect of MDM2 and XPO1 co-inhibition (Figure 2I).
PC14586-reactivated p53 does not bind to anti-apoptotic BCL-2 proteins: rationale for concomitant BCL-2 inhibition
A recent study showed that p53 interacts with BCL-2 and competes with BAX for the same binding pocket37. To better understand the mechanism underlying the limited apoptogenic activity of PC14586, we re-probed the membrane in Figure 1B with a BCL-2 antibody. As expected, BCL-2 was pulled down with p53 in TP53-WT cells, and nutlin-3a increased p53 and p53-bound BCL-2 (Figure 3A, left panel). However, neither Y220C-p53 nor PC14586-reactivated conformational p53-WT (with levels comparable to p53 in WT cells) binds to BCL-2 (Figure 3A, right panel). Results suggest, as illustrated (Figure 3B), that in TP53-WT cells, nutlin-3a stabilizes p53, which not only induces the expression of pro-apoptotic proteins like BAX, but also directly interacts with BCL-2, antagonizing its anti-apoptotic function, leading to apoptosis induction. In TP53-Y220C cells, PC14586 reactivates p53, but to a much lesser degree than nutlin-3a in TP53-WT cells, likely through p53-induced MDM2 that limits p53 transcriptionally upregulating pro-apoptotic proteins. Importantly, PC14586 reactivated-p53 does not bind to and antagonize BCL-2. This finding is further supported by BCL-2 co-IP assays showing that in TP53-WT cells, VEN or nutlin-3a decreased BAX-bound BCL-2; whereas in TP53-Y220C cells, only VEN, but not PC14586 did so (Figure 3C).
Figure 3. PC14586-reacativated p53 does not bind to BCL-2 proteins and PC14586 synergizes with VEN.
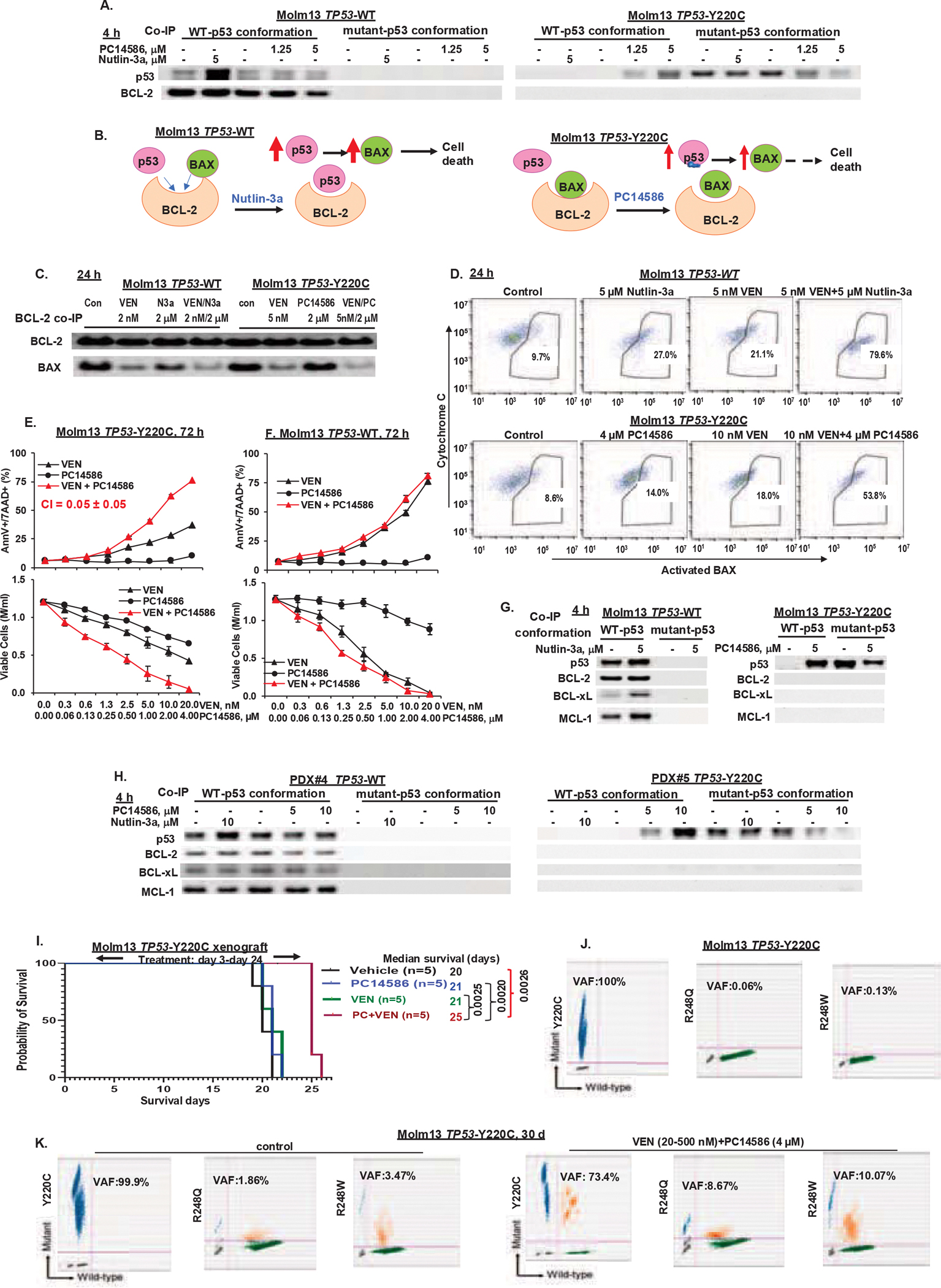
A. TP53-WT or -Y220C Molm13 cells were treated with nutlin-3a (5 μM) or PC14586 (4 μM) for 4 h. p53-BCL-2 interaction was determined by pulling down p53 with antibodies selective for the p53-WT- or -mutant conformation in the treated cells, followed by Western blotting for p53 or BCL-2. B. The proposed mechanism of action of PC14586 in TP53-Y220C cells. Nutlin-3a in TP53-WT cells is used as a control. C and D. TP53-WT Molm13 cells were treated with VEN, nutlin-3a or both and TP53-Y220C cells were treated with VEN, PC14586, or both at the indicated doses for 24 h. BCL-2 was pulled down by IP and BCL-2 bound BAX (co-IP) were determined by Western blotting (C). BAX activation and cytochrome C levels were determined by flow cytometry (D). E-F. TP53-Y220C (E) or -WT (F) Molm13 cells were treated with PC14586, VEN, or both for 72 h. Cell death and viable cells were determined by flow cytometry. G-H. TP53-WT or -Y220C Molm13 cells (G) or PDX cells (H) were treated with nutlin-3a or PC14586 for 4 h. p53-BCL-2, -BCL-xL, or -MCL-1 interaction was determined by co-IP followed by Western blotting. I. Survival curves for NSG mice bearing TP53-Y220C Molm13 xenografts and treated with PC14586, VEN, or both. J. Detection of TP53 mutations by ddPCR in Molm13 TP53-Y220C cells. K. Detection of TP53 mutations by ddPCR in TP53-Y220C Molm13 cells treated in vitro with PC14586 (4 μM) plus VEN (initially 20 nM and increased to 500 nM over time) or in untreated control cells for 30 days. Con, control; PC, PC14586; M/ml, million/ml; d, day; AnnV, annexin V; 7AAD, 7-aminoactinomycin D. Cell death and viability experiments were performed in triplicate; results were expressed as means±standard errors of the means.
To determine whether BCL-2 inhibition could functionally substitute for the lack of binding of reactivated p53 to BCL-2, we treated TP53-WT Molm13 cells with VEN, nutlin-3a, or both and TP53-Y220C Molm13 cells with VEN, PC14586, or both and determined BAX activation. In TP53-WT Molm13 cells, nutlin-3a sufficiently activated BAX, which was greatly enhanced when combined with VEN (Figure 3D, top); while in TP53-Y220C Molm13 cells, PC14586 had minimal effects on BAX activation, but in combination with VEN markedly increased BAX activation (Figure 3D, bottom) and apoptosis induction (CI=0.05±0.05, Figure 3E). In TP53-WT Molm13 cells treated with PC14586 and VEN, the effect was largely due to VEN and minimally enhanced by the PC14586/VEN combination (Figure 3F). The combination was also highly synergistic in TP53-Y220C OCI-AML2 cells (Figure S3A). Remarkably, p53-WT also binds to BCL-xL and MCL-1, whereas PC14586-reactivated p53 did not (Figure 3G). To validate this finding, we treated PDX#4 (TP53-WT) and PDX#5 (TP53-Y220C, VAF 50%) cells (Table S1) with nutlin-3a or PC14586. Co-IP experiments revealed that p53-WT (PDX#4) interacts with BCL-2, BCL-xL, and MCL-1, while PC14586-converted conformational p53-WT (PDX#5) lacks these interactions even with high levels of conformational p53-WT (Figure 3H). Hence, lack of the transcription-independent BCL-2 family inactivation limits PC14586 apoptogenic activity in TP53-Y220C AML cells, which can be compensated by combinations with BH3 memetics.
We evaluated the efficacy of PC14586 alone and PC14586 plus VEN in vivo in a TP53-Y220C Molm13 xenograft model. Mouse body weights are shown ( Figure S3B). In this extremely aggressive disease model, untreated mice died within 3 weeks. Neither PC14586 nor VEN had any efficacies, but their combination significantly extended survival by 25% (P=0.0026) (Figure 3I).
We previously observed expansions of TP53-mutant clones in TP53-WT cells during prolonged MDM2 inhibition52 and this was also observed in clinical trials of AML patients treated with MDM2 inhibitors7,8. DdPCR detected extremely low levels of TP53-R248Q (0.06%) and TP53-R248W (0.13%) mutations in TP53-Y220C Molm13 cells (Figure 3J). The same mutations (TP53-R248Q, 0.09% and TP53-R248W, 0.05%) were also detected in TP53-WT controls (Figure S3C) from which the TP53-Y220C cells were generated. These low levels of mutations normally would not be noticed in the short-duration in vitro experiments. In an in vitro experiment mimicking the in vivo xenograft experiment, we treated TP53-Y220C cells with PC14586, VEN, and both for 30 days and determined the levels of each mutation. In the combination-treated cells, the TP53-Y220C decreased, but both TP53-R248W and TP53-R248Q were enriched (Figure 3K), which may partially explain the diminished activity of PC14586 plus VEN in vivo and support the selectivity of PC14586 for TP53-Y220C mutation. PC14586 or VEN alone had minimal effect on these mutations under the condition (Figure S3D).
PC14586 in combination with MDM2, XPO1, or BCL-2 inhibition synergistically induces apoptosis in TP53-Y220C AML blasts and stem/progenitor cells but has minimal toxicity in NBM stem/progenitor cells
We treated AML samples (n=3) (Table S1) with PC14586. PDX#1 carries TP53-Y220C (VAF 48%), TP53-P151A (VAF 47%), RAS, and other mutations. Although p53-WT conformation was induced (Figure S4A), the cells were resistant to PC14586 (Figure 4A). PC14586 induced apoptosis in CD45+ AML blasts and CD34+/CD34+CD38− stem/progenitor cells from Pt#2 (Figure 4B) with TP53-Y220C (VAF 77%) as sole TP53 mutation and Pt#3 (Figure S4B) with TP53-Y220C (VAF 45.7%)/TP53-G105fs (frame shift, VAF 39.7%). Combinations of PC14586 with MDM2, XPO1, or BCL-2 inhibitors synergistically induced the death of AML blasts and stem/progenitor cells (CI=0.60±0.40 to CI<0.001) not only in the PC14586 resistant PDX#1, but also in Pt#2 and Pt#3 (Figure 4A, B and Figure S4B) and further decreased viable cells (Figure S5A) with a greater than log10 cell kill in all samples. Importantly, for clinical translation, PC14586 or the combinations had minimal activity in NBM and BM stem/progenitor cells by cell death or clonogenic assays (Figure 4C and Figure S5B).
Figure 4. PC14586 with concomitant MDM2, XPO1, or BCL-2 inhibition synergistically induces cell death in AML blasts and stem/progenitor cells from patients with the TP53-Y220C mutation but has limited toxicity in NBM cells and stem/progenitor cells.
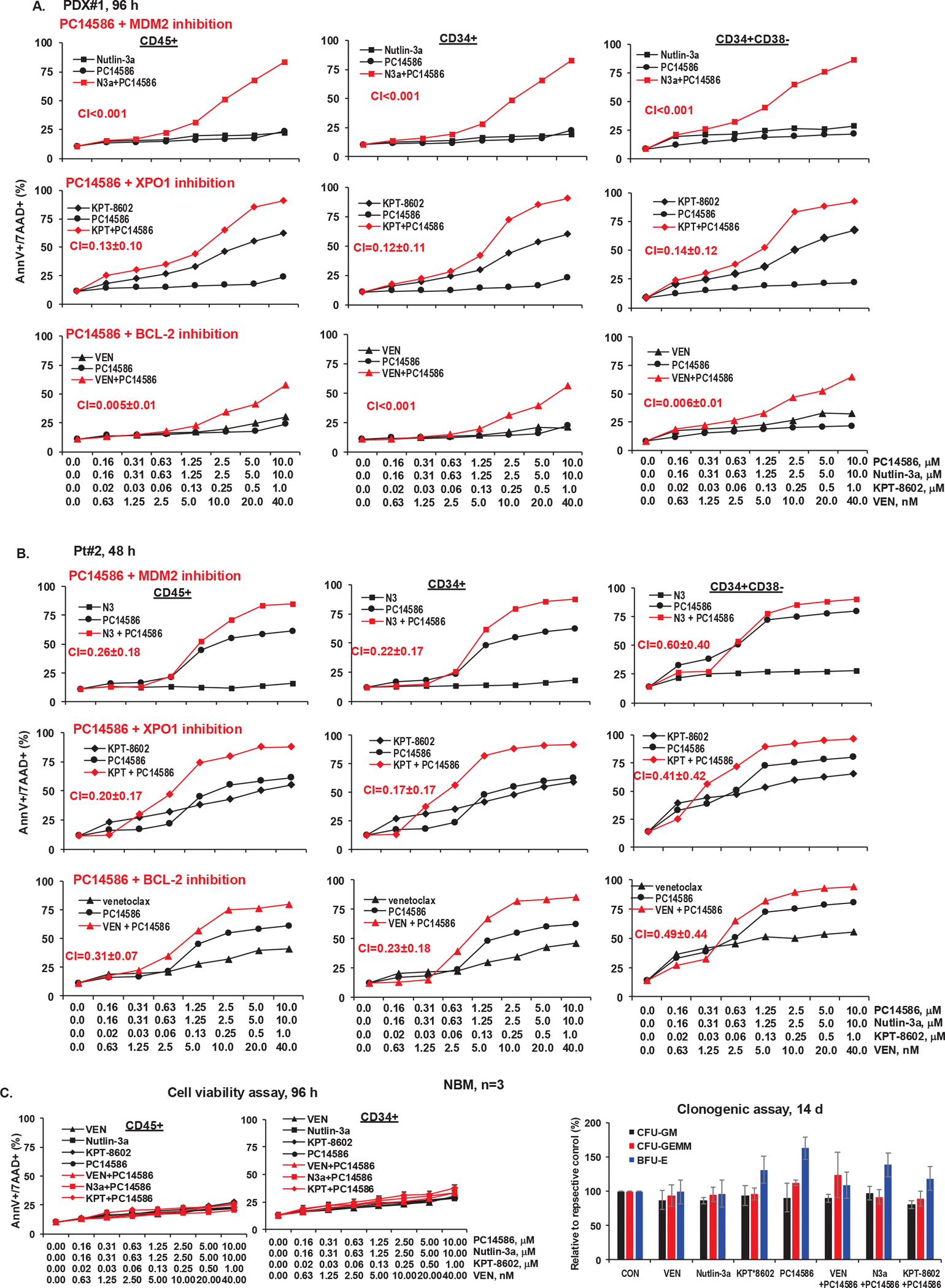
A-C, Cells from PDX#1 (A), Pt#2 (B), and NBM (n=3) (C) were treated with PC14586 alone or in combination with the indicated concentrations of nutlin-3a, KPT-8602, or VEN for the indicated times. Cell death was determined by flow cytometry in CD45+, CD34+, and CD34+CD38− cells. For NBM cells, cell death (left) and colony forming units (CFU) of cells treated with VEN (40 nM), nutlin-3a (10 μM), KPT-8602 (1 μM), PC14586 (10 μM), and each agent in combinations with PC14586 (right) are shown. N3a or N3, nutlin-3a; KPT, KPT-8602; AnnV, annexin V; 7AAD, 7-aminoactinomycin D.
Combination of PC14586 with MDM2 inhibition has antileukemia activity in vivo in TP53-Y220C AML PDX model
We treated NSG mice engrafted with PDX#1 cells carrying TP53-Y220C/TP53-P151A and non-TP53 co-mutations (Table S1) with PC14586, RG7388, or both. We knew that this model, the first and only established PDX carrying TP53-Y220C AML to date, was not an optimal model with which to assess PC14586 efficacy but thought it potentially useful to assess combinatorial efficiencies and understand mechanisms of action. Mouse body weights did not significantly change compared to controls (Figure S6). PC14586 decreased circulating blasts after 6- and 8-week treatment (Figure 5A) and spleen weights but not spleen size (Figure 5B) or BM blasts (Figure 5C) after 4 weeks. The combination significantly reduced circulating blasts after 4-, 6-, and 8-weeks, compared to control or either agent alone (Figure 5A) and was more effective in reducing spleen sizes/weights and in reducing BM blasts (Figure 5B, C). PC14586 monotherapy increased the median survival from 94 to 104 days, which did not reach statistical significance (P=0.057). The combination significantly extended survival to 111 days (P=0.0041) (Figure 5D).
Figure 5. PC14586 in combination with RG7388 has therapeutic efficacy in a PDX model in vivo.

NSG mice harboring PDX cells with TP53-Y220C and TP53-P151A mutations and other co-mutations were treated with vehicle, PC14586, RG7388, or both from d-38 to d-129 (RG7388 was stopped from d-97 to d-111). A. Circulating blasts after 4-week (left), 6-week (middle), or 8-weerk (right) treatment were determined by flow cytometric measurement of huCD45+ cells in PB. B. Spleen sizes (left) and weights(right) after 4-week treatment. C. BM blasts after 4-week treatment. D. Mouse survival. E-G. CyTOF analysis of BM cells collected after 4-week treatment. E. Treatment effects on various cell populations. F. Cell clustering based on cell surface markers (left and middle) and the effects of treatment on selected cell populations (right). G. tsne blots illustrate the effects of treatment on huCD45+ cells and p53-mutant in huCD45+ cells. RG, RG7388; PC, PC14586; d, day; Pop, population.
CyTOF analysis of BM cells after 4-week combinatorial treatment revealed little changes in phenotypical stem/progenitor populations (Figure 5E) but noticeable decreases in populations A (CD34+CD38+CD123+Tim3+CD33+), especially populations A1 (CD34+CD38+CD123+Tim3+highCD33+), and B (CD33+CD38+) and enrichment in population C (CD33+CD34+) (Figure 5F). Staining for huCD45 and p53-mutant protein showed (Figure 5G) that population A had relatively high and B had low levels of p53-mutant conformation in controls, and that PC14586 and RG7388 combination greatly decreased p53-mutant and leukemia cells in these populations, especially population A1. Population C, which also expressed p53-mutant conformation was relatively infrequent in controls, but combination treatments greatly increased this population, likely due to TP53-P151A mutation.
To understand resistance mechanisms, we performed scDNA-seq of BM cells collected from moribund mice treated with vehicle or PC14586. We detected two TP53 (Y220C and P151A) and two NRAS mutations (Q61R and Q61L). Compared to controls, cells that survived PC14586 treatment had proportionally higher TP53-P151A and NRAS-Q61R and lower TP53-Y220C and NRAS-Q61L (Figure 6A). Cells were clustered into four clones based on TP53 and NRAS mutations (Figure 6B). Quantitative analysis of the clone distribution showed that clones 1 and 2, both carrying TP53-P151A, persisted with PC14586: clone 1, which co-carried NRAS-Q61L, was largely unchanged, whereas clone 2, which co-carried NRAS-Q61R, expanded >10-fold (Figure 6C). Clones 3 and 4 both harbored TP53-Y220C and TP53-P151A. Clone 4, which co-carried NRAS-Q61L, was partially reduced, whereas clone 3, which co-carried NRAS-Q61R, was largely unchanged (Figure 6C). Data support that PC14586 selectively targets p53-Y220C and suggest that co-mutations affect the efficacy of PC14586. In this case, cells with NRAS-Q61R had survival and growth advantages over those with NRAS-Q61L.
Figure 6. ScDNA-seq of BM cells collected from vehicle- and PC14586-treated mice at moribund stage.

NSG mice harboring PDX cells with TP53-Y220C and TP53-P151A mutations and other co-mutations (#1) were treated with vehicle or PC14586. BM cells were collected from moribund mice in each group and subjected to scDNA-seq using the MissionBio platform. A. Heatmap showing the frequencies of TP53-Y220C, TP53-P151A, NRAS-Q61R, and NRAS-Q61L mutations in BM cells from vehicle- or PC14586-treated mice. Arrows indicate whether the mutations in PC14586-treated mice were increased or decreased compared to vehicle-treated mice. B. Characterizations of clones. C. Fish plot showing clonal evolution (left) and clonal distribution (right).
Discussion
Conceptually, reactivation of p53-WT in TP53-mutant cells not only reduces p53-mutant proteins but, importantly, also restores p53 functions in tumor cells and reinstates immunity suppressed by TP53-mutant cells in the tumor microenvironment. However, this approach has been attempted for several decades53 without success. PC14586 was shown to convert p53-Y220C into the p53-WT protein conformation and demonstrated preliminary antitumor activity in solid tumor patients in an ongoing clinical trial (NCT04585750)32–34. This is the first study to investigate the efficacy and mechanisms of action of a mutation-specific p53 reactivator in AML. We demonstrate that PC14586 converts p53-Y220C to p53-WT conformation and activates its transcriptional activity but induces less apoptosis in TP53-Y220C AML cells than MDM2 inhibition in isogenic TP53-WT cells. This reduced effect is due to 1) lower transcriptional activity: newly generated MDM2 and XPO1-mediated nuclear export limit p53’s effectiveness and 2) lack of transcription-independent mechanisms: reactivated-p53 cannot bind and inactivate antiapoptotic BCL-2 proteins. The findings highlight the importance of both transcriptional and non-transcriptional p53 function in apoptosis induction. Consequently, PC14586 synergizes with agents targeting MDM2, XPO1, and BCL-2. The combination of PC14586 and VEN should be easily translatable into the clinic.
We previously showed that p53 negatively regulates c-Myc43. We observed that PC14586 and more so PC14586 plus MDM2 inhibitor suppressed c-Myc signaling, which may contribute to the enhanced apoptogenic activity in TP53-Y220C AML.
p53 is known to directly interact with and antagonize BCL-2 and BCL-xL, which sufficiently induces apoptosis36,37,54. p53 indirectly antagonizes MCL-1 by activating BAK, resulting in the disruption of the BAK-MCL-1 complex55. Here, we report that p53 directly interacts with not only BCL-2 and BCL-xL but also MCL-1, whereas PC14586-reactivated p53 does not. The structural/functional differences between native and newly generated p53 remain to be elucidated.
PC14586 lacked activities in leukemia cells with TP53-WT, -KO, or other TP53 mutations and in PDX cells with TP53-Y220C/TP53-P151A co-mutations in vitro. It had no activity against a TP53-P151A mutant subclone and therefore limited activity in TP53-Y220C/P151A mutant cells in vivo, supporting the exclusivity of the TP53-Y220C mutation as a biomarker for patient selection. We recently reported that ~25% of TP53-Y220C AML/MDS have only this mutation31. Owing to its specificity, PC14586 may select/expand non-Y220C TP53-mutant cells, much like MDM2 inhibition does in TP53-WT AML7,8,52. ScDNA-seq analysis of subclones with TP53-Y220C/TP53-P151A co-mutations that survived PC14586 treatment revealed that non-TP53 co-mutations, such as NRAS, also affect response to PC14586. This is further supported by the finding that TP53-Y220C OCI-AML2 cells (no known kinase mutations) are relatively more sensitive than TP53-Y220C Molm13 cells (with FLT3-ITD) to PC14586-induced apoptosis. Analyses of additional samples and, ultimately, clinical trials are needed to determine the degree to which non-TP53 co-mutations affect the efficacy of PC14586 in TP53-Y220C AML and if co-targeting MDM2, XPO1, or BCL-2 synergistically advances cell death by enhancing p53 transcriptional activities or by BH3 mimetic-mediated antagonizing of BCL-2. A clinical trial of PC14586 in TP53-Y220C AML/MDS has recently been initiated (NCT06616636).
This study establishes the concept of reactivating p53-WT conformation in p53-mutant AML, identifies its limitations, and develops mechanism-based effective combinations for patients with TP53-Y220C leukemia, who lack therapeutic options. The concept should give hope to patients with other TP53 mutations. Advances in medicinal chemistry, protein modeling, and AI-assisted drug design may lead to the development of other agents targeting one or more TP53 mutations. In addition, the frequent co-mutations in TP53-mutant AML, such as NRAS, may also become increasingly targetable. We are establishing AML PDX models with TP53-Y220C as a sole TP53 mutation and will monitor clinical trials with single-cell technologies to further investigate the efficacy and mechanisms of PC14586 responses/resistance and develop mechanism-based combination therapies, including combinations targeting non-TP53 co-mutations.
One of the limitations of this study was that we did not address p53-WT-mediated immune effects, as we used immunodeficient murine models. Whether PC14586-reactivated p53 exerts immune-stimulatory functions remain unknown, although it is likely, given the profound effects of activated p53-WT on the immune response56. Another limitation is the unsolved mechanism underlying the lack of binding of reactivated p53 to BCL-2. Further in-depth studies are needed.
Supplementary Material
Supplemental Methods, Table S1, Table S2, Figures S1 to Figure S6
Key points.
PC14586 refolds Y220C-mutant p53 into a wild-type (WT)-like conformation and activates p53 targets but induces limited cell death in AML
We identified factors limiting the apoptogenic efficacy of the refolded p53-WT, which can be enhanced by co-targeting MDM2, XPO1, or BCL-2
Acknowledgements:
This work was supported in part by research funding from PMV Pharmaceuticals (to M.A. and B.Z.C.), the Leukemia & Lymphoma Society (to B.Z.C), the Paul and Mary Haas Chair in Genetics (to M.A.), and the National Institutes of Health through MD Anderson’s Cancer Center Support Grant (P30CA016672, supporting the Flow Cytometry and Cellular Imaging Facility and Advanced Technology Genomics Core).
We thank MissionBio for helping with scDNA-seq and data analysis and Joe Munch in MD Anderson’s Research Medical Library for editing the manuscript.
Footnotes
Conflict of interest disclosure: B.Z.C. and M.A. received research funding from PMV, A.P.K. and M.V.P. are employees of PMV, and A.L. is on the board directors of PMV and a consultant for the science of p53 and for Chugai Pharma for monoclonal antibodies.
Data and code availability:
RNA-Seq data have been uploaded to NCBI/SRA and the BioProject accession number is PRJNA1200638. The data will be public available after publication of the manuscript.ScDNA-seq was carried out by MissionBio. Data will be provided upon request.Original Western blotting images will be shared by the lead contact upon request.
Referencies
- 1.Hou HA, Chou WC, Kuo YY, et al. TP53 mutations in de novo acute myeloid leukemia patients: longitudinal follow-ups show the mutation is stable during disease evolution. Blood Cancer J. 2015;5:e331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stengel A, Kern W, Haferlach T, Meggendorfer M, Fasan A, Haferlach C. The impact of TP53 mutations and TP53 deletions on survival varies between AML, ALL, MDS and CLL: an analysis of 3307 cases. Leukemia. 2017;31(3):705–711. [DOI] [PubMed] [Google Scholar]
- 3.Kadia TM, Jain P, Ravandi F, et al. TP53 mutations in newly diagnosed acute myeloid leukemia: Clinicomolecular characteristics, response to therapy, and outcomes. Cancer. 2016;122(22):3484–3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Short NJ, Montalban-Bravo G, Hwang H, et al. Prognostic and therapeutic impacts of mutant TP53 variant allelic frequency in newly diagnosed acute myeloid leukemia. Blood Adv. 2020;4(22):5681–5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N Engl J Med. 2020;383(7):617–629. [DOI] [PubMed] [Google Scholar]
- 6.DiNardo CD, Tiong IS, Quaglieri A, et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood. 2020;135(11):791–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daver NG, Dail M, Garcia JS, et al. Venetoclax and idasanutlin in relapsed/refractory AML: a nonrandomized, open-label phase 1b trial. Blood. 2023;141(11):1265–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DiNardo CD, Olin R, Wang ES, et al. Phase 1 dose escalation study of the MDM2 inhibitor milademetan as monotherapy and in combination with azacitidine in patients with myeloid malignancies. Cancer Med. 2024;13(14):e70028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nishikawa S, Iwakuma T. Drugs Targeting p53 Mutations with FDA Approval and in Clinical Trials. Cancers (Basel). 2023;15(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Topatana W, Juengpanich S, Li S, et al. Advances in synthetic lethality for cancer therapy: cellular mechanism and clinical translation. J Hematol Oncol. 2020;13(1):118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang X, Simon R. Identification of potential synthetic lethal genes to p53 using a computational biology approach. BMC Med Genomics. 2013;6:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu J, Cao J, Topatana W, et al. Targeting mutant p53 for cancer therapy: direct and indirect strategies. J Hematol Oncol. 2021;14(1):157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bukhari AB, Lewis CW, Pearce JJ, Luong D, Chan GK, Gamper AM. Inhibiting Wee1 and ATR kinases produces tumor-selective synthetic lethality and suppresses metastasis. J Clin Invest. 2019;129(3):1329–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alexandrova EM, Yallowitz AR, Li D, et al. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature. 2015;523(7560):352–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carter BZ, Mak PY, Muftuoglu M, et al. Epichaperome inhibition targets TP53-mutant AML and AML stem/progenitor cells. Blood. 2023;142(12):1056–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adams CM, Mitra R, Xiao Y, et al. Targeted MDM2 Degradation Reveals a New Vulnerability for p53-Inactivated Triple-Negative Breast Cancer. Cancer Discov. 2023;13(5):1210–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang W, Kim SH, El-Deiry WS. Small-molecule modulators of p53 family signaling and antitumor effects in p53-deficient human colon tumor xenografts. Proc Natl Acad Sci U S A. 2006;103(29):11003–11008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tian X, Ahsan N, Lulla A, et al. P53-independent partial restoration of the p53 pathway in tumors with mutated p53 through ATF4 transcriptional modulation by ERK1/2 and CDK9. Neoplasia. 2021;23(3):304–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hernandez Borrero L, Dicker DT, Santiago J, et al. A subset of CB002 xanthine analogs bypass p53-signaling to restore a p53 transcriptome and target an S-phase cell cycle checkpoint in tumors with mutated-p53. Elife. 2021;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang S, Zhou L, Hong B, et al. Small-Molecule NSC59984 Restores p53 Pathway Signaling and Antitumor Effects against Colorectal Cancer via p73 Activation and Degradation of Mutant p53. Cancer Res. 2015;75(18):3842–3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zandi R, Selivanova G, Christensen CL, Gerds TA, Willumsen BM, Poulsen HS. PRIMA-1Met/APR-246 induces apoptosis and tumor growth delay in small cell lung cancer expressing mutant p53. Clin Cancer Res. 2011;17(9):2830–2841. [DOI] [PubMed] [Google Scholar]
- 22.Bykov VJ, Issaeva N, Shilov A, et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med. 2002;8(3):282–288. [DOI] [PubMed] [Google Scholar]
- 23.Chen S, Wu JL, Liang Y, et al. Arsenic Trioxide Rescues Structural p53 Mutations through a Cryptic Allosteric Site. Cancer Cell. 2021;39(2):225–239. [DOI] [PubMed] [Google Scholar]
- 24.Joerger AC, Fersht AR. Structure-function-rescue: the diverse nature of common p53 cancer mutants. Oncogene. 2007;26(15):2226–2242. [DOI] [PubMed] [Google Scholar]
- 25.Guiley KZ, Shokat KM. A Small Molecule Reacts with the p53 Somatic Mutant Y220C to Rescue Wild-type Thermal Stability. Cancer Discov. 2023;13(1):56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou S, Chai D, Wang X, et al. AI-powered discovery of a novel p53-Y220C reactivator. Front Oncol. 2023;13:1229696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng Q, Wang P, Liang C, et al. Abstract 5940: JAB-30355: A highly potent, orally bioavailable p53-Y220C reactivator. Cancer Research. 2024. [Google Scholar]
- 28.Liu M, Geng K, Lu B, Xia Y, Yang F. Abstract 7275: GS-P-328, a brain-penetrant small molecule p53 Y220C reactivator for tumors harboring p53 Y220C mutation. Cancer Research. 2024;84(6_Supplement):7275–7275. [Google Scholar]
- 29.Vu BT, Dominique R, Fahr BJ, et al. Discovery of Rezatapopt (PC14586), a First-in-Class, Small-Molecule Reactivator of p53 Y220C Mutant in Development. ACS Medicinal Chemistry Letters. 2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bouaoun L, Sonkin D, Ardin M, et al. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum Mutat. 2016;37(9):865–876. [DOI] [PubMed] [Google Scholar]
- 31.Gener-Ricos G, Bewersdorf JP, Loghavi S, et al. TP53 Y220C mutations in patients with myeloid malignancies. Leuk Lymphoma. 2024:1–5. [DOI] [PubMed] [Google Scholar]
- 32.Puzio-Kuter AM, Xu L, McBrayer MK, et al. Restoration of the Tumor Suppressor Function of Y220C-Mutant p53 by Rezatapopt, a Small Molecule Reactivator. Cancer Discov. 2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dumble M, Xu L, Dominique R, et al. PC14586: The first orally bioavailable small molecule reactivator of Y220C mutant p53 in clinical development. Cancer Res. 2021;81:LB006. [Google Scholar]
- 34.Dumbrava EE, Johnson ML, Tolcher AW, et al. First-in-human study of PC14586, a small molecule structural corrector of Y220C mutant p53, in patients with advanced solid tumors harboring a TP53 Y220C mutation. Journal of Clinical Oncology. 2022;40. [Google Scholar]
- 35.Peuget S, Zhou X, Selivanova G. Translating p53-based therapies for cancer into the clinic. Nat Rev Cancer. 2024. [DOI] [PubMed] [Google Scholar]
- 36.Kojima K, Konopleva M, McQueen T, O'Brien S, Plunkett W, Andreeff M. Mdm2 inhibitor Nutlin-3a induces p53-mediated apoptosis by transcription-dependent and transcription-independent mechanisms and may overcome Atm-mediated resistance to fludarabine in chronic lymphocytic leukemia. Blood. 2006;108(3):993–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wei H, Wang H, Wang G, et al. Structures of p53/BCL-2 complex suggest a mechanism for p53 to antagonize BCL-2 activity. Nat Commun. 2023;14(1):4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kojima K, Konopleva M, Samudio IJ, et al. MDM2 antagonists induce p53-dependent apoptosis in AML: implications for leukemia therapy. Blood. 2005;106(9):3150–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andreeff M, Kelly KR, Yee K, et al. Results of the Phase I Trial of RG7112, a Small-Molecule MDM2 Antagonist in Leukemia. Clin Cancer Res. 2016;22(4):868–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kojima K, Konopleva M, Samudio IJ, Schober WD, Bornmann WG, Andreeff M. Concomitant inhibition of MDM2 and Bcl-2 protein function synergistically induce mitochondrial apoptosis in AML. Cell Cycle. 2006;5(23):2778–2786. [DOI] [PubMed] [Google Scholar]
- 41.Pan R, Ruvolo V, Mu H, et al. Synthetic Lethality of Combined Bcl-2 Inhibition and p53 Activation in AML: Mechanisms and Superior Antileukemic Efficacy. Cancer Cell. 2017;32(6):748–760.e746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kojima K, Kornblau SM, Ruvolo V, et al. Prognostic impact and targeting of CRM1 in acute myeloid leukemia. Blood. 2013;121(20):4166–4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nishida Y, Ishizawa J, Ayoub E, et al. Enhanced TP53 reactivation disrupts MYC transcriptional program and overcomes venetoclax resistance in acute myeloid leukemias. Sci Adv. 2023;9(48):eadh1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boettcher S, Miller PG, Sharma R, et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science. 2019;365(6453):599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Studeny M, Marini FC, Champlin RE, Zompetta C, Fidler IJ, Andreeff M. Bone marrow-derived mesenchymal stem cells as vehicles for interferon-beta delivery into tumors. Cancer Res. 2002;62(13):3603–3608. [PubMed] [Google Scholar]
- 46.Carter BZ, Mak PY, Tao W, et al. Targeting MCL-1 dysregulates cell metabolism and leukemia-stroma interactions and resensitizes acute myeloid leukemia to BCL-2 inhibition. Haematologica. 2020;23(10):260331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carter BZ, Mak PY, Tao W, et al. Maximal Activation of Apoptosis Signaling by Cotargeting Antiapoptotic Proteins in BH3 Mimetic-Resistant AML and AML Stem Cells. Mol Cancer Ther. 2022;21(6):879–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thijssen R, Diepstraten ST, Moujalled D, et al. Intact TP-53 function is essential for sustaining durable responses to BH3-mimetic drugs in leukemias. Blood. 2021;137(20):2721–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jacamo R, Chen Y, Wang Z, et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-κB mediates chemoresistance. Blood. 2014;123(17):2691–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carter BZ, Tao W, Mak PY, et al. Menin inhibition decreases Bcl-2 and synergizes with venetoclax in NPM1/FLT3-mutated AML. Blood. 2021;138(17):1637–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. [DOI] [PubMed] [Google Scholar]
- 52.Nishida Y, Montoya RH, Morita K, et al. Clonal Expansion of Mutant p53 Clones By MDM2 Inhibition in Acute Myeloid Leukemias. Blood. 2020;136(Supplement 1):27–28. [Google Scholar]
- 53.Santini V, Stahl M, Sallman DA. TP53 Mutations in Acute Leukemias and Myelodysplastic Syndromes: Insights and Treatment Updates. Am Soc Clin Oncol Educ Book. 2024;44(3):e432650. [DOI] [PubMed] [Google Scholar]
- 54.Mihara M, Erster S, Zaika A, et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 2003;11(3):577–590. [DOI] [PubMed] [Google Scholar]
- 55.Leu JI, Dumont P, Hafey M, Murphy ME, George DL. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat Cell Biol. 2004;6(5):443–450. [DOI] [PubMed] [Google Scholar]
- 56.Blagih J, Buck MD, Vousden KH. p53, cancer and the immune response. J Cell Sci. 2020;133(5). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-Seq data have been uploaded to NCBI/SRA and the BioProject accession number is PRJNA1200638. The data will be public available after publication of the manuscript.ScDNA-seq was carried out by MissionBio. Data will be provided upon request.Original Western blotting images will be shared by the lead contact upon request.