

ABSTRACT
Model‐informed drug development (MIDD) entails applying quantitative approaches to assist with decision‐making during drug development and has been used for dose optimization, to inform clinical trial design, and to support clinical trial waivers. With increasing cost and competitiveness in drug development, the use of tools that improve efficiency, like MIDD, is increasingly crucial. A unique case for the successful application of MIDD approaches from early Phase 1 through postapproval for the upadacitinib development program is described herein. Upadacitinib is an orally administered selective Janus kinase inhibitor, which is approved for rheumatoid arthritis, psoriatic arthritis, atopic dermatitis, axial spondylarthritis, nonradiographic axial spondyloarthritis, ulcerative colitis, and Crohn's disease for adults, in addition to recent approvals for polyarticular juvenile idiopathic arthritis and pediatric patients with psoriatic arthritis. Applications and impact of modeling and simulation approaches for informing key development decisions are presented to highlight the success of using MIDD for the clinical development of upadacitinib. The lessons learned can provide a framework for the clinical development of other drugs.
1. Introduction
Model‐informed drug development (MIDD) entails applying quantitative models to assist with decision‐making during drug development and has been used for dose optimization, to inform clinical trial design, and to support waiver of clinical studies [1]. MIDD has gained widespread acceptance and adoption by sponsors and regulatory agencies over the last two decades [1]. Incorporation of MIDD into programs for drug development has been beneficial for sponsors through efficiency, alignment, and learning and clarity [2]. With increasing cost and competitiveness in the drug development environment, the use of tools that increase efficiency, like MIDD, is more important than ever [3].
A recent drug development program that used MIDD was upadacitinib. It is an orally administered selective Janus kinase (JAK) inhibitor, which is approved for rheumatoid arthritis (RA), psoriatic arthritis (PsA), atopic dermatitis (AD), axial spondylarthritis (AS), nonradiographic axial spondyloarthritis, ulcerative colitis (UC), Crohn's disease (CD), and giant cell arteritis for adults. Pediatric indications include AD for adolescents 12 years and older, PsA and polyarticular juvenile idiopathic arthritis for children 2 years and older (approved in the US only) [4]. Upadacitinib is available as an extended‐release (ER) once daily (QD) tablet ranging in strength from 15 mg to 45 mg and an oral solution formulation for pediatric patients [5].
This review describes how MIDD has been used from Phase 1 through post‐approval for the development of upadacitinib. Figure 1 shows a timeline of modeling activities that were performed for upadacitinib to help with dose selection going into Phase 2 and Phase 3 as well as for bridging formulations, special population assessment, pediatric development, and supporting postapproval manufacturing changes. Figure 2 shows the different factors that were considered prior to conducting each clinical study and the respective analyses and how these led to increased drug development efficiency, such as time and cost savings through waivers and fewer required studies, and enabled faster availability of the drug to patients.
FIGURE 1.
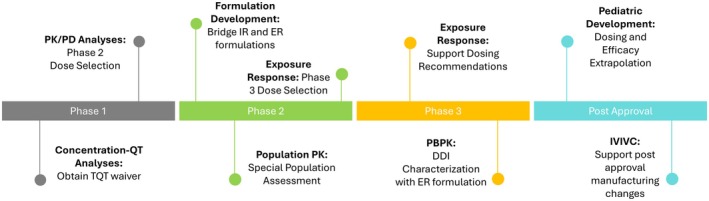
Timeline of modeling activities. DDI, drug–drug interaction; ER, extended release; IR, immediate release; PBPK, physiologically based pharmacokinetic; TQT, thorough QT clinical study.
FIGURE 2.

Planning for modeling and simulation across the phases of upadacitinib development. BA, bioavailability; BE, bioequivalence; BID, twice daily; DDI, drug–drug interaction; ECG, electrocardiogram; HV, healthy volunteer; IVIVC, in vitro–in vivo correlation; JAK, Janus kinase; MAD, multiple ascending dose; PBPK, physiologically based pharmacokinetic; PD, pharmacodynamic; PK, pharmacokinetic; QD, once daily; SAD, single ascending dose; UPA, upadacitinib.
2. Phase 1
2.1. PK/PD Analyses Based on Biomarker Data
Upadacitinib demonstrated reversible and concentration‐dependent inhibition of interleukin (IL)‐6‐induced pSTAT3 (as a measure of JAK1 activity) and IL‐7‐induced pSTAT5 (as a measure of JAK1/3 activity) in samples from healthy subjects and subjects with RA treated with different upadacitinib doses. Ex vivo pharmacodynamic (PD) assay results showed a greater selectivity of upadacitinib on JAK1 versus JAK3 compared to another JAK inhibitor, tofacitinib, confirming higher in vitro potency of upadacitinib against JAK1 compared to JAK3 [6]. Maximizing potency for JAK1, while minimizing the inhibitory effects on other JAK isoforms, was hypothesized as a potential approach to improve risk–benefit profiles of JAK inhibitors in the treatment of RA compared to less selective JAK inhibitors [7]. These analyses demonstrated that the approved dose of tofacitinib in RA (5 mg twice daily [BID]) provides comparable effects on IL‐6‐induced pSTAT3 to 3 mg BID of upadacitinib (immediate‐release [IR] formulation). In this Phase 1 study, pharmacokinetic (PK)/PD analysis used the within‐study control of tofacitinib to help benchmark and guide upadacitinib doses to be studied in Phase 2 (Figure 2).
2.2. Concentration–QT Analyses
In order to inform the QT prolongation potential of upadacitinib, the upadacitinib development team used data from the Phase 1 single‐ and multiple‐ascending dose studies to demonstrate a lack of QT prolongation potential for upadacitinib at the plasma exposures associated with doses higher than the ones being used in RA Phase 3 studies [8]. Additionally, the QT analysis used the change in QT following food consumption as a positive control in the absence of a pharmacological positive control (e.g., moxifloxacin) in the study to demonstrate assay sensitivity. These analyses were discussed with regulatory agencies at the end of Phase 2 interactions and accepted in lieu of a standalone, resource‐intensive thorough QT (TQT) study.
3. Phase 2
3.1. Early Assessment of Special Populations Pharmacokinetics Using Population Pharmacokinetic Modeling
At the end of Phase 2 prior to conducting dedicated organ impairment studies (which were conducted in parallel to Phase 3), population pharmacokinetic analyses of data from Phase 2 studies were used to evaluate the impact of intrinsic (e.g., renal impairment) factors on the pharmacokinetics of upadacitinib. Differences in renal function (mild or moderate renal impairment) did not result in clinically relevant effects on upadacitinib exposures [9]. Additionally, there was no relationship between aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels and upadacitinib clearance in this analysis. Although this assessment provided insight on the lack of a significant trend with clearance within the range of AST and ALT levels in the data set, it was limited due to the exclusion of patients with significant AST and ALT elevations from the Phase 2 clinical trials. Overall, these analyses provided early assessment of the effects of organ impairment prior to the availability of data from standalone Phase 1 studies in subjects with renal and hepatic impairment, which helped inform the enrollment criteria particularly with regard to renal function for subjects in the pivotal Phase 3 trials. Subjects with mild and moderate renal impairment were still included in Phase 1 renal impairment study [10] along with a severe renal impairment group to better inform the overall association between renal function and upadacitinib exposures.
3.2. Formulation Development: Bridging IR and ER Formulations and Phase 3 Dose Selection
Phase 2 studies in patients with RA and CD for upadacitinib were conducted using mainly BID regimens of the IR capsule formulation [11, 12], while there was a preference to advance into Phase 3 trials with a more convenient QD dosing using an ER formulation. Population PK analyses using PK data from the IR formulation in Phase 1 and 2 studies as well as from the ER formulation in a pilot Phase 1 study was used to bridge the IR and ER formulations and select ER doses which would achieve comparable daily exposures of upadacitinib to the target BID IR doses based on Phase 2 (Figure 2). Additionally, simulations were conducted to predict the efficacy with the QD ER formulations based on the developed exposure‐response models using the full time‐course data from Phase 2 studies [11, 12]. These analyses enabled proceeding into large registrational Phase 3 trials with a QD regimen using an ER formulation and with unique doses without prior assessment of these regimens in Phase 2 clinical trials. The analyses were used to inform and de‐risk the decision of advancing with QD doses using the ER formulation directly into Phase 3 trials. This saved significant development time and resources which would have been spent on conducting separate Phase 2 trials using the planned ER QD regimens prior to evaluating them in large Phase 3 trials. Furthermore, exposure‐response analyses were used to support Phase 3 dose selection in all approved indications as well as for the ongoing development programs in hidradenitis suppurativa, non‐segmental vitiligo, and systemic lupus erythematosus.
4. Phase 3
4.1. Population Pharmacokinetics and Exposure–Response Analyses
Population PK and exposure–response analyses for efficacy and safety for all approved indications supported the marketed dosing recommendations as well as dosing recommendations in special populations or with concomitant medications. Phase 3 data provided a large enough dataset to characterize the effects of covariates and specific disease characteristics on the PK and exposure‐response relationships (Figure 2) [13, 14, 15, 16, 17, 18]. In the AD program (which enrolled adolescents in the pivotal adult Phase 3 trials), population PK analyses were important to characterize upadacitinib PK in adolescents, which helped inform the appropriateness of administering the same adult dose to adolescents.
4.2. Physiologically Based Pharmacokinetic (PBPK) Modeling
Early in development, Phase 1 drug‐drug interaction (DDI) studies assessing the impact of strong cytochrome P450 (CYP) 3A4 modulators (inducer or inhibitor) were conducted with the upadacitinib IR formulation [19]. With the change to an ER formulation, PBPK modeling was used to bridge DDI liabilities to the ER QD formulation. This analysis was included in the upadacitinib marketing authorization applications and was used to inform the label language without the need to conduct additional DDI studies with the ER QD formulation [20].
5. Postapproval Manufacturing Changes
A robust nonlinear level A in vitro–in vivo correlation (IVIVC) that meets the Food and Drug Administration (FDA) and European Medicines Agency (EMA) and other major regulatory agencies validation criteria for both internal and external predictability was established for upadacitinib ER formulation [21]. This model was built based on in vitro dissolution and in vivo PK data from a dedicated bioavailability study with formulations containing varying levels of hydroxypropyl methylcellulose (HPMC).
Upadacitinib IVIVC and exposure–response analyses for efficacy and safety in RA patients were used to conduct clinical trial simulations to support the clinical relevance of dissolution specifications for the upadacitinib ER tablet formulation [22]. The results demonstrated that a formulation at the lower dissolution boundary is predicted to have noninferior efficacy, and a formulation at the upper dissolution boundary is predicted to have similar safety to the target ER formulation. Utilization of the IVIVC model has supported several scale‐up and postapproval change (SUPAC) level 3 changes. Development of this model prior to the original New Drug Application was critical in saving resources by avoiding the need to conduct bioequivalence studies to support such postapproval changes.
5.1. Pediatric Drug Development
A model‐informed approach, based on population PK analyses of adult data and the assumption of typical allometric values for body weight (exponents of 0.75 for CL and 1 for volume parameters), was employed for selecting the initial pediatric doses for upadacitinib. The initial doses were evaluated in Phase 1 pediatric studies for upadacitinib in patients with polyarticular‐course juvenile idiopathic arthritis (pcJIA) and AD [23]. Both studies included an adaptive design, where a preplanned analysis was performed to update the PK model and assess the appropriateness of the initial pediatric doses using emerging data from the studies. This analysis prompted a dose revision and helped inform the dose selection for multiple Phase 3 pediatric studies.
In addition, a model‐informed approach was utilized to support the extrapolation of upadacitinib efficacy from adult patients with RA and PsA to pediatric patients with pcJIA and PsA, respectively, in lieu of Phase 3 clinical trials [24]. This approach involved population PK analyses of data in both adults and pediatrics and simulation of efficacy leveraging existing adult exposure–response models and real‐world pediatric patient databases. The analyses demonstrated that the proposed pediatric doses of upadacitinib were predicted to provide noninferior efficacy in pcJIA and pediatric PsA to that in adult RA and PsA, respectively, resulting in upadacitinib being available to pediatric patients with polyarticular JIA and PsA several years earlier than the traditional approach through one or more Phase 3 trials in the pediatric populations.
6. Discussion
The MIDD paradigm was critical to enable decision‐making, accelerate development, waive clinical trials, and assess the benefit–risk profile of upadacitinib during development and postapproval. The use of modeling and simulation approaches at the different stages of upadacitinib development led to significant cost savings, risk mitigation, and earlier availability of the drug to patients with approvals in multiple adult and pediatric indications. This case study is applicable to other drug development programs where there is increasing competitiveness and cost, bringing additional focus to leveraging all available tools to improve efficiency, facilitate decision‐making, reduce cost, and accelerate drug availability [3]. Several important considerations should be assessed during MIDD implementation in drug development. These include the validity of modeling assumptions, the adequacy of available data, and the ability to communicate technical analyses clearly to cross‐functional teams in order to inform and impact drug development strategies. For example, in pediatric extrapolation, a thorough understanding of the diseases and mechanisms of action of the study drug is required to demonstrate similarities between the adult and pediatric populations including disease pathophysiology and response to treatment [25].
To enable a meaningful and successful implementation of the MIDD paradigm in drug development, it is imperative for drug developers to incorporate the collection of adequate data, particularly in early clinical trials, so that the development of robust models can be achieved and can inform decisions and even waive clinical trials. One challenge in this aspect is to gain cross‐functional alignment within the organization to prioritize spending such resources early in development and to highlight how the collected data have the potential to result in significant benefits in future development. As shown in Figure 2 for the upadacitinib development program, the collection of early triplicate ECGs in Phase 1 trials, the generation of early in vitro and in vivo data for different formulations to enable IVIVC development, the collection of adequate PK and biomarker data in early clinical trials, and the collection of adequate PK data in later phase trials were all critical to enable the application of modeling and simulation approaches described. This point is becoming more crucial as the increased cost of drug development naturally makes companies consider limiting costs in trials, for example, not collecting samples for biomarkers, triplicate ECGs, or even PK in early trials. However, this early apparent cost saving comes with a substantial cost later, as the inadequate data collection in early trials may not enable the development of robust and informative models later. Drug developers, in such suboptimal scenarios, would carry significant cost and uncertainty into later stages of development and may have to conduct additional late‐stage, large, and costly clinical trials due to the inability to fully leverage MIDD approaches. The examples highlighted herein from the upadacitinib development further highlight the need for adequate prospective planning and the value of early collection of data in clinical trials in maximizing the potential application and value of modeling and simulation approaches in drug development programs.
Conflicts of Interest
The authors are employees of AbbVie and may hold AbbVie stock or stock options.
Acknowledgments
Medical writing support was provided by Mia DeFino, MS, ELS, CMPP, a freelance medical writer under contract with AbbVie.
Bhatnagar S., Stodtmann S., Qian Y., Marroum P., Liu W., and Mohamed M.‐E. F., “Model‐Informed Paradigm in Drug Development—An End‐To‐End Case Study From Upadacitinib Development,” Clinical and Translational Science 18, no. 8 (2025): e70295, 10.1111/cts.70295.
Funding: This study was funded by AbbVie Inc. AbbVie contributed to the study design, research, and interpretation of data, and the writing, reviewing, and approving of the publication.
References
- 1. Wang Y., Zhu H., Madabushi R., Liu Q., Huang S. M., and Zineh I., “Model‐Informed Drug Development: Current US Regulatory Practice and Future Considerations,” Clinical Pharmacology and Therapeutics 105 (2019): 899–911. [DOI] [PubMed] [Google Scholar]
- 2. Galluppi G. R., Brar S., Caro L., et al., “Industrial Perspective on the Benefits Realized From the FDA'S Model‐Informed Drug Development Paired Meeting Pilot Program,” Clinical Pharmacology and Therapeutics 110 (2021): 1172–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. IQVIA Institute for Human Data Science , “Global Trends in R&D 2024: Activity, P., and Enablers,” (2024), www.iqviainstitute.org. A.f.
- 4. AbbVie Inc ., “RINVOQ (Upadacitinib Extended‐Release Tablets) [US Package Insert],” (2022).
- 5. Mohamed M. F., Bhatnagar S., Parmentier J. M., Nakasato P., and Wung P., “Upadacitinib: Mechanism of Action, Clinical, and Translational Science,” Clinical and Translational Science 17 (2024): e13688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mohamed M. F., Beck D., Camp H. S., and Othman A. A., “Preferential Inhibition of JAK1 Relative to JAK3 by Upadacitinib: Exposure‐Response Analyses of Ex Vivo Data From 2 Phase 1 Clinical Trials and Comparison to Tofacitinib,” Journal of Clinical Pharmacology 60 (2020): 188–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Norman P., “Selective JAK Inhibitors in Development for Rheumatoid Arthritis,” Expert Opinion on Investigational Drugs 23 (2014): 1067–1077. [DOI] [PubMed] [Google Scholar]
- 8. Mohamed M. F., Zeng J., Jiang P., Hosmane B., and Othman A. A., “Use of Early Clinical Trial Data to Support Thorough QT Study Waiver for Upadacitinib and Utility of Food Effect to Demonstrate ECG Assay Sensitivity,” Clinical Pharmacology and Therapeutics 103 (2018): 836–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Klunder B., Mohamed M. F., and Othman A. A., “Population Pharmacokinetics of Upadacitinib in Healthy Subjects and Subjects With Rheumatoid Arthritis: Analyses of Phase I and II Clinical Trials,” Clinical Pharmacokinetics 57 (2018): 977–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mohamed M. F., Trueman S., Feng T., Anderson J., Marbury T. C., and Othman A. A., “Characterization of the Effect of Renal Impairment on Upadacitinib Pharmacokinetics,” Journal of Clinical Pharmacology 59 (2019): 856–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mohamed M. F., Klunder B., Lacerda A. P., and Othman A. A., “Exposure‐Response Analyses for Upadacitinib Efficacy and Safety in the Crohn's Disease CELEST Study and Bridging to the Extended‐Release Formulation,” Clinical Pharmacology and Therapeutics 107 (2020): 639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mohamed M. F., Klunder B., Camp H. S., and Othman A. A., “Exposure‐Response Analyses of Upadacitinib Efficacy in Phase 2 Trials in Rheumatoid Arthritis and Basis for Phase 3 Dose Selection,” Clinical Pharmacology and Therapeutics 106, no. 6 (2019): 1319–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mohamed M. F., Zeng J., Marroum P. J., Song I. H., and Othman A. A., “Pharmacokinetics of Upadacitinib With the Clinical Regimens of the Extended‐Release Formulation Utilized in Rheumatoid Arthritis Phase 3 Trials,” Clinical Pharmacology in Drug Development 8 (2019): 208–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nader A., Mohamed M. E. F., Winzenborg I., et al., “Exposure‐Response Analyses of Upadacitinib Efficacy and Safety in Phase II and III Studies to Support Benefit‐Risk Assessment in Rheumatoid Arthritis,” Clinical Pharmacology and Therapeutics 107 (2020): 994–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Muensterman E., Engelhardt B., Gopalakrishnan S., Anderson J. K., and Mohamed M. F., “Upadacitinib Pharmacokinetics and Exposure‐Response Analyses of Efficacy and Safety in Psoriatic Arthritis Patients—Analyses of Phase III Clinical Trials,” Clinical and Translational Science 15 (2022): 267–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ponce‐Bobadilla A. V., Stodtmann S., Eckert D., Zhou W., Liu W., and Mohamed M. E., “Pharmacokinetics and Exposure‐Response Analyses of Upadacitinib in Patients With Moderate to Severe Ulcerative Colitis‐ Analyses of Induction and Maintenance Clinical Trials,” 17th Congress of ECCO (European Crohn's and Colitis Organisation) (2022), vol. S1, i355–i357.
- 17. Bhatnagar S., Eckert D., Stodtmann S., et al., “Population Pharmacokinetics and Exposure‐Response Analyses for Efficacy and Safety of Upadacitinib in Patients With Axial Spondyloarthritis,” Clinical and Translational Science 17 (2024): e13733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ismail M., Doelger E., Eckert D., et al., “Population Pharmacokinetic and Exposure‐Response Modelling to Inform Upadacitinib Dose Selection in Adolescent and Adult Patients With Atopic Dermatitis,” British Journal of Clinical Pharmacology 89 (2023): 3139–3151. [DOI] [PubMed] [Google Scholar]
- 19. Mohamed M. F., Jungerwirth S., Asatryan A., Jiang P., and Othman A. A., “Assessment of Effect of CYP3A Inhibition, CYP Induction, OATP1B Inhibition, and High‐Fat Meal on Pharmacokinetics of the JAK1 Inhibitor Upadacitinib,” British Journal of Clinical Pharmacology 83 (2017): 2242–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Food and Drug Administration & Center for Drug Evaluation and Research , “Application Number 211675Orig1s000. Product Clinical Pharmacology and BioPharmaceutics Review(s) for Upadacitinib Extended Release Tablets,” (2019).
- 21. Mohamed M. F., Trueman S., Othman A. A., Han J. H., Ju T. R., and Marroum P., “Development of In Vitro‐In Vivo Correlation for Upadacitinib Extended‐Release Tablet Formulation,” AAPS Journal 21 (2019): 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mohamed M. F., Winzenborg I., Othman A. A., and Marroum P., “Utility of Modeling and Simulation Approach to Support the Clinical Relevance of Dissolution Specifications: A Case Study From Upadacitinib Development,” AAPS Journal 24 (2022): 39. [DOI] [PubMed] [Google Scholar]
- 23. Qian Y., Raymundo E. M., Hao S., et al., “Pharmacokinetics, Safety, Tolerability, and Exploratory Efficacy of Upadacitinib in Children With Severe Atopic Dermatitis,” Clinical Therapeutics 46 (2024): 733–741. [DOI] [PubMed] [Google Scholar]
- 24. Qian Y., Schlachter L., Eckert D., et al., “Extrapolation of Upadacitinib Efficacy in Juvenile Idiopathic Arthritis Leveraging Pharmacokinetics, Exposure‐Response Models, and Real‐World Patient Data,” Clinical Pharmacology and Therapeutics 116 (2024): 1635–1645. [DOI] [PubMed] [Google Scholar]
- 25. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research & (CBER) , “C.f.B.E.a.R. E11A Pediatric Extrapolation: Guidance for Industry,” (2024).