

Abstract
The CRISPR-Cas system has transformed our ability to edit and modify genomes in eukaryotic cells, offering unmatched precision and broad applicability. By utilizing a programmable RNA protein complex to introduce targeted double-strand breaks, the CRISPR-Cas system enables the correction of pathogenic mutations and the modulation of gene function with unprecedented efficiency. Its broad applicability spans the correction of inherited genetic defects through homology-directed repair to the disruption of deleterious alleles via non-homologous end joining. In this review, we first outline the molecular architecture and mechanistic basis of CRISPR-Cas9 and then consider its latest applications in modeling, drug screening, small-molecule-mediated editing, and treating hereditary, autoimmune, and oncological diseases. Emphasis is placed on the generation of disease-relevant cellular and animal models and on the potential of CRISPR-Cas9-mediated gene therapy to address hitherto intractable disorders. Finally, we discuss current challenges including off-target activity, gene editing efficiency, delivery constraints, and immunogenicity and highlight emerging strategies to overcome these hurdles and broaden the clinical impact of CRISPR-Cas systems.
Keywords: MT: RNA/DNA Editing, CRISPR-Cas9, DNA repair, gene therapy, drug screening, cell engineering, genetic editing efficiency
Graphical abstract
This review summarizes the genome editing mechanism and therapeutic applications of CRISPR-Cas9 systems. It highlights recent advances in disease modeling, drug screening, and treatment of genetic, autoimmune, and oncologic disorders, while addressing key challenges and emerging strategies to enhance clinical translation.
Introduction
CRISPR and their associated Cas proteins have emerged as transformative tools in molecular biology, enabling precise genome editing in many different ways.1,2 Although originally characterized as an adaptive immune system in bacteria and archaea, CRISPR-Cas9 has transcended its native role to revolutionize fields ranging from functional genomics to translational medicine.3 Pivotal observations of mysterious repeat sequences in the Escherichia coli genome in 1987 laid the foundation for this paradigm shift, yet it was not until the early 2000s that these loci, now known as CRISPR arrays, were recognized as evidences of past viral encounters and the crucible of a heritable defense system.4 The subsequent discovery of CRISPR-associated genes and the elucidation of Cas9’s RNA-guided endonuclease activity, transformed our understanding of microbial immunity and predicted the system’s extraordinary potential for programmable genome editing.5
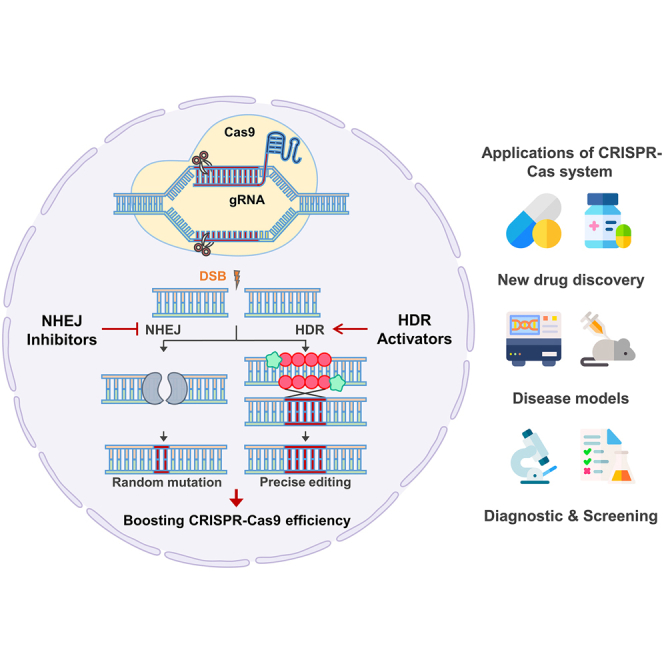
By utilizing a single effector protein and a customizable guide RNA (gRNA), CRISPR-Cas9 surpasses earlier engineered nucleases such as zinc-finger nucleases and transcription activator-like effector nucleases in simplicity and flexibility.6 This efficient architecture has catalyzed rapid adoption across diverse organisms and cell types, facilitating both loss-of-function screens and precise gene corrections. Central to its diverse function is the cellular response to Cas9-induced double-strand breaks (DSBs), which are resolved by competing repair pathways, primarily error-prone non-homologous end joining (NHEJ) and high-fidelity homology-directed repair (HDR).7,8 Although NHEJ-mediated insertion or deletion events have proven invaluable for gene disruption, the therapeutic aspiration of precise gene correction mandates the enhancement of HDR efficiency in mammalian cells, where this pathway is intrinsically restricted by cell-cycle constraints and enzymatic competition.
Early efforts to bias repair outcomes employed pharmacological inhibitors such as SCR7 to transiently impede DNA ligase IV, shifting the balance toward HDR.9,10 Complementary strategies have since emerged to recruit HDR machinery directly to DSB sites, including fusions of Cas9 to end-resection factors like CtIP or Mre11-Rad50-Nbs1 (MRN) complex-recruiting domains.11 Parallel advances in small-molecule modulation, notably the emergence of RS-1 as a RAD51 activator, have further amplified HDR frequencies up to 6-fold in diverse model systems.10,12 However, these approaches are tempered by concerns of off-target effects, genome-wide perturbations, and cell type specificity, underscoring the need for refined control over both nuclease activity and repair dynamics.
Conversely, suppression of NHEJ components such as Ku70/80 and 53BP1 via small molecules or dominant-negative Cas9 fusions offers an orthogonal route to precision editing by forestalling rapid end ligation and allowing resection-dependent repair.10,13,14 These dual paradigms of HDR enhancement and NHEJ suppression have demonstrated synergistic gains in editing fidelity, yet their broad application in therapeutic contexts remains constrained by challenges in delivery, immunogenicity, and locus-specific control.
Beyond the repair landscape, the therapeutic translation of CRISPR-Cas9 depends on overcoming immunological barriers to bacterial nucleases, optimizing delivery vehicles for clinical-grade applications, and mitigating nuclease promiscuity through high-fidelity variants and chemical guide modifications.15 The field has responded with innovations such as lipid nanoparticle formulations for ribonucleoprotein delivery, epitope-engineered Cas9 orthologs with reduced antigenicity, and inducible anti-CRISPR modules for temporal regulation of editing.16
In this review, we discuss recent advances in HDR-promoting and NHEJ-suppressing strategies, describe emerging solutions to CRISPR-Cas9’s systemic weaknesses, and explore next-generation screening modalities that leverage diverse Cas effector platforms for drug discovery. Furthermore, we discuss key challenges associated with the therapeutic application of CRISPR and outline potential strategies to overcome them. Overall, this review underscores the promising potential of CRISPR-based approaches to enable more effective and disease-specific gene therapies.
History of the CRISPR-Cas9 system
The discovery of the CRISPR-Cas9 system was not a single feature but rather the result of a series of scientific advances that unfolded over several decades. The first clues emerged in 1987, when a group of researchers identified a series of unusual repetitive sequences in the genome of Escherichia coli.4,17 Although the function of these sequences was unclear at the time, similar patterns were later found in a wide range of bacterial and archaeal genomes, drawing growing interest from the scientific community (Figure 1).
Figure 1.

Historical overview of CRISPR technologies
CRISPR sequences were first identified in Escherichia coli in 1987 and subsequently observed across various prokaryotes between 1993 and 2005. The discovery of Cas genes in 2002 and the presence of viral DNA fragments within CRISPR spacers in 2005 provided insights into its potential role in adaptive immunity, which was experimentally validated in 2007. In 2013, two independent groups repurposed the CRISPR-Cas9 system as a programmable genome-editing tool. The first clinical application of CRISPR-Cas9 occurred in China in 2016. Over the past 3 years, multiple CRISPR-based therapies have entered clinical stages. In recognition of their pioneering work in developing CRISPR-Cas9 as a genome-editing technology, Emmanuelle Charpentier and Jennifer Doudna were awarded the Nobel Prize in Chemistry in 2020.
In the mid-1990s, a significant step came forward when a researcher identified similar repetitive elements in an archaeal species and hypothesized that these structures might play a role in a prokaryotic immune system.17,18 These specific sequences included fragments of viral DNA integrated into the host genome. This bold idea began to gain traction in the early 2000s as more evidence emerged linking these sequences to resistance against viral infection.18
The concept of CRISPR as an adaptive immune system was experimentally validated in 2007, when a key study showed that certain bacteria could acquire resistance to phages by incorporating new DNA fragments into their CRISPR loci.19 This marked the first clear demonstration that CRISPR systems could provide heritable, sequence-specific immunity.
The term CRISPR was formally introduced in the early 2000s by researchers who also identified a group of genes consistently located near CRISPR sequences20 (Figure 1). These genes, later called Cas (CRISPR-associated) genes, were found to encode proteins that play essential roles in the function of the CRISPR system.20,21 Among these, Cas9 stood out for its DNA-cutting ability, which was later linked to specific conserved nuclease motifs.
Another key advance came with the discovery of RNA molecules that guide Cas proteins to their DNA targets. Scientists identified small RNAs, now known as CRISPR RNAs (crRNAs), which are processed from long precursor transcripts containing both repeats and viral-derived spacer sequences.20,22 Further research revealed the presence of a second RNA molecule, known as trans-activating crRNA (tracrRNA), which is required for the proper processing of crRNAs and the activation of Cas9. A major turning point occurred when researchers demonstrated that crRNA and tracrRNA could be fused into a single-guide RNA (sgRNA), creating a simplified yet highly effective system for directing Cas9 to specific DNA sequences21,23,24 (Figure 1).
These foundational discoveries paved the way for the CRISPR-Cas9 system to be developed into a powerful genome-editing tool. Its simplicity requiring only a Cas9 protein and a customizable gRNA made it more accessible and adaptable than previous genome-editing technologies like ZFNs and TALENs.6,25 Since then, CRISPR-Cas9 has revolutionized genetic engineering and has become a cornerstone of modern biotechnology, with widespread applications in basic science, medicine, agriculture, and beyond.26
Components and functions of the Cas9 complex
The CRISPR-Cas9 structure, particularly that of Streptococcus pyogenes Cas9 (SpCas9), underlies its remarkable precision and flexibility in genome-editing applications.3 As a multidomain endonuclease, SpCas9 executes site-specific DNA cleavage through the coordinated actions of distinct architectural modules.3,19 Key to this function is the division of the protein into two functional lobes: the recognition (REC) lobe and the nuclease (NUC) lobe.27 The REC lobe, comprising the REC1 and REC2 domains, facilitates gRNA binding and conformational regulation, enabling the surveillance complex to achieve high-fidelity recognition of the DNA substrate. REC1 constitutes the largest subdomain within the REC lobe and plays a pivotal role in stabilizing the gRNA-DNA heteroduplex27,28,29 (Figure 2). Structural analyses reveal that REC1 forms an extended helical bundle that cradles the gRNA backbone, allowing the RNA to adopt a catalytically competent conformation.29 This domain also mediates extensive interactions with the spacer region of the sgRNA, thereby enhancing the specificity of base pairing with the complementary target strand.29,30
Figure 2.

Schematic overview of the Cas9-gRNA complex and its mechanism of action in the presence of target DNA
Compartments of the CRISPR-Cas9 complex bound to the target DNA and guide RNA (gRNA). Cas9 consists of two regions, called the recognition (REC) lobe and the nuclease (NUC) lobe. The REC lobe, comprising the REC1 and REC2 domains, is responsible for nucleic acid recognition. The NUC lobe contains the HNH and RuvC domains and a C-terminal region containing a PAM-interaction domain. The HNH domain and the RuvC domain cleave the target DNA double strand, forming a duplex with crRNA and the other DNA strand, respectively.
In contrast, the REC2 domain, though smaller in size and exhibiting structural variability across Cas9 orthologs, functions primarily as a conformational modulator30 (Figure 2). Upon target recognition, REC2 undergoes significant rearrangement that transmits allosteric signals to the NUC lobe, thereby coupling RNA recognition to catalytic activation.30,31 Notably, REC2 deletion mutants retain residual DNA cleavage activity, indicating that while this domain is not strictly essential for catalysis, it contributes to the kinetic efficiency and structural integrity of the ribonucleoprotein complex.31,32 Recent cryoelectron microscopy studies suggest that REC2 also participates in sensing mismatches between the gRNA and target DNA, implicating it in the fidelity checkpoint mechanism of Cas9.33
Anchored to the REC lobe is the NUC lobe, which harbors the dual nuclease domains, RuvC and HNH, responsible for the endonucleolytic cleavage of the DNA strands34 (Figure 2). The RuvC domain, named after the bacterial Holliday junction resolvase RuvC, adopts an RNase H-like fold and catalyzes cleavage of the non-target DNA strand.34 It relies on a conserved DEDDh motif to coordinate divalent metal ions, typically Mg2+, essential for phosphodiester bond hydrolysis.35 RuvC executes its function in a metal-dependent manner, cleaving the non-complementary strand approximately three nucleotides upstream of the protospacer adjacent motif (PAM) (Figure 2). The HNH domain, in contrast, cleaves the complementary strand of the DNA duplex and is named for its conserved histidine-asparagine-histidine catalytic triad.36 Structurally, it comprises a ββα-metal fold characteristic of nucleases that utilize a single divalent ion for catalysis.36 Importantly, the HNH domain exhibits target-dependent activation. It remains catalytically inactive until precise base pairing between the gRNA and target DNA is established.36,37 This dependency is central to the proofreading capacity of Cas9, ensuring that off-target effects are minimized through conformational gating mechanisms. Collectively, the HNH domain aligns with the target strand and cleaves the DNA in a coordinated fashion with RuvC, resulting in a blunt-ended DSB.
The precise targeting of Cas9 is further dictated by the PAM, a short conserved DNA sequence immediately downstream of the target site.28 For SpCas9, this sequence is typically 5′-NGG-3′, where “N” can be any nucleotide.38 The PAM is not recognized by the gRNA but rather by the PAM-interacting domain situated within the NUC lobe. Structural interrogation of the PAM-binding pocket reveals a constellation of arginine and glutamine residues that insert into the DNA minor groove to interrogate the presence of the canonical NGG motif38,39 (Figure 2). This interaction serves two purposes: first, it enables Cas9 to distinguish self from non-self DNA by ensuring that only foreign sequences flanked by a PAM are targeted, and second, it triggers local DNA strand unwinding to facilitate R-loop formation.39,40 The thermodynamic asymmetry introduced by the PAM facilitates directional strand invasion by the gRNA and stabilizes the RNA-DNA hybrid through continuous stacking interactions.40
Collectively, the interdependent functionalities of the REC1, REC2, HNH, RuvC, and PAM-interacting domains orchestrate a highly regulated sequence of molecular events that culminate in programmable DNA cleavage (Figure 2). The process begins with PAM surveillance and gRNA-guided target binding, which induces conformational shifts in the REC lobe, particularly within REC2.35,37,38,39 This structural rearrangement licenses the activation of the HNH and RuvC domains, which execute strand-specific cleavage in a temporally coordinated manner.32,37 The evolutionary convergence of these domains into a single polypeptide allows for exceptional modularity, providing a framework that has been readily adapted for diverse biotechnological applications.21 Indeed, protein engineering efforts have exploited the structural plasticity of the REC lobe to enhance target specificity, while mutational studies of RuvC and HNH have yielded catalytically dead or nickase variants instrumental for base editing and transcriptional modulation.32 Understanding the mechanistic interplay among these domains not only illuminates the natural logic of CRISPR-mediated immunity but also expands the toolkit for precise genome manipulation.
NHEJ pathway in DSB repair
Although DSBs can be remedied via either NHEJ or HDR, NHEJ predominates in mammalian cells regardless of the cell cycle phase.41 The repair process initiates with the rapid recruitment of the Ku heterodimer, comprising Ku70 (∼70 kDa) and Ku80 (∼80 kDa), which forms a pre-assembled ring that encircles the duplex DNA immediately at the DSB ends.41 This interaction not only protects the DNA termini from exonucleolytic degradation but also functions as a scaffold for the sequential assembly of the NHEJ machinery (Figure 3).
Figure 3.

The mechanism and outcome of two major DNA repair pathways, NHEJ and HDR, used by CRISPR-Cas9-induced DSBs
Cas9 ribonucleoprotein introduces a site-specific DSB, which is primarily repaired by either the error-prone NHEJ pathway or the high-fidelity HDR pathway. In the NHEJ pathway (left), the Ku70/Ku80 heterodimer recognizes and binds to DSB ends, recruiting 53BP1 to protect the ends from resection. DNA-PK stabilizes the complex and promotes synapsis of the DNA ends. XRCC4 recruits DNA ligase IV to ligate the DNA ends, often resulting in insertions or deletions. In the HDR pathway (right), the MRN complex comprising MRE11, RAD50, and NBS1 is recruited to DSBs and facilitates end resection, which is further promoted by CtIP. Single-stranded DNA is coated with RPA, followed by replacement with RAD51 to form a nucleoprotein filament. This filament invades the homologous donor template, forming a D-loop that serves as a platform for DNA synthesis and precise repair. Exo1 and the Dna2/BLM complex contribute to extended end resection, further promoting strand invasion and repair accuracy. For efficient strand invasion and integration, the donor DNA must share homology with the target sequence, and it is often resected at both ends to enable proper alignment and recombination with the host genome.
Key to the pathway are several distinct sub-pathways that are determined by the nature of the DNA ends. First, blunt end ligation occurs in cases where DSBs generate blunt ends, with the Ku complex facilitating the recruitment of X-ray repair cross-complementing protein 4 (XRCC4) and DNA ligase IV.41,42 These factors form a stable ligation complex that directly rejoins the DNA termini with minimal processing. Second, nuclease-dependent processing occurs when DSBs display 5′ or 3′ overhangs (commonly referred to as sticky ends), prompting the Ku complex to recruit DNA-dependent protein kinase catalytic subunit (DNA-PKcs). DNA-PKcs, upon binding to the Ku-DNA complex, promotes the formation of an enzymatically active complex that engages artemis.43,44 Artemis endonuclease is then activated to trim the overhangs, thereby generating ends amenable to ligation by XRCC4-DNA ligase IV, often in conjunction with accessory factors such as XRCC4-like factor and paralog of XRCC444 (Figure 3). Third, polymerase-dependent filling and microhomology involves the processing of mismatched 3′ overhangs, which may alternatively engage template-dependent polymerases, such as Pol μ and Pol λ.45,46 These polymerases, recruited via the Ku-DNA interaction, facilitate the generation of terminal microhomology that promotes end joining through a mechanism that sometimes involves limited nucleolytic processing by artemis in coordination with DNA-PKcs.44
The orchestration of these proteins ensures that, despite occasional insertions or deletions (InDels) introduced during processing, the NHEJ pathway remains a critical, template-independent mechanism for preserving genomic stability.47 Notably, some studies have underscored that ablation of core components such as Ku80 or XRCC4 abolishes even the subset of error-free repair events, highlighting their indispensable roles.47,48
HDR pathway in DSB repair
HDR is a highly accurate mechanism for resolving DSBs, predominantly active during the S and G2 phases of the cell cycle when a sister chromatid is available as a template.49 Upon the occurrence of a DSB, the MRN complex rapidly recognizes and binds to the break site, thereby initiating the end resection process by degrading the 5′ termini to produce short 3′ single-stranded DNA (ssDNA) overhangs50 (Figure 3). Almost simultaneously, the MRN complex recruits the ATM kinase to the damage site, where ATM phosphorylates a number of substrates including CtIP.51 This phosphorylation event activates CtIP, which in turn interacts with BRCA1 to form a stable assembly with the MRN complex; the combined action of this BRCA1/MRN/CtIP unit extends the resection beyond the initial short overhangs, yielding long 3′ ssDNA tracts that are essential for the downstream repair process51,52 (Figure 3). These extended ssDNA regions are then rapidly coated by replication protein A (RPA), which serves to stabilize the ssDNA and prevent the formation of secondary structures that could impede proper repair.53,54 The RPA-coated ssDNA is subsequently subjected to a critical handoff mediated by BRCA2, which facilitates the replacement of RPA with RAD51. The loading of RAD51 onto the ssDNA forms a nucleoprotein filament that is central to the search for a homologous template, as this filament actively invades an intact duplex of DNA, most commonly the sister chromatid, forming a displacement loop (D-loop)55 (Figure 3). This RAD51 nucleoprotein filament not only facilitates the homology search but also stabilizes the D-loop structure and protects the invading strand from nucleolytic degradation, thereby ensuring efficient and accurate strand invasion and extension. Within this invasion structure, DNA polymerases are recruited to extend the 3′ end of the invading strand using the homologous sequence as a template. Following strand invasion, the repair process can proceed by two closely related sub-pathways: in the classical DSB repair pathway, the formation of joint molecules that include Holliday junctions occurs, which are later processed by resolvases to yield either crossover or non-crossover products, thereby potentially exchanging genetic material between sister chromatids.53,54,55 Conversely, the synthesis-dependent strand annealing pathway involves a limited period of DNA synthesis after strand invasion, after which the nascent strand dissociates from the donor template and anneals back to its original complementary strand, invariably resulting in a non-crossover outcome.54,56,57 In both sub-pathways, after the synthesis step, any remaining gaps are filled in, and the nicks are sealed by ligases, thereby restoring the integrity and continuity of the DNA molecule with high fidelity. The coordinated action of these proteins, spanning from the MRN complex and ATM through CtIP, BRCA1, RPA, BRCA2, and RAD51, ensures that HDR provides a precise repair mechanism, effectively maintaining genomic stability while minimizing the risk of mutation.10,56 Furthermore, recent findings demonstrate that exogenous overexpression of RAD51 not only facilitates HDR but also enhances CRISPR-Cas9-mediated genome editing efficiency by increasing the frequency of on-target DSBs, likely through the stabilization of R-loop structures and promotion of Cas9-gRNA complex formation at target sites.12
Enhancing CRISPR-Cas9 precision through HDR pathway extension
To enable CRISPR-Cas9-mediated genome editing, the generation of DSBs is an essential requirement. Therefore, efficient DNA repair processes following DSB induction constitutes a critical determinant of the accuracy of genome editing. Following the generation of DSBs, the endogenous cellular machinery predominantly engages either the NHEJ pathway or the high-fidelity HDR pathway to resolve the lesion. Despite its accuracy, HDR is inherently less efficient in somatic cells due to cell cycle constraints and pathway competition, limiting its utility in therapeutic and research contexts that demand precision.8,14,56,58 To address these limitations, several strategies have emerged to promote HDR over NHEJ, thereby improving the efficacy and fidelity of CRISPR-Cas9-mediated gene editing.
One of the most effective strategies to enhance HDR involves modulating the activity of key DNA repair proteins. RAD51, a critical strand exchange protein central to HDR, facilitates homology search and strand invasion, forming nucleoprotein filaments on resected ssDNA. Overexpression of RAD51 has been shown to significantly increase knockin and knockout efficiencies in eukaryotic cells (Table 1).10,59,60 Furthermore, the combination of RAD51 overexpression with pharmacological inhibitors of the NHEJ pathway has demonstrated synergistic effects. For example, SCR7, an inhibitor of DNA ligase IV, a core enzyme in NHEJ, disrupts re-ligation of DSB ends and shifts repair dynamics in favor of HDR. In human embryonic kidney (HEK293T) cells, co-administration of RAD51 and SCR7 resulted in a notable increase in knockin efficiency, with a concomitant reduction in R-loop accumulation, suggesting an interplay between transcription-associated DNA structures and repair pathway choice.9,10,61 This dual strategy exemplifies the utility of both promoting HDR machinery and simultaneously suppressing competing repair processes.
Table 1.
Small molecules classified as NHEJ inhibitors and HDR activators based on their mechanism in enhancing CRISPR-Cas9 genome editing
| Target protein | Small molecule | Observed effects |
|---|---|---|
| NHEJ inhibitor | ||
| DNA-PKcs | AZD7648 | inhibits DNA-PKcs, suppressing NHEJ initiation |
| DNA-PKcs | NU7441 | blocks DNA-PKcs catalytic activity, reducing NHEJ |
| DNA-PKcs | KU-0060648 | dual PI3K/DNA-PKcs inhibitor, impairs NHEJ signaling |
| DNA ligase IV | SCR7 | inhibits DNA ligase IV, prevents end ligation in NHEJ |
| MRE11 | mirin | inhibits MRE11, affecting resection and NHEJ resolution |
| Ku complex | STL127705 | blocks Ku70/80 interaction, reduces NHEJ recruitment |
| DNA ligase IV | SCR130 | targets ligase IV, blocks final NHEJ ligation step |
| DNA-PKcs | M3814 | selective DNA-PKcs inhibitor, suppresses NHEJ |
| Calmodulin | W7 | inhibits calmodulin, indirectly alters NHEJ dynamics |
| HDR activator | ||
| RAD51 | RAD51 | key recombinase in strand invasion during HDR |
| RAD18 | RAD18 | E3 ubiquitin ligase, facilitates post-replication HDR |
| RAD51 | RS-1 | allosteric activator of RAD51, enhances HDR strand pairing |
| β3-AR | L755507 | promotes HDR gene expression |
| ATM | resveratrol | activates ATM, enhances DSB end resection and HDR |
| NAE | MLN4924 | inhibits neddylation, upregulates HDR-related genes |
| GBF1 | brefeldin A | disrupts ER-Golgi transport, biases repair toward HDR |
| HDAC (class I) | entinostat | inhibits HDACs, opens chromatin to favor HDR machinery |
Small molecules are categorized based on their effects on either the NHEJ or HDR pathway. Molecules listed as NHEJ inhibitors (e.g., AZD7648, SCR7) function by suppressing critical proteins involved in end joining, such as DNA-PKcs and ligase IV, thereby favoring HDR. HDR activators (e.g., RS-1, resveratrol) promote strand invasion, end resection, or chromatin accessibility, enhancing the efficiency of precise repair. Mechanistic effects are derived from biochemical and cell-based studies using CRISPR-Cas9 systems.
Beyond protein overexpression and NHEJ inhibition, small-molecule modulators have emerged as potent enhancers of HDR. RS-1, a RAD51-stimulatory compound, binds and stabilizes RAD51-ssDNA filaments, enhancing strand invasion and D-loop formation (Table 1).10,62,63 Experimental data indicate that RS-1 can amplify HDR activity by 3- to 6-fold in multiple human cell lines and even in rabbit embryos when co-delivered with CRISPR-Cas9 components.10 The use of RS-1 is particularly advantageous due to its cell-permeable nature and transient action, which reduces concerns regarding long-term genomic instability.
In addition to pharmacological modulation, recent advancements have focused on re-engineering the Cas9 protein to bias repair outcomes toward HDR. Fusion of Cas9 to HDR-promoting protein domains such as CtIP, UL12, and MRN-recruiting motifs has shown promising results.11,64,65 CtIP, a critical nuclease involved in DNA end resection, enhances the generation of ssDNA overhangs conducive to RAD51 loading.11,64,65 Fusion constructs like Cas9-CtIP or MS2-CtIP significantly elevated the HDR/NHEJ ratio by up to 6-fold in HEK293 cells.11 Similarly, UL12, a domain derived from HSV-1 alkaline nuclease that recruits the MRN complex, when tethered to Cas9, augmented HDR efficiency 2-fold.64 These chimeric proteins integrate DSB induction with simultaneous recruitment of repair facilitators, presenting an elegant solution to boost precision editing.
Further expanding on HDR enhancement, genetic fusion of Cas9 with ubiquitin-binding domains from Rad18 or RNF169 has shown to upregulate BRCA1 and DNA end resection processes.58,66,67 This fusion strategy not only augments HDR but also suppresses NHEJ by interfering with 53BP1 recruitment, thereby improving the HDR/NHEJ ratio up to 6-fold in certain human cell lines.10,14 A novel approach leveraging dominant-negative RNF168, which lacks the RING domain, also demonstrated up to 7-fold improvement in error-free editing.66 These developments underscore the therapeutic potential of HDR-favoring constructs, particularly in ex vivo applications where delivery and expression can be tightly controlled.
Despite the growing repertoire of HDR-enhancing tools, one of the critical challenges lies in their lack of specificity to targeted cell types or editing loci.68,69 Many of these approaches, while potent, may cause undesired genome-wide perturbations or increase the risk of chromosomal rearrangements due to excessive end resection or prolonged repair activity.69,70 Nevertheless, these strategies hold great promise for applications in regenerative medicine, functional genomics, and gene therapy, particularly when confined to ex vivo contexts such as patient-derived stem cells or organoids.71,72
Taken together, enhancing HDR efficiency represents a foundation in advancing the precision of CRISPR-Cas9 genome editing. Through a combination of RAD51 modulation, small-molecule treatments such as RS-1 and SCR7, and innovative protein engineering strategies, researchers are increasingly able to overcome the inherent limitations of HDR in mammalian cells.9,10,11,50,62 As CRISPR-Cas technology progresses, future efforts will likely focus on integrating these tools into delivery systems that ensure cell type specificity, temporal control, and minimized genomic perturbation, paving the way for safe and efficient clinical applications of gene editing technologies.
Enhancing CRISPR-Cas9 precision: Strategies to suppress NHEJ
In many cell types, NHEJ acts as the default repair mechanism, quickly ligating DNA ends with minimal regard for sequence accuracy. Consequently, while CRISPR-Cas9 can efficiently introduce InDels, achieving targeted sequence replacement or gene correction requires overcoming the cellular bias toward NHEJ.73
A growing body of research has therefore focused on shifting the DSB repair balance toward HDR by directly or indirectly inhibiting NHEJ components. Pharmacological and genetic strategies targeting key mediators of NHEJ such as DNA-PKcs, Ku70/80, 53BP1, and SHROOM1 have emerged as promising avenues for enhancing CRISPR-mediated precision editing (Table 1).13,14,43,44,74 One of the earliest efforts in this direction involved the small molecule SCR7, which inhibits DNA ligase IV, a core NHEJ enzyme.10 Treatment with SCR7 has been shown to significantly improve the efficiency of HDR-mediated editing in mammalian cells, highlighting the utility of transiently disrupting NHEJ to favor accurate repair outcomes.10,75
Further evidence for this approach comes from studies targeting Ku proteins, which rapidly recognize DSBs and recruit downstream NHEJ factors.48 Ku70/80 form a heterodimer that binds to DNA ends and initiates the NHEJ cascade. The multifunctionality of Ku including its DNA-binding and scaffolding properties makes it a crucial early checkpoint in the repair pathway.76 Interference with Ku function, either through small molecules or dominant-negative constructs, has been demonstrated to increase HDR rates by preventing NHEJ initiation and allowing greater access to resection-dependent HDR machinery.76
The suppression of 53BP1, a key NHEJ-promoting factor that inhibits DNA end resection, has also emerged as a potent enhancer of CRISPR precision. Genetic knockout or knockdown of 53BP1 shifts the balance toward HDR, enabling more efficient homology-based gene insertion (Table 1).14,77 This concept was further advanced through the development of Cas9 fusion proteins, where a dominant-negative form of 53BP1 is tethered to the Cas9 enzyme itself.77 These fusions localize the NHEJ-inhibitory effect precisely to the site of editing, reducing off-target consequences and increasing local HDR activity.56 This strategy demonstrates a sophisticated regulation of repair dynamics, customized for CRISPR applications, by restricting NHEJ exclusively to the target genomic site.
In parallel, suppression of less-characterized NHEJ regulators has yielded surprising improvements in editing fidelity. One notable example is SHROOM1, a cytoskeletal regulator whose silencing unexpectedly enhanced HDR efficiency both in vitro and in vivo.74,78 Although the mechanistic link between SHROOM1 and DSB repair remains incompletely understood, its suppression appears to influence nuclear architecture or chromatin accessibility in ways that favor homology-based repair.79 This finding expands the list of potential NHEJ targets and suggests that manipulating non-canonical pathways may further enhance CRISPR outcomes.
Combining multiple strategies to suppress NHEJ and promote HDR can produce synergistic effects. The co-application of RAD51, a recombinase that facilitates strand invasion during HDR, and SCR7 has been shown to significantly boost editing precision.9,10,53 In particular, this combination was reported to mitigate R-loop accumulation at DSB sites, which otherwise impedes repair progression.80 These results indicate that modulating both HDR enhancers and NHEJ inhibitors simultaneously can adjust the cellular progression for efficient and accurate genome editing.
The interplay between NHEJ suppression and HDR enhancement is complex and context dependent, influenced by cell type, cell cycle phase, and chromatin. However, the cumulative evidence underscores a central principle: transient and localized inhibition of NHEJ can unlock the full potential of CRISPR-Cas9 for precise genome manipulation.30,65 As genome editing moves toward therapeutic applications, especially in human cells where HDR is tightly regulated, these strategies will be indispensable for achieving clinically relevant levels of precision and safety.65,73 Future efforts will likely focus on refining these approaches to minimize toxicity, avoid global DNA repair disruption, and enable tunable control over editing outcomes. The integration of NHEJ-inhibitory strategies with next-generation Cas9 variants and delivery systems holds promise for expanding the reach of CRISPR-based therapies.
Application of CRISPR-Cas technology in next-generation screening for novel drug development
The advent of CRISPR-based screening has revolutionized early-stage drug discovery, enabling systematic interrogation of gene function at unprecedented scale and precision. Since 2020, innovations across Cas9, Cas12, and Cas13 platforms have yielded versatile assays for identifying novel targets, explaining drug mechanisms and predicting resistance pathways (Figure 4).81 In this part, we explain the evolution of these technologies from pooled loss- and gain-of-function libraries to high-content screens and inducible systems highlighting how each modality contributes to the discovery of hit drugs, what unique advantages it confers, and where challenges remain.
Figure 4.

Applications of the CRISPR-Cas system
This diagram illustrates the diverse applications of CRISPR-Cas genome-editing technology across multiple fields. These include (1) functional genomics (e.g., gene knockout and regulatory element studies), (2) disease modeling using animal or cell-based systems, (3) drug target identification and validation, (4) synthetic lethality screening for cancer vulnerabilities, (5) gene therapy and personalized medicine approaches, and (6) agricultural and biotechnology innovations such as crop improvement and livestock engineering.
Pooled CRISPR-Cas9 knockout and activation libraries remain the backbone of functional drug screens.81 Initial genome-wide Cas9 knockout studies adapted well-curated gRNA collections to identify vulnerabilities in cancer cell lines under pharmacological pressure, revealing both established and unexpected resistance genes.81,82 Enhancements in gRNA design and delivery have improved coverage and on-target efficiency, such as custom libraries focusing on druggable gene families distinguished by screening throughput, while drug-enabled analysis empowered detection of subtle chemogenetic interactions by modeling drug-gene synergy from CRISPR depletion data.82,83 Parallel efforts have coupled Cas9 libraries with high-content readouts, automated imaging, and single-cell profiling to capture complex phenotypes beyond survival, such as changes in cell morphology, signaling states, or subcellular localization.84 Such multidimensional data have uncovered modulators of drug uptake and intracellular trafficking inaccessible to viability only assays.
Functional genomic screening using CRISPR-Cas9 libraries has emerged as a powerful, unbiased approach for identifying potential druggable targets.81,83,84,85,86 For instance, a genome-wide CRISPR-Cas9 knockout screen using the human GeCKO v.2 library, which contains 65,386 sgRNAs targeting 19,052 protein-coding genes, identifies phosphoglycerate dehydrogenase (PHGDH) as a critical mediator of sorafenib resistance in hepatocellular carcinoma (HCC) cells. Genetic ablation of PHGDH enhances sorafenib-induced apoptosis in HCC cells, while pharmacological inhibition using NCT-503 synergistically suppresses HCC tumor growth in vivo.84 Similarly, murine SMB21 cells, a representative model for sonic hedgehog-driven medulloblastoma (SHH-MB), are screened using the Brie CRISPR-Cas9 knockout library, which provides 78,637 sgRNAs targeting 19,674 murine genes. This genome-scale approach effectively identifies DNA methyltransferase 1 (DNMT1) as a novel therapeutic target in sonic hedgehog (SHH)-dependent tumors. Genetic inhibition of DNMT1 significantly reduces tumor growth and prolongs survival in an SHH-MB mouse model by suppressing the SHH signaling pathway.83
Domain-focused CRISPR-Cas9 screening is performed using a customized sgRNA library targeting 192 chromatin regulatory domains in murine RN2 acute myeloid leukemia (AML) cells. Instead of genome-wide coverage, the domain-focused library comprises sgRNAs targeting functional protein domains, including bromodomains, methyltransferases, demethylases, acetyltransferases, deacetylases, and ATPase regions within each protein of interest. This targeted negative-selection screening identifies six known drug targets, such as BRD4, DOT1L, EHMT1, EHMT2, EZH2, and KDM1A, as well as 19 additional candidates essential for AML cell survival.81,85
Moreover, an in vivo CRISPR-Cas9 knockout screen in an orthotopic patient-derived xenograft model of pancreatic ductal adenocarcinoma (PDAC) utilizes a custom library of 8,031 sgRNAs targeting 619 epigenetic regulators. This screen aims to identify chromatin regulators whose inhibition creates synthetic lethality when PDAC cells are treated with gemcitabine, a standard chemotherapy agent for PDAC. Protein arginine methyltransferase gene 5 (PRMT5) emerges as a promising druggable candidate. Genetic depletion or pharmacological inhibition of PRMT5 significantly enhances gemcitabine-induced cytotoxicity by reducing RPA levels and HDR activity, causing DNA damage accumulation and cell death. Consequently, combinatorial treatment leads to synergistic tumor reduction in PDAC models in vivo.86 Collectively, these studies underscore the effectiveness of CRISPR-based screening approaches in discovering novel drug targets and identifying potent combinatorial therapeutic strategies in clinically relevant cancer models.
Complementing Cas9 knockout, CRISPR-Cas12 and Cas13 systems have recently been repurposed for drug screening with distinct modalities. Cas12a (Cpf1) enzymes, with their T-rich PAM requirement and ability to mediate multiplexed editing from a single transcript, have facilitated streamlined combinatorial screens targeting gene families or pathway modules.87,88 Meanwhile, catalytically inactive dead Cas13 fused to fluorescent reporters enables live-cell tracking of RNA dynamics, permitting focused screens of splicing regulators and noncoding RNA elements that modulate drug sensitivity.89 Beyond functional genomics, collateral cleavage activity of Cas12 and Cas13 has been developed for ultrasensitive detection of small molecules and biomarkers in diagnostic assays. For instance, ligand-responsive aptamer modules can gate Cas13 activity, translating metabolite binding into a fluorescent signal suitable for high-throughput compound profiling.87,88,89 These CRISPR-based biosensors, adaptable to microfluidic formats, promise to accelerate phenotypic screening in disease-relevant contexts, including pathogen drug interactions.
Recent advances have also addressed temporal control and context specificity.90 Inducible Cas9 platforms incorporate destabilizing domains or small-molecule-responsive switches to restrict editing to defined windows, reducing compensatory adaptations and lethal off-target effects during long-term screens.91 Such systems have been used to map stage-specific dependencies in differentiation models and in vivo tumor xenografts, enabling identification of drug targets that would be missed in constitutive screens.90,91 Likewise, integration of CRISPR screening with 3D organoid and co-culture models has begun to bridge the gap between cell-line-based assays and physiological complexity: Cas9 and Cas12a libraries delivered by optimized lentiviral or ribonucleoprotein methods have dissected microenvironmental influences on drug response, uncovering context-dependent vulnerabilities in cancer stem cells and immune tumor interactions.71,73,81,91
These CRISPR-based platforms offer several compelling advantages over traditional approaches. First, gRNA libraries allow for rapid and repeated targeting across genomic space, ranging from genome wide to pathway focused screens, without requiring the generation of large chemical libraries or cumbersome protein reagents.81 Second, the inherent expandability of pooled formats, coupled with next-generation sequencing readouts, delivers high statistical power and quantitative effect sizes, enabling detection of both strong and subtle modulators of drug action.81,83 Third, multiplexing capabilities whether via Cas12a’s crRNA arrays or dual guide designs permit combinatorial screening to uncover synthetic lethal interactions and drug-target synergies in a single experiment.87,92,93 Finally, the compatibility of CRISPR with diverse phenotypic readouts, including transcriptomic and proteomic endpoints, provides mechanistic insight far beyond binary viability outcomes, guiding downstream medicinal chemistry and target validation efforts.81,83
Looking ahead, the convergence of CRISPR technologies with emerging modalities such as base and prime editing, single-cell multiomics profiling, and AI-driven library design promises to further enhance screening precision and interpretability.94 Improved delivery vehicles, including engineered viral capsids and lipid nanoparticles, will expand CRISPR screens into primary cells and patient-derived models, better capturing clinical heterogeneity. Multiplexed biosensors that couple Cas effector collateral activity to barcoded reporters could enable simultaneous monitoring of dozens of molecular events in real time. In addition, integration of CRISPR perturbations with high-throughput structural and chemical proteomics may reveal not only which targets affect drug response but also how small molecules modulate complex interactomes.93,94 By addressing current technical bottlenecks and embracing these innovations, CRISPR-based drug screening stands poised to accelerate the translation of genomic insights into safer, more effective therapeutics.
CRISPR-Cas9-mediated immunotherapy and cancer treatment research
CRISPR-Cas9-based genome-editing technologies are being applied across diverse fields, including autoimmune, oncologic pathologies, and various areas of gene therapy research. In autoimmune diseases, where loss of self-tolerance precipitates tissue damage, CRISPR-mediated editing of key regulatory genes has provided mechanistic insights and novel therapeutic avenues. For example, in systemic lupus erythematosus (SLE), introduction of a C103A point mutation into the deubiquitinase domain of TNFAIP3 (A20) in human monocyte-like U937 cells disrupted A20’s ability to limit nuclear factor κB signaling.95 The resulting phenotype characterized by heightened neutrophil extracellular trap formation, increased citrullinated peptides, and a pro-inflammatory cytokine milieu mirrored the hyperactive immune state observed in patients with SLE.95 Likewise, CRISPR-Cas9-mediated knockdown of CXorf21, an X-linked gene implicated in the female predisposition to SLE, markedly reduced tumor necrosis factor alpha and interleukin-6 secretion in human immune cell lines, demonstrating its role in amplifying innate immune activation.96 Together, these studies emphasize how CRISPR-driven functional genomics can both validate pathogenic targets and inform strategies to restore tolerance.
Building upon gene discovery, CRISPR-engineered cellular therapies have generated promising results in preclinical and early clinical settings. Chimeric antigen receptor (CAR) T cells targeting CD19, traditionally used against B cell malignancies, have induced long-lasting remission in refractory SLE and rheumatoid arthritis by depleting autoreactive B cells and resetting immune homeostasis.97 More recently, CTX112, a CRISPR therapeutics autoimmune cocktail that concurrently disrupts regnase-1 and TGFBR2 in CAR-T cells demonstrated enhanced anti-inflammatory properties in a phase 1 trial for SLE initiated in early 2024. Preliminary data reveal robust disease control with minimal adverse events, and efforts are underway to extend this platform to systemic sclerosis and idiopathic inflammatory myopathies. In parallel, preclinical CRISPR targeting of TL1A, HDAC7, or IFNG in murine inflammatory bowel disease models attenuated colonic inflammation and reduced pro-inflammatory cytokine production.97 Similarly, knockout of activation-induced cytidine deaminase (also known as RAD51) in pancreatic β cells highlighted pathways driving β cell autoimmunity in type 1 diabetes, suggesting avenues to preserve β cell mass.98
In oncology, CRISPR-Cas9 is reshaping adoptive immunotherapy by enabling sophisticated editing of T cells, natural killer (NK) cells, and macrophages to overcome tumor-induced immunosuppression. Deletion of PDCD1 (PD-1) in CAR-T cells has conferred resistance to tumor microenvironment-driven exhaustion; when combined with simultaneous disruption of the endogenous T cell receptor, these edited cells generated sustained in vivo persistence exceeding 100 days, drove durable tumor regressions, and exhibited minimal off-target effects.99 To facilitate universal off-the-shelf therapies, CRISPR-mediated knockout of TRAC prevents graft-versus-host disease, while B2M disruption abrogates human leukocyte antigen (HLA) class I expression, reducing allorejection; exogenous expression of non-classical HLA-E further shields edited cells from NK cell-mediated elimination.100 Dual immune checkpoint disruption strategies, exemplified by CTX112’s additional knockout of regnase-1 and TGFBR2 in tandem with CAR insertion, yielded up to 10-fold enhancement of antitumor activity in preclinical models. Early clinical cohorts of relapsed or refractory B cell malignancies treated with these double-edited CAR-T cells have shown durable remissions and an acceptable safety profile. Beyond T cells, CRISPR editing of NK cells to delete inhibitory receptors or enhance cytokine expression is underway, and macrophages are being reprogrammed to resist tumor-derived immunosuppressive cues, expanding the repertoire of CRISPR-enabled cellular therapies.100
Lethal weaknesses and emerging solutions in CRISPR-Cas systems
The CRISPR-Cas system has revolutionized genome engineering by enabling naive, programmable DNA targeting and cleavage. However, its application in basic research and therapeutic development has been tempered by several critical limitations, including off-target mutagenesis, immunogenicity, delivery inefficiencies, and constrained editing modalities.68,93 These liabilities not only undermine editing precision and safety but also hinder clinical translation.
Since its adaptation from bacterial adaptive immunity, CRISPR-Cas9 has been lauded for its simplicity and flexibility. Yet, early interest revealed that Cas9’s reliance on RNA-DNA base pairing does not guarantee absolute fidelity.69 Genome-wide profiling has uncovered pervasive off-target cleavage at loci bearing partial sequence homology, a phenomenon attributed to tolerance for mismatches in the protospacer region and PAM-adjacent flexibility.101 These off-target events can trigger insertions, deletions, or chromosomal rearrangements, risking functional disruption of tumor suppressors or activation of oncogenes, thereby posing safety concerns in a therapeutic approach.93 Compounding this, the DSBs induced by Cas nucleases recruit endogenous DNA repair pathways, primarily NHEJ, which operates with low fidelity and often yields heterogeneous mutational spectra.7,41,58 Even HDR, when supplied with exogenous templates, competes poorly with NHEJ, limiting precise allele replacement efficiency.64
Another major hurdle stems from host immune recognition. As a prokaryotic protein, Cas9 can provoke both innate and adaptive immune responses.5,20 Preexisting antibodies and T cell memory against Cas9 variants (e.g., SpCas9) have been detected in human sera, raising the specter of inflammatory cytokine release, vector clearance, and diminished editing efficacy.5,20 Furthermore, the use of viral vectors such as adeno-associated virus can provoke additional immunogenicity and restrict repeat dosing. Beyond humoral immunity, pattern recognition receptors may sense foreign nucleic acids (e.g., gRNAs), activating interferon pathways and further complicating in vivo applications.102,103
Effective delivery of the CRISPR machinery remains a formidable challenge. The large size of Cas proteins (∼4.2 kb coding sequence for SpCas9) approaches or exceeds the packaging limits of many viral vectors, necessitating split-Cas systems or smaller orthologs, which often compromise activity or specificity.102,103 Nonviral modalities including lipid nanoparticles, gold nanoparticles, and polymeric carriers offer promise for transient delivery of Cas ribonucleoprotein complexes, reducing off-target and immunogenic problems.16,104 However, achieving tissue-specific targeting, endosomal escape, and efficient cellular uptake continues to impede robust in vivo editing.78 In the field of solid tumors, for example, the dense extracellular matrix and heterogeneous vascularization can further obstruct nanoparticle penetration, limiting therapeutic approach.91
Collectively, these weaknesses, nuclease promiscuity, error-prone repair, immunogenicity, and delivery constraints, delineate the current frontier of CRISPR-Cas translational research. A detailed mechanistic understanding of each liability has catalyzed the development of orthogonal solutions. To enhance specificity, rational mutagenesis and directed evolution have yielded high-fidelity Cas9 variants (e.g., eSpCas9, SpCas9-HF1) featuring diminished nonspecific DNA interactions, which reduce off-target cleavage by orders of magnitude without appreciable loss of on-target activity.105,106 Chemical modifications of gRNAs such as 2′-O-methyl and phosphorothioate linkages have been shown to stabilize the RNA, improve nuclease loading, and further suppress off-target activity.107
Complementing nuclease engineering, the discovery and application of anti-CRISPR proteins provide sequential control over editing events. Endogenous to phage-infected bacteria, these small proteins bind to Cas9 and inhibit its catalytic function. Transient co-delivery or inducible expression of anti-CRISPR (Acr) modules enables on-demand editing windows, reducing unintended DSB accumulation and off-target succession.108 Similarly, combining Cas9 to small-molecule-responsive degrons permits external modulation of nuclease stability, offering another layer of control over editing duration.109
Strategies to modulate DNA repair have also emerged. Small-molecule inhibitors of NHEJ factors (e.g., DNA-PK inhibitor such as AZD7648) shift the balance toward HDR, increasing precise editing yields. Co-delivery of HDR-promoting proteins, like RAD51 or HDR stimulators (e.g., RAD18), further enhances template-mediated repair in mammalian cells.10,12,58 In Saccharomyces cerevisiae, genetic ablation of competing repair pathways and targeted recruitment of HDR complexes have achieved near-complete precise editing, underscoring the potential of repair manipulation as a universal strategy.110
To challenge immunogenicity, researchers have explored human-derived and engineered Cas orthologs with reduced antigenicity. For instance, Cas12a from Acidaminococcus sp. exhibits distinct PAM requirements and lower immunogenic epitopes, offering alternative editing platforms.111 Deimmunization via epitope deletion or sequence humanization has yielded Cas9 variants with attenuated antibody recognition while preserving function.111 Concurrently, non-viral delivery of ribonucleoproteins or mRNA, hereby avoiding prolonged Cas expression, minimizes immune stimulation and facilitates repeated dosing.
Finally, advances in nanomedicine are redefining delivery paradigms. Lipid nanoparticle formulations optimized for endosomal escape and organ targeting have demonstrated efficient in vivo liver editing in preclinical models, achieving therapeutic correction of metabolic disorders with minimal off-target issues.104 Hybrid systems combining peptide ligands, DNA aptamers, and responsive polymer matrices are under development to confer cell type specificity and improve pharmacokinetics, setting the stage for precision therapeutics.78
Conclusions
CRISPR-Cas system weaknesses, off-target activity, error-prone repair, immunogenicity, and delivery obstacles, reflect the inherent tension between microbial defense mechanisms and mammalian therapeutic demands. Yet, through synergistic engineering of nucleases, guides, regulators, and carriers, current advancements are driving the evolution of genome editing into a stage marked by enhanced safety, precision, and clinical applicability. As these complicated strategies mature, they will underpin the next generation of CRISPR-based interventions, transforming the promise of gene therapy into widespread reality.
Acknowledgments
This work was supported by National Research Foundation of Korea (NRF) funded by the Korea government (MSIT) (no. RS-2023-00247618), Chungcheongbuk-do RISE (Regional Innovation System & Education) grants funded by the Ministry of Education and Chungcheongbuk-do, and the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Ministry of Science, ICT & Future Planning (NRF-2022M3A9J4079468).
Author contributions
S.-J.P., G.E.L., S.M.C., and E.-H.C. contributed equally to this manuscript. S.-J.P., G.E.L., and E.-H.C. completed literature review, investigation, wrote the original draft, and designed the figures. E.-H.C. conceived the study, revised the draft, and provided funding acquisition and supervision. All the authors approved the submission.
Declaration of interests
The authors declare no competing interests.
References
- 1.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Redman M., King A., Watson C., King D. What is CRISPR/Cas9? Arch. Dis. Child. Educ. Pract. Ed. 2016;101:213–215. doi: 10.1136/archdischild-2016-310459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khoshandam M., Soltaninejad H., Mousazadeh M., Hamidieh A.A., Hosseinkhani S. Clinical applications of the CRISPR/Cas9 genome-editing system: Delivery options and challenges in precision medicine. Genes Dis. 2024;11:268–282. doi: 10.1016/j.gendis.2023.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gostimskaya I. CRISPR-Cas9: A History of Its Discovery and Ethical Considerations of Its Use in Genome Editing. Biochemistry. 2022;87:777–788. doi: 10.1134/S0006297922080090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rath D., Amlinger L., Rath A., Lundgren M. The CRISPR-Cas immune system: biology, mechanisms and applications. Biochimie. 2015;117:119–128. doi: 10.1016/j.biochi.2015.03.025. [DOI] [PubMed] [Google Scholar]
- 6.Gupta R.M., Musunuru K. Expanding the genetic editing tool kit: ZFNs, TALENs, and CRISPR-Cas9. J. Clin. Investig. 2014;124:4154–4161. doi: 10.1172/JCI72992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xue C., Greene E.C. DNA Repair Pathway Choices in CRISPR-Cas9-Mediated Genome Editing. Trends Genet. 2021;37:639–656. doi: 10.1016/j.tig.2021.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liao H., Wu J., VanDusen N.J., Li Y., Zheng Y. CRISPR-Cas9-mediated homology-directed repair for precise gene editing. Mol. Ther. Nucleic Acids. 2024;35 doi: 10.1016/j.omtn.2024.102344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu Z., Shi Z., Guo X., Jiang B., Wang G., Luo D., Chen Y., Zhu Y.S. Ligase IV inhibitor SCR7 enhances gene editing directed by CRISPR-Cas9 and ssODN in human cancer cells. Cell Biosci. 2018;8 doi: 10.1186/s13578-018-0200-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park S.J., Park S.J., Kwon Y.W., Choi E.H. Synergistic combination of RAD51-SCR7 improves CRISPR-Cas9 genome editing efficiency by preventing R-loop accumulation. Mol. Ther. Nucleic Acids. 2024;35 doi: 10.1016/j.omtn.2024.102274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Charpentier M., Khedher A.H.Y., Menoret S., Brion A., Lamribet K., Dardillac E., Boix C., Perrouault L., Tesson L., Geny S., et al. CtIP fusion to Cas9 enhances transgene integration by homology-dependent repair. Nat. Commun. 2018;9 doi: 10.1038/s41467-018-03475-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park S.J., Yoon S., Choi E.H., Hyeon H., Lee K., Kim K.P. Elevated expression of exogenous RAD51 enhances the CRISPR/Cas9-mediated genome editing efficiency. BMB Rep. 2023;56:102–107. doi: 10.5483/BMBRep.2022-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fan Y., Li J., Wei W., Fang H., Duan Y., Li N., Zhang Y., Yu J., Wang J. Ku80 gene knockdown by the CRISPR/Cas9 technique affects the biological functions of human thyroid carcinoma cells. Oncol. Rep. 2019;42:2486–2498. doi: 10.3892/or.2019.7348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jayavaradhan R., Pillis D.M., Goodman M., Zhang F., Zhang Y., Andreassen P.R., Malik P. CRISPR-Cas9 fusion to dominant-negative 53BP1 enhances HDR and inhibits NHEJ specifically at Cas9 target sites. Nat. Commun. 2019;10 doi: 10.1038/s41467-019-10735-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang S.W., Gao C., Zheng Y.M., Yi L., Lu J.C., Huang X.Y., Cai J.B., Zhang P.F., Cui Y.H., Ke A.W. Current applications and future perspective of CRISPR/Cas9 gene editing in cancer. Mol. Cancer. 2022;21 doi: 10.1186/s12943-022-01518-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duan L., Ouyang K., Xu X., Xu L., Wen C., Zhou X., Qin Z., Xu Z., Sun W., Liang Y. Nanoparticle Delivery of CRISPR/Cas9 for Genome Editing. Front. Genet. 2021;12 doi: 10.3389/fgene.2021.673286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ishino Y., Krupovic M., Forterre P. History of CRISPR-Cas from Encounter with a Mysterious Repeated Sequence to Genome Editing Technology. J. Bacteriol. 2018;200 doi: 10.1128/JB.00580-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Terns R.M., Terns M.P. CRISPR-based technologies: prokaryotic defense weapons repurposed. Trends Genet. 2014;30:111–118. doi: 10.1016/j.tig.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero D.A., Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 20.Barrangou R., Marraffini L.A. CRISPR-Cas systems: Prokaryotes upgrade to adaptive immunity. Mol. Cell. 2014;54:234–244. doi: 10.1016/j.molcel.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang F., Doudna J.A. CRISPR-Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017;46:505–529. doi: 10.1146/annurev-biophys-062215-010822. [DOI] [PubMed] [Google Scholar]
- 22.Karvelis T., Gasiunas G., Miksys A., Barrangou R., Horvath P., Siksnys V. crRNA and tracrRNA guide Cas9-mediated DNA interference in Streptococcus thermophilus. RNA Biol. 2013;10:841–851. doi: 10.4161/rna.24203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hille F., Charpentier E. CRISPR-Cas: biology, mechanisms and relevance. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2016;371 doi: 10.1098/rstb.2015.0496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhattacharya S., Satpati P. Insights into the Mechanism of CRISPR/Cas9-Based Genome Editing from Molecular Dynamics Simulations. ACS Omega. 2023;8:1817–1837. doi: 10.1021/acsomega.2c05583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hillary V.E., Ceasar S.A. A Review on the Mechanism and Applications of CRISPR/Cas9/Cas12/Cas13/Cas14 Proteins Utilized for Genome Engineering. Mol. Biotechnol. 2023;65:311–325. doi: 10.1007/s12033-022-00567-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu H., Li C., Gao C. Applications of CRISPR-Cas in agriculture and plant biotechnology. Nat. Rev. Mol. Cell Biol. 2020;21:661–677. doi: 10.1038/s41580-020-00288-9. [DOI] [PubMed] [Google Scholar]
- 27.Nethery M.A., Korvink M., Makarova K.S., Wolf Y.I., Koonin E.V., Barrangou R. CRISPRclassify: Repeat-Based Classification of CRISPR Loci. CRISPR J. 2021;4:558–574. doi: 10.1089/crispr.2021.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vink J.N.A., Baijens J.H.L., Brouns S.J.J. PAM-repeat associations and spacer selection preferences in single and co-occurring CRISPR-Cas systems. Genome Biol. 2021;22 doi: 10.1186/s13059-021-02495-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Makarova K.S., Koonin E.V. Annotation and Classification of CRISPR-Cas Systems. Methods Mol. Biol. 2015;1311:47–75. doi: 10.1007/978-1-4939-2687-9_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee J.K., Jeong E., Lee J., Jung M., Shin E., Kim Y.H., Lee K., Jung I., Kim D., Kim S., Kim J.S. Directed evolution of CRISPR-Cas9 to increase its specificity. Nat. Commun. 2018;9 doi: 10.1038/s41467-018-05477-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nishimasu H., Ran F.A., Hsu P.D., Konermann S., Shehata S.I., Dohmae N., Ishitani R., Zhang F., Nureki O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156:935–949. doi: 10.1016/j.cell.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palermo G., Chen J.S., Ricci C.G., Rivalta I., Jinek M., Batista V.S., Doudna J.A., McCammon J.A. Key role of the REC lobe during CRISPR-Cas9 activation by 'sensing', 'regulating', and 'locking' the catalytic HNH domain. Q. Rev. Biophys. 2018;51 doi: 10.1017/S0033583518000070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakagawa R., Hirano H., Omura S.N., Nety S., Kannan S., Altae-Tran H., Yao X., Sakaguchi Y., Ohira T., Wu W.Y., et al. Cryo-EM structure of the transposon-associated TnpB enzyme. Nature. 2023;616:390–397. doi: 10.1038/s41586-023-05933-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huai C., Li G., Yao R., Zhang Y., Cao M., Kong L., Jia C., Yuan H., Chen H., Lu D., Huang Q. Structural insights into DNA cleavage activation of CRISPR-Cas9 system. Nat. Commun. 2017;8 doi: 10.1038/s41467-017-01496-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang F., Zhou K., Ma L., Gressel S., Doudna J.A. STRUCTURAL BIOLOGY. A Cas9-guide RNA complex preorganized for target DNA recognition. Science. 2015;348:1477–1481. doi: 10.1126/science.aab1452. [DOI] [PubMed] [Google Scholar]
- 36.Huang H., Yuan H.S. The conserved asparagine in the HNH motif serves an important structural role in metal finger endonucleases. J. Mol. Biol. 2007;368:812–821. doi: 10.1016/j.jmb.2007.02.044. [DOI] [PubMed] [Google Scholar]
- 37.Zuo Z., Liu J. Structure and Dynamics of Cas9 HNH Domain Catalytic State. Sci. Rep. 2017;7 doi: 10.1038/s41598-017-17578-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller S.M., Wang T., Randolph P.B., Arbab M., Shen M.W., Huang T.P., Matuszek Z., Newby G.A., Rees H.A., Liu D.R. Continuous evolution of SpCas9 variants compatible with non-G PAMs. Nat. Biotechnol. 2020;38:471–481. doi: 10.1038/s41587-020-0412-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao L., Koseki S.R.T., Silverstein R.A., Amrani N., Peng C., Kramme C., Savic N., Pacesa M., Rodríguez T.C., Stan T., et al. PAM-flexible genome editing with an engineered chimeric Cas9. Nat. Commun. 2023;14 doi: 10.1038/s41467-023-41829-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pacesa M., Loeff L., Querques I., Muckenfuss L.M., Sawicka M., Jinek M. Publisher Correction: R-loop formation and conformational activation mechanisms of Cas9. Nature. 2023;623:E10. doi: 10.1038/s41586-023-06779-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mao Z., Bozzella M., Seluanov A., Gorbunova V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle. 2008;7:2902–2906. doi: 10.4161/cc.7.18.6679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chang H.H.Y., Pannunzio N.R., Adachi N., Lieber M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017;18:495–506. doi: 10.1038/nrm.2017.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Franco S., Murphy M.M., Li G., Borjeson T., Boboila C., Alt F.W. DNA-PKcs and Artemis function in the end-joining phase of immunoglobulin heavy chain class switch recombination. J. Exp. Med. 2008;205:557–564. doi: 10.1084/jem.20080044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chang H.H.Y., Lieber M.R. Structure-Specific nuclease activities of Artemis and the Artemis: DNA-PKcs complex. Nucleic Acids Res. 2016;44:4991–4997. doi: 10.1093/nar/gkw456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sfeir A., Symington L.S. Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem. Sci. 2015;40:701–714. doi: 10.1016/j.tibs.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meyer D., Fu B.X.H., Heyer W.D. DNA polymerases δ and λ cooperate in repairing double-strand breaks by microhomology-mediated end-joining in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA. 2015;112:E6907–E6916. doi: 10.1073/pnas.1507833112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kosicki M., Allen F., Steward F., Tomberg K., Pan Y., Bradley A. Cas9-induced large deletions and small indels are controlled in a convergent fashion. Nat. Commun. 2022;13 doi: 10.1038/s41467-022-30480-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schulte-Uentrop L., El-Awady R.A., Schliecker L., Willers H., Dahm-Daphi J. Distinct roles of XRCC4 and Ku80 in non-homologous end-joining of endonuclease- and ionizing radiation-induced DNA double-strand breaks. Nucleic Acids Res. 2008;36:2561–2569. doi: 10.1093/nar/gkn094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li G., Yang X., Luo X., Wu Z., Yang H. Modulation of cell cycle increases CRISPR-mediated homology-directed DNA repair. Cell Biosci. 2023;13 doi: 10.1186/s13578-023-01159-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lamarche B.J., Orazio N.I., Weitzman M.D. The MRN complex in double-strand break repair and telomere maintenance. FEBS Lett. 2010;584:3682–3695. doi: 10.1016/j.febslet.2010.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu X., Chen J. DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol. Cell Biol. 2004;24:9478–9486. doi: 10.1128/MCB.24.21.9478-9486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.You Z., Bailis J.M. DNA damage and decisions: CtIP coordinates DNA repair and cell cycle checkpoints. Trends Cell Biol. 2010;20:402–409. doi: 10.1016/j.tcb.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bhat K.P., Cortez D. RPA and RAD51: fork reversal, fork protection, and genome stability. Nat. Struct. Mol. Biol. 2018;25:446–453. doi: 10.1038/s41594-018-0075-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Choi E.H., Yoon S., Koh Y.E., Seo Y.J., Kim K.P. Maintenance of genome integrity and active homologous recombination in embryonic stem cells. Exp. Mol. Med. 2020;52:1220–1229. doi: 10.1038/s12276-020-0481-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fasching C.L., Cejka P., Kowalczykowski S.C., Heyer W.D. Top3-Rmi1 dissolve Rad51-mediated D loops by a topoisomerase-based mechanism. Mol. Cell. 2015;57:595–606. doi: 10.1016/j.molcel.2015.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu M., Rehman S., Tang X., Gu K., Fan Q., Chen D., Ma W. Methodologies for Improving HDR Efficiency. Front. Genet. 2018;9 doi: 10.3389/fgene.2018.00691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Scully R., Panday A., Elango R., Willis N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019;20:698–714. doi: 10.1038/s41580-019-0152-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nambiar T.S., Billon P., Diedenhofen G., Hayward S.B., Taglialatela A., Cai K., Huang J.W., Leuzzi G., Cuella-Martin R., Palacios A., et al. Stimulation of CRISPR-mediated homology-directed repair by an engineered RAD18 variant. Nat. Commun. 2019;10:3395. doi: 10.1038/s41467-019-11105-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Choi E.H., Yoon S., Park K.S., Kim K.P. The Homologous Recombination Machinery Orchestrates Post-replication DNA Repair During Self-renewal of Mouse Embryonic Stem Cells. Sci. Rep. 2017;7 doi: 10.1038/s41598-017-11951-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Choi E.H., Yoon S., Kim K.P. Combined Ectopic Expression of Homologous Recombination Factors Promotes Embryonic Stem Cell Differentiation. Mol. Ther. 2018;26:1154–1165. doi: 10.1016/j.ymthe.2018.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vartak S.V., Raghavan S.C. Inhibition of nonhomologous end joining to increase the specificity of CRISPR/Cas9 genome editing. FEBS J. 2015;282:4289–4294. doi: 10.1111/febs.13416. [DOI] [PubMed] [Google Scholar]
- 62.Song J., Yang D., Xu J., Zhu T., Chen Y.E., Zhang J. RS-1 enhances CRISPR/Cas9- and TALEN-mediated knock-in efficiency. Nat. Commun. 2016;7 doi: 10.1038/ncomms10548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lucas C.G., Redel B.K., Chen P.R., Spate L.D., Prather R.S., Wells K.D. Effects of RAD51-stimulatory compound 1 (RS-1) and its vehicle, DMSO, on pig embryo culture. Reprod. Toxicol. 2021;105:44–52. doi: 10.1016/j.reprotox.2021.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reuven N., Adler J., Broennimann K., Myers N., Shaul Y. Recruitment of DNA Repair MRN Complex by Intrinsically Disordered Protein Domain Fused to Cas9 Improves Efficiency of CRISPR-Mediated Genome Editing. Biomolecules. 2019;9 doi: 10.3390/biom9100584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wen W., Zhang X.B. CRISPR-Cas9 gene editing induced complex on-target outcomes in human cells. Exp. Hematol. 2022;110:13–19. doi: 10.1016/j.exphem.2022.03.002. [DOI] [PubMed] [Google Scholar]
- 66.Bashir S., Dang T., Rossius J., Wolf J., Kühn R. Enhancement of CRISPR-Cas9 induced precise gene editing by targeting histone H2A-K15 ubiquitination. BMC Biotechnol. 2020;20 doi: 10.1186/s12896-020-00650-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu Y., Zou R.S., He S., Nihongaki Y., Li X., Razavi S., Wu B., Ha T. Very fast CRISPR on demand. Science. 2020;368:1265–1269. doi: 10.1126/science.aay8204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guo C., Ma X., Gao F., Guo Y. Off-target effects in CRISPR/Cas9 gene editing. Front. Bioeng. Biotechnol. 2023;11 doi: 10.3389/fbioe.2023.1143157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang Y., Xu J., Ge S., Lai L. CRISPR/Cas: Advances, Limitations, and Applications for Precision Cancer Research. Front. Med. 2021;8 doi: 10.3389/fmed.2021.649896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu W., Li L., Jiang J., Wu M., Lin P. Applications and challenges of CRISPR-Cas gene-editing to disease treatment in clinics. Precis. Clin. Med. 2021;4:179–191. doi: 10.1093/pcmedi/pbab014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Driehuis E., Clevers H. CRISPR/Cas 9 genome editing and its applications in organoids. Am. J. Physiol. Gastrointest. Liver Physiol. 2017;312:257–265. doi: 10.1152/ajpgi.00410.2016. [DOI] [PubMed] [Google Scholar]
- 72.Valenti M.T., Serena M., Carbonare L.D., Zipeto D. CRISPR/Cas system: An emerging technology in stem cell research. World J. Stem Cell. 2019;11:937–956. doi: 10.4252/wjsc.v11.i11.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kwon T., Ra J.S., Lee S., Baek I.J., Khim K.W., Lee E.A., Song E.K., Otarbayev D., Jung W., Park Y.H., et al. Precision targeting tumor cells using cancer-specific InDel mutations with CRISPR-Cas9. Proc. Natl. Acad. Sci. USA. 2022;119 doi: 10.1073/pnas.2103532119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhao Z., Zhang H., Xiong T., Wang J., Yang D., Zhu D., Li J., Yang Y., Sun C., Zhao Y., Xi J.J. Suppression of SHROOM1 Improves In Vitro and In Vivo Gene Integration by Promoting Homology-Directed Repair. Int. J. Mol. Sci. 2020;21 doi: 10.3390/ijms21165821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li G., Zhang X., Zhong C., Mo J., Quan R., Yang J., Liu D., Li Z., Yang H., Wu Z. Small molecules enhance CRISPR/Cas9-mediated homology-directed genome editing in primary cells. Sci. Rep. 2017;7 doi: 10.1038/s41598-017-09306-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fell V.L., Schild-Poulter C. Ku regulates signaling to DNA damage response pathways through the Ku70 von Willebrand A domain. Mol. Cell Biol. 2012;32:76–87. doi: 10.1128/MCB.05661-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Panier S., Boulton S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014;15:7–18. doi: 10.1038/nrm3719. [DOI] [PubMed] [Google Scholar]
- 78.Taha E.A., Lee J., Hotta A. Delivery of CRISPR-Cas tools for in vivo genome editing therapy: Trends and challenges. J. Contr. Release. 2022;342:345–361. doi: 10.1016/j.jconrel.2022.01.013. [DOI] [PubMed] [Google Scholar]
- 79.Nambiar T.S., Baudrier L., Billon P., Ciccia A. CRISPR-based genome editing through the lens of DNA repair. Mol. Cell. 2022;82:348–388. doi: 10.1016/j.molcel.2021.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Crossley M.P., Bocek M., Cimprich K.A. R-Loops as Cellular Regulators and Genomic Threats. Mol. Cell. 2019;73:398–411. doi: 10.1016/j.molcel.2019.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chan Y.T., Lu Y., Wu J., Zhang C., Tan H.Y., Bian Z.X., Wang N., Feng Y. CRISPR-Cas9 library screening approach for anti-cancer drug discovery: overview and perspectives. Theranostics. 2022;12:3329–3344. doi: 10.7150/thno.71144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Joung J., Konermann S., Gootenberg J.S., Abudayyeh O.O., Platt R.J., Brigham M.D., Sanjana N.E., Zhang F. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 2017;12:828–863. doi: 10.1038/nprot.2017.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tsiami F., Lago C., Pozza N., Piccioni F., Zhao X., Lülsberg F., Root D.E., Tiberi L., Kool M., Schittenhelm J., et al. Genome-wide CRISPR-Cas9 knockout screens identify DNMT1 as a druggable dependency in sonic hedgehog medulloblastoma. Acta Neuropathol. Commun. 2024;12 doi: 10.1186/s40478-024-01831-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wei L., Lee D., Law C.T., Zhang M.S., Shen J., Chin D.W.C., Zhang A., Tsang F.H.C., Wong C.L.S., Ng I.O.L., et al. Genome-wide CRISPR/Cas9 library screening identified PHGDH as a critical driver for Sorafenib resistance in HCC. Nat. Commun. 2019;10 doi: 10.1038/s41467-019-12606-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shi J., Wang E., Milazzo J.P., Wang Z., Kinney J.B., Vakoc C.R. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat. Biotechnol. 2015;33:661–667. doi: 10.1038/nbt.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wei X., Yang J., Adair S.J., Ozturk H., Kuscu C., Lee K.Y., Kane W.J., O'Hara P.E., Liu D., Demirlenk Y.M., et al. Targeted CRISPR screening identifies PRMT5 as synthetic lethality combinatorial target with gemcitabine in pancreatic cancer cells. Proc. Natl. Acad. Sci. USA. 2020;117:28068–28079. doi: 10.1073/pnas.2009899117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Leta S., Chibssa T.R., Paeshuyse J. CRISPR-Cas12/Cas13: Bibliometric analysis and systematic review of its application in infectious disease detection. J. Infect. Public Health. 2024;17:741–747. doi: 10.1016/j.jiph.2024.03.003. [DOI] [PubMed] [Google Scholar]
- 88.Singh M., Bindal G., Misra C.S., Rath D. The era of Cas12 and Cas13 CRISPR-based disease diagnosis. Crit. Rev. Microbiol. 2022;48:714–729. doi: 10.1080/1040841X.2021.2025041. [DOI] [PubMed] [Google Scholar]
- 89.Tang H., Peng J., Peng S., Wang Q., Jiang X., Xue X., Tao Y., Xiang L., Ji Q., Liu S.M., et al. Live-cell RNA imaging using the CRISPR-dCas13 system with modified sgRNAs appended with fluorescent RNA aptamers. Chem. Sci. 2022;13:14032–14040. doi: 10.1039/d2sc04656c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Carlson-Stevermer J., Kelso R., Kadina A., Joshi S., Rossi N., Walker J., Stoner R., Maures T. CRISPRoff enables spatio-temporal control of CRISPR editing. Nat. Commun. 2020;11 doi: 10.1038/s41467-020-18853-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Naeem M., Hazafa A., Bano N., Ali R., Farooq M., Razak S.I.A., Lee T.Y., Devaraj S. Explorations of CRISPR/Cas9 for improving the long-term efficacy of universal CAR-T cells in tumor immunotherapy. Life Sci. 2023;316 doi: 10.1016/j.lfs.2023.121409. [DOI] [PubMed] [Google Scholar]
- 92.Do P.T., Nguyen C.X., Bui H.T., Tran L.T.N., Stacey G., Gillman J.D., Zhang Z.J., Stacey M.G. Demonstration of highly efficient dual gRNA CRISPR/Cas9 editing of the homeologous GmFAD2-1A and GmFAD2-1B genes to yield a high oleic, low linoleic and α-linolenic acid phenotype in soybean. BMC Plant Biol. 2019;19 doi: 10.1186/s12870-019-1906-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Uddin F., Rudin C.M., Sen T. CRISPR Gene Therapy: Applications, Limitations, and Implications for the Future. Front. Oncol. 2020;10 doi: 10.3389/fonc.2020.01387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lee M. Deep learning in CRISPR-Cas systems: a review of recent studies. Front. Bioeng. Biotechnol. 2023;11 doi: 10.3389/fbioe.2023.1226182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sukocheva O.A., Neganova M.E., Aleksandrova Y., Burcher J.T., Chugunova E., Fan R., Tse E., Sethi G., Bishayee A., Liu J. Signaling controversy and future therapeutical perspectives of targeting sphingolipid network in cancer immune editing and resistance to tumor necrosis factor-α immunotherapy. Cell Commun. Signal. 2024;22 doi: 10.1186/s12964-024-01626-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Harris V.M., Koelsch K.A., Kurien B.T., Harley I.T.W., Wren J.D., Harley J.B., Scofield R.H. Characterization of cxorf21 Provides Molecular Insight Into Female-Bias Immune Response in SLE Pathogenesis. Front. Immunol. 2019;10 doi: 10.3389/fimmu.2019.02160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rangel-Peláez C., Martínez-Gutiérrez L., Tristán-Manzano M., Callejas J.L., Ortego-Centeno N., Martín F., Martín J. CD19 CAR-T cell therapy: a new dawn for autoimmune rheumatic diseases? Front. Immunol. 2024;17 doi: 10.3389/fimmu.2024.1502712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sheehy D.F., Quinnell S.P., Vegas A.J. Targeting Type 1 Diabetes: Selective Approaches for New Therapies. Biochemistry. 2019;58:214–233. doi: 10.1021/acs.biochem.8b01118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Page A., Chuvin N., Valladeau-Guilemond J., Depil S. Development of NK cell-based cancer immunotherapies through receptor engineering. Cell. Mol. Immunol. 2024;21:315–331. doi: 10.1038/s41423-024-01145-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kamali E., Rahbarizadeh F., Hojati Z., Frödin M. CRISPR/Cas9-mediated knockout of clinically relevant alloantigenes in human primary T cells. BMC Biotechnol. 2021;21 doi: 10.1186/s12896-020-00665-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gleditzsch D., Pausch P., Müller-Esparza H., Özcan A., Guo X., Bange G., Randau L. PAM identification by CRISPR-Cas effector complexes: diversified mechanisms and structures. RNA Biol. 2019;16:504–517. doi: 10.1080/15476286.2018.1504546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang Y., Jiang H., Li M., Xu Z., Xu H., Chen Y., Chen K., Zheng W., Lin W., Liu Z., et al. Delivery of CRISPR/Cas9 system by AAV as vectors for gene therapy. Gene. 2024;927 doi: 10.1016/j.gene.2024.148733. [DOI] [PubMed] [Google Scholar]
- 103.Hanlon K.S., Kleinstiver B.P., Garcia S.P., Zaborowski M.P., Volak A., Spirig S.E., Muller A., Sousa A.A., Tsai S.Q., Bengtsson N.E., et al. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun. 2019;10 doi: 10.1038/s41467-019-12449-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wei T., Cheng Q., Min Y.L., Olson E.N., Siegwart D.J. Systemic nanoparticle delivery of CRISPR-Cas9 ribonucleoproteins for effective tissue specific genome editing. Nat. Commun. 2020;11 doi: 10.1038/s41467-020-17029-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Matsumoto D., Matsugi E., Kishi K., Inoue Y., Nigorikawa K., Nomura W. SpCas9-HF1 enhances accuracy of cell cycle-dependent genome editing by increasing HDR efficiency, and by reducing off-target effects and indel rates. Mol. Ther. Nucleic Acids. 2024;35 doi: 10.1016/j.omtn.2024.102124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kleinstiver B.P., Pattanayak V., Prew M.S., Tsai S.Q., Nguyen N.T., Zheng Z., Joung J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–495. doi: 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hendel A., Bak R.O., Clark J.T., Kennedy A.B., Ryan D.E., Roy S., Steinfeld I., Lunstad B.D., Kaiser R.J., Wilkens A.B., et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015;33:985–989. doi: 10.1038/nbt.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Deng X., Sun W., Li X., Wang J., Cheng Z., Sheng G., Wang Y. An anti-CRISPR that represses its own transcription while blocking Cas9-target DNA binding. Nat. Commun. 2024;15 doi: 10.1038/s41467-024-45987-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gangopadhyay S.A., Cox K.J., Manna D., Lim D., Maji B., Zhou Q., Choudhary A. Precision Control of CRISPR-Cas9 Using Small Molecules and Light. Biochemistry. 2019;58:234–244. doi: 10.1021/acs.biochem.8b01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.McGlincy N.J., Meacham Z.A., Reynaud K.K., Muller R., Baum R., Ingolia N.T. A genome-scale CRISPR interference guide library enables comprehensive phenotypic profiling in yeast. BMC Genom. 2021;22 doi: 10.1186/s12864-021-07518-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jacobsen T., Liao C., Beisel C.L. The Acidaminococcus sp. Cas12a nuclease recognizes GTTV and GCTV as non-canonical PAMs. FEMS Microbiol. Lett. 2019;366 doi: 10.1093/femsle/fnz085. [DOI] [PMC free article] [PubMed] [Google Scholar]