

Abstract
Background
Bispecific T cell engager (BiTE), such as blinatumomab, has demonstrated significant clinical success in treating hematological malignancies like B cell acute lymphoblastic leukemia and non-Hodgkin's lymphoma. However, the application of BiTEs in solid tumors has proven challenging, primarily due to the lack of targetable tumor antigens and the immunologically “cold” nature of the tumor microenvironment, which limits immune system activation.
Methods
We developed a novel oncolytic virus (OV) platform by engineering a chimeric vaccinia virus to express either a truncated non-signaling CD19 antigen (CD19t) or truncated B cell maturation antigen (BCMAt) on the surface of infected tumor cells. Here, we advance a combinatorial platform using an OV to redirect CD19-targeted or BCMA-targeted T cell engagers (TCEs) to drive antitumor responses against multiple solid tumors.
Results
We found that OV-infected tumor cells in combination with TCEs significantly improved tumor cell killing against solid tumor models, with efficacy comparable to that of chimeric antigen receptor T cells. This combination approach enhanced antitumor responses using in vivo human tumor xenograft models and promoted more effective elimination of solid tumor cells than either therapy alone. Our studies highlight OVs combined with clinically approved TCEs as a readily translatable, tumor-agnostic, off-the-shelf strategy to effectively target solid tumors.
Conclusions
Our findings demonstrate that the combination of OV and TCEs offers a promising strategy to drive antitumor immune responses against solid tumors. This approach represents a novel and universal platform currently in phase 1 clinical trial combining TCE therapy with oncolytic virotherapy, overcoming antigen heterogeneity and immunological barriers for the effective treatment of solid tumors.
Keywords: Oncolytic virus, Chimeric antigen receptor - CAR, Bispecific T cell engager - BiTE, Combination therapy, Immunotherapy
WHAT IS ALREADY KNOWN ON THIS TOPIC.
WHAT THIS STUDY ADDS
This study presents a novel application of OV and bispecific T cell engagers (TCEs) providing a potent and adaptable strategy to deliver a tumor antigen and recruit and activate endogenous T cells against solid cancers. Unlike previous strategies that have combined OV with CAR T cells, this combination leverages OV-mediated expression of a tumor target to redirect polyclonal bystander T cells, thus eliminating the need for CAR T cell manufacturing. This new combinatorial enables a universal off-the-shelf approach that can be used across diverse cancer patient populations without the need for personalized cell manufacturing.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY.
These findings represent a significant advancement in the field of solid cancer treatment by introducing a readily scalable, universal off-the-shelf immunotherapy that integrates the complementary advantages of OV and TCE. This approach establishes a foundational platform for further clinical development of OV-based strategies, not only for delivering tumor-associated antigens that may be targeted by TCE, but also for other immunomodulatory payloads with the potential to enhance antitumor efficacy and improve clinical outcomes.
Background
A universal tumor-agnostic therapy remains elusive in the field of cancer immunotherapy. There have been impressive patient-specific immunotherapies with recent Food and Drug Administration (FDA) approvals including chimeric antigen receptor (CAR) T cell therapies. Limitations include the autologous nature of these cells and the length of time required for cellular manufacturing. In contrast to autologous CAR T cells, the benefits of bispecific T cell engagers (TCEs) are their off-the-shelf availability and the ability to acutely tune targeting with dosing regimens.1 2 TCEs have emerged as a promising immunotherapy strategy for the treatment of B-cell hematological malignancies. Blinatumomab, an FDA-approved TCE carrying CD19 and CD3 single-chain variable fragments (scFvs) that drives endogenous T-cell mediated immune responses against malignant cells, has shown durable clinical responses for the treatment of B-cell acute lymphoblastic leukemia and non-Hodgkin’s lymphoma.3 4 Challenges facing solid tumor therapies using CAR T cells and TCE therapies are driven by a lack of amenable and targetable tumor antigens.5 6 The shared expression of solid tumor antigens on normal tissue and their heterogeneous, and non-uniform, expression patterns on tumors limits the potential for effective and durable antitumor responses.7 8 Many solid tumors are also immunologically “cold” and limit T cell trafficking and antitumor functionality, a phenomenon uncommonly observed in hematological malignancies.9,11 Thus, improved and more accessible immunotherapies remain to be explored.
Oncolytic viruses (OVs) have emerged as a promising off-the-shelf treatment modality for various tumor types. OVs are tumor-specific viruses that have desirable immunogenic properties with the capacity for transgene delivery to tumors.12 We recently developed a novel combination therapy that enabled redirection of CD19-CAR T cells to solid tumors by exploiting the transgene delivery potential of a vaccinia-based OV to selectively infect and drive tumor-specific expression of a truncated non-signaling variant of CD19 (CD19t).13 The parent version of the chimeric OV used here has shown safety and antitumor activity in several preclinical models.14 15 Here, we build a fully off-the-shelf therapeutic approach using these OVs in combination with TCEs. We demonstrate robust cell surface CD19t expression on multiple tumor types infected with OV carrying the CD19t-encoding gene (OV19t), which promoted activation and tumor cell killing by T cells when treated with CD19-TCE. Importantly, this approach is not limited to CD19 and can be used with other tumor antigens including B cell maturation antigen (BCMA). We demonstrate potent antitumor activity with this combination using multiple in vivo tumor models, including efficacy in animals bearing multiple tumors following local OV administration to a single tumor site. In summary, we develop a tumor-agnostic combinatorial therapy leveraging CD19 and BCMA as proof-of-concept antigens for the combination of OVs and TCEs to target solid tumors.
Methods
Study design
In this study, we evaluated a combination approach using a novel OV to induce de novo cell surface expression of tumor antigen targets on solid tumors, which in combination with off-the-shelf bispecific TCEs, redirects endogenous T cell-mediated antitumor immunity. Our oncolytic vaccinia virus (VACV), CF33 carrying a truncated CD19 (CD19t) (OV19t) was previously developed in combination with CD19-CAR T cells.13 We also expanded this platform using CF33 carrying a truncated BCMA (BCMAt) (OV encoding BCMAt (OVBCMAt)), for combination with BCMA-targeting agents. All in vitro assays were performed with at least duplicate samples and were repeated in at least three independent experiments. In vivo studies were performed using 6–8 weeks old NSG, using at least three mice per group for all studies, and four to nine mice were included within each group for all therapeutic and survival studies to ensure statistical power and evenly distributed tumor sizes across groups at treatment initiation. The health condition of mice was monitored daily by the Department of Comparative Medicine at City of Hope with euthanasia applied according to the American Veterinary Medical Association Guidelines. Investigators were not blinded when monitoring mouse survival. All studies were performed under approved protocols of the Institutional Animal Care and Use Committee and the institutional review board.
Cell lines
Human triple-negative breast cancer cell line MDA-MB-468 (American Type Culture Collection (ATCC); HTB-132) was cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% fetal bovine serum (FBS; HyClone) and 1× antibiotic/antimycotic (AA; Gibco), supplemented with 25 mM Hepes (Irvine Scientific) and 2 mM L-glutamine (Thermo Fisher Scientific; complete DMEM). Both MDA-MB-468-CD19t and MDA-MB-468-human epidermal growth factor receptor 2 (HER2) cell lines were cultured as mentioned above. Human pancreatic cancer cell line Capan-1 (ATCC, HTB-79) was cultured in Iscove’s modified Dulbecco’s medium containing 20% FBS and 1× AA. Human pancreatic cancer cell line Panc-1 (ATCC, CRL-1469) was cultured in Roswell Park Memorial Institute (RPMI) containing 10% FBS and 1× AA. Human ovarian cancer cell line OV90 (ATCC CRL-11732) was cultured in 1:1 vol of MCDB 105 medium (Sigma-Aldrich) and medium 199 (Gibco) containing 20% FBS and 1× AA. Human head and neck carcinoma line UM-SCC-47 (EMD Millipore) was cultured in DMEM containing 10% FBS, 1× AA, and 1× non-essential amino acids. Human prostate cancer cell line DU145 (ATCC, HTB-81) was cultured in RPMI containing 10% FBS and 1× AA. Human glioblastoma cell line U251T (gift from W Debinski, Wake Forest School of Medicine) was cultured in complete DMEM. Human embryonic kidney cell line 293T (ATCC CRL-3216) and human fibrosarcoma cell line HT1080 (ATCC CCL-121) were cultured in complete DMEM. African green monkey kidney fibroblasts (CV-1; ATCC CCL-70) were cultured in DMEM containing 10% FBS and 1× AA. CV-1 cells were used for both amplification and titration of orthopoxviruses.
Generation of recombinant chimeric orthopoxvirus expressing human CD19t and BCMAt
To generate a shuttle vector containing the human (hCD19t) CD19t expression cassette with the VACV Synthetic Early Promoter (PSE), the hCD19t complementary DNAs (cDNAs) were PCR amplified from the plasmids hCD19t-2A-IL2-pHIV7 and mCD19t-epHIV7 using Q5 High-Fidelity 2X Master Mix (New England Biolabs, Ipswich, Massachusetts, USA) and the following primers: 5′-GCG GTC GAC CAC CAT GCC ACC TCC TCG CCT CCT CTT CTT CCT CCT CTT CCTC-3′ and 5′-GCG GGA TCC ATA AAA ATT AAT TAA TCA TCT TTT CCT CCT CAG GAC CAG GGC TCT TTG AAG ATG-3′. The PCR fragment was digested with Sal I and Bam HI and cloned into the same-cut p33NCTK-SE-hNIS replacing hNIS to yield p33NCTK-SE-hCD19t and p33NCTK-SE-mCD19t. The hCD19t and mCD19t cDNAs in p33NCTK-SE-hCD19t and p33NCTK-SE-mCD19t were confirmed by sequencing. CV-1 cells were infected with CF33 at a multiplicity of infection (MOI) of 0.1 for 1 hour and then transfected with p33NCTK-SE-hCD19t and p33NCTK-SE-mCD19t by using jetPRIME in vitro DNA and small interfering RNA transfection reagent (Polyplus-transfection, New York, New York, USA). Two days after infection, infected and transfected cells were harvested, and recombinant viruses (OV19t) were selected and plaque purified as described previously.13
For the construction of a recombinant OVBCMAt, BCMA lacking the intracellular domain, the CRISPR/Cas9 technology was used to assist with the selection of recombinant virus, as previously described16 and purchased from IDT. The oligo (5’-GTTATAGTAGCCGCACTCG-3’) was used to synthesize single guide RNA (sgRNA) using the EnGen sgRNA Synthesis Kit (Cat#E3322S; NEB) following the manufacturer’s instruction. Next, ∼70% confluent HEK293 cells in a 24-well plate were infected with CF33 virus at an MOI of 0.1. Two hours after infection, the medium was changed with fresh DMEM medium supplemented with 10% FBS, and the cells were transfected with ribonucleoprotein complex of sgRNA and Cas9 together with the template for homologous recombination, that is, the J2R-shuttle plasmid containing BCMAt cassette. Lipofectamine CRISPRMAX transfection reagent (Cat#CMAX00008; Thermo Fisher) was used for transfection; 1250 ng of Cas9 and 240 ng sgRNA was used per well. The following day, cell lysate was collected and subjected to three rounds of freeze-thaw cycles. The lysate was then used to infect CV1 cells in six well plates and methylcellulose overlay was applied. Two days after incubation, plaques were picked and PCR verified. One more round of plaque purification was performed prior to amplification and purification of the virus on sucrose gradient. The transgene in the recombinant virus was sequence verified.
DNA constructs
MDA-MB-468 cells were engineered to express hCD19t by transduction with epHIV7 lentivirus carrying the human CD19t gene under the control of the EF1 promoter. This same process was used to engineer the expression of HER2 in MDA-MB-468. The human CD19-28z CAR lentiviral construct with truncated human epidermal growth factor receptor (hEGFRt) separated by a T2A ribosome skip sequence was used as previously described.17 18 The human BCMA-28z CAR lentiviral construct with hEGFRt separated by a T2A ribosome skip sequence was generated using BCMA scFv (clone C11D5.3; US20120082661A1), extracellular spacer domain including the 129-amino acid CH2-deleted version (ΔCH2) of the IgG4 Fc spacer, and intracellular costimulatory signaling domain containing 28z with a CD28 transmembrane domain. The human HER2-41BBz CAR lentiviral construct previously described19 was modified to remove the CD19t domain. For T cell trafficking studies, the firefly luciferase (ffluc) gene was cloned into an epHIV7 lentivirus construct and then used to transduce human T cells cultured as previously described.20
Human T cell enrichment, lentivirus production and transduction, and ex vivo expansion
T cell isolation, lentivirus production and transduction, and ex vivo expansion of untransduced (UTD) and CAR T cells were performed as previously described.20 UTD human T cells in all studies were processed in parallel with CAR T cells.
T cell engagers
Blinatumomab (CD19-TCE) (US7112324B1), and teclistamab (BCMA-TCE) (WO2017031104A1) were obtained as clinical discard from City of Hope Pharmacy. Where indicated, blinatumomab biosimilar was used (ProteoGenix; Reference #PX-TA1067). Concentrations for in vitro studies and doses for in vivo studies are detailed below.
Extracellular staining and flow cytometry
Flow cytometric analysis was performed as previously described.13 20 Tumor cells and T cells were discriminated using CD45 (PerCP, BD Biosciences) for all in vitro studies. T cell activation was determined by using antibodies against CD69 and 4-1BB (CD137) (BD Biosciences). HER2 expression on tumor cells was determined using a HER2 antibody (PE, BD Biosciences). Tumor cells were identified using an antibody recognizing Ep-CAM (APC, BioLegend) for all in vivo studies. CD19t and BCMAt expression following virus infection were determined using an antibody against CD19 (PE-Cy7, BD Pharmingen) and BCMA (PE, BioLegend) for all in vitro and in vivo studies, respectively. For the detection of TCE on the surface of T cells, biotinylated Protein-L (GenScript) and a secondary streptavidin PE (BD Biosciences) antibody were used. Samples were then washed twice, stained with 4′,6-diamidino-2-phenylindole (DAPI) for viability, and processed on the MACSQuant Analyzer V.10 or V.16 (Miltenyi Biotec). Data were analyzed with FlowJo software (V.10, Tree Star).
OV transduction and T cell functional assays
For OV19t transduction and tumor killing assays, tumor targets plated with varying MOIs of OV19t were co-cultured with UTD T cells or peripheral blood mononuclear cells (PBMCs) at varying effector T cell-to-tumor cell ratios along with TCE concentrations of either 0, 20, 100, or 500 ng/mL. Cocultures were maintained in complete X-VIVO (Lonza) and in the absence of exogenous cytokines in round-bottom 96-well tissue culture-treated plates (Corning) for 1–3 days and analyzed by using flow cytometry as described above. Tumor cell killing by T cells with TCE was calculated by comparing CD45− cell counts relative to the killing observed by T cells without TCE from the same healthy donor in the absence of OV. For T cell activation assays, T cells and tumor targets were cocultured at an effector T cell-to-tumor cell ratio of 1:1 along with varying MOIs of OV in complete X-VIVO in the absence of exogenous cytokines in 96-well plates for 1–3 days and analyzed by using flow cytometry for specific markers of T cell activation.
For the preloading of TCE onto T cells, T cells and PBMCs were thawed and rested overnight in X-VIVO containing 10% FBS and interleukin (IL)-2 (100 U/mL) and IL-15 (0.5 ng/mL) cytokines. T cells and PBMCs (3×106 cells/mL) were then incubated on ice for 30 min with 10 µg/mL, 25 µg/mL, 50 µg/mL or 100 µg/mL TCE. Following incubation, T cells were then washed with phosphate-buffered saline (PBS) and co-cultured with tumor cell targets with varying MOIs of OV. T cell activation and killing was determined as previously mentioned.
For OV transduction of MDA-MB-468-HER2 tumor killing assays, tumor targets plated with varying MOIs of OV19t were co-cultured with UTD T cells or HER2-CAR T cells at an effector to tumor target ratio of 1:5 (5,000–20,000) and maintained in X-VIVO containing 10% FBS. Wells were seeded with either 100% MDA-MB-468, 100% MDA-MB-468-HER2, or 80% MDA-MB-468 and 20% MDA-MB-468-HER2 (80/20). TCE was added at a concentration of 100 ng/mL when indicated. Following 24 hours, wells were rechallenged with 3× the number of MDA-MB-468P tumor cells per well (60,000 cells). Cocultures were maintained for 1–2 days as described above and then analyzed using flow cytometry. For wells rechallenged a second time, an additional round of 3× MDA-MB-468 (60,000 cells) was added to all wells. 72 hours after the second rechallenge, co-cultures were processed and analyzed using flow cytometry as mentioned above.
For OVBCMAt transduction and tumor killing assays, similar protocols were used as described above BCMA-TCE, at a concentration of either 1, 10, or 100 µg/mL in culture.
Cytokine ELISA
Tumor cells and T cells were plated into 96-well round-bottom plates (Costar) along with varying MOIs of OV19t or OVBCMAt in the presence or absence of TCE (CD19 or BCMA-targeting), respectively. After incubations at 37°C for 24, 48, or 72 hours, supernatants were collected and analyzed according to the human interferon (IFN) or IL-2 ELISA Ready-SET-Go! (eBioscience) manufacturer’s protocol. Plates were read at 450 nm and 570 nm using the Cytation 3 Cell Imaging Multi-Mode Reader and Gen5 Microplate Reader and Imager Software (BioTek).
In vivo studies
All animal experiments were performed under protocols approved by the City of Hope Institutional Animal Care and Use Committee. For human tumor xenograft studies, MDA-MB-468 and MDA-MB-468-CD19t cells (5×106 cells per mouse) were prepared in PBS and injected subcutaneously into the flank of female NSG mice. Tumor growth was monitored 2–3 times per week by caliper measurement. Once tumor volumes reached about 100–300 mm3, OV19t virus was prepared and diluted in PBS (pH 7.4) and intratumorally administered at 106 plaque-forming units (pfu) per MDA-MB-468 tumor-bearing mice. Two days post-OV19t treatment, PBMCs were isolated from leukapheresis products obtained from a consented research participant (healthy donor) under protocols approved by the City of Hope (COH) Internal Review Board (IRB) using density gradient centrifugation over Ficoll-Paque (GE Healthcare) followed by multiple washes in PBS/EDTA (Milteny Biotec). PBMCs were then counted, washed, and prepared in PBS (pH 7.4) for intravenous tail vein injection (10×106 cells per mouse). Four days post T cell engraftment, mice were treated with TCE (8 µg/ms in PBS) intravenously for 5 consecutive days (Monday–Friday) and then again for three consecutive days the following week (Monday–Wednesday) for a total of 8 treatments (64 µg total per mouse). Mice were euthanized 2 weeks post-OV19t treatment in accordance with our animal safety guidelines.
For in vivo studies with mice bearing MDA-MB-468-CD19t or U251T-CD19t tumors as CD19t+controls, treatment with PBMCs and TCE coincided with the same schedule as described above, in the absence of OV treatment.
For in vivo T cell trafficking studies, mice were engrafted subcutaneously with either MDA-MB-468 or MDA-MB-468-CD19t and treated with OV19t. Two days post-OV19t treatment, ffluc-expressing T cells were thawed, washed, and prepared in PBS (pH 7.4) for intravenous tail vein injection (10×106 cells per mouse). TCE treatment followed as previously described. ffluc-T cell trafficking was monitored 2–3 times a week by noninvasive optical imaging (LagoX). Mice were imaged after intraperitoneal injection of 150–250 uL of d-luciferin potassium salt (PerkinElmer) suspended in PBS (pH 7.4, 4.29 mg per mouse). Flux was then analyzed with Living Image software (Aura).
For in vivo studies assessing the systemic effects of the combinatorial OV and TCE therapy, MDA-MB-468, MDA-MB-468-CD19t U251T, and U251T-CD19t cells were prepared in PBS and injected subcutaneously into the right and left flanks of female NSG mice (5×106 cells per tumor). Tumor growth on both flanks was monitored 2–3 times per week by caliper measurement. Once tumor volumes reached about 60–300 mm3, OV19t was prepared and diluted in PBS (pH 7.4) and intratumorally administered to either the right (MDA-MB-468) or left (U251T) flank tumor only at 106 or 103 pfu per tumor. Two days post-OV19t treatment, PBMCs were prepared as described above and administered via intravenous tail vein injection (10×106 cells per mouse). 1–4 days post-PBMC engraftment, mice were treated with CD19-TCE (8 µg/ms in PBS) or BCMA-TCE (25 µg/ms in PBS) intravenously for 5 consecutive days (Monday–Friday) and then again for three consecutive days the following week (Monday–Wednesday) for a total of eight treatments (64 or 200 µg total per mouse, respectively). Following the initial OV treatment, tumor volumes on both flanks were monitored by caliper measurement 2–3 times per week.
Immunohistochemistry
Tumor tissue was fixed for up to 3 days in 4% paraformaldehyde (Boston BioProducts) and stored in 70% ethanol until further processing. Immunohistochemistry was performed by the Pathology Core at the City of Hope. Briefly, paraffin-embedded sections (10 µm) were stained with H&E (Sigma Aldrich) and anti-VACV (Abcam). Images were obtained using the NanoZoomer 2.0-HT digital slide scanner and the associated NDP.view2 software (Hamamatzu).
Statistical analysis
Data are presented as means±SEM, unless otherwise stated. Statistical comparisons between groups were performed using the unpaired two-tailed Student’s t test to calculate p value, unless otherwise stated.
Data availability
All raw data for both the in vitro and in vivo studies are available on request from the corresponding author.
Results
CD19-CD3 TCE redirects human T cells to target solid tumors infected with OV19t
For these studies, we used the oncolytic chimeric orthopoxvirus carrying CD19t (OV19t) as previously described.13 We first assessed whether the CD19t delivered to tumors via OV could activate non-targeting T cells in the presence of blinatumomab (CD19-CD3 TCEs (CD19-TCEs) (figure 1a). MDA-MB-468 human triple-negative breast cancer tumor cells were infected with varying MOI (0, 0.00625, 0.0125, 0.025, 0.05, 0.1, and 1) of OV19t and treated with 100 ng/mL CD19-TCE. Tumor cells were then co-cultured with human PBMC-derived non-targeting T cells at an effector-to-target (E:T) ratio of 1:1. At 48 hours, T cell activation (CD69, CD137) and secretion of IFNγ and IL-2 were induced in the presence of 100 ng/mL CD19-TCE when co-cultured with OV19t-infected tumor cells in an MOI-dependent manner (figure 1b–d).
Figure 1. CD19-TCE redirects in vitro activation and cytotoxicity of human T cells against tumor cells infected with OV19t. (a) Schematic of combination therapy using OV19t to introduce CD19t to solid tumor cells allowing tumor cell-targeting by peripheral blood mononuclear cells (PBMC) with the addition of CD19-TCE. Control refers to wells that contain PBMCs only. (b–c) Quantification of CD69 (b) and CD137 (c) expression on human T cells following 24 hours co-culture with tumor cells at an effector:target (E:T) ratio of 1:1 with or without CD19-TCE and the indicated multiplicity of infection (MOI) of OV19t. n=2 per condition and each experiment was repeated three times. (d) IFNy (left) and IL-2 (right) production measured by ELISA in supernatants collected from co-cultures with or without CD19-TCE with indicated MOI of OV19t after 24 hours. n=2 per condition and each experiment was repeated three times. (e) Tumor cell killing assay of MDA-MB-468 tumor cells visualized by phase-contrast microscopy at 5× magnification with or without CD19-TCE in the presence of human T cells or CD19-CAR T cells and indicated MOI of OV19t. MDA-MB-468-CD19t cells were used as a positive control. Representative images are shown from one experiment which has been repeated three times. (f) Quantification of MDA-MB-468 cell killing assessed by flow cytometry. Tumor cells were co-cultured with human T cells with or without CD19-TCE and indicated MOI of OV19t for 24 (top), 48 (middle), and 72 (bottom) hours. (g) CD19t percentage on MDA-MB-468 tumor cells from (f) after 24 (left), 48 (middle), and 72 (right) hours. n=2 per condition and each experiment was repeated three times. P values indicate differences between CD19-TCE groups with PBMCs and PBMCs only determined using unpaired Student’s t-test (*p<0.05, **p<0.01, and ***p<0.005; ns=not significant). CAR, chimeric antigen receptor; IFNγ, interferon gamma; IL, interleukin; OV19T, oncolytic virus carrying the CD19t-encoding gene; TCE, T cell engager.

We then performed in vitro cell killing assays with tumor cells infected with OV19t at MOIs of 0, 0.0125, 0.5, and 1 in combination with 0, 20, or 100 ng/mL CD19-TCE. MDA-MB-468 tumor cells stably expressing CD19t via lentiviral transduction (MDA-MB-468-CD19t) were used as a positive control. We observed robust tumor cell killing of MDA-MB-468 tumor cells infected with OV19t and co-cultured with non-targeting T cells in the presence of either 20 or 100 ng/mL CD19-TCE when compared with OV19t-infected tumors alone (figure 1e). Tumor cell killing was quantified using flow cytometry. At each time point (24 hours, 48 hours, and 72 hours), we observed greater killing of tumor cells infected with OV19t in the presence of CD19-TCE compared with respective controls without CD19-TCE (figure 1f). T cell-mediated tumor cell killing when co-cultured with CD19-TCE was confirmed with MDA-MB-468-CD19t (online supplemental figure 1).
We confirmed the activity of this combination comparing the FDA-approved blinatumomab to research grade CD19-TCE (ProteoGenix), which demonstrated similar in vitro activity (online supplemental figure 2a-c). Therefore, research grade CD19-TCE was used for all subsequent studies. Similar tumor cell killing was observed against the OV90 cell line infected with OV19t and co-cultured with non-targeting T cells in the presence of either 20 or 100 ng/mL CD19-TCE when compared with OV19t-infected tumors alone (online supplemental figure 3). Importantly, CD19t expression was significantly reduced in tumor cells with CD19-TCE, showing on-target activity (figure 1g). Additional solid tumor cell lines including PC3 (prostate), U87 (glioma), and SNU-16 (gastric) were tested using this combination therapy and demonstrated similar advantages in activation, cytokine secretion, tumor cell killing, and targeting of CD19t expressing cells (online supplemental figure 4). Taken together, our data suggest that CD19-TCE redirects T cell-mediated activation and antitumor activity against OV19t-infected tumor cells.
Development of a novel target for delivery by OV to be combined with teclistamab, an FDA-approved BCMA-TCE
To expand on potential targets that can be used within this combinatorial strategy, we developed a new oncolytic chimeric orthopoxvirus expressing a novel BCMAt target under the control of a synthetic early promoter that allowed for immediate surface target expression. BCMAt was inserted into the J2R locus encoding thymidine kinase as was done for CD19t (figure 2a). We demonstrated the ability of OVBCMAt to effectively deliver and generate cell surface expression of BCMAt on multiple solid tumor cell lines (figure 2b). To evaluate the antitumor activity of OVBCMAt in combination with BCMA-CAR T cells in vivo, we treated NSG mice bearing subcutaneous MDA-MB-468 tumors with a single intratumoral injection of 106 pfu OVBCMAt. 7 days after OVBCMAt treatment, tumors were harvested and demonstrated high expression of BCMAt (online supplemental figure 5a). Therefore, mice were systemically treated with BCMA-CAR T cells on day 7 following OVBCMAt treatment and tumor volumes were monitored. Mice treated with OVBCMAt alone delayed tumor growth as expected, but we observed marked tumor regression with the combination of OVBCMAt and BCMA-CAR T cells (online supplemental figure 5b).
Figure 2. OV effectively delivers BCMAt to solid tumors and redirects activation and cytotoxicity of BCMA-CAR T cells and human T cells in the presence of BCMA-TCE in vitro. (a) Illustration of vaccinia OV with BCMAt incorporated under the control of the synthetic early promoter inserted into the J2R locus and replacing the tk gene (OVBCMAt). (b) Quantification of BCMAt expression on various solid tumor cell lines 24 (left) and 48 hours (right) after exposure to indicated MOI of OVBCMAt. n=2 per condition and each experiment was repeated two times. (c–e) Quantification of CD137 expression (left), tumor cell killing (middle), and BCMAt expression (right) assessed by flow cytometry after 24 hours of (c) MDA-MB-468, (d) SNU-16, or (e) OV90 tumor cells co-cultured with or without BCMA-TCE in the presence of human T cells or BCMA-CAR T cells and indicated MOI of OVBCMAt. n=2 per condition and each experiment was repeated three times. P values indicate differences between BCMA-TCE groups with PBMCs and PBMCs only determined using unpaired Student’s t-test (*p<0.05, **p<0.01, and ***p<0.005; ns=not significant). BCMAt, truncated B cell maturation antigen; CAR, chimeric antigen receptor; MOI, multiplicity of infection; OV, oncolytic virus, OVBCMAt, OV carrying the BCMAt-encoding gene; PBMC, peripheral blood mononuclear cell; TCE, T cell engager; tk, thymidine kinase; UTD, untransduced.
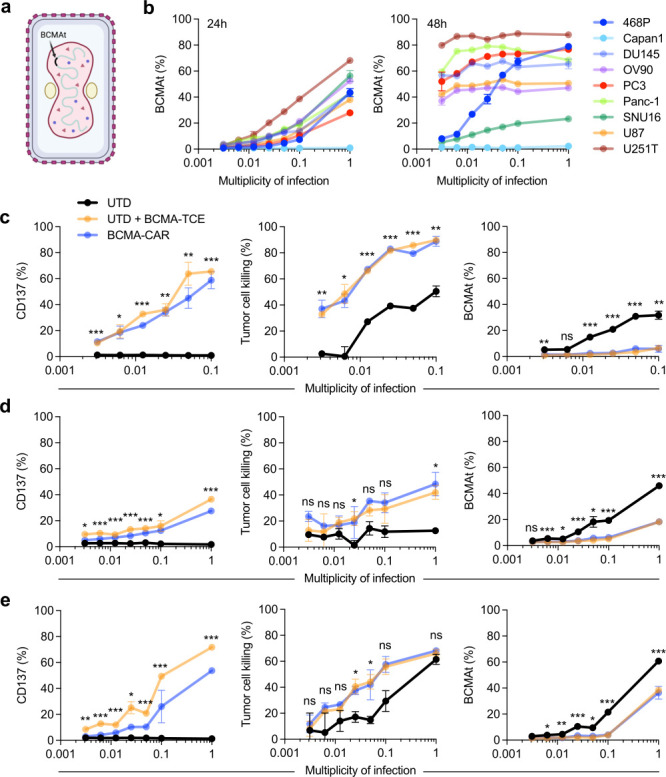
Using OVBCMAt, we next assessed whether BCMAt delivered to tumors activated BCMA-CAR T cells or non-targeting T cells in the presence of teclistamab (BCMA-TCE). MDA-MB-468 triple-negative breast cancer tumor cells were infected with varying MOIs of OVBCMAt, and BCMA-TCE was added at a concentration of 100 µg/mL. These tumor cells were then co-cultured with BCMA-CAR T cells or non-targeting T cells at an E:T ratio of 1:1 for 24 hours. Cell surface expression of 4-1BB (CD137) was used as a marker of T cell activation and quantified via flow cytometry. At 24 hours, BCMA-CAR T cells and non-targeting T cells in combination with BCMA-TCE demonstrated activation against OVBCMAt infected tumor cells in an MOI-dependent manner compared with respective controls without BCMA-TCE (figure 2c). To further quantify the T cell activation in the presence of BCMA-TCE, supernatants from co-cultures were collected to evaluate T cell-dependent cytokine production. At 24 hours, IFNγ and IL-2 secretion were detected in an MOI-dependent manner (online supplemental figure 6). We quantified killing of tumor cells when co-cultured with OVBCMAt and BMCA-CAR T cells or non-targeting T cells with BCMA-TCE using flow cytometry. At 24 hours, we observed enhanced killing of tumor cells infected with OVBCMAt and co-cultured with non-targeting T cells in the presence of BCMA-TCE (figure 2c). In addition, we validated the ability of BCMA-TCE to specifically redirect non-targeting T cells to BCMAt-expressing tumor cells following OVBCMAt expression and found that the percent BCMAt expression was significantly reduced in wells with teclistamab (figure 2c). Additionally, we tested this combination in vitro against two other solid tumor cell lines SNU16 (gastric) and OV90 (ovarian). Following 24 hours co-culture, we demonstrated enhanced activation, tumor cell killing, and BCMAt targeting in the presence of BCMA-TCE similar to BCMA-CAR T cells (figure 2d–e).
OV19t promotes tumor infiltration of non-targeting T cells following CD19-TCE treatment
OV has been previously shown to augment infiltration of T cells and other immune cells into tumors inducing endogenous antitumor immunity.21 Therefore, we assessed the tumor trafficking of engrafted T cells lentivirally transduced to stably express ffluc (Tc-ffluc) following OV19t infection with or without CD19-TCE treatment in MDA-MB-468 xenograft model. We treated NSG mice bearing subcutaneous MDA-MB-468 tumors with a single intratumoral injection of 106 pfu OV19t as previously described.13 Two days after OV19t treatment, we engrafted Tc-ffluc with 5×106 cells per mouse. 4 days after Tc-ffluc transfer, mice were treated intravenously with CD19-TCE (8 µg/dose) or PBS for 5 consecutive days (Monday–Friday) and then 3 consecutive days (Monday–Wednesday) the following week for a total of 64 µg/mouse (figure 3a). The combination of OV19t, Tc-ffluc, and CD19-TCE again exhibited significant tumor regression (figure 3b). Interestingly, OV19t and CD19-TCE combination treatment induced significantly higher influx of non-targeting T cells compared with PBMC-ffluc alone or PBMC-ffluc with OV19t alone (figure 3c–d). These data suggest that CD19-TCE redirects T cells to target CD19t, enabling tumor infiltration of endogenous T cells to target and kill tumors expressing CD19t induced by OV19t infection.
Figure 3. Tumor infiltration of non-targeting T cells expressing ffluc (Tc-ffluc) in combination with CD19-TCE and OV19t in a human xenograft MDA-MB-468 tumor model. (a) Schematic of subcutaneous MDA-MB-468 tumor-bearing NSG mice (5×106 cells on day 0) treated with intratumoral (IT) OV19t (1×106 pfu) on day 32, engrafted with intravenous (IV) Tc-ffluc (1×107 cells) on day 34, and treated with IV CD19-TCE (8 µg/dose, 8 times) on days 38–42 and 45–47. (b) NSG mice were subcutaneously injected with MDA-MB-468 (5×106 cells) on day 0. Tumor-bearing mice were treated with IT OV19t (1×106 pfu) on day 32, engrafted with IV Tc-ffluc (1×107 cells) on day 34, and treated with IV CD19-TCE (100 µg/dose, 8 times) on days 38–42 and 45–47. Tumor volumes are shown as means±SEM (n=5). P value indicates differences between OV19t+Tc-ffluc treated groups against OV19t+Tc + CD19-TCE treated group at endpoint determined using unpaired Student’s t-test (**p<0.01). (c) Representative flux imaging of mice on indicated days after engraftment with Tc-ffluc only (left), OV19t with Tc-ffluc (middle), and OV19t with Tc-ffluc and CD19-TCE (right). (d) Quantification of T cell flux at the tumor site. P values indicate differences between OV19t and Tc-ffluc treated group against OV19t, Tc-ffluc, and CD19-TCE treated group over multiple days determined using unpaired Student’s t-test (*p<0.05 and ***p<0.005). ffluc, firefly luciferase; OV19T, oncolytic virus carrying the CD19t-encoding gene; PBMC, peripheral blood mononuclear cell; pfu, plaque-forming unit; TCE, T cell engager.

Antitumor efficacy with the combination of OV and TCE in human solid tumor xenograft models
To evaluate the antitumor activity of OV19t in combination with CD19-TCE in vivo, we treated NSG mice bearing subcutaneous MDA-MB-468 tumors with a single intratumoral injection of 106 pfu OV19t. Two days after OV19t treatment, we collected PBMCs from a healthy human donor and engrafted with 5×106 cells per mouse. 4 days after PBMC transfer, mice were treated intravenously with CD19-TCE (8 µg/dose) or PBS for 5 consecutive days (Monday–Friday) and then 3 consecutive days (Monday–Wednesday) the following week for a total of 64 µg/mouse (figure 4a). Over the course of 2 weeks, MDA-MB-468 tumor-bearing mice treated with PBMCs alone or PBMCs and CD19-TCE showed no tumor control. Notably, mice treated with OV19t and PBMCs or OV19t and CD19-TCE slowed tumor growth as expected with OV19t alone, but we observed marked tumor regression with the combination of OV19t, PBMCs, and CD19-TCE (figure 4b, online supplemental figure 7). Similarly, antitumor responses were validated using OVBCMAt in combination with BCMA-TCE in vivo (figure 4c,d, online supplemental figure 8). These studies highlight the therapeutic efficacy of combining OV and TCE in preclinical models.
Figure 4. In vivo efficacy of combination therapy of TCE and OV expressing a target in a human xenograft tumor models. (a) Schematic of subcutaneous MDA-MB-468 tumor-bearing NSG mice (5×106 cells on day 0) treated with IT OV19t (1×106 pfu) on day 32, engrafted with IV PBMC (1×107 cells) on day 34, and treated with IV CD19-TCE (8 µg/dose, 8 times) on days 38–42 and 45–47. (b) Tumor volumes are shown as mean±SEM (n≥5 per group). P values indicate differences between OV19t and PBMC or CD19-TCE treated groups against OV19t, PBMC, and CD19-TCE treated groups determined using unpaired Student’s t-test (***p<0.005). (c) Schematic of subcutaneous MDA-MB-468 tumor-bearing NSG mice (5×106 cells on day 0) treated with IT OVBCMAt (1×106 pfu) on day 38, engrafted with IV PBMC (1×107 cells) on day 40, and treated with IV BCMA-TCE (25 µg/dose, 8 times) on days 41–43 and 46–50. (d) Tumor volumes are shown as mean±SEM (n≥8 per group). P values indicate differences between OV19t and PBMC or BCMA-TCE treated groups against OV19t, PBMC, and BCMA-TCE treated groups determined using unpaired Student’s t-test (***p<0.005). (e) Schematic of dual subcutaneous MDA-MB-468 tumor-bearing NSG mice on opposite flanks (5×106 cells on day 0) where only the right flank tumor was treated with IT OV19t (1×106 pfu) on day 34, engrafted with IV PBMC (1×107 cells) on day 34, and treated with either IV CD19-TCE (8 µg/dose, 8 doses) or IV BCMA-TCE (25 µg/dose, 8 doses) on days 40–44 and 47–49. (f) Tumor volumes of OV treated right flank tumor and (g) untreated left flank tumor are shown as mean±SEM (n≥6 per group). P values indicate differences between OV19t, PBMC, and BCMA-TCE treated groups against OV19t, PBMC, and CD19-TCE treated groups determined using unpaired Student’s t-test (***p<0.005). (h) Schematic of dual subcutaneous U251T tumor-bearing NSG mice on opposite flanks (5×106 cells on day 0) where only the left flank tumor was treated with IT OV19t (1×103 pfu) on day 35, engrafted with IV PBMC (1×107 cells) on day 37, and treated with either IV CD19-TCE (8 µg/dose, 8 doses) or IV BCMA-TCE (25 µg/dose, 8 doses) on days 38–42 and 45–47. (i) Tumor volumes of OV-treated left flank tumor and (j) untreated right flank tumor are shown as mean±SEM (n≥6 per group). P values indicate differences between OV19t, PBMC, and BCMA-TCE treated groups against OV19t, PBMC, and CD19-TCE treated groups determined using unpaired Student’s t-test (***p<0.005). BCMA, B cell maturation antigen; IT, intratumoral; IV, intravenous; OV, oncolytic virus; OV19T, OV carrying the CD19t-encoding gene; OVBCMAt, OV carrying the truncated BCMA-encoding gene; PBMC, peripheral blood mononuclear cell; pfu, plaque-forming unit; TCE, T cell engager.

Systemic antitumor effects using the combination of OV and TCE in a dual tumor xenograft model
To evaluate the potential for targeting non-OV-injected tumors with the combination, we treated NSG mice bearing dual subcutaneous MDA-MB-468 tumors on opposing flanks (left and right side) with a single intratumoral injection of 106 pfu OV19t to the right tumor only (figure 4e). MDA-MB-468 tumor-bearing mice treated with PBMCs alone, PBMCs with BCMA-TCE, and PBMCs with CD19-TCE in the absence of OV19t showed no tumor control. Importantly, in mice treated with OV19t to only one tumor site, we observed antitumor responses in both tumor sites in the presence of PBMCs and CD19-TCE. Other treatment groups demonstrated predictable antitumor responses only at the OV-treated site (figure 4f,g, online supplemental figures 9,10). Both the right and left flank subcutaneous tumors were harvested on day 58 (24 days post-OV19t treatment). Immunohistochemistry analysis of the right flank (OV-treated) tumor demonstrated equivalent VACV presence in BCMA-TCE or CD19-TCE treated groups. However, treatment with OV and CD19-TCE resulted in significant increases in VACV presence in the untreated tumor site (online supplemental figure 11). To further confirm the CD19 specificity of the observed efficacy, we treated MDA-MB-468 tumors (lentivirally transduced to stably express CD19t, termed MDA-MB-468-CD19t) on opposing flanks with either CD19-TCE or BCMA-TCE as a non-targeting TCE control. Both the right and left tumors demonstrated tumor regression in mice treated with CD19-TCE, but not in groups treated with BCMA-TCE (online supplemental figure 12). This experiment was repeated using U251T as the tumor model and observed similar antitumor efficacy on both the treated and non-OV-injected tumors (figure 4h–j). These data demonstrate the potent antitumor efficacy of the OV19t and CD19-TCE combination approach, and the potential for targeting multifocal disease following intratumoral administration of OV.
Antitumor activity of CD19-TCE preloaded T cells against OV19t-infected solid tumor cells in vitro
To evaluate if CD19-TCE preloaded onto a non-targeting T cell has the same antitumor efficacy as T cells exposed to CD19-TCE in culture, we co-cultured MDA-MB-468 tumor cells infected with varying MOIs of OV19t and non-targeting T cells. To appropriate wells, 100 ng/mL CD19-TCE and 1:1 E:T with non-targeting T cells, 1:1 E:T of non-targeting T cells preloaded with 10 µg/mL CD19-TCE, or CD19-CAR T cells were added in a tumor killing assay. CD19-TCE preloading was confirmed prior to co-culture with tumor targets by evaluating binding of protein L and blocking of anti-CD3 antibody binding using flow cytometry. We demonstrated reduction of anti-CD3 antibody binding while confirming approximately 95–97% protein L binding on T cells preloaded with 10 µg/mL and thus continued all preloading experiments with a concentration of 10 µg/mL (figure 5a). Additionally, we performed a time course study over 72 hours to determine CD19-TCE binding, activation, and exhaustion following preloading of T cells. Anti-CD3 antibody binding increased (data not shown) most likely due to proliferation of T cells with newly expressed CD3 molecules following activation while protein L expression remained high over 72 hours (figure 5b left). After 4 hours of culturing following preloading of T cells with CD19-TCE, there was an initial increase in activation marker (CD137) but decreased over 72 hours (figure 5b right). There was a slight increase in immune checkpoint markers (TIM3, LAG3, and PD-1) at 24 hours, but decreased over 72 hours (online supplemental figure 13).
Figure 5. HER2-CAR T cells combined with CD19-TCE direct activation and cytotoxicity against HER2-positive and HER2-negative tumor cells infected with OV19t. (a) Protein L and CD3 expression on T cells preloaded with CD19-TCE (10 µg/mL) for 1 hour. (b) Protein L and CD137 expression on T cells at 0.5, 4, 24, 48, and 72 hours following CD19-TCE preloading (10 µg/mL). (c) Quantification of CD137 expression (left), tumor cell killing (middle), and CD19t expression (right) on CD19-TCE preloaded human T cells following 24 hours co-culture with MDA-MB-468 tumor cells at an E:T ratio of 1:1 with or without CD19-TCE and indicated MOI of OV19t. n=2 per condition and each experiment was repeated two times. (d) Schematic of tumor cell rechallenge killing assay (top). MDA-MB-468 tumor cells with or without HER2 expression (1:4 ratio) were plated and infected with OV19t at indicated MOI. Human T cells or HER2-CAR T cells were added with or without CD19-TCE and rechallenged with non-HER2 expressing MDA-MB-468 cells following 24 hours incubation. CD137 expression, tumor cell killing, CD19t expression, and T cell counts were quantified using flow cytometry (left to right). The graphs shown represent analysis following 48 hours after the first rechallenge. n=2 per condition and each experiment was repeated two times. (e) Assay was performed as demonstrated in (d) with an additional rechallenge 48 hours after the first rechallenge, and CD137 expression, tumor cell killing, CD19t expression, and T cell counts (left to right) at the lowest MOI (0.003125) of OV19t were quantified using flow cytometry. n=2 per condition and each experiment was repeated two times. P values indicate differences between OV19t and HER2-CAR treated groups and OV19t, HER2-CAR, and CD19-TE treated groups determined using unpaired Student’s t-test (*p<0.05, **p<0.01, and ***p<0.005; ns=not significant). CAR, chimeric antigen receptor; E:T, effector-to-target; HER2, human epidermal growth factor receptor 2; MOI, multiplicity of infection; OV19T, oncolytic virus carrying the CD19t-encoding gene; TCE, T cell engager; UTD, untransduced.

After 24 hours in culture with OV19t infected MDA-MB-468 tumor cells, we observed an increase in CD137 of preloaded non-targeting T cells when compared with non-targeting T cells with CD19-TCE added in culture and CD19-CAR T cells when assessed by flow cytometry (figure 5c left). CD137 expression remained higher among preloaded T cells at 48 hours as well (online supplemental figure 14a). We quantified T cell killing ability against MDA-MB-468 tumor cells infected with OV19t using flow cytometry. At 24 and 48 hours, we observed greater killing of non-targeting T cells with CD19-TCE in culture, CD19-TCE preloaded non-targeting T cells, and CD19-CAR T cells than OV19t with non-targeting T cells alone (figure 5c middle and online supplemental figure 14b). Furthermore, targeting of CD19t expression following OV19t infection by CD19-TCE was confirmed when comparing expression to co-cultured wells in the absence of CD19-TCE (figure 5c right and online supplemental figure 14c). This data suggests that the presence of CD19-TCE, both pre-loaded onto T cells or suspended in culture with T cells, and CD19-CAR T cells elicit comparable antitumor activity against OV19t infected solid tumors in vitro.
Addressing solid tumor antigen heterogeneity by combining HER2 targeting CAR T cells with CD19-TCE against a tumor cancer cell line with low levels of HER2 expression following OV19t
To determine whether CD19-TCE in combination with OV19t could further improve the targeting of a CAR T cell onto a tumor with heterogenous expression patterns of an existing CAR antigen, we co-cultured MDA-MB-468 triple-negative breast cancer cells with 80% of the cells belonging to the parental line and 20% of cells expressing the CAR targetable HER2 antigen (MDA-MB-468-HER2 was lentivirally transduced to stably express HER2). We then treated wells with varying MOIs of OV19t and added HER2 CAR T cells with or without CD19-TCE. These cells were co-cultured for 1–6 days, which were rechallenged as shown in figure 5d. At 24 hours, OV19t and HER2 CAR T cells showed comparable killing to OV19t with HER2 CAR T cells and CD19-TCE while CD137 expression was significantly higher in the presence of CD19-TCE (online supplemental figure 15a). At 48 hours, CD19-TCE in combination with HER2 CAR T cells demonstrated higher killing and activation but similar CD19t targeting (online supplemental figure 1b). When wells were rechallenged with antigen-negative MDA-MB-468 tumor cells (not expressing HER2), the combinatorial treatment of OV19t, HER2 CAR T cells, and CD19-TCE showed additive and synergistic tumor cell killing when compared with OV19t and HER2 CAR T cells alone (figure 5d). Wells were then rechallenged once more with antigen-negative MDA-MB-468 tumor cells and again showed improved tumor cell killing when a combinatorial approach using OV19t, HER2 CAR T cells, and CD19-TCE. We demonstrated greater tumor cell killing, CD19t targeting, and T cell count at the lowest MOI (0.003125) (figure 5e). This data suggests that using OV19t to deliver a CD19-TCE-targetable antigen can be combined with existing CAR T cells to elicit dual-targeting of heterogeneous tumors.
Discussion
We previously developed a novel combination therapy for solid tumors by delivering a CD19t antigen with an OV followed by CD19-CAR T cells to mitigate antigen heterogeneity and elicit an immunologically warm tumor microenvironment (TME).13 Since our original report, other groups have demonstrated similar approaches to redirecting CAR or other therapeutic modalities to target de novo tumor antigens using OV.22,25 In our approach, tumor antigens encoded by the OV are expressed under the control of an early promoter, enabling rapid antigen presentation. This strategy is designed to potentially minimize early immune-mediated clearance of OV while facilitating antigen-directed mechanisms that enhance viral dissemination within the TME. We now demonstrate the ability of OV to deliver CD19t or BCMA to solid tumors that enable targeting with bispecific TCEs to induce endogenous T cell activation and antitumor responses in vitro and in vivo. We further demonstrate that local OV treatment combined with TCE allows for targeting of multiple tumors that are not directly injected with OV in two xenograft tumor models. While the mechanism of action warrants further investigation, our findings suggest improved spread of OV to untreated tumors following the combination therapy due to increased viral release from the CD19-TCE targeted tumors that have been infected with OV19t. The current findings support a fully off-the-shelf therapeutic combination with immediate clinical applications. Further, we demonstrate that this paradigm can be advanced with two clinically active TCEs, with the prospect of additional tumor antigens that may be exploited for this combinatorial immunotherapy strategy.
Use of TCE rather than a CAR T cell carries several key advantages. Given the cost and length of time required to generate patient-specific CD19-CAR T cells, we reasoned that the availability of off-the-shelf immunotherapies overcomes time restraints for patients undergoing treatment. With current manufacturing practices and capacities, the demand for CAR T cells exceeds supply and is still viewed as a niche technology that not all sites can perform.26 Moreover, TCEs such as blinatumomab or teclistamab provide a safety advantage over CAR T cells due to the nature of their short in vivo half-life and dosing strategies.27 28 This treatment can then be suspended or delayed if needed, with the potential for reversing unfavorable immune-related adverse events. This could expand readily to other targets where TCEs are being developed, including CD20, HER2, PSMA, and others. Importantly, the proof-of-concept phase one trial for the combination therapy using OV19t and blinatumomab is underway (NCT06063317).
Numerous trials are currently being investigated for the treatment of solid tumors with CAR T cells, targeting solid tumors that express HER2, GD2, PSMA, Claudin 18.2, and others.29 While the benefits of these approaches have yet to be fully clinically investigated, our off-the-shelf approach OV and TCE for the treatment of solid tumors may be used in combination with existing CAR T cell therapies. To that end, we show in vitro that CAR T cells can be combined with OV and TCE to improve tumor cell killing. One potential application of this combination is the ex vivo preloading of CAR T cells with CD19-TCE, enabling their use alongside OV19t without the need for direct administration of TCEs to patients. Further studies, including in vivo assessment, are warranted to better understand how this combination with existing CAR T cell strategies can be employed to overcome tumor antigen heterogeneity.
A limitation of the current study is the lack of testing in immunocompetent mouse models. While we were able to perform syngeneic studies in our prior work combining OV19t with CD19-CAR T cells, issues with access to the murine versions of CD19-TCEs and BCMA-TCEs limited our ability to perform such studies in the current work. These studies will be of interest given the biodistribution profile of TCEs compared with CAR T cells, the ability to agonize endogenous T cells with anti-CD3, and a model for which safety concerns might be addressed. Importantly, a recent study demonstrated the safety of CD19-TCE in syngeneic tumor models where CD19 was stably expressed on tumor cells.30 While blinatumomab has been widely used in clinic for the treatment of hematological malignancies, reports of various toxicities with targeting CD19 in patients, including cytokine release syndrome and neurotoxicities,31 suggest that this combinatorial strategy could employ alternate antigen targets, such as BCMA, which may have a more favorable safety profile.28 To that end, we generated OV carrying a novel BCMAt molecule and demonstrated the ability to redirect teclistamab to OVBCMAt-infected tumors. The use of either OV19t or OVBCMAt in this combination may be dictated by several features including disease setting, for instance in glioblastoma, where mitigation of potential neurological side effects would be highly desirable. Importantly, the initial decision to target these two B-cell-related antigens was multi-fold, as compared with other known tumor antigens including HER2, one may envision a benefit of targeting B cells in cancer as they may contribute to suppressing antitumor immunity.32 Furthermore, immunocompetent mouse models may provide a more accurate representation of the antitumor efficacy observed in the non-OV treated tumors. The viral spread to the contralateral untreated tumors seen in the current studies may not occur at lower OV doses or in less-permissive tumor types. While we have shown that numerous solid tumor cell lines are susceptible to OV infection, some remain resistant, which may limit therapeutic benefits of this combination. Our ongoing phase one clinical trial evaluating OV19t in combination with the CD19-TCE will provide further insight into the variability of OV infection across patient populations. These findings may inform future patient screening strategies to optimize therapeutic activity.
In summary, we have successfully developed a clinically viable, tumor-agnostic, combinatorial immunotherapy using OV to deliver TCE targets for the off-the-shelf treatment of solid tumors.
Supplementary material
Acknowledgements
We thank the staff members of the following cores at the Beckman Research Institute at City of Hope Comprehensive Cancer Center: Animal Facility, Small Animal Imaging, Pathology, and Center for Biomedicine and Genetics (CBG) for their excellent technical assistance. Work performed was supported by the National Cancer Institute of the National Institutes of Health under grant number P30CA033572 (COH) and P30CA014089 (USC). We also thank the staff members at the City of Hope Pharmacy. Research reported in this publication was supported by an Imugene Sponsored Research Agreement to City of Hope (PI: SJP), the Fiterman Family Foundation fund, and the Doug and Rhonda Collier Foundation fund. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Funding: Research reported in this publication was supported by a Sponsored Research Agreement from Imugene (PI: SJP). Work performed was supported by the National Cancer Institute of the National Institutes of Health under grant number P30CA033572 (COH) and P30CA014089 (USC). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Provenance and peer review: Not commissioned; externally peer reviewed.
Patient consent for publication: Not applicable.
Ethics approval: This study involves human participants and was approved by Internal Review Board (IRB) 09025 for the collection of de-identified healthy donor discard apheresis kits was deemed NHSR, and thus consent was not required.
Data availability statement
Data are available upon reasonable request.
References
- 1.Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015;93:290–6. doi: 10.1038/icb.2014.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goebeler M-E, Bargou RC. T cell-engaging therapies - BiTEs and beyond. Nat Rev Clin Oncol. 2020;17:418–34. doi: 10.1038/s41571-020-0347-5. [DOI] [PubMed] [Google Scholar]
- 3.Kantarjian H, Stein A, Gökbuget N, et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N Engl J Med. 2017;376:836–47. doi: 10.1056/NEJMoa1609783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bargou R, Leo E, Zugmaier G, et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science. 2008;321:974–7. doi: 10.1126/science.1158545. [DOI] [PubMed] [Google Scholar]
- 5.Stern LA, Jonsson VD, Priceman SJ. CAR T Cell Therapy Progress and Challenges for Solid Tumors. Cancer Treat Res. 2020;180:297–326. doi: 10.1007/978-3-030-38862-1_11. [DOI] [PubMed] [Google Scholar]
- 6.Hamieh M, Mansilla-Soto J, Rivière I, et al. Programming CAR T Cell Tumor Recognition: Tuned Antigen Sensing and Logic Gating. Cancer Discov. 2023;13:829–43. doi: 10.1158/2159-8290.CD-23-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 2021;11:69. doi: 10.1038/s41408-021-00459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen N, Li X, Chintala NK, et al. Driving CARs on the uneven road of antigen heterogeneity in solid tumors. Curr Opin Immunol. 2018;51:103–10. doi: 10.1016/j.coi.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murad JP, Tilakawardane D, Park AK, et al. Pre-conditioning modifies the TME to enhance solid tumor CAR T cell efficacy and endogenous protective immunity. Mol Ther. 2021;29:2335–49. doi: 10.1016/j.ymthe.2021.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonaventura P, Shekarian T, Alcazer V, et al. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front Immunol. 2019;10:168. doi: 10.3389/fimmu.2019.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Young RM, Engel NW, Uslu U, et al. Next-Generation CAR T-cell Therapies. Cancer Discov. 2022;12:1625–33. doi: 10.1158/2159-8290.CD-21-1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2015;14:642–62. doi: 10.1038/nrd4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park AK, Fong Y, Kim S-I, et al. Effective combination immunotherapy using oncolytic viruses to deliver CAR targets to solid tumors. Sci Transl Med. 2020;12:559.:eaaz1863. doi: 10.1126/scitranslmed.aaz1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim S-I, Park AK, Chaurasiya S, et al. Recombinant Orthopoxvirus Primes Colon Cancer for Checkpoint Inhibitor and Cross-Primes T Cells for Antitumor and Antiviral Immunity. Mol Cancer Ther. 2021;20:173–82. doi: 10.1158/1535-7163.MCT-20-0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang A, Zhang Z, Chaurasiya S, et al. Development of the oncolytic virus, CF33, and its derivatives for peritoneal-directed treatment of gastric cancer peritoneal metastases. J Immunother Cancer. 2023;11:e006280. doi: 10.1136/jitc-2022-006280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whelan JT, Singaravelu R, Wang F, et al. CRISPR-mediated rapid arming of poxvirus vectors enables facile generation of the novel immunotherapeutic STINGPOX. Front Immunol. 2022;13:1050250. doi: 10.3389/fimmu.2022.1050250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X, Naranjo A, Brown CE, et al. Phenotypic and Functional Attributes of Lentivirus-modified CD19-specific Human CD8+ Central Memory T Cells Manufactured at Clinical Scale. J Immunother. 2012;35:689–701. doi: 10.1097/CJI.0b013e318270dec7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X, Popplewell LL, Wagner JR, et al. Phase 1 studies of central memory-derived CD19 CAR T-cell therapy following autologous HSCT in patients with B-cell NHL. Blood. 2016;127:2980–90. doi: 10.1182/blood-2015-12-686725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Priceman SJ, Tilakawardane D, Jeang B, et al. Regional Delivery of Chimeric Antigen Receptor-Engineered T Cells Effectively Targets HER2+ Breast Cancer Metastasis to the Brain. Clin Cancer Res. 2018;24:95–105. doi: 10.1158/1078-0432.CCR-17-2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Priceman SJ, Gerdts EA, Tilakawardane D, et al. Co-stimulatory signaling determines tumor antigen sensitivity and persistence of CAR T cells targeting PSCA+ metastatic prostate cancer. Oncoimmunology. 2018;7:e1380764. doi: 10.1080/2162402X.2017.1380764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ribas A, Dummer R, Puzanov I, et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell. 2017;170:1109–19. doi: 10.1016/j.cell.2017.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aalipour A, Le Boeuf F, Tang M, et al. Viral Delivery of CAR Targets to Solid Tumors Enables Effective Cell Therapy. Mol Ther Oncolytics. 2020;17:232–40. doi: 10.1016/j.omto.2020.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y, Zheng Y, Deng T, et al. Oncolytic herpes simplex virus delivery of dual CAR targets of CD19 and BCMA as well as immunomodulators to enhance therapeutic efficacy in solid tumors combined with CAR T cell therapy. Front Oncol. 2022;12:1037934. doi: 10.3389/fonc.2022.1037934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taha Z, Crupi MJF, Alluqmani N, et al. Complementary dual-virus strategy drives synthetic target and cognate T-cell engager expression for endogenous-antigen agnostic immunotherapy. Nat Commun. 2024;15:7267. doi: 10.1038/s41467-024-51498-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang AQ, Hostetler A, Chen LE, et al. Universal redirection of CAR T cells against solid tumours via membrane-inserted ligands for the CAR. Nat Biomed Eng. 2023;7:1113–28. doi: 10.1038/s41551-023-01048-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Papathanasiou MM, Stamatis C, Lakelin M, et al. Autologous CAR T-cell therapies supply chain: challenges and opportunities? Cancer Gene Ther. 2020;27:799–809. doi: 10.1038/s41417-019-0157-z. [DOI] [PubMed] [Google Scholar]
- 27.Goebeler ME, Bargou R. Blinatumomab: a CD19/CD3 bispecific T cell engager (BiTE) with unique anti-tumor efficacy. Leuk Lymphoma. 2016;57:1021–32. doi: 10.3109/10428194.2016.1161185. [DOI] [PubMed] [Google Scholar]
- 28.Moreau P, Garfall AL, van de Donk NWCJ, et al. Teclistamab in Relapsed or Refractory Multiple Myeloma. N Engl J Med. 2022;387:495–505. doi: 10.1056/NEJMoa2203478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Daei Sorkhabi A, Mohamed Khosroshahi L, Sarkesh A, et al. The current landscape of CAR T-cell therapy for solid tumors: Mechanisms, research progress, challenges, and counterstrategies. Front Immunol. 2023;14:1113882. doi: 10.3389/fimmu.2023.1113882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Belmontes B, Sawant DV, Zhong W, et al. Immunotherapy combinations overcome resistance to bispecific T cell engager treatment in T cell-cold solid tumors. Sci Transl Med. 2021;13:608.:eabd1524. doi: 10.1126/scitranslmed.abd1524. [DOI] [PubMed] [Google Scholar]
- 31.Jain T, Litzow MR. No free rides: management of toxicities of novel immunotherapies in ALL, including financial. Blood Adv. 2018;2:3393–403. doi: 10.1182/bloodadvances.2018020198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tan R, Nie M, Long W. The role of B cells in cancer development. Front Oncol. 2022;12:958756. doi: 10.3389/fonc.2022.958756. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data for both the in vitro and in vivo studies are available on request from the corresponding author.
Data are available upon reasonable request.