

Abstract
The direct oxidation of methane (CH4) to methanol (CH3OH) remains a formidable challenge due to the inertness of CH4 and the tendency of CH3OH to overoxidize. Here, we report Pt–Cu nanoalloys encapsulated within hydrophobic, acid-free silicalite-1 (S-1) zeolite that breaks activity–selectivity limits in CH4 oxidation to CH3OH. The best catalyst exhibits a CH3OH productivity of 134 mol of CH3OH per mol of Pt per hour and a selectivity of 95% at 150 °C. Kinetic and spectroscopic studies revealed a sequential oxidation mechanism: CH4 is first oxidized to methyl hydroperoxide (CH3OOH) by in situ generated hydrogen peroxide, which subsequently converts to CH3OH. The catalytic reaction proceeds with an apparent activation energy of only 42 kJ/mol, the lowest reported to date. The outstanding performance arises from the synergy of the Pt–Cu alloy sites and the hydrophobic pore of S-1. Pt–Cu alloy sites specifically generate oxidizing species and selectively form CH3OH, which was not achieved by single metal catalysts and other bimetallic catalysts. A confined hydrophobic, acid-free environment enables rapid extraction of the CH3OH from the reaction field and thereby prevents overoxidation. These findings highlight how precious control over both the composition and the local environment of Pt–Cu nanoalloys can markedly enhance the catalytic oxidation of CH4 to CH3OH.


Introduction
Methane (CH4), the main component of natural gas, is an abundant and inexpensive feedstock with significant potential for sustainable energy and chemical conversion. , Among the various transformation pathways, the selective oxidation of CH4 to methanol (CH3OH) is particularly attractive, as CH3OH serves as a versatile platform molecule for fuels and chemical feedstock. However, commercial CH3OH production still relies on a two-step process involving energy-intensive steam reforming followed by catalytic CH3OH synthesis, which requires high temperatures and large-scale infrastructure. Direct conversion of CH4 to CH3OH remains highly challenging due to the strong C–H bond of CH4 (∼435 kJ/mol) and the inherent instability of CH3OH under oxidative conditions, often leading to overoxidation to undesirable byproducts such as formic acid (HCOOH) and CO2. Various catalytic systems, including metal-containing zeolites, have been explored using oxidants such as hydrogen peroxide (H2O2), , oxygen (O2), − water (H2O), , or nitrous oxide, , yet achieving both high CH3OH selectivity and productivity remains elusive.
In the presence of a biological reductant, typically dihydronicotinamide adenine dinucleotide, the reductive activation of O2 is a general strategy employed by enzymatic systems such as methane monooxygenase to achieve selective oxidation. Inspired by these biological processes, a CO-assisted mechanism for selective CH4 oxidation on heterogeneous catalysts has recently attracted considerable attention. By utilizing CO as a reductant in aqueous media, O2 activation is promoted, leading to the in situ generation of H2O2 and hydroxyl radicals (OH•). − Using these reactive oxygen species as the oxidants, the active metal sites catalytically oxidize CH4 to organic oxygenates while facilitating the rapid release of the oxygenates into the water solvent. A wide range of heterogeneous catalysts containing single atoms or nanoparticles of noble metals have been reported for the CO-assisted oxidation of CH4. These include Rh, ,,− Ru, Ir, ,, Pd, Au, ,, and Pt-based catalysts. To enhance the CH3OH selectivity, several key strategies have been identified. These include choosing an appropriate active metal, , controlling the metal particle size and loading, ,, tailoring the local structure or environment, , adding a second metal complex (e.g., CuCl2) as a homogeneous catalyst, , removing Brønsted acid sites from the catalyst surface, , and inducing hydrophobicity through surface modification. , Nevertheless, achieving a high CH3OH productivity while maintaining high selectivity remains challenging; existing heterogeneous catalysts cannot perform better than a CH3OH productivity of 73 mol of CH3OH per mol of noble metal (NM) per hour (Table S1). To overcome this limitation, coaddition of an excess amount of Cu-based homogeneous catalysts is needed.
Here, we report that precisely designing a nanoalloy catalyst confined within a solid acid-free hydrophobic reaction space allows us to break through the existing activity–selectivity limits in the CO-assisted oxidation of CH4 to CH3OH. We focus on Pt–Cu alloy nanoparticles encapsulated within the hydrophobic micropores of silicalite-1 zeolite (PtCu@S-1). Pt–Cu alloys are well-known for their high activity in C–H oxidation, and a recent study has demonstrated that Pt is a particularly useful element in CO-assisted oxidation mechanisms. Furthermore, alloying Pt with Cu, which can stabilize methoxy intermediates, is expected to further enhance CH3OH selectivity. Silicalite-1 is a pure silica zeolite having a hydrophobic nature and exhibits a relatively high transport efficiency for CH3OH compared to water. Therefore, using the hydrophobic micropores of S-1 as the reaction nanospace, CH3OH, once synthesized by the active sites, can be quickly expelled from the micropores, reducing the side reaction rate and thereby enhancing CH3OH selectivity. An additional advantage of S-1 is its lack of Brønsted acid sites, which effectively suppresses acid-catalyzed pathways responsible for the formation of undesirable byproducts such as HCOOH and CH3COOH. ,, Taken together, these insights suggest that PtCu@S-1 could overcome the activity–selectivity limits faced by earlier systems. Although the precise encapsulation of alloy nanoparticles inside micropores of S-1 has long been challenging, recent advances in one-step hydrothermal synthesis have made this approach feasible. − Despite these advances, its potential for selective CH4 oxidation has not been explored.
The purpose of this study is to demonstrate the significance of encapsulating specific alloy nanoparticles within a solid acid-free hydrophobic nanospace. First, a series of model catalysts are designed and characterized, including (1) Pt–Cu alloy nanoparticles with varying Pt/Cu molar ratios encapsulated in S-1, (2) Pt–Cu alloy nanoparticles encapsulated in MFI zeolites with different Si/Al molar ratios (to introduce varying amounts of Brønsted acid sites), and (3) Pt–Cu alloy nanoparticles supported on the external surface of S-1. Next, the catalytic activity and selectivity of these materials in CO-assisted oxidation of CH4 are evaluated. Based on the obtained data, structure–function relationships are clarified. Finally, through controlled kinetic and spectroscopic experiments using the best catalyst, the reaction mechanism is investigated. By comparison of the performance of PtCu@S-1 with that of analogous Pt-only or Cu-only catalysts, the synergistic interplay between Pt and Cu sites within the micropores of S-1 is clarified.
Results and Discussion
Synthesis and Characterization
Pt–Cu alloy nanoparticles encapsulated in S-1 (PtCu@S-1) were synthesized via a one-step hydrothermal method, as illustrated in Figure A. − Briefly, a zeolite synthesis gel containing ethylenediamine complexes of Pt and Cu was subjected to static hydrothermal treatment at 170 °C for 3 days to crystallize the zeolite. The resulting material was then calcined under flowing H2 at 500 °C to form the alloy within the micropores and subsequently decompose organic structure-directing agents, tetrapropylammonium cations (TPA+) that occupy the zeolite channels. Details of the synthesis procedure are provided in the Supporting Information (Materials and Methods section and Table S2). Simultaneous thermogravimetry and differential thermal analysis, as well as textural properties determined by N2-adsorption isotherms, confirmed complete removal of TPA+ (Figure S1 and Table S3), in agreement with the relevant literature, , and the liberated, metal-encapsulated micropores are accessible for catalysis. The calcination process did not change the concentration of defects (Si–OH) influencing the hydrophobicity of S-1 zeolite, as evidenced by 29Si magic-angle spinning nuclear magnetic resonance spectra (Figure S2). The catalyst synthesized with a Pt:Cu molar ratio of 1:1 is denoted as Pt1Cu1@S-1. For comparison, a Pt-only encapsulated catalyst (Pt@S-1) and a Cu-only encapsulated catalyst (Cu@S-1) were synthesized using the same one-step method but with only the Pt or Cu ethylenediamine complex, respectively. Additionally, an S-1 supported Pt–Cu nanoalloy catalyst was also prepared by impregnating S-1 with Pt and Cu (1:1) followed by reduction. This externally supported catalyst is denoted as Pt1Cu1/S-1. The chemical compositions of mother gels and resultant catalysts determined by inductively coupled plasma–optical emission spectroscopy are summarized in Tables S2 and S3.
1.

(A) Schematic representation of the one-step hydrothermal synthesis used to encapsulate Pt–Cu alloy nanoparticles within the S-1 framework. Legend: black, C; blue, N; red, O; light blue, Cu; yellow, Pt; white, H; gray frame, MFI-type zeolite. (B) XRD profiles. (C, D) XPS of Pt 4f and Cu 2p regions. (E) Representative HAADF-STEM images of each catalyst.
Powder X-ray diffraction (XRD) patterns (Figure B) confirmed that all catalysts exhibit the characteristic XRD patterns of the MFI-type zeolite, indicating the successful crystallization of the MFI-type zeolite. No diffraction lines corresponding to Pt or Cu metal phases were observed for any of the catalysts, suggesting that metal species were highly dispersed. To assess the distribution of Pt and Cu, X-ray photoelectron spectroscopy (XPS) was performed on these catalysts (Figure C,D). XPS, being surface-sensitive (probe depth of ∼3 nm), readily detects metal species located on the external surface of the zeolite. ,, Actually, the impregnated Pt1Cu1/S-1 catalyst provided clear Pt 4f and Cu 2p signals. In contrast, the one-step hydrothermally synthesized catalysts (Pt1Cu1@S-1, Pt@S-1, and Cu@S-1) exhibited negligible signals; therefore, the fraction of metals located on the external surface of S-1 is extremely low, suggesting that in these catalysts, the Pt and Cu species are predominantly encapsulated within the zeolite micropores.
High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) provided direct evidence of the metal nanoparticle locations (Figure E). Additional HAADF-STEM images are available in the Supporting Information (Figures S3–S6). In the Pt1Cu1/S-1 catalyst, abundant 2–5 nm metal nanoparticles were observed on the external surface of S-1, consistent with the XPS results. Lattice fringes corresponding to Pt–Cu alloy nanocrystals were also visible (see Figure S3 in the Supporting Information), confirming that Pt and Cu form an alloy. In contrast, the catalysts prepared by the one-step hydrothermal synthesis method (PtCu@S-1, Pt@S-1, and Cu@S-1) showed virtually no metal nanoparticles on the external surface of S-1; instead, uniformly dispersed nanoparticles were observed within the zeolite micropores. These observations, together with the XPS data, demonstrate that the one-step hydrothermal synthesis method effectively encapsulates Pt and Cu within the S-1 micropores. The average particle sizes were 1.2 ± 0.4 nm for Pt1Cu1@S-1, 0.9 ± 0.4 nm for Pt@S-1, 2.0 ± 0.8 nm for Cu@S-1, and 3.0 ± 1.3 nm for Pt1Cu1/S-1 (Figure S7).
X-ray absorption fine structure (XAFS) spectroscopy was used to probe the local atomic structure and electronic state of Pt and Cu in the encapsulated Pt–Cu alloys. Figure A shows the Pt LIII-edge X-ray absorption near-edge structure (XANES) spectra. The white-line intensity for Pt1Cu1@S-1 was lower than that for Pt@S-1 and was close to that of a metallic Pt foil, suggesting that Pt exists in a reduced form as a result of interaction with Cu. In comparison, Pt@S-1 exhibited a white-line intensity that was higher than that of Pt1Cu1@S-1, indicating that Pt is present predominantly in a more oxidized state. It is expected that the absence of Cu allowed Pt atoms to interact more strongly with the zeolite framework oxygen, thereby stabilizing partially oxidized Pt species. Figure B displays the Cu K-edge XANES spectra. Compared to Cu@S-1, the absorption edge of Pt1Cu1@S-1 was shifted to lower energy, indicating that Cu is also in a lower oxidation state (more reduced) due to alloying with Pt. Moreover, the XANES spectrum of Pt1Cu1@S-1 could not be reproduced by a linear combination of reference spectra for Cu@S-1, Cu foil, Cu2O, and CuO (Figure S8), implying the formation of a Cu species having a unique local environment. The spectral feature observed for Pt1Cu1@S-1 is consistent with that reported for Pt–Cu alloy nanoparticles, , thereby supporting the formation of Pt–Cu nanoalloys within the micropores of S-1. Focusing on Cu@S-1, the absorption edge was shifted toward higher energy relative to Cu foil and appears closer to that of Cu2O. This suggests that, because the Cu species interact directly with the zeolite framework oxygen rather than with Pt, more oxidized Cu species predominated compared with those in Pt1Cu1@S-1.
2.

(A, B) Pt LIII-edge and Cu K-edge XANES spectra. (C, D) Pt LIII-edge and Cu K-edge FT-EXAFS spectra. (E) Pt LIII and (F) Cu K-edge WT-EXAFS of Pt1Cu1@S-1. Reference data: (G) Pt foil and (H) Cu foil. (I) Curve-fitting of the Pt LIII-edge EXAFS function for Pt1Cu1@S-1 assuming two contributions: Pt–Cu and Pt–Pt backscattering. (J) Inverse FT of the best fit EXAFS function with the experimental FT-EXAFS spectrum.
The extended X-ray absorption fine structure (EXAFS) was analyzed to further confirm the Pt–Cu alloys. The Fourier transform EXAFS (FT-EXAFS) spectra for the Pt LIII-edge and Cu K-edge are shown in Figure C,D. Pt1Cu1@S-1 exhibits a prominent backscattering peak at a radial distance shorter than the Pt–Pt distance in Pt foil but longer than the Cu–Cu distance in Cu foil. This observation supports the presence of a Pt–Cu bond with a distance intermediate between those of Pt–Pt and Cu–Cu. This assignment is further supported by wavelet-transformed EXAFS (WT-EXAFS) as shown in Figure E–H. At the Pt LIII-edge of Pt1Cu1@S-1, a lobe-shaped feature was observed that cannot be explained solely by Pt–Pt backscattering, in contrast to the Pt foil. This is attributed not only to Pt–Pt backscattering but also to Pt–Cu backscattering, which contributes to the lobe localized in the low-k (∼6 Å) and low-R (∼2.1 Å) regions. Similarly, at the Cu K-edge, a complex spectral shape was observed that cannot be explained by Cu–Cu backscattering in Cu foil alone. While lobes with maxima of WT coefficient appear at (k, R) positions similar to those of Cu–Cu backscattering, the lobes extend into the high-R (>2.3 Å) region in the high-k range (>8 Å–1). This clearly indicates the involvement of heavy-element-derived backscattering, specifically Cu–Pt backscattering. To quantify the local structure, EXAFS curve-fitting was performed (Figure I,J). The fitting confirmed that two scattering paths (Pt–Cu and Pt–Pt) are necessary to reproduce the Pt1Cu1@S-1 data, further validating the alloy nature. The best fit bond lengths were 2.64 Å for Pt–Cu and 2.71 Å for Pt–Pt (see Table S4), in agreement with literature values for Pt–Cu nanoalloys, , corroborating that Pt–Cu alloy nanoparticles are formed inside the S-1 micropores. In contrast, prominent Pt–O and Cu–O backscattering features were observed for Pt@S-1 and Cu@S-1, respectively, suggesting strong interactions between the metals and lattice oxygens.
CO adsorption Fourier transform infrared spectroscopy (CO-FTIR) was employed to analyze the surface sites of the encapsulated Pt–Cu alloy nanoparticles and their monometallic analogues. Since the position, intensity, and full width at half-maximum (fwhm) of the νCO bands for adsorbed CO species reflect the nature of the active sites on the catalyst surface, this technique provides detailed structural information about the active sites. ,,,,, After CO adsorption, the CO gas supply was turned off, and the system was purged with Ar. Time-resolved measurements were carried out during the purge process to monitor both irreversibly and reversibly adsorbed CO on the catalyst surface (Figure A–D). For Pt@S-1, the νCO band (band I) was observed at 2063–2060 cm–1, characteristic of CO adsorbed on atop Pt sites; this band shifted slightly to lower energy during the Ar purge. For Pt1Cu1@S-1, band I was observed at 2049–2044 cm–1, which was 14–16 cm–1 lower than that observed in Pt@S-1. This energy difference shows that Pt sites in the Pt–Cu alloy are more electron-rich than in the Pt-only catalyst. The excess charge originates from electron donation by neighboring Cu atoms, whose lower electronegativity drives electron transfer to Pt and, in turn, weakens the C–O bond of the CO species adsorbed on the Pt sites. Similar effects have been reported in previous studies of Pt–Cu alloy catalysts. ,, Additionally, the νCO band (band II) was transiently detected at 2116 cm–1 during the Ar purge; this band II, which can be assigned to the CO species adsorbed on atop Cu sites, diminished during the purge process. Notably, as band II (Cu–CO) decayed, band I (Pt–CO) grew in intensity, shifted further to lower energy, and became sharper (narrower fwhm). The variations in the band intensities correlated (Figure E,F), indicating that bands I and II are related to the Pt and Cu sites in close proximity; a bimetallic ensemble site was further evidenced. In contrast, for the supported catalyst (Pt1Cu1/S-1), qualitatively similar bands I and II were observed but with much lower intensity because of a lower surface area (i.e., larger particles). The time-dependent behavior of band I in PtCu/S-1 also differed from that in Pt1Cu1@S-1, suggesting that the geometries of Pt–Cu alloy sites are different. On the other hand, Cu@S-1 exhibited only a very weak band II before purging, suggesting that Cu nanoparticles bind CO weakly.
3.

(A–D) Time-resolved CO-FTIR spectra under Ar purge following CO adsorption. The characteristics of the change in band intensity and position over time are indicated by arrows. (E) Time course of intensities for νCO bands at 2116 and 2044 cm–1. To eliminate the contribution of the signals of gas-phase CO, a scan range (12–60 spectrum numbers) where almost all gas-phase CO is removed from the system is shown. (F) Correlation plot between intensities of νCO bands at 2116 and 2044 cm–1. (G, H) CO-FTIR spectra of PtCu@S-1 with different Cu/Pt molar ratios and Pt3Cu1@MFI with different Si/Al molar ratios. These spectra were collected after CO adsorption and subsequent Ar purge. For comparison, the absorbance of the spectra was normalized to the highest intensity.
Having established the structural characteristics of the Pt–Cu encapsulated catalysts, variants were synthesized to probe the impact of the alloy composition and framework aluminum (acid sites). In short, PtCu@S-1 catalysts with higher or lower Cu content (denoted Pt3Cu1@S-1 and Pt1Cu3@S-1, corresponding to Pt:Cu precursor ratios of 3:1 and 1:3, respectively) and Pt3Cu1@MFI catalysts with Si/Al molar ratios of 274, 181, and 127 were synthesized. Chemical compositions of mother gels and resultant catalysts are summarized in Tables S2 and S3. All of these catalysts exhibited XRD patterns consistent with the MFI zeolite structure (Figure S9) and showed negligible XPS signatures of Pt and Cu (Figure S10), confirming the successful encapsulation of Pt–Cu nanoalloys in each case. HAADF-STEM directly observed the encapsulated nanoparticles with average diameters in the 1.2–1.4 nm range (Figures S11–S16), similar to the 1.2 nm size estimated for Pt1Cu1@S-1. CO-FTIR spectroscopy further verified that these variants all form Pt–Cu alloy sites (Figure G,H). In PtCu@S-1, as the Cu/(Pt + Cu) ratio increased, the νCO band corresponding to CO on Pt sites (band I after Ar purge) shifted to lower energy, indicating that Pt sites become increasingly electron-rich with higher Cu content, consistent with enhanced alloying effects. Meanwhile, the transient Cu–CO band (band II) observed during the purge exhibited a slight blueshift with a higher Cu content (Figure S17A). A correlation between the positions of bands I and II was observed across the different alloy compositions (Figure S17B), suggesting that the geometric and electronic structures of the alloy sites vary systematically with composition. On the other hand, introducing aluminum into the zeolite framework (decreasing Si/Al molar ratio) led to a blueshift of the Pt–CO band I, implying that the presence of framework Al (and the associated Brønsted acid sites) makes the Pt sites more electrophilic due to the confinement effects of micropores containing positively charged acid sites.
Al incorporation into the zeolite framework influences not only the electronic state of the encapsulated Pt–Cu nanoparticles but also the polarity of the surrounding pore space. To assess how framework Al concentration modulates the hydrophobicity of the zeolite cages that host the Pt–Cu alloy nanoparticles, we carried out continuous-flow CH3OH breakthrough experiments: a dilute CH3OH/Ar stream was passed through each catalyst bed at ambient temperature, and the effluent CH3OH concentration (C) relative to the inlet concentrations (C 0) was recorded over time, yielding the breakthrough profiles (Figure A). The time required for the effluent to reach C/C 0 = 0.05 was defined as the breakthrough time; these values were estimated for all catalysts and are compared in Figure B. A purely siliceous S-1 framework encapsulating Pt–Cu nanoparticles led to the earliest breakthrough, whereas breakthrough for the Pt3Cu1@MFI catalysts occurred progressively later as the Si/Al ratio decreases from 274 to 184 and 127. Quantitatively, the breakthrough time increased from 223–258 s for the siliceous catalysts to 340, 364, and 421 s for MFI with Si/Al = 274, 184, and 127, respectively. Because the three PtCu@S-1 catalysts differ only in alloy stoichiometry yet display nearly identical breakthrough times, host composition rather than the metal ratio governs CH3OH affinity, confirming that hydrophobicity is dictated by the concentration of framework Al. In short, the hydrophobicity of the reaction environment can be tuned by adjusting the framework Al content without compromising the encapsulation of the Pt–Cu alloy nanoparticles.
4.

(A) CH3OH breakthrough curves for PtCu@S-1 with different Cu/Pt molar ratios and Pt3Cu1@MFI with different Si/Al molar ratios. The horizontal dashed line marks the breakthrough threshold (C/C 0 = 0.05). (B) Comparison of the breakthrough times.
Accordingly, continuous modulation was achieved in both the structure and the electronic state of the Pt–Cu alloy species and their surrounding environment. The library of well-defined catalysts provides a solid foundation for investigating how composition and environment affect catalytic performance in CO-assisted oxidation of CH4.
Catalytic Performance and Structure–Function Relationship
The catalytic performance of the aforementioned catalysts for the CO-assisted oxidation of CH4 to CH3OH was investigated. Activity assays were carried out in an aqueous medium at 150 °C for 1 h, using 20 bar CH4, 5 bar CO, and 3 bar O2 as reactant gases. Following the reaction, the liquid products were analyzed by 1H NMR spectroscopy. Representative spectra are available in Figure S18. It was found that C1 and C2 oxygenates were formed and their productivities and selectivities varied significantly with the catalyst employed.
Figure A compares the catalytic performance of PtCu@S-1 catalysts with varying alloy compositions as well as the monometallic Pt@S-1 and Cu@S-1. As the Cu content in the alloy increased (moving from Pt@S-1 to Pt3Cu1@S-1 to Pt1Cu1@S-1), the CH3OH productivity increased dramatically from 0.10 mmol/gcat/h (Pt@S-1) to 1.30 mmol/gcat/h (Pt3Cu1@S-1) and further to 1.44 mmol/gcat/h (Pt1Cu1@S-1). Along with this ∼14-fold increase in activity, the CH3OH selectivity improved from 31% (with Pt alone) to 94 and 95% for the 3:1 and 1:1 Pt–Cu alloys, respectively. However, an excess of Cu was detrimental: the Pt1Cu3@S-1 catalyst (with a 1:3 ratio) gave a lower CH3OH productivity (0.5 mmol/gcat/h) and a slightly reduced selectivity (87% CH3OH). Meanwhile, Cu@S-1 (which contains no Pt) showed no measurable activity for CH4 oxidation under these conditions. These results clearly demonstrate that both Pt and Cu are required in roughly equal proportions to achieve high activity and selectivity, a strong indication of synergy between Pt and Cu in the alloy. This is also true when the productivity is normalized by moles of Pt, further supporting the optimal composition (Table S5). The synergy observed here aligns with our design hypothesis that a Pt–Cu alloy can outperform the individual metals: neither Pt nor Cu alone was effective, but together, they achieved a remarkable outcome.
5.

Structure–function relationship in the CO-assisted oxidation of CH4 over Pt–Cu nanoalloy-encapsulated zeolites. (A) Effect of the Cu/Pt molar ratio on oxygenate productivity and CH3OH selectivity. (B) Influence of noble metal and alloy composition within S-1. For bimetallic systems, a molar ratio of 1:1 was employed as Pt1Cu1@S-1. (C) Comparison of encapsulated and supported catalysts. (D) Effect of the Si/Al molar ratio. Reaction conditions: catalyst loading, 5 mg; solvent, 15 mL of H2O; reaction gas, 20 bar CH4 + 5 bar CO + 3 bar O2; temperature, 150 °C; reaction time, 1 h.
To verify that the Pt–Cu combination is indeed unique in promoting selective CH4 oxidation, a series of analogous bimetallic and monometallic catalysts using other metals was additionally synthesized and tested. These one-step hydrothermally prepared catalysts included Rh@S-1, RhCu@S-1, Pd@S-1, PdCu@S-1, Ir@S-1, IrCu@S-1, and PtFe@S-1, PtCo@S-1, and PtNi@S-1. The performance of these catalysts is summarized in Figure B. Only the Pt–Cu alloys (PtCu@S-1) exhibited high CH3OH productivity and selectivity. Catalysts containing only a single noble metal (Pt, Rh, Pd, or Ir) showed modest productivity (0.2–0.7 mmol/gcat/h) with poor selectivities (16–31% CH3OH), primarily yielding overoxidation products, HCOOH. The various bimetallic combinations (PtFe, PtCo, RhCu, PdCu, and IrCu) did show some degree of activity, but their methanol selectivities were only 7–40%. In fact, many of these bimetallics preferentially produced HCOOH, indicating that they did not solve the overoxidation issue. Only the Pt–Cu alloy encapsulated in S-1 achieved both high CH3OH productivity and high selectivity. Notably, for one of the least effective catalysts (RhCu@S-1), which exhibited activity comparable to that of PtCu@S-1 but predominantly yielded HCOOH, STEM and XPS analyses confirmed the successful encapsulation of Rh–Cu alloy nanoparticles (Figures S19–S21). This means the inferior performance of Rh–Cu alloy nanoparticles was not due to a failure in synthesis or lack of alloy formation, but rather an inherent difference in how Rh–Cu vs Pt–Cu function. These screening results demonstrate that the Pt–Cu pairing is uniquely effective for this reaction, validating our focus.
Next, the encapsulated Pt–Cu nanoalloy catalyst was compared with analogous catalysts in which Pt–Cu nanoalloys are supported on the external surface of S-1, to assess the significance of the zeolite confinement effect. Additionally, a high-surface SiO2-supported Pt–Cu catalyst with an average particle size of 1.5 nm was also prepared and used as the reference (Figures S22 and S23). Figure C compares the performance of Pt1Cu1@S-1 with Pt1Cu1/S-1 and Pt1Cu1/SiO2. The encapsulated Pt1Cu1@S-1 catalyst exhibited a CH3OH productivity of 1.44 mmol/gcat/h with 95% CH3OH selectivity, whereas both supported catalysts displayed a CH3OH productivity of less than 1/10th of this value (approximately 0.14 mmol/gcat/h) and a CH3OH selectivity of only 7%. The main product of the supported catalyst was HCOOH. This indicates that once CH3OH was formed, most of it was overoxidized to HCOOH. These results highlight that encapsulation within the zeolite micropores is critical for achieving high CH3OH selectivity. The hydrophobic micropores of S-1 facilitate the rapid expulsion of CH3OH, synthesized by the encapsulated Pt–Cu alloy nanoparticles, into the aqueous solvent, preventing further oxidation to HCOOH. In contrast, on open surfaces such as external S-1 and SiO2, CH3OH remains in prolonged contact with active sites, leading to continuous oxidation to HCOOH. What must not be overlooked here is that the overall formation rate of C1 oxygenates varies little among the three catalysts. The average nanoparticle diameters are 1.2 ± 0.4 nm for Pt1Cu1@S-1 and 1.5 ± 0.5 nm for Pt1Cu1/SiO2, whereas Pt1Cu1/S-1 contains significantly larger particles with a diameter of 3.0 ± 1.3 nm, implying substantial differences in the number of exposed metal sites. Yet, all three catalysts displayed almost identical total C1 oxygenate formation rates. This finding suggests that the particle size is not the decisive factor governing catalytic activity.
The effect of Brønsted acid sites in the zeolite (introduced by framework Al) on the catalytic performance was also investigated. Figure D compares PtCu@MFI catalysts with varying Si:Al molar ratios. When using purely siliceous S-1 (Si/Al = ∞, no acid sites), the catalyst gave 1.44 mmol/gcat/h CH3OH with 95% selectivity. Productivity increases when the ratio of Si/Al is 274, but CH3OH selectivity decreases due to the further oxidation of CH3OH. As the Si/Al molar ratio further decreases, productivity decreases, and excessive oxidation reactions also reduce the selectivity of CH3OH. These findings indicate that Brønsted acid sites (associated with Al in the framework) negatively impact selectivity, likely by promoting the further oxidation of CH3OH to HCOOH via acid-catalyzed pathways; acid-free support is essential for maximizing CH3OH selectivity, consistent with previous studies showing that the removal of acid sites or proton in aqueous solvent improves CH3OH selectivity. , Furthermore, incorporating framework Al renders the zeolite channels more hydrophilic, hampering the prompt desorption of CH3OH from the reaction environment (Figure ). The resulting longer residence time favors sequential CH3OH oxidation and therefore diminishes the CH3OH selectivity.
Figure summarizes the CH3OH productivity (mol/molNM/h) and CH3OH selectivity (%) of the best catalyst developed in this study (Pt1Cu1@S-1) with those of previously reported catalysts. − ,,,,, In the literature systems, it has been challenging to enhance activity while maintaining a high selectivity (>90%). The maximum CH3OH productivity is limited to approximately 73 mol/molNM/h reported for Au/MOR (0.07 wt % Au loading). In contrast, the Pt1Cu1@S-1 catalyst achieved a CH3OH productivity of 134 molmethanol/molNM/h with 95% CH3OH selectivity, breaking the activity–selectivity limit. This remarkable catalytic performance underscores the importance of integrating the distinct catalytic functions of Pt and Cu within a confined, acid-free, hydrophobic environment that rapidly removes CH3OH and prevents overoxidation.
6.

Comparison of Pt1Cu1@S-1 with literature catalysts in terms of CH3OH productivity and selectivity.
Catalyst recyclability was assessed over two consecutive batch runs (Figure S24). After reuse, the catalyst exhibited a 1.1–1.6-fold increase in the C1–C2 oxygenase formation rate but a moderate decline in CH3OH selectivity: 95% in the first run versus 78% for the second cycle (with no pretreatment) and 52% for the second (H2) cycle (prereduced with H2 at 500 °C). In CO-assisted oxidation of light alkanes, performance changes are frequently linked to nanoparticle sintering; we previously observed rapid Pt aggregation on r-TiO2 under analogous conditions, resulting in activity loss. Consistent with this precedent, HAADF–STEM reveals a modest particle growth from 1.2 ± 0.4 to 1.5 ± 0.5 nm (Figures S25 and S26), while XPS detects no Pt or Cu on the external zeolite surface (Figure S27), confirming that incipient sintering occurs within the micropores of S-1. Complementary CO-FTIR spectroscopy further revealed the irreversible surface structure changes associated with aggregation (Figure S28). Slight differences were detected in the XANES spectra of the catalysts before and after the reaction (Figure S29A,B), supporting the irreversible subtle changes in surface structure shown by CO-FTIR. On the other hand, k 3-weighted FT-EXAFS showed that the Pt–Cu and Pt–Pt backscattering peaks were retained after the reaction, which were comparable to those of the fresh catalysts (Figure S29C,D), suggesting that no extensive structural changes, such as dealloying, occurred during the reaction. Thus, the moderate decrease in CH3OH selectivity is more plausibly attributed to fine-scale surface modifications than to an overall change in the bulk structure of the alloy. Although further improvements in long-term durability, especially enhanced resistance to aggregation, are still necessary, the extremely high initial activity and selectivity of the Pt–Cu nanoparticles confined within the hydrophobic S-1 micropores validate our design strategy and provide a compelling platform for deeper mechanistic exploration.
Mechanistic Insights
Having established the superior performance of the PtCu@S-1 catalyst, the reaction mechanism was subsequently investigated to elucidate how the Pt–Cu synergy operates. Focusing on the Pt1Cu1@S-1 catalyst, a series of kinetic experiments was conducted by varying the reactant partial pressures and reaction temperatures while simultaneously investigating potential reaction intermediates.
Figure illustrates how the oxygenates productivity (mmol/gcat/h) and CH3OH selectivity (%) vary with CO or O2 pressures. Starting from the standard conditions (20 bar CH4 + 5 bar CO + 3 bar O2 at 150 °C), the CO partial pressure was varied between 0 and 7 bar while keeping CH4 at 20 bar and O2 at 3 bar (Figure A). Separately, the O2 partial pressure was varied between 0 and 6 bar with CH4 at 20 bar and CO at 5 bar (Figure B). When either CO or O2 was omitted, CH3OH was not formed, confirming that both coreactants are essential for the reaction. This observation is consistent with the CO-assisted oxidation mechanism proposed in earlier studies. −
7.
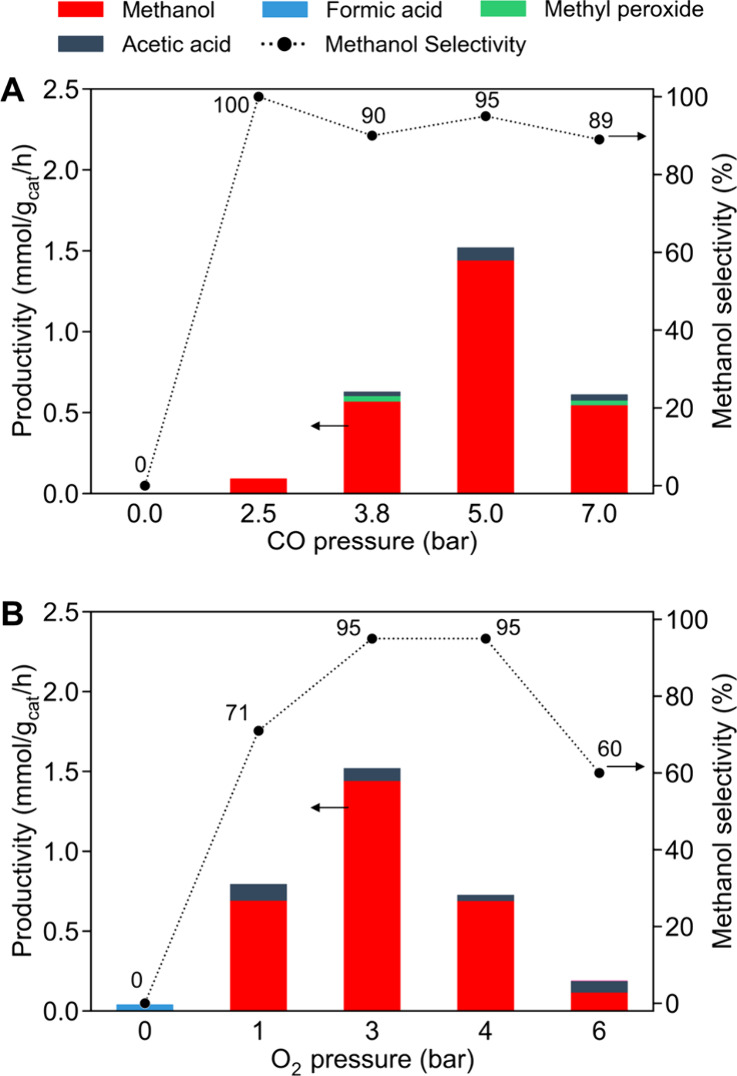
Effect of (A) CO and (B) O2 partial pressures on oxygenate productivity and CH3OH selectivity. Reaction conditions: catalyst loading, 5 mg; solvent, 15 mL of H2O; reaction gas, 20 bar CH4 + 0–7 bar CO + 3 bar O2 for (A) or 20 bar CH4 + 5 bar CO + 0–6 bar O2 for (B); temperature, 150 °C; reaction time, 1 h.
When CO was introduced, CH3OH production began once the CO pressure reached a threshold value. Specifically, as the CO pressure increased from 0 up to about 2.5 bar, CH3OH formation started with 100% selectivity. Further increasing the CO pressure from 2.5 to ∼5 bar led to a rapid rise in the CH3OH productivity, reaching a maximum of 1.44 mmolCH3OH/gcat/h. Interestingly, increasing the CO pressure beyond this optimum caused the overall oxygenate yield to decrease, suggesting that excessive CO inhibits the reaction. Excess CO may saturate active sites, hindering O2 activation and reducing oxidant availability. In other words, an optimal CO concentration is needed to effectively generate the active oxidizing species without poisoning the catalyst. It is known that CO helps maintain metal active sites in a low-valent (reduced) state, which is beneficial for sustained activity; , however, too much CO could simply compete with CH4 or O2 binding. Therefore, an optimal CO concentration is vital. Importantly, CH3OH selectivity remained very high (>89%) over a broad range of CO pressures (2.5–7 bar). Even at the highest CO pressures, a significant shift toward CH3COOH formation was not observed, demonstrating robust selectivity for CH3OH.
In the case of O2, an optimal behavior was also observed. Starting from 0 bar O2, the CH3OH productivity increased as O2 pressure was raised to ∼3 bar, reaching a maximum value of oxygenate productivity of 1.44 mmolmethanol/gcat/h with a 95% CH3OH selectivity. However, further increasing the O2 pressure to 4–6 bar decreased activity and lowered CH3OH selectivity to ∼60%. This indicates that an excess of O2 promotes the overoxidation of CH3OH to undesired products. Additionally, at high O2 partial pressure, the ability of CO to maintain the catalyst in a reduced state may be overwhelmed, leading to oxidative deactivation of active sites and a lower overall activity. These observations highlight that achieving high CH3OH yields requires careful balance in the supply of CO and O2.
To gain insight into the reaction pathway, the dependence of the reaction temperature on the product distribution was investigated. Figure A presents the selectivity for various products as a function of temperature (100–150 °C) over Pt1Cu1@S-1. At lower temperatures (e.g., 100–125 °C), a significant fraction of methyl hydroperoxide (CH3OOH) was observed in the postreaction solution, whereas at higher temperatures (≥135 °C), the CH3OOH signal diminished and more CH3OH was seen. Notably, the increase in CH3OOH selectivity at lower temperatures was correlated with a decrease in CH3OH selectivity. At 135–150 °C, a small amount (∼4 to 5%) of CH3COOH was detected, but no other liquid products were found. The temperature-dependent covariation of CH3OOH and CH3OH indicates that CH3OOH functions as an intermediate species in the pathway leading to CH3OH formation rather than as a terminal byproduct that does not further participate in the reaction sequence. In other words, at lower temperatures, CH3OOH accumulates because its conversion to CH3OH is slower, whereas at higher temperatures, CH3OOH is more rapidly converted to CH3OH, increasing the CH3OH selectivity. This mechanistic interpretation is consistent with our previous report on CO-assisted oxidation of ethane, where an ethyl hydroperoxide intermediate was implicated in the selective formation of ethanol. The implication for the present system is that CH4 is first oxidized to CH3OOH, which then decomposes or reacts further to yield CH3OH. To further substantiate this mechanistic hypothesis, a time-dependent selectivity profile was collected at 100 °C (Figure S30). After 3 h, the selectivity for CH3OOH decreased to 18%, while that for CH3OH increased to 82%. No other C1–C2 oxygenates were detected during this period, confirming the sequential conversion of CH3OOH to CH3OH under these conditions. When the reaction was extended to 6 h, the selectivities for CH3COOH and CH3CHO increased to 11 and 3%, respectively. This sequential conversion is expected to be attributed to the secondary reaction of the accumulated CH3OH with CO in the closed batch system.
8.

(A) Product selectivity at low temperatures. Reaction conditions: catalyst, 5 mg; solvent, 15 mL of H2O; reaction gas, 20 bar CH4 + 5 bar CO + 3 bar O2; reaction temperature, 100, 115, 125, 135, and 150 °C; reaction time, 1 h. (B) Comparison of product selectivities between the standard CH4 + CO+ O2 and CH4 + CO + H2O2 systems. Reaction conditions: catalyst, 5 mg; solvent, 15 mL of H2O or 16 mM H2O2 aq.; reaction gas, 20 bar CH4 + 5 bar CO + 3 bar O2 for the CH4 + CO + O2 system or 20 bar CH4 + 5 bar CO for the CH4 + CO + H2O2 system; temperature, 100 °C; reaction time, 1 h. (C) Arrhenius plot based on the productivity of total oxygenates. The dashed line represents the linear least-squares regression, and the corresponding fitting equation including the calculated activation energy is displayed within the figure. (D) Comparison of E a between the present catalyst and previously reported catalysts. ,,
In the low-temperature regime, the productivity of total oxygenates produced a linear Arrhenius plot (Figure C). From the slope, the apparent activation energy (E a) was estimated as 42 kJ/mol. This value is the lowest among the literature data regarding the CO-assisted oxidation of CH4 (Figure D). ,, The unusually low E a in our system aligns with its exceptional activity (Figure ) and provides further evidence of beneficial cooperation between Pt and Cu within hydrophobic micropores of S-1.
One key question is what oxidizing species are responsible for the initial conversion of CH4 to CH3OOH. A plausible hypothesis is that H2O2 is generated in situ from O2 and a source of H2 and that thus-formed H2O2 oxidizes CH4 to CH3OOH. To verify this hypothesis, the amount of H2O2 present in the aqueous solution after the reaction with CO and O2 was quantified using a Ce4+/Ce3+ titration method. Because H2O2 readily undergoes self-decomposition, the reaction temperature was set to a relatively low temperature (100 °C) to minimize its loss. The formation of H2O2 was evidenced by a distinct color change in the indicator (Figure S31), and its productivity was determined to be 4.6 × 10–2 mmol/gcat/h, which is on a similar order of magnitude as the oxygenate productivity observed in the CH4 + CO + O2 system at 100 °C (∼0.17 mmol/gcat/h). This relatively low H2O2 productivity may be attributed to its self-decomposition; it is expected that under CH4 reaction conditions, any H2O2 formed would be immediately consumed by CH4 oxidation, so the steady-state concentration of H2O2 remains low, thereby diminishing the impact of its decomposition.
To confirm that in situ generated H2O2 can indeed serve as the oxidant promoting CH4 oxidation, a controlled experiment was conducted in which O2 under standard CH4 + CO + O2 conditions was replaced by H2O2, keeping all other reaction parameters identical. Product selectivities were then compared between those of the standard CH4 + CO + O2 system and the CH4 + CO + H2O2 system. As shown in Figure B, both of the conditions gave similar selectivity, both producing primarily CH3OH and CH3OOH in comparable ratios. This result supports the above hypothesis that H2O2 produced in situ during a standard reaction (in the presence of CH4, CO, and O2) indeed acts as the oxidant in this selective oxidation process.
A key remaining question is the origin of hydrogen for H2O2 generation in this system. One plausible route is the water-gas shift (WGS) reaction: CO + H2O → CO2 + H2, which can occur under the present catalytic conditions in the presence of water as the solvent. We verified the formation of H2 gas from CO and H2O (solvent) over the Pt1Cu1@S-1 catalyst using gas chromatography (Figure S32), where the reaction was conducted at 150 °C, i.e., the same temperature employed to boost oxidation of CH4 to CH3OH. The measured H2 productivity was 1.0 mmol/gcat/h, which was almost comparable with that of total oxygenates (Table S5), thereby supporting the idea that the H2 generated via the WGS reaction supplies the hydrogen required for in situ H2O2 production.
Accordingly, the complete proposed reaction mechanism is illustrated in Figure . In this mechanism, CO reacts with water (the solvent) on the catalyst to generate H2, which then react with O2 to form H2O2. Finally, H2O2 oxidizes CH4 to yield CH3OOH, which subsequently decomposes into CH3OH. This mechanism aligns with the observation that both CO and O2 are required to catalyze the partial oxidation of CH4 to CH3OH (refer to Figure ).
9.

Proposed mechanism of the CO-assisted oxidation of CH4 to CH3OH catalyzed by PtCu@S-1.
An alternative mechanistic proposal in the literature is that OH• generated from H2O2 or other sources are the actual species that convert CH4 to form CH3OH. To verify whether such a radical mechanism operates in our system, electron spin resonance (ESR) experiments were performed using a radical trap (5,5-dimethyl-1-pyrroline N-oxide, DMPO). If OH• were formed during the reaction, then they would adduct with DMPO to form a DMPO–OH spin adduct, which can be detected as a characteristic quartet signal in the ESR spectrum. When the CH4 + CO + O2 reaction was conducted in the presence of aqueous DMPO, a weak 1:2:2:1 quartet signal derived from the DMPO–OH adduct was indeed detected, though its intensity was very low (Figure S33, red line). An OH• productivity of only ∼0.4 × 10–4 mmol/gcat/h was revealed by fitting analysis, which is over three orders of magnitude lower than the oxygenate productivity (∼0.17 mmol/gcat/h) at the same temperature. It is indicated by this extremely low OH• productivity that even if all detected OH• were involved in the reaction, their contribution would be negligible. Furthermore, even when a large excess of the OH• scavenger (30 mmol/gcat of Na2SO3, which is ∼1000 times the detected amount of OH•) was added, no significant change in productivity and selectivity was observed (Figure S34). By contrast, a previously reported OH•-mediated pathway in the Au/MOR catalyst system was inhibited by 63% after the addition of only 4 mmol/gcat of Na2SO3. Therefore, the negligible effect of the OH• scavenger and the extremely low productivity of OH•, observed in the present study, suggest that an OH•-mediated mechanism does not govern CH4 oxidation over Pt1Cu1@S-1.
Conclusions
We have demonstrated that encapsulating Pt–Cu nanoalloys within a hydrophobic, acid-free S-1 matrix enables low-temperature CH4 oxidation to CH3OH with a record productivity and nearly 100% selectivity. Comparative studies with model catalysts highlight the importance of precisely tuning alloy compositions and reaction environments of the encapsulated nanoalloys for optimizing activity and selectivity. The mechanistic insights into how hydrophobic, acid-free micropores promote CH3OH desorption and supprese overoxidation demonstrate the high potential of catalyst design that synergistically couple metal functionality with tailored pore architecture for efficient CH3OH production. Our findings suggest that further refinements of alloy composition and pore engineering can yield even greater control over product selectivity and productivity. In particular, establishing a synthesis protocol that embeds nanoparticles with the target alloy composition inside zeolite pores while simultaneously minimizing the density of Si–OH defects, which govern framework hydrophobicity, will be increasingly important. Because the present study did not completely eliminate these Si–OH groups (about 10% of the Q3 band was observed in 29Si MAS NMR, Figure S2), further improvements in hydrophobicity and therefore in catalytic performances can be anticipated by reducing the defect density. Furthermore, alloying the encapsulated nanoalloys with elements that improve their durability would address the stability limitations identified in this study. These strategies can be extended to other C–H bond activation processes. Furthermore, the results obtained here not only deepen our understanding of how CH4 can be directly converted to CH3OH but also inspire a broader exploration of hierarchical multifunctional catalysts for industrially relevant oxidation reactions. They are thus expected to open a promising avenue for more sustainable, economically viable routes to oxygenated fuels and chemicals, promoting continued advances in catalytic science to meet global energy and environmental challenges.
Supplementary Material
Acknowledgments
This work was supported by the Japan Society for the Promotion of Science (JSPS) Grant-in-Aid for Scientific Research (B) (nos. 22H01866 and 23H01759) and JSPS Grant-in-Aid for Transformative Research Areas (B) (no. 22H05045). XAFS measurements were performed at the synchrotron radiation facilities of AichiSR. Part of the results of this work were obtained by using research equipment shared in MEXT Project for promoting public utilization of advanced research infrastructure (program for supporting construction of core facilities) grant number JPMXS04411025. A part of this study was supported by the Joint Usage/Research Center for Catalysis.
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.5c07414.
Experimental details, additional catalytic data, additional characterization data, and comparison with the literature catalysts (PDF)
The authors declare no competing financial interest.
References
- Schwach P., Pan X., Bao X.. Direct Conversion of Methane to Value-Added Chemicals over Heterogeneous Catalysts: Challenges and Prospects. Chem. Rev. 2017;117:8497–8520. doi: 10.1021/acs.chemrev.6b00715. [DOI] [PubMed] [Google Scholar]
- Ravi M., Ranocchiari M., van Bokhoven J. A.. The Direct Catalytic Oxidation of Methane to Methanol-A Critical Assessment. Angew. Chem., Int. Ed. 2017;56:16464–16483. doi: 10.1002/anie.201702550. [DOI] [PubMed] [Google Scholar]
- Dummer N. F., Willock D. J., He Q., Howard M. J., Lewis R. J., Qi G., Taylor S. H., Xu J., Bethell D., Kiely C. J., Hutchings G. J.. Methane Oxidation to Methanol. Chem. Rev. 2023;123:6359–6411. doi: 10.1021/acs.chemrev.2c00439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond C., Forde M. M., Ab Rahim M. H., Thetford A., He Q., Jenkins R. L., Dimitratos N., Lopez-Sanchez J. A., Dummer N. F., Murphy D. M., Carley A. F., Taylor S. H., Willock D. J., Stangland E. E., Kang J., Hagen H., Kiely C. J., Hutchings G. J.. Direct catalytic conversion of methane to methanol in an aqueous medium by using copper-promoted Fe-ZSM-5. Angew. Chem., Int. Ed. 2012;51:5129–5133. doi: 10.1002/anie.201108706. [DOI] [PubMed] [Google Scholar]
- Yu T., Li Z., Lin L., Chu S., Su Y., Song W., Wang A., Weckhuysen B. M., Luo W.. Highly Selective Oxidation of Methane into Methanol over Cu-Promoted Monomeric Fe/ZSM-5. ACS Catal. 2021;11:6684–6691. doi: 10.1021/acscatal.1c00905. [DOI] [Google Scholar]
- Narsimhan K., Iyoki K., Dinh K., Román-Leshkov Y.. Catalytic Oxidation of Methane into Methanol over Copper-Exchanged Zeolites with Oxygen at Low Temperature. ACS Cent. Sci. 2016;2:424–429. doi: 10.1021/acscentsci.6b00139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sushkevich V. L., Palagin D., Ranocchiari M., van Bokhoven J. A.. Selective Anaerobic Oxidation of Methane Enables Direct Synthesis of Methanol. Science. 2017;356:523–527. doi: 10.1126/science.aam9035. [DOI] [PubMed] [Google Scholar]
- Sun L., Wang Y., Wang C., Xie Z., Guan N., Li L.. Water-Involved Methane-Selective Catalytic Oxidation by Dioxygen over Copper Zeolites. Chem. 2021;7:1557–1568. doi: 10.1016/j.chempr.2021.02.026. [DOI] [Google Scholar]
- Koishybay A., Shantz D. F.. Water Is the Oxygen Source for Methanol Produced in Partial Oxidation of Methane in a Flow Reactor over Cu-SSZ-13. J. Am. Chem. Soc. 2020;142:11962–11966. doi: 10.1021/jacs.0c03283. [DOI] [PubMed] [Google Scholar]
- Zhang H., Han P., Wu D., Du C., Zhao J., Zhang K. H. L., Lin J., Wan S., Huang J., Wang S., Xiong H., Wang Y.. Confined Cu-OH Single Sites in SSZ-13 Zeolite for the Direct Oxidation of Methane to Methanol. Nat. Commun. 2023;14:7705. doi: 10.1038/s41467-023-43508-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder B. E. R., Vanelderen P., Bols M. L., Hallaert S. D., Böttger L. H., Ungur L., Pierloot K., Schoonheydt R. A., Sels B. F., Solomon E. I.. The Active Site of Low-Temperature Methane Hydroxylation in Iron-Containing Zeolites. Nature. 2016;536:317–321. doi: 10.1038/nature19059. [DOI] [PubMed] [Google Scholar]
- Xiao P., Wang L., Toyoda H., Wang Y., Nakamura K., Huang J., Osuga R., Nishibori M., Gies H., Yokoi T.. Revealing Active Sites and Reaction Pathways in Direct Oxidation of Methane over Fe-Containing CHA Zeolites Affected by the Al Arrangement. J. Am. Chem. Soc. 2024;146:31969–31981. doi: 10.1021/jacs.4c11773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinberg C. E., Lippard S. J.. Dioxygen Activation in Soluble Methane Monooxygenase. Acc. Chem. Res. 2011;44:280–288. doi: 10.1021/ar1001473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan J., Li M., Allard L., Lee S., Flytzani-Stephanopoulos M.. Mild Oxidation of Methane to Methanol or Acetic Acid on Supported Isolated Rhodium Catalysts. Nature. 2017;551:605–608. doi: 10.1038/nature24640. [DOI] [PubMed] [Google Scholar]
- Li M., Shan J., Giannakakis G., Ouyang M., Cao S., Lee S., Allard L. F., Flytzani-Stephanopoulos M.. Single-Step Selective Oxidation of Methane to Methanol in the Aqueous Phase on Iridium-Based Catalysts. Appl. Catal., B. 2021;292:120124. doi: 10.1016/j.apcatb.2021.120124. [DOI] [Google Scholar]
- Wang W., Zhou W., Tang Y., Cao W., Docherty S. R., Wu F., Cheng K., Zhang Q., Copéret C., Wang Y.. Selective Oxidation of Methane to Methanol over Au/H-MOR. J. Am. Chem. Soc. 2023;145:12928–12934. doi: 10.1021/jacs.3c04260. [DOI] [PubMed] [Google Scholar]
- Yu X., Mao J., Wu B., Wei Y., Sun Y., Zhong L.. Boosting Direct Oxidation of Methane with Molecular Oxygen at Low Temperature over Rh/ZSM-5 Catalyst. ChemCatChem. 2023;15:e202300077. doi: 10.1002/cctc.202300077. [DOI] [Google Scholar]
- Oda A., Kimura Y., Ichino K., Yamamoto Y., Kumagai J., Lee G., Sawabe K., Satsuma A.. Rutile TiO2-Supported Pt Nanoparticle Catalysts for the Low-Temperature Oxidation of Ethane to Ethanol. J. Am. Chem. Soc. 2024;146:20122–20132. doi: 10.1021/jacs.4c04381. [DOI] [PubMed] [Google Scholar]
- Tang Y., Li Y., Fung V., Jiang D.-E., Huang W., Zhang S., Iwasawa Y., Sakata T., Nguyen L., Zhang X., Frenkel A. I., Tao F. F.. Single Rhodium Atoms Anchored in Micropores for Efficient Transformation of Methane under Mild Conditions. Nat. Commun. 2018;9:1231. doi: 10.1038/s41467-018-03235-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moteki T., Tominaga N., Tsunoji N., Yokoi T., Ogura M.. Impact of the Zeolite Cage Structure on Product Selectivity in CO-Assisted Direct Partial Oxidation of Methane over Rh Supported AEI-, CHA-, and AFX-Type Zeolites. Chem. Lett. 2021;50:1597–1600. doi: 10.1246/cl.210250. [DOI] [Google Scholar]
- Moteki T., Tominaga N., Ogura M.. Mechanism Investigation and Product Selectivity Control on CO-Assisted Direct Conversion of Methane into C1 and C2 Oxygenates Catalyzed by Zeolite-Supported Rh. Appl. Catal., B. 2022;300:120742. doi: 10.1016/j.apcatb.2021.120742. [DOI] [Google Scholar]
- Oda A., Horie M., Murata N., Sawabe K., Satsuma A.. Highly Efficient CO-Assisted Conversion of Methane to Acetic Acid over Rh-Encapsulated MFI Zeolite Prepared Using RhCl3 Molten Salt. Catal. Sci. Technol. 2022;12:5488–5494. doi: 10.1039/D2CY01471H. [DOI] [Google Scholar]
- Gu F., Qin X., Pang L., Zhang R., Peng M., Xu Y., Hong S., Xie J., Wang M., Han D., Xiao D., Guo G., Wang X., Wang Z., Ma D.. Acid-Promoted Selective Oxidation of Methane to Formic Acid over Dispersed Rhodium Catalysts under Mild Conditions. ACS Catal. 2023;13:9509–9514. doi: 10.1021/acscatal.3c01743. [DOI] [Google Scholar]
- Li H., Xiong C., Fei M., Ma L., Zhang H., Yan X., Tieu P., Yuan Y., Zhang Y., Nyakuchena J., Huang J., Pan X., Waegele M. M., Jiang D.-E., Wang D.. Selective Formation of Acetic Acid and Methanol by Direct Methane Oxidation Using Rhodium Single-Atom Catalysts. J. Am. Chem. Soc. 2023;145:11415–11419. doi: 10.1021/jacs.3c03113. [DOI] [PubMed] [Google Scholar]
- Moteki T., Tominaga N., Ogura M.. CO-assisted Direct Methane Conversion into C1and C2 Oxygenates over ZSM-5 Supported Transition and Platinum Group Metal Catalysts Using Oxygen as an Oxidant. ChemCatChem. 2020;12:2957–2961. doi: 10.1002/cctc.202000168. [DOI] [Google Scholar]
- Li H., Fei M., Troiano J. L., Ma L., Yan X., Tieu P., Yuan Y., Zhang Y., Liu T., Pan X., Brudvig G. W., Wang D.. Selective Methane Oxidation by Heterogenized Iridium Catalysts. J. Am. Chem. Soc. 2023;145:769–773. doi: 10.1021/jacs.2c09434. [DOI] [PubMed] [Google Scholar]
- Moteki T., Tominaga N., Ogura M.. CO-Assisted Partial Oxidation of Methane and Selective C1 Oxygenate Formation Using Iridium-ZSM-5 Catalyst. Bull. Chem. Soc. Jpn. 2024;97:uoae053. doi: 10.1093/bulcsj/uoae053. [DOI] [Google Scholar]
- Xu W., Liu H.-X., Hu Y., Wang Z., Huang Z.-Q., Huang C., Lin J., Chang C.-R., Wang A., Wang X., Zhang T.. Metal-oxo Electronic Tuning via in Situ CO Decoration for Promoting Methane Conversion to Oxygenates over Single-atom Catalysts. Angew. Chem., Int. Ed. 2024;63:e202315343. doi: 10.1002/anie.202315343. [DOI] [PubMed] [Google Scholar]
- Qi G., Davies T., Nasrallah A., Sainna M. A., Howe A. G. R., Lewis R., Quesne M., Catlow C., Willock D., He Q., Bethell D., Howard M. J., Murrer B., Harrison B., Kiely C., Zhao X., Deng F., Xu J., Hutchings G.. Au-ZSM-5 Catalyses the Selective Oxidation of CH4 to CH3OH and CH3COOH Using O2 . Nat. Catal. 2022;5:45–54. doi: 10.1038/s41929-021-00725-8. [DOI] [Google Scholar]
- Wu B., Yin H., Ma X., Liu R., He B., Li H., Zeng J.. Highly Selective Synthesis of Acetic Acid from Hydroxyl-mediated Oxidation of Methane at Low Temperatures. Angew. Chem., Int. Ed. 2025;64:e202412995. doi: 10.1002/anie.202412995. [DOI] [PubMed] [Google Scholar]
- Yin H., Wu B., Ma X., Su G., Han M., Lin H., Liu X., Li H., Zeng J.. CO-Assisted Methane Oxidation into Oxygenates over Surface Platinum-Titanium Alloyed Layers. Nano Lett. 2024;24:5002–5009. doi: 10.1021/acs.nanolett.4c00786. [DOI] [PubMed] [Google Scholar]
- Gu F., Qin X., Li M., Xu Y., Hong S., Ouyang M., Giannakakis G., Cao S., Peng M., Xie J., Wang M., Han D., Xiao D., Wang X., Wang Z., Ma D.. Selective Catalytic Oxidation of Methane to Methanol in Aqueous Medium over Copper Cations Promoted by Atomically Dispersed Rhodium on TiO2 . Angew. Chem., Int. Ed. 2022;61:e202201540. doi: 10.1002/anie.202201540. [DOI] [PubMed] [Google Scholar]
- Wang L., Jin J., Li W., Li C., Zhu L.-L., Zhou Z., Zhang L., Zhang X., Yuan L.. Highly Selective Catalytic Oxidation of Methane to Methanol Using Cu–Pd/Anatase. Energy Environ. Sci. 2024;17:9122–9133. doi: 10.1039/D4EE02671C. [DOI] [Google Scholar]
- Narsimhan K., Michaelis V. K., Mathies G., Gunther W. R., Griffin R. G., Román-Leshkov Y.. Methane to Acetic Acid over Cu-Exchanged Zeolites: Mechanistic Insights from a Site-Specific Carbonylation Reaction. J. Am. Chem. Soc. 2015;137:1825–1832. doi: 10.1021/ja5106927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z., Wang L., Zuidema E., Mondal K., Zhang M., Zhang J., Wang C., Meng X., Yang H., Mesters C., Xiao F.-S.. Hydrophobic Zeolite Modification for in Situ Peroxide Formation in Methane Oxidation to Methanol. Science. 2020;367:193–197. doi: 10.1126/science.aaw1108. [DOI] [PubMed] [Google Scholar]
- Fujisaki H., Ishizuka T., Kotani H., Shiota Y., Yoshizawa K., Kojima T.. Selective Methane Oxidation by Molecular Iron Catalysts in Aqueous Medium. Nature. 2023;616:476–481. doi: 10.1038/s41586-023-05821-2. [DOI] [PubMed] [Google Scholar]
- Tahsini N., Yang A.-C., Streibel V., Werghi B., Goodman E. D., Aitbekova A., Bare S. R., Li Y., Abild-Pedersen F., Cargnello M.. Colloidal Platinum-Copper Nanocrystal Alloy Catalysts Surpass Platinum in Low-Temperature Propene Combustion. J. Am. Chem. Soc. 2022;144:1612–1621. doi: 10.1021/jacs.1c10248. [DOI] [PubMed] [Google Scholar]
- Newton M. A., Knorpp A. J., Sushkevich V. L., Palagin D., van Bokhoven J. A.. Active Sites and Mechanisms in the Direct Conversion of Methane to Methanol Using Cu in Zeolitic Hosts: A Critical Examination. Chem. Soc. Rev. 2020;49:1449–1486. doi: 10.1039/C7CS00709D. [DOI] [PubMed] [Google Scholar]
- Chen H., Li Y., Yang W.. Preparation of Silicalite-1 Membrane by Solution-Filling Method and Its Alcohol Extraction Properties. J. Membr. Sci. 2007;296:122–130. doi: 10.1016/j.memsci.2007.03.021. [DOI] [Google Scholar]
- Sun Q., Wang N., Zhang T., Bai R., Mayoral A., Zhang P., Zhang Q., Terasaki O., Yu J.. Zeolite-Encaged Single-Atom Rhodium Catalysts: Highly-Efficient Hydrogen Generation and Shape-Selective Tandem Hydrogenation of Nitroarenes. Angew. Chem., Int. Ed. 2019;58:18570–18576. doi: 10.1002/anie.201912367. [DOI] [PubMed] [Google Scholar]
- Zhang X., He N., Liu C., Guo H.. Pt–Cu Alloy Nanoparticles Encapsulated in Silicalite-1 Molecular Sieve: Coke-Resistant Catalyst for Alkane Dehydrogenation. Catal. Lett. 2019;149:974–984. doi: 10.1007/s10562-019-02671-4. [DOI] [Google Scholar]
- Sun Q., Wang N., Fan Q., Zeng L., Mayoral A., Miao S., Yang R., Jiang Z., Zhou W., Zhang J., Zhang T., Xu J., Zhang P., Cheng J., Yang D.-C., Jia R., Li L., Zhang Q., Wang Y., Terasaki O., Yu J.. Subnanometer Bimetallic Platinum-Zinc Clusters in Zeolites for Propane Dehydrogenation. Angew. Chem., Int. Ed. 2020;59:19450–19459. doi: 10.1002/anie.202003349. [DOI] [PubMed] [Google Scholar]
- Wei X., Cheng J., Li Y., Cheng K., Sun F., Zhang Q., Wang Y.. Bimetallic Clusters Confined inside Silicalite-1 for Stable Propane Dehydrogenation. Nano Res. 2023;16:10881–10889. doi: 10.1007/s12274-023-5953-y. [DOI] [Google Scholar]
- Shi W., Oda A., Yamamoto Y., Harada S., Ohtsu T., Sawabe K., Satsuma A.. Encapsulated Platinum-Tin Nanoparticles in Silicalite-1 Zeolite for Methylcyclohexane Dehydrogenation. ACS Sustain. Chem. Eng. 2025;13:3608–3621. doi: 10.1021/acssuschemeng.4c09762. [DOI] [Google Scholar]
- Lee Y.-S., Lim K.-Y., Chung Y.-D., Whang C.-N., Jeon Y.. XPS Core-Level Shifts and XANES Studies of Cu-Pt and Co-Pt Alloys. Surf. Interface Anal. 2000;30:475–478. doi: 10.1002/1096-9918(200008)30:1<475::AID-SIA817>3.0.CO;2-W. [DOI] [Google Scholar]
- Foucher A. C., Yang S., Rosen D. J., Huang R., Pyo J. B., Kwon O., Owen C. J., Sanchez D. F., Sadykov I. I., Grolimund D., Kozinsky B., Frenkel A. I., Gorte R. J., Murray C. B., Stach E. A.. Synthesis and Characterization of Stable Cu-Pt Nanoparticles under Reductive and Oxidative Conditions. J. Am. Chem. Soc. 2023;145:5410–5421. doi: 10.1021/jacs.2c13666. [DOI] [PubMed] [Google Scholar]
- Deng Z., Gong Z., Gong M., Wang X.. Multiscale Regulation of Ordered PtCu Intermetallic Electrocatalyst for Highly Durable Oxygen Reduction Reaction. Nano Lett. 2024;24:3994–4001. doi: 10.1021/acs.nanolett.4c00583. [DOI] [PubMed] [Google Scholar]
- Oda A., Ichihashi K., Yamamoto Y., Ohtsu T., Shi W., Sawabe K., Satsuma A.. Pt Single Atom Alloyed Sub-1 Nm Thick Fe Overlayer on Supported Cu Nanoparticles for Methylcyclohexane Dehydrogenation. J. Mater. Chem. A. 2024;12:22655–22667. doi: 10.1039/D4TA03453H. [DOI] [Google Scholar]
- Oda A., Ogawa N., Yamamoto Y., Sawabe K., Satsuma A.. Atom-to-Nm Scale Engineering of PtCo Alloy Catalysts over Supported Co Nanoparticles for Advanced Toluene Hydrogenation Efficiency in Hydrogen Storage Applications. ACS Catal. 2025;15:3191–3202. doi: 10.1021/acscatal.4c07206. [DOI] [Google Scholar]
- Sun G., Zhao Z.-J., Mu R., Zha S., Li L., Chen S., Zang K., Luo J., Li Z., Purdy S. C., Kropf A. J., Miller J. T., Zeng L., Gong J.. Breaking the Scaling Relationship via Thermally Stable Pt/Cu Single Atom Alloys for Catalytic Dehydrogenation. Nat. Commun. 2018;9:4454. doi: 10.1038/s41467-018-06967-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin R., Peng M., Li A., Deng Y., Jia Z., Huang F., Ling Y., Yang F., Fu H., Xie J., Han X., Xiao D., Jiang Z., Liu H., Ma D.. Low Temperature Oxidation of Ethane to Oxygenates by Oxygen over Iridium-Cluster Catalysts. J. Am. Chem. Soc. 2019;141:18921–18925. doi: 10.1021/jacs.9b06986. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.