

Abstract
Methylmercury (MeHg), an environmental pollutant, reaches the human body predominantly through contaminated fish consumption, potentially leading to severe neurological disorders. Upon ingestion MeHg reaches the brain and selectively accumulates in astrocytes. The activation of nuclear factor erythroid 2-related factor 2 (Nrf2) has been identified as a key early response to MeHg-induced oxidative injury, positioning it as a potential therapeutic target. However, recent studies suggest that Nrf2 activation alone may not be sufficient to mitigate MeHg toxicity, indicating the existence of other protective mechanisms. The signal transducer and activator of transcription 3 (STAT3) signaling pathway, known for its role in cell growth and survival, has emerged as a potential player in redox homeostasis. In this study, we investigated the role of STAT3 in acute (≤ 24 h) MeHg-induced neurotoxicity. MeHg exposure induced STAT3 expression in C8-D1A astrocytic cells. Our data demonstrated that pharmacological inhibition of STAT3 using AG490 or C188-9 exacerbated MeHg-induced cell death and compromised antioxidant responses. Furthermore, to fully characterize the role of STAT3 in oxidative stress, we used two different antioxidants, N-acetylcysteine (NAC) and Trolox. Conversely, reactive oxygen species (ROS)-scavenging antioxidants partially ameliorated STAT3 activation, suggesting that MeHg-induced STAT3 activation is mediated, at least in part, by mechanisms independent of ROS. Our findings suggest that STAT3 contributes to neuroprotection against MeHg exposure in astrocytes and is, at least in part, regulated by the increase in ROS levels within these cells.
Supplementary Information
The online version contains supplementary material available at 10.1007/s11064-025-04507-7.
Keywords: Methylmercury, Astrocytes, Oxidative stress, Neurotoxicity, Inflammation
Introduction
Mercury is naturally found within Earth’s crust [1]. For certain species like wild piscivorous fish, mammals, and birds, toxicological impacts include altered growth, reproduction, neurodevelopment, learning capacity, and behavioral abnormalities that raise mortality [2, 3]. Inorganic mercury is deposited into water bodies by attaching to airborne particles in various forms of precipitation. Microscopic organisms in water bodies combine mercury with carbon to form organic mercury (Hg). The most common type of organic mercury, methylmercury (MeHg), is found in the environment and is highly toxic [4].
MeHg can directly interact with brain and fetal cells by crossing the blood-brain and placental barriers [5]. In the gastrointestinal tract, MeHg forms a complex with L-Cysteine (CH3Hg-Cys) by imitating the amino acid methionine, enabling it to simply pass through the blood-brain barrier [6]. MeHg has a high affinity to sulfhydryl (-SH) protein groups, making it difficult to eliminate the compound from cells [7]. The kidneys and the nervous systems are particularly susceptible to MeHg poisoning [8].
The key mechanisms involved in MeHg-induced toxicity include increased reactive oxygen species (ROS), biomarkers of oxidative stress, and superoxide dismutase activities. The activation of antioxidant molecules may defend the central nervous system (CNS) from these neurotoxic symptoms of MeHg [9, 10]. Previously, upregulation of the nuclear factor erythroid 2-related factor 2 (Nrf2) and the downstream genes have been implicated in MeHg toxicity [11]. However, many studies demonstrate the involvement of different redox signaling in MeHg-induced neurotoxicity, including the mitogen-activated protein kinase (MAPK) cascade and Rho-associated coiled coil-forming protein kinase (ROCK) signaling [12, 13], suggesting the involvement of multiple pathways to combat MeHg toxicity.
The seven members of the signal transducer and activator of transcription (STAT) proteins (STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6) serve as innovative therapeutic targets for the treatment of diseases [14, 15]. STAT3 is crucial for cell cycle, cell proliferation, cell apoptosis, and the development of tumors [16]. It is activated by phosphorylation and translocated to the nucleus to participate in the regulation of gene expression. STAT3 can be regulated by upstream signaling molecules, such as Janus kinase (JAK). It then translocates to the nucleus of cells and binds to target DNA to regulate the expression of downstream proteins [14]. STAT3 acts as a downstream response protein for multiple cytokines and modulates cellular proliferation and intercellular interactions [17]. Due to its critical role in a variety of biological processes, including angiogenesis, differentiation, anti-apoptosis, proliferation, and inflammatory response, this signal transducer is the subject of extensive research.
Notably, the STAT3 signaling pathway has been poorly studied in the context of MeHg toxicity. Low concentrations of MeHg enhance ciliary neurotrophic factor (CNTF)-evoked STAT3 phosphorylation in human neuroblastoma SH-SY5Y and mouse cortical neural progenitor cells (NPCs) [18]. Additionally, persistent MeHg exposure via drinking water triggers STAT3 phosphorylation in the hypothalamus of mice [19], while prolonged exposure to MeHg-albumin conjugates increases STAT3 phosphorylation in microglial N9 cells [20]. Noteworthy, the hypothalamic neuronal cell line GT1-7 provided the first evidence of a mechanistic correlation between MeHg-induced oxidative stress and STAT3 signaling [21]. Interestingly, MeHg has also been associated with amyotrophic lateral sclerosis (ALS)-like disease [22, 23], which is characterized by elevated STAT3 protein levels [24]. Despite these findings, the precise role of STAT3 activation in MeHg toxicity, specifically within astrocytes, remains unclear.
Astrocytes play a critical role in the nervous system, maintaining neuronal homeostasis, regulating neurotransmitter levels, supporting blood-brain barrier integrity, and modulating oxidative stress responses. STAT3 is a critical regulator of astrocyte function, controlling both reactive astrogliosis and astrogenesis [25]. While astrocytes are essential for nervous system health, they are major targets of MeHg toxicity in the central nervous system. Notably, astrocytes accumulate more MeHg than neurons. This accumulation disrupts glutamate homeostasis and induces oxidative stress, ultimately compromising neuronal function [26].
Although studies have documented MeHg-induced STAT3 phosphorylation in diverse cell types, including NPCs, hypothalamic neurons, and microglia [18, 20, 21], a definitive link between STAT3 activation and MeHg-mediated astrocytic damage is lacking. This knowledge gap is particularly concerning given the crucial role astrocytes play in maintaining CNS homeostasis and their established susceptibility to MeHg exposure. To address the specific role of STAT3 in the astrocytic response to MeHg, we employed an in vitro approach using C8-D1A astrocytic cells. To dissect the STAT3 pathway’s contribution, we utilized pharmacological inhibitors: AG490, a selective inhibitor of the JAK2 tyrosine kinase [27], and C188-9, a small molecule inhibitor of STAT3 that binds to the phosphotyrosyl peptide binding site in the STAT3 Src homology 2 (SH2) domain, preventing STAT3 activation [28]. To further elucidate the role of STAT3 in oxidative stress, we treated the cells with two different antioxidants, N-acetylcysteine (NAC) and Trolox. We hypothesized that MeHg exposure leads to STAT3 phosphorylation in astrocytes, potentially linking STAT3 activation to antioxidant defense mechanisms. Conversely, we predicted that disrupting STAT3 signaling would exacerbate MeHg toxicity by increasing ROS production and promoting cellular death.
Materials and methods
Cell Culture
Mouse astrocytic neuronal C8-D1A cell line was obtained from the American Type Cell Culture Collection (ATCC, CRL-2541) and grown in Dulbecco’s modified Eagles’ medium (DMEM) (Gibco™, 11995040) supplemented with 10% heat-inactivated fetal bovine serum (Gibco™, 10438034) and 1% penicillin/streptomycin (Gibco™, 15140148). Cells were maintained at 37 °C with 5% CO2.according to the ATCC mammalian tissue culture protocol and sterile techniques. All experiments were performed using C8-D1A astrocytic cells between passages 10 and 30 to ensure phenotypic consistency. Independent experiments using different cell batches were conducted to ensure biological reproducibility.
Methylmercury Exposure
Cells were subcultured and treated with methylmercury (II) chloride (MeHg) (Sigma-Aldrich, 442534) at 0 or 0.5, 0.1, 1, 5, 10, and 20 µM for 1, 3, 6, and 24 h unless otherwise specified. These concentrations were chosen based on our group’s previous studies [29, 30] and on literature reports. Published studies using astrocytes demonstrated that 10 µM MeHg exposure for 24 h caused an accumulation of 124ng Hg/mg protein [31]. This accumulation is compatible with the reported Hg concentration in the brain from human exposures reported to cause toxic effects [32, 33].
For the experiments with the co-exposure of MeHg and STAT3 inhibitors or antioxidants, cells were treated with 0 or 10 µM for 1, 3, 6, and 24 h unless otherwise specified.
STAT3 Inhibitor Treatments
Cells were pre-treated for 1 h with JAK2 inhibitor AG490 (Tyrphostin B42 or 2-cyano-3-(3,4-dihydroxyphenyl)-N-(phenylmethyl)-2-propenamideor), Santa Cruz Biotechnology, sc-202046) at 0, 10, 50, or 100 µM, followed by co-treatment with 0 or 10 µM MeHg at specified times.
To evaluate if the effect was specific of STAT3 or other STATs regulated by JAK2, we also inhibited STAT3 using STAT3 Inhibitor XIII, C188-9 (CalbioChem, 573128). Cells were pre-treated for 1 h with C188-9 at 0, 3, or 30 µM, followed by co-treatment with 0 or 10 µM MeHg at specified times.
Antioxidant Treatments
Cells were treated with the antioxidants N-acetylcysteine (NAC, Sigma-Aldrich, A8199) and Trolox (Santa Cruz Biotechnology, sc-200810) to determine whether STAT3 activation is altered by oxidative stress conditions. C8-D1A cells were pre-treated for 1 h with 0, 25, or 100 µM Trolox, a vitamin E derivative, or 1 or 5 mM of NAC, a precursor to cysteine. Cells were then co-treated with 0 or 10 µM MeHg.
Lactate Dehydrogenase Cytotoxicity Assay
C8-D1A cells were plated in a 96-well plate. Lactate dehydrogenase (LDH) released from the cells was used to measure MeHg cytotoxicity. The released LDH oxidizes lactate to generate NADH, which reacts with iodonitrotetrazolium (INT) to form the red-colored formazan salt (Smith, Wunder et al. 2011). 24 h after seeding, cells were pre-treated for 1 h with assigned concentrations of STAT3 inhibitors or antioxidants, followed by co-exposure with MeHg. LDH release was measured in the medium using the CyQUANT LDH Cytotoxicity Assay Kit (Invitrogen, C20301), following manufacturer’s instructions. Briefly, 50 µL of the medium from each well was transferred to a new plate and 50 µL of LDH reaction mixture was added to the new plate. The plate was incubated for 30 min under a fume hood protected from light, followed by the addition of 50 µL of stop solution. Absorbance was measured at 490 nm and 680 nm using a microplate reader.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide Cell Viability Assay
Cell viability was measured with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Thermo Fisher Scientific, M6394). After 1 h of pre-treatment with STAT3 inhibitors or antioxidants, followed by MeHg exposure at specified times and concentrations, the medium was removed. Then, 100 µL of 0.5 µg/µL MTT in Hank’s balanced salt solution (HBSS) (Gibco™, 14175095) was added, and cells were incubated at 37℃ with 5% CO2 for one hour and 30 min. Following incubation, the supernatant was removed, and MTT was solubilized in dimethyl sulfoxide (DMSO) (Sigma Aldrich, D8418). Absorbance was measured at 540 nm using a microplate reader.
Reactive Oxygen Species Production
Cellular reactive oxygen species (ROS) production was assessed using a general oxidative stress indicator CM-H2DCFDA (Thermo Fisher Scientific, C6827). Cells were seeded in a black 96-well microplate and labeled with 5 µM CM-H2DCFDA. Cells were washed with HBSS, followed by treatment with STAT3 inhibitor or antioxidants for 1 h and then MeHg exposure. Fluorescence was measured at 30 min, 1, 3, 6, and 24 h after MeHg addition at excitation 495 nm and emission 525 nm using a microplate reader.
Mitochondrial ROS production was visualized using MitoSOX (Thermo Fisher Scientific, M36008). Cells were seeded in a black 96-well microplate and labeled with 500 nM MitoSOX diluted in HBSS. Cells were treated with STAT3 inhibitors for 1 h, followed by exposure to 10 µM MeHg. Fluorescence was measured at 30 min, 1, 3, and 6 h after addition of MeHg using a microplate reader.
Glutathione Assay
Total glutathione (GSH) and reduced GSH levels were measured using the SensoLyte®Total GSH Assay kit (AnnaSpec, AS-72153). C8-D1A cells were pre-treated for 1 h with STAT3 inhibitors, followed by co-exposure with 10 µM MeHg for 24 h at 37 °C with 5% CO2. Cells were collected and washed in phosphate buffer solution (PBS) (Gibco™, 10,010–023). Then, cells were lysed using freeze-thaw cycles, and protein concentration was measured using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, 23,225). GSH was measured following the manufacturer’s protocol. GSH levels were measured at 405 nm using SpectraMax iD3 microplate reader (Molecular Devices, USA). Values are expressed in mg/mL protein.
Interleukin 6 Quantification in Culture Medium
Cells were pre-treated 1 h with STAT3 inhibitors, followed by 10 µM MeHg co-exposure for 16 h. Interleukin 6 (IL-6) concentrations were measured by enzyme-linked immunosorbent assay (ELISA) kit (DuoSet mouse IL-6, R&D systems, DY-406), following manufacturer’s protocol.
Western Blot
Cells were sub-cultured into 60 mm culture dishes. For whole cell protein extracts, cold radioimmunoprecipitation assay (RIPA) buffer (Sigma Aldrich, R0278) was added with phosphatase inhibitor cocktails 2 and 3 (Sigma Aldrich, P5726 and P0044, respectively) and Halt™ protease inhibitor cocktail (Thermo Fisher Scientific, 78437). The lysates were sonicated and centrifuged. The supernatant was collected, and protein concentration was measured using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, 23225). Samples were dissolved in Laemmli sample buffer (Bio-Rad, #1610737) and boiled at 100℃ for 5 min. 20 µg of protein was resolved by 4–20% sodium dodecyl sulfate-polyacrylamide gel (SDS-PAGE, Bio-Rad #4561096). The gels were then transferred to a nitrocellulose membrane and incubated for one hour with 5% bovine serum albumin (BSA, Fisher Scientific, BP1600) in Tris buffer saline (Bio-Rad, #1706435)- 0.1% tween-20 (Promega, H5151) (TBS-T) at room temperature to block non-specific binding, followed by incubation with primary antibodies diluted in 5% BSA in TBS-T buffer overnight at 4℃. The blots were washed three times and incubated with selected horseradish peroxidase-conjugated secondary antibody. Protein bands were detected with a chemiluminescent method (Thermo Fisher Scientific 34579 or 37071). ImageJ software (NIH) was used to analyze the bands. For whole lysates, densitometry was normalized to β-actin as a housekeeping protein. Antibodies used included: mouse anti-β-actin (Sigma Aldrich, A1978), rabbit anti-phospho-STAT3 (Tyr705) (Cell Signaling Technology, #9145), mouse anti-STAT3 (Cell Signaling Technology, #9139), rabbit anti-Nrf2 (Cell Signaling Technology, #12721), mouse anti-KEAP1 (R&D systems, MAB3024), rabbit anti-SOD2 (Cell Signaling Technology, #813141), goat anti-PTP1B (R&D systems, AF3954), and mouse anti-HO1 (Cell Signaling Technology, #86806).
Quantitative Real time PCR
Cells were cultured into 60 mm culture dishes. Total RNA was isolated using the RNeasy Mini kit (Qiagen, 74104), following the manufacturer’s instructions. RNA concentration was quantified with a Nanodrop 2000 Spectrophotometer (Thermo Fisher Scientific, ND2000) and then reverse-transcribed to cDNA using a High-Capacity cDNA Reverse Transcription kit (Applied Biosystems, 4368814) according to the manufacturer’s instructions. Gene expression was determined using TaqMan Gene Expression assay (Applied Biosystems). TaqMan probes are listed in Table 1. Gene expression was determined using the 2 − ΔΔCt method [34], and data were normalized using Gapdh ID: Mn99999915 as a housekeeping gene.
Table 1.
Taqman probes for qPCR analysis
| Gene Symbol | Reference Sequence | Official Full Name |
|---|---|---|
| Hmox1 | Mm00516005_m1 | heme oxygenase 1 |
| Il-18 | Mm00434226_m1 | interleukin 18 |
| Il-6 | Mm00446190_m1 | interleukin 6 |
| Nlrp3 | Mm00840904_m1 | NLR family, pyrin domain containing 3 |
| Nqo1 | Mm0125356ss1_m1 | NAD(P)H dehydrogenase, quinone 1 |
| Nfe2l2 | Mm00477784_m1 | nuclear factor, erythroid derived 2, like 2 |
| Slc7a11 | Mm00442530_m1 | solute carrier family 7 member 11 |
| Socs3 | Mm00545913_s1 | suppressor of cytokine signaling 3 |
| Sod2 | Mm01313000_m1 | superoxide dismutase 2 |
| Stat3 | Mm01219775_m1 | signal transducer and activator of transcription 3 |
| Tgfb1 | Mm01178820_m1 | transforming growth factor, beta 1 |
| Tnf | Mm00443259_g1 | tumor necrosis factor |
| Gapdh | Mm99999915_g1 | glyceraldehyde-3-phosphatase dehydrogenase |
Statistical Analysis
Statistical analyses were conducted using IBM Statistical Package for the Social Sciences (SPSS) software version 29.0.2.0. Graphical representations were created using GraphPad Prism software version 10.4.1. No sample size calculations or outliers’ tests were performed, and no data point was excluded. Normality was assessed using the Shapiro-Wilk test and the Kolmogorov-Smirnov test.
The effects of MeHg on cytotoxicity, cell viability, ROS production, protein expression, and gene expression were assessed using one-way analysis of variance (ANOVA), followed by Bonferroni’s post hoc test. In cases where normality was not achieved, the data were analyzed with the non-parametric Kruskal-Wallis test, followed by Dunn’s post hoc test adjusted by Bonferroni correction.
The effects of MeHg on GSH levels were analyzed using Mann-Whitney U test.
The effects of STAT3 inhibitors in the presence or absence of MeHg on cytotoxicity, cell viability, oxidative stress, GSH levels, IL-6, protein expression, and gene expression were assessed using two-way ANOVA, followed by Bonferroni’s post hoc test. F values were indicated as follows: FMeHg (methylmercury significant effect), FAG (AG490 inhibitor significant effect), FC188 (C188-9 inhibitor significant effect), FIntA (Methylmercury x AG490 inhibitor interaction significant effect), and FIntC (Methylmercury x C188-9 inhibitor interaction significant effect).
The effects of antioxidants on MeHg-exposed cells regarding cytotoxicity, cell viability, oxidative stress, GSH levels, protein expression, and gene expression were analyzed using two-way ANOVA, followed by Bonferroni’s post hoc test. F values were indicated as follows: FMeHg (methylmercury significant effect), FNAC (NAC significant effect), FTx (Trolox significant effect), FIntN (Methylmercury x NAC interaction significant effect), and FIntT (Methylmercury x Trolox interaction significant effect).
For the two-way ANOVA analyses, when data did not meet normality assumptions, a logarithmic or square root transformation was used to achieve a closer approximation to normality. All data are reported as mean ± standard deviation (SD), and statistical significance was defined as p < 0.05.
Results
Exposure To MeHg Induces Mortality and Stress Oxidative in C8-D1A Astrocytic Cells
C8-D1A astrocytic cells were exposed to various concentrations of MeHg, and cytotoxicity was assessed at different time points using an LDH assay. After 24 h of exposure, the highest concentration (20 µM MeHg) significantly increased LDH release (H(6) = 19.926, p = 0.003; post hoc p = 0.006 compared to non-exposed cells) (Fig. 1A). No significant effects were observed at shorter exposure times for any of the concentration tested (Sup. Figure 1A, B, C).
Fig. 1.

MeHg reduced cell viability and induced oxidative stress in C8-D1A astrocytic cells. Astrocytic C8-D1A cells were treated with MeHg (0, 0.1, 0.5, 1, 5, 10, or 20 µM). Cytotoxicity (A) and cell viability (B) were measured at 24 h using LDH and MTT assays, respectively. Total ROS production was measured at 3 h using the CM-H2DCFDA probe (C), and mitochondrial ROS production was measured using the MitoSOX probe (D). Data are presented as mean ± SD. Statistical significance was determined using one-way ANOVA followed by Bonferroni’s post-hoc analysis, or with the Kruskal-Wallis test followed by Dunn’s post hoc test, adjusted by Bonferroni correction when normality was not achieved. p < 0.05 was considered statistically significant, * denotes a significant difference
To corroborate these results, cell viability was assessed with the MTT assay. Similar to the results from the LDH assay, after 24 h of exposure, MeHg significantly reduced cell viability in a concentration dependent manner (H(6) = 18.351, p = 0.005) (Fig. 1B). Lower concentrations and shorter exposure periods did not result in significant effects on cell viability (Sup. Figure 1D, E, F).
MeHg is known to induce oxidative stress in various cell types [35]. To assess this, we measure ROS production using CM-H2DCFDA probe, which is a general marker of intracellular ROS, in C8-D1A astrocytic cells. Twenty µM MeHg increased ROS production at 3 h of exposure (H(6) = 21.927, p = 0.001; post hoc p = 0.006 compared to non-exposed cells) (Fig. 1C). This concentration (20 µM MeHg) also increased ROS production at 30 min (H(6) = 12.897, p = 0.045; post hoc p = 0.013 compared to non-exposed cells) (Sup. Figure 1G) and 6 h (H(6) = 27.918, p < 0.001; post hoc p = 0.002 compared to non-exposed cells) (Sup. Figure 1I). Interestingly, exposure to 10 µM MeHg also significantly increased ROS production at 3 h (post hoc p = 0.031 compared to non-exposed cells) (Fig. 1C). and at 6 h (post hoc p = 0.014 compared to non-exposed cells) (Sup. Figure 1I).
MeHg disrupts mitochondrial function, leading to increased ROS production, particularly superoxide, within the mitochondria [35, 36]. To investigate this, we assessed mitochondrial superoxide production in C8-D1A astrocytic cells exposed to MeHg using MitoSOX Red. MeHg significantly increased mitochondrial superoxide production at 3 h (H(6) = 33.564, p < 0.001) (Fig. 1D). Similar results were observed at 30 min of exposure (H(6) = 25.446, p < 0.001) (Sup. Figure 1 J), 1 h (H(6) = 23.903, p < 0.001) (Sup. Figure 1 K), and 6 h (H(6) = 22.712, p < 0.001) (Sup. Figure 1 L). Notably, a concentration of 5 µM MeHg was sufficient to induce a significant increase in superoxide production as early as 30 min.
To further assess the impact of MeHg on oxidative stress, we next evaluate intracellular glutathione (GSH) levels. It is well-established that MeHg induces cellular GSH depletion, which consequently reduces antioxidant capacity and exacerbates oxidative stress [37–39]. However, after 6 h of exposure to 10 µM MeHg we did not observe significant changes in total GSH levels (Sup. Figure 1 M).
Exposure to MeHg Induces Antioxidant Enzymes Expression in C8-D1A Astrocytic Cells
Under oxidative stress conditions, cellular defense mechanisms against oxidative, electrophilic, and environmental stressors are activated. MeHg activates the kelch like ECH associated protein 1 (KEAP1)/Nrf2 signaling pathway, increasing Nrf2 expression in the nucleus and upregulating the transcription of Nrf2-target genes, including Heme oxygenase 1 (HO1), NAD(P)H: quinone-oxidoreductase (NQO1), and solute carrier family 7 member 11 (SLC7a11) [29, 40–42]. In our study, MeHg significantly induced Nrf2 protein levels in both time-dependent (H(5) = 18.378, p = 0.003) (Fig. 2A, E) and concentration-dependent manner (H(6) = 24.884, p < 0.001) (Sup. Figure 2A, E). However, MeHg did not affect KEAP1 protein expression (Fig. 2B, E, and Sup. Figure 2B, E). To confirm the activation of this pathway, we measured HO-1 protein expression. MeHg significantly increased HO-1 protein levels in time-dependent manner (H(5) = 20.680, p < 0.001) (Fig. 2C, E), although no significant effects were observed when C8-D1A cells were exposed to different concentrations of MeHg for 3 h (Sup. Figure 2C, E). Notably, MeHg did not affect protein levels of Superoxide dismutase 2 (SOD2), a mitochondrial antioxidant enzyme (Fig. 2D, E, and Sup. Figure 2D, E).
Fig. 2.

MeHg induced the expression of antioxidant enzymes in C8-D1A astrocytic cells. Astrocytic C8-D1A cells were treated with MeHg (0, 0.1, 0.5, 1, 5, 10, or 20 µM). Western blot analysis was used to measure changes in the antioxidant proteins Nrf2 (A), KEAP1 (B), HO-1 (C), and SOD2 (D) in C8-D1A cells exposed to 10 µM MeHg for 30 min, 1, 3, 6, or 24 h. (E) shows representative densitometry images. Hmox1 (F), Nqo1 (G), Sod2 (H), and Slc7a11 (I) gene expression were measured using qPCR in cells exposed to 10 µM MeHg for 1, 3, or 6 h. Data are presented as mean ± SD. Statistical significance was determined using one-way ANOVA followed by Bonferroni’s post-hoc analysis, or with the Kruskal-Wallis test followed by Dunn’s post hoc test, adjusted by Bonferroni correction when normality was not achieved. p < 0.05 was considered statistically significant, * denotes a significant difference
To further characterize the effects of MeHg on KEAP1/Nrf2 pathway, we assessed the gene expression of Nrf2-target antioxidant genes by qPCR in C8-D1A astrocytic cells exposed to 10 µM MeHg for up to 6 h. MeHg significantly induced Hmox1 gene expression (H(3) = 17.147, p < 0.001) (Fig. 2F) and Nqo1 gene expression (F(3,20) = 3.930, p = 0.023) (Fig. 2G) in a time-dependent manner, with significant effects observed at 6 h. However, no changes were detected in Sod2 (Fig. 2H) and Slc7a11 (Fig. 2I) mRNA expression.
Exposure to MeHg Induces STAT3 Signaling Pathway in C8-D1A Astrocytic Cells
Our group has previously demonstrated that MeHg induces the STAT3 signaling pathway in hypothalamic cells [19, 21]. Next, we assessed whether MeHg increases STAT3 activation in C8-D1A astrocytic cells. Western blot analysis demonstrated that MeHg increased the ratio of phosphorylated STAT3 to total STAT3 (pSTAT3/STAT3) in a concentration-dependent (F(6,35) = 4.724, p = 0.001) (Fig. 3A, C) and time-dependent manner (F(5,24) = 5.799, p = 0.001) (Fig. 2D, F).
Fig. 3.

MeHg induced STAT3 activation in C8-D1A astrocytic cells. For the concentration curve, C8-D1A astrocytic cells were treated with MeHg (0, 0.1, 0.5, 1, 5, 10, or 20 µM) for 3 h. The ratio of phosphorylated to total STAT3 (A) was calculated, and PTP1B (B) protein levels were measured using western blot analysis. (C) shows representative densitometry images. For the time curve, C8-D1A astrocytic cells were treated with 10 µM MeHg for 30 min, 1, 3, 6, or 24 h. The ratio of phosphorylated to total STAT3 (D) was calculated, and PTP1B (E) protein levels were measured using western blot analysis. (F) shows representative densitometry images. Stat3 (G), Socs3 (H), and Il-6 (I) gene expression were measured using qPCR in cells exposed to 10 µM MeHg for 1, 3, or 6 h. Data are presented as mean ± SD. Statistical significance was determined using one-way ANOVA followed by Bonferroni’s post-hoc analysis, or with the Kruskal-Wallis test followed by Dunn’s post hoc test, adjusted by Bonferroni correction when normality was not achieved. p < 0.05 was considered statistically significant. * denotes a significant difference
Protein Tyrosine Phosphatase 1B (PTP1B) acts as a negative regulator of the Janus Kinase 2 (JAK2)/ STAT3 signaling pathway by dephosphorylating JAK2 and STAT3 [43, 44]. However, MeHg exposure did not significantly alter PTP1B protein levels in C8-D1A astrocytic cells (Fig. 3B, C, E, F).
To further characterize MeHg-induced activation of the STAT3 signaling pathway, we evaluated the mRNA expression of various STAT3 target genes in C8-D1A astrocytic cells exposed to 10 µM MeHg for up to 6 h. MeHg significantly induced the gene expression of Stat3 (H(3) = 10.700, p = 0.013) (Fig. 3G) and Socs3 (H(3) = 13.555, p = 0.004) (Fig. 3H) in a time-dependent manner. Notably, significant Stat3 gene induction by MeHg was observed at 6 h (post hoc p = 0.033 compared to non-exposed cells) (Fig. 2G), while significant induction of Socs3 mRNA expression was observed at 1 h (post hoc p = 0.004 compared to non-exposed cells) and at 6 h (post hoc p = 0.036 compared to non-exposed cells) (Fig. 3H).
Given the involvement of STAT3 in inflammatory processes [45], we examined the gene expression of Interleukin 6 (Il-6) in C8-D1A cells exposed to 10 µM MeHg for up to 6 h. However, no significant changes in Il-6 gene expression were detected (Fig. 3I). Similarly, exposure to MeHg did not significantly alter the expression of other inflammatory genes, including NLR family pyrin domain containing 3 (Nlrp3), Interleukin 18 (Il-18), Tumor Necrosis Factor (Tnf), and Transforming Growth Factor Beta 1 (Tgfb1) (Sup. Figure 21 F, G, H, I).
Inhibition of STAT3 Induces Death in C8-D1A Astrocytic Cells
To assess the role of STAT3 in MeHg toxicity, we used a pharmacological inhibition strategy using two different STAT3 inhibitors. This approach allowed us to discern the role of JAK2/STAT3 pathway in MeHg-induced toxicity.
AG490 selectively blocks JAK2, inhibiting the tyrosine phosphorylation of STAT3 [27]. The impact of STAT3 inhibition on cytotoxicity and cell survival was examined. C8-D1A astrocytic cells were pre-treated for 1 h with 0, 10, 50, or 100 µM AG490, followed by co-exposure to 10 µM MeHg. Cytotoxicity was measured using an LDH assay at 3, 6, and 24 h (Fig. 4A, B, C). No significant effects were detected at 3–6 h. However, at 24 h, MeHg exposure significantly increased cytotoxicity in C8-D1A astrocytic cells across all concentrations of the inhibitor (FMeHg(1,40) = 45.065, p < 0.001) (Fig. 4C). AG490 did not cause LDH-assay assessed cytotoxicity at any time tested (Fig. 4A, B, C).
Fig. 4.

Inhibition of STAT3 phosphorylation exacerbated mortality in C8-D1A astrocytic cells. Cells were pretreated with 0, 10, 50, or 100 µM AG490, followed by co-treatment with 0 or 10 µM MeHg. Cytotoxicity (LDH release) was measured using LDH at 3 h (A), 6 h (B), and 24 h (C) of MeHg addition. Cell viability was assessed using MTT at 3 h (D), 6 h (E), and 24 h (F) of MeHg addition. Data are presented as mean ± SD. Statistical significance was determined using two-way ANOVA followed by Bonferroni’s post-hoc analysis. When data did not meet the assumptions of normality, a logarithmic or square root transformation was employed before conducting the two-way ANOVA. p < 0.05 was considered statistically significant. * denotes a significant difference
Cell viability was assessed using an MTT assay at 3, 6, and 24 h (Fig. 4D, E, F). MeHg exposure significantly decreased cell viability at 3 h (FMeHg(1,30) = 12.340, p = 0.001) (Fig. 4D) and 24 h (FMeHg(1,40) = 35.563, p < 0.001) (Fig. 4F). Additionally, at 24 h, AG490 exacerbated cell death (FAG(1,40) = 5.979, p = 0.002) (Fig. 4F).
C188-9 is a small-molecule inhibitor of STAT3. It directly inhibits STAT3 by binding to the phosphotyrosyl peptide binding site within the STAT3 SH2 domain [28, 46]. Using LDH and MTT assays, we assessed the role of STAT3 in cell survival. Consistent with our previously shown data on cell exposure to MeHg alone at different concentrations (Fig. 1 and Sup. Figure 1), no effects of MeHg were observed at early time points in the LDH assay (Fig. 5A, B) or the MTT assay (Fig. 5D, E). At 24 h, MeHg significantly increased LDH release (FMeHg(1,42) = 33.035, p < 0.001) (Fig. 5C) and decreased tetrazolium salt (MTT) reduction (FMeHg(1,42) = 17.669, p < 0.001) (Fig. 5F). Interestingly, the lower concentration (3 µM) of C188-9 decreased LDH release at 3 h (FC188(2,30) = 7.159, p = 0.003; post hoc p = 0.046 compared to non-exposed cells), though this effect was not observed with the highest concentration (30 µM) (Fig. 5A). Notably, this effect disappears at 6 h, and the lethality of the high concentration becomes significant (FC188(2,30) = 5.973, p = 0.007; post hoc p = 0.005 compared to non-exposed cells) (Fig. 5B), and it still persistent at 24 h (FC188(2,42) = 8.339, p < 0.001; post hoc p = 0.002 compared to non-exposed cells) (Fig. 5C). In turn, the LDH assay detected an increase in mortality at early time points (6 h), the MTT assay showed a significant decrease in cell viability in the cells treated with the higher concentration (30 µM) of C188-9 at 24 h (FC188(2,42) = 6.830, p = 0.003; post hoc p = 0.003 compared to non-exposed cells) (Fig. 5F).
Fig. 5.

Blocking STAT3 SH2 domain increases mortality in C8-D1A astrocytic cells. Cells were pretreated with 0, 3, or 30 µM C188-9, followed by co-treatment with 0 or 10 µM MeHg. Cytotoxicity (LDH release) was measured using LDH at 3 h (A), 6 h (B), and 24 h (C) of MeHg addition. Cell viability was assessed using MTT at 3 h (D), 6 h (E), and 24 h (F) of MeHg addition. Data are presented as mean ± SD. Statistical significance was determined using two-way ANOVA followed by Bonferroni’s post-hoc analysis. When data did not meet the assumptions of normality, a logarithmic or square root transformation was employed before conducting the two-way ANOVA. p < 0.05 was considered statistically significant. * denotes a significant difference
LDH assay measures lactate dehydrogenase (LDH) release, an indicator of compromised membrane integrity. Conversely, the MTT assay assesses metabolic activity within mitochondria. Differences observed between LDH and MTT results suggest that STAT3 inhibition may exacerbate MeHg toxicity through different mechanisms depending on the inhibitor. Therefore, disruption of JAK2/STAT3 pathway mediates death potentially through mitochondrial dysfunction, while direct inhibition of STAT3 induces death associated with membrane integrity alterations. Moreover, C188-9 may exert a transient, potentially protective effect against cell death at an early point with lower concentration.
Inhibition of STAT3 Phosphorylation Increases MeHg-induced ROS Production in C8-D1A Astrocytic Cells
The effect of STAT3 inhibition on MeHg-induced intracellular ROS production was also measured. First, we measured overall cellular ROS production using the CM-H2DCFDA probe. MeHg exposure resulted in a significant elevation of ROS levels at 6 h (FMeHg(1,52) = 4.345, p = 0.042) (Fig. 6D), and this effect persisted at 24 h (FMeHg(1,40) = 32.916, p < 0.001) (Fig. 6E). Interestingly, AG490 treatment induced ROS production in a concentration- dependent manner at 24 h (FAG(3,40) = 5.518, p = 0.003) (Fig. 6E), aggravating MeHg toxicity.
Fig. 6.

Inhibition of STAT3 phosphorylation increases oxidative stress in C8-D1A astrocytic cells. Cells were pretreated with 0, 10, 50, or 100 µM AG490 (A-E) or with 0, 3, or 30 µM C188-9 (F-J), followed by co-treatment with 0 or 10 µM MeHg. ROS production was measured using CM-H2DCFDA probe after MeHg addition at 30 min (A, F), 1 h (B, G), 3 h (C, H), 6 h (D, I), and 24 h (E, J). Data are presented as mean ± SD. Statistical significance was determined using two-way ANOVA followed by Bonferroni’s post-hoc analysis. When data did not meet the assumptions of normality, a logarithmic or square root transformation was employed before conducting the two-way ANOVA. p < 0.05 was considered statistically significant. * denotes a significant difference
When cells were pre-treated with C188-9 and then co-treated with MeHg, significant MeHg effects were detected at 1 h (FMeHg(1,78) = 5.313, p = 0.024) (Fig. 6G), 3 h (FMeHg(1,78) = 6.058, p = 0.016) (Fig. 6H), 6 h (FMeHg(1,78) = 10.483, p = 0.002) (Fig. 6I), and 24 h (FMeHg(1,42) = 49.003, p < 0.001) (Fig. 6J). Surprisingly, the C188-9 inhibitor did not affect total cellular ROS production.
Next, we assessed mitochondrial oxidative stress specifically using MitoSOX. Our data revealed that MeHg significantly increased mitochondrial superoxide levels at 30 min (FMeHg(1,30) = 5.047, p = 0.032) (Fig. 7A), with the effect persisting at 1 h (FMeHg(1,30) = 30.345, p < 0.001) (Fig. 7B), 3 h (FMeHg(1,30) = 75.958, p < 0.001) (Fig. 7C), and 6 h (FMeHg(1,30) = 93.996, p < 0.001) (Fig. 7D). Notably, AG490 increased superoxide production at 30 min (FAG(2,30) = 5.460, p = 0.009) (Fig. 7A) and at 1 h (FAG(2,30) = 7.336, p = 0.003) (Fig. 7B).
Fig. 7.

Inhibition of STAT3 phosphorylation increases mitochondrial oxidative stress in C8-D1A astrocytic cells. Cells were pretreated with 0, 10, 50, or 100 µM AG490 (A-D) or with 0, 3, or 30 µM C188-9 (F-H), followed by co-treatment with 0 or 10 µM MeHg. Mitochondrial superoxide production was assessed using mitoSOX fluorescent staining, after MeHg addition at 30 min (A, E), 1 h (B, F), 3 h (C, G), and 6 h (D, H). Data are presented as mean ± SD. Statistical significance was determined using two-way ANOVA followed by Bonferroni’s post-hoc analysis. When data did not meet the assumptions of normality, a logarithmic or square root transformation was employed before conducting the two-way ANOVA. p < 0.05 was considered statistically significant. * denotes a significant difference
When cells were treated with C188-9 and MeHg the toxic compound significantly increased ROS production at 3 h (FMeHg(1,30) = 31.500, p < 0.001) (Fig. 7G), and 6 h (FMeHg(1,30) = 37.267, p < 0.001) (Fig. 7H). Interestingly, a significant interaction between MeHg and C188-9 was observed at 1 h (FIntC(2,30) = 3.383, p = 0.047) (Fig. 7F). At this time point, MeHg-increased ROS levels (post hoc p = 0.005 compared to non-exposed cells) were significantly rescued at the higher (30 µM) C188-9 concentration (post hoc p = 0.004 compared with MeHg).
These findings suggest that altered STAT3 signaling contributes to modulated ROS production. AG490 potentially enhanced oxidative stress, in part through the disruption of mitochondrial homeostasis. Surprisingly, C188-9 seems to have some protective effects against MeHg-induced oxidative stress. This observation strengthens the hypothesis that STAT3 plays a key role in maintaining cellular redox balance.
Inhibition of STAT3 Phosphorylation Decreased GSH
To better understand the role of STAT3 in oxidative stress, we assessed the effects of STAT3 inhibitors on GSH levels in cells exposed to MeHg. GSH is a low molecular weight thiol antioxidant compound that protects cells from oxidative stress by neutralizing ROS [38]. One of the MeHg toxic mechanisms is the depletion of GSH levels through chemical interaction between MeHg and GSH, or by impairing the GSH synthesis [38]. C8-D1A astrocytic cells were pre-treated with AG490 or C188-9, and then co-expose with 10 µM MeHg for 24 h. At the 10 µM MeHg exposure level, no significant effect of MeHg on GSH levels were detected (Fig. 8). In contrast, total GSH levels were depleted in cells treated with AG490 (FAG(2,10) = 21.183, p < 0.001) (Fig. 8A). Interestingly, a significant interaction between MeHg and AG490 inhibitor was detected when the reduced GSH levels were measured (FIntA(1,10) = 8.680, p = 0.015) (Fig. 8B), where MeHg exposure sensitized cells to the GSH depleting effects of AG490 (post hoc p < 0.001 compare with MeHg). When GSH levels were represented as the GSH/GSSG ratio, a significant interaction between MeHg and AG490 was observed ((FIntA(1,10) = 6.165, p = 0.032), showing that this ratio increased in cells treated with AG490 ((post hoc p = 0.008 compared with non-treated cells). This increase was averted in cells exposed to MeHg (post hoc p = 0.012, compared with cells treated with AG490 and exposed to MeHg) (Fig. 8C). This change in GSH levels may explain, at least in part, the AG490-induced ROS levels (Figs. 6 and 7) observed in cells exposed to MeHg, suggesting a role for JAK2/STAT3 in redox homeostasis. Interestingly, no effects of the C188-9 inhibitor on total or reduced GSH levels were detected (Fig. 8C, D, F).
Fig. 8.

Inhibition of STAT3 phosphorylation exacerbates GSH depletion in C8-D1A astrocytic cells. Cells were pretreated with 0 or 100 µM AG490 (A, B, C) or with 0 or 30 µM C188-9 (D, E, F), followed by co-treatment with 0 or 10 µM MeHg. Total GSH (A, D) reduced GSH (B, C), and GSH/GSSG ratio were measured at 24 h after adding MeHg. Data are presented as mean ± SD. Statistical significance was determined using two-way ANOVA followed by Bonferroni’s post-hoc analysis. When data did not meet the assumptions of normality, a logarithmic or square root transformation was employed before conducting the two-way ANOVA. p < 0.05 was considered statistically significant. * denotes a significant difference
STAT3 Inhibition Induces Aberrant Expression of Antioxidant Defense Enzymes in C8-D1A Astrocytic Cells
To fully elucidate the role of STAT3 in cellular defense against oxidative stress, cells were pre-treated 1 h with 0, 10, 50, or 100 µM AG490 or 0, 3, or 30 µM C188-9, followed by co-treatment with 0 or 10 µM MeHg at different times. Protein expression was then assessed using immunoblot. First, we determined the effects on STAT3 activation by assessing the ratio phosphorylated STAT3 to total STAT3.
In cells pre-treated with AG490 and co-exposed with MeHg, the toxic compound increased the phosphorylated STAT3 to total STAT3 ratio at 1 h (FMeHg(1,56) = 190.833, p < 0.001) (Sup. Figure 3 A, C), 3 h (FMeHg(1,24) = 49.268, p < 0.001) (Sup. Figure 3D, H), and 6 h (FMeHg(1,24) = 16.708, p < 0.001) (Sup. Figure 3I, K). However, the effects of AG490 were evident at 24 h, where it decreased the phosphorylated STAT3 to total STAT3 ratio at the highest concentration tested (100 µM) (FAG(2,36) = 14.789, p < 0.001; post hoc p < 0.001 compared to non-expose cells) (Fig. 9A, D).
Fig. 9.

STAT3 inhibition induces aberrant antioxidant enzyme expression in C8-D1A astrocytic cells. Cells were pretreated with 0, 10, or 100 µM AG490 or with 0 or 30 µM C188-9, followed by 24 h of co-treatment with 0 or 10 µM MeHg. Protein levels of phosphorylated STAT3 to total STAT3 ratio (pSTAT3/STAT3) (A, E), HO-1 (B, F), and Nrf2 (C, G) were measured by western blot. Representative densitometry images are shown in (D, H). Data are presented as mean ± SD. Statistical significance was determined using two-way ANOVA followed by Bonferroni’s post-hoc analysis. When data did not meet the assumptions of normality, a logarithmic or square root transformation was employed before conducting the two-way ANOVA. P < 0.05 was considered statistically significant. * denotes a significant difference
In cells treated with C188-9 and co-exposed to MeHg, significant MeHg effects were detected following 24 h of exposure (FMeHg(1,30) = 4.704, p = 0.038) (Fig. 9E, H). This effect was also evident at 3 h (FMeHg(1,24) = 11.653, p = 0.002) (Sup. Figure 4 A, E). The highest C188-9 concentration (30 µM) significantly decreased the ratio phosphorylated STAT3 to total STAT3, indicating decreased STAT3 activation at 24 h (FC188(2,30) = 12.513, p < 0.001; post hoc p < 0.001 compared with non-exposed cells) (Fig. 9E, H). This effect began to be evident at 6 h, although it did not reach statistical significance (FC188(2,12) = 3.868, p = 0.051) (Sup. Figure 4 F, H).
We also examined protein tyrosine phosphatase 1B (PTP1B), a negative regulator of JAK2/STAT3 signaling pathway [47]. Neither MeHg nor AG490 or C188-9 treatment affected PTP1B protein expression (Sup. Figure 4E, H, and Sup. Figure 5B, E).
Heme oxygenase-1 (HO-1), a key antioxidant enzyme regulated by the Nrf2 pathway, is responsible for heme group degradation [48]. MeHg exposure increased HO-1 protein levels at 24 h (FMeHg(1,36) = 11.095, p = 0.002) (Fig. 9B, D). Similar effects were observed at 3 h (FMeHg(1,24) = 5.550, p = 0.027) (Sup. Figure 3 F, H), and at 6 h (FMeHg(1,24) = 11.291, p = 0.003) (Sup. Figure 3 J, K). Contrary to what was observed with the phosphorylated STAT3 to total STAT3 ratio, AG490 increased HO-1 protein levels as early as 1 h at the 50 µM concentration (FAG(2,56) = 2.917, p = 0.042; post hoc p = 0.036 compared to non-exposed cells) (Sup. Figure 3B, C), suggesting a direct link between activation of the JAK2 pathway and the Nrf2 signaling pathway. Similar to the 1 h treatment, AG490 increased HO-1 protein expression in a concentration-dependent manner at 3 h (FAG(2,24) = 13.013, p < 0.001) (Sup. Figure 3 F, H), and at 6 h (FAG(2,24) = 42.454, p < 0.001) (Sup. Figure 3 J, K). At 24 h, only the highest concentration (100 µM) significantly increased HO-1 protein levels (FAG(2,36) = 8.330, p = 0.001; post hoc p < 0.001 compared to non-exposed cells) (Fig. 9B, D), indicating sustained HO-1 activation by the AG490 inhibitor.
In cells pre-treated with C188-9, MeHg significantly increased HO-1 expression at 24 h (FMeHg(1,30) = 12.250, p = 0.001) (Fig. 9F, H). This activation was also observed after 6 h of exposure (FMeHg(1,12) = 22.242, p < 0.001) (Sup. Figure 4G, H). Interestingly, 30 µM C188-9 enhanced the HO-1 protein levels at 3 h (FC188(2,24) = 12.601, p < 0.001; post hoc p < 0.001 compared to non-exposed cells) (Sup. Figure 4 C, E) and 6 h (FC188(2,12) = 38.053, p < 0.001; post hoc p < 0.001 compared to non-exposed cells) (Sup. Figure 4G, H).
To gain further insight into the mechanism by which HO-1 is expressed, we quantified Nrf2 protein levels, the transcription factor responsible for HO-1 gene expression. In cells pre-treated with AG490 and co-exposed to MeHg, Nrf2 expression was significantly upregulated at 24 h post-exposure (FMeHg(1,18) = 10.882, p = 0.004) (Fig. 9C, D). A similar effect was also observed at the earlier 3-hour time point (FMeHg(1,24) = 17.026, p < 0.001) (Sup. Figure 3G, H). No differences are detected at 24 h of exposure (Fig. 9G, H).
However, in cells pre-treated with C188-9, 3 h after co-exposed to MeHg Nrf2 significantly increased (FMeHg(1,24) = 7.462, p = 0.012) (Sup. Figure 4D, E). However, Notably, no effects oAG490 or C188-9 inhibitors were detected in any of the time points measured.
To confirm that MeHg-induced STAT3 activation, we measured the expression of some STAT3-target genes using qPCR, such as Stat3 and Socs3. In cells pre-treated 1 h with the AG490 inhibitor and co-exposed to MeHg, no effects of AG490 were detected on Stat3 mRNA expression. However, MeHg significantly increased its expression at 3 h of exposure (FMeHg(1,30) = 25.387, p < 0.001) (Fig. 10A). At 3 h, MeHg also significantly increased Socs3 mRNA expression (FMeHg(1,30) = 35.156, p < 0.001) (Fig. 10B). This increase was sustained at 24 h (FMeHg(1,18) = 8.605, p = 0.009) (Fig. 10F), suggesting that MeHg induced STAT3 transcriptional activities. AG490 decreased Socs3 gene expression, with significant effects observed at 24 h (FAG(2,18) = 5.440, p = 0.014) (Fig. 10F).
Fig. 10.

STAT3 inhibition induces aberrant antioxidant gene expression in C8-D1A astrocytic cells. Cells were pretreated with 0, 10, or 100 µM AG490 or with 0 or 30 µM C188-9, followed by 24 h of co-treatment with 0 or 10 µM MeHg. Gene expression of Stat3 (A, C) and Socs3 (B, D) at 3 h, and Stat3 (E, I), Socs3 (F, J), Hmox1 (G, K), and Slc7a11 (H, L) at 24 h was measured by qPCR. Data are presented as mean ± SD. Statistical significance was determined using two-way ANOVA followed by Bonferroni’s post-hoc analysis. When data did not meet the assumptions of normality, a logarithmic or square root transformation was employed before conducting the two-way ANOVA. p < 0.05 was considered statistically significant. * denotes a significant difference
When STAT3 activity was inhibited with C188-9, no effects of C188-9 inhibitor were detected on Stat3 mRNA expression. However, MeHg significantly increased Stat3 gene expression at 3 h (FMeHg(1,30) = 35.706, p < 0.001) (Fig. 10C) and at 24 h (FMeHg(1,18) = 5.618, p = 0.029) (Fig. 10I). MeHg also induced Socs3 mRNA at 3 h (FMeHg(1,30) = 59.075, p < 0.001) (Fig. 10D) and at 24 h (FMeHg(1,18) = 15.694, p < 0.001) (Fig. 10J). The C188-9 inhibitor decreased Socs3 mRNA expression at 3 h (FC188(2,30) = 5.257, p = 0.011) (Fig. 10D) and at 24 h (FC188(2,18) = 10.592, p < 0.001) (Fig. 10J), indicating that STAT3 transcriptional activity was inhibited.
To further confirm that the inhibition of STAT3 mediates the activation of antioxidant defense, we measured the gene expression of various antioxidant genes using qPCR. As expected, at 24 h, MeHg significantly increased Hmox1 mRNA expression (FMeHg(1,18) = 32.115, p < 0.001) (Fig. 10G), and AG490 further activated its expression (FAG(2,18) = 15.764, p < 0.001) (Fig. 10G). Notably, these effects were observed at 3 h, where MeHg significantly increased Hmox1 gene expression (FMeHg(1,30) = 20.727, p < 0.001) (Sup. Figure 5 A) and this effect was enhanced by AG490 treatment (FAG(2,30) = 24.767, p < 0.001) (Sup. Figure 5 A). At 24 h, we also assessed Slc7a11 gene expression. Although MeHg did not induce its expression, the lowest concentration (10 µM) of AG490 upregulated it (FAG(2,18) = 4.524, p = 0.026; post hoc p = 0.0028 compared to non-exposed cells) (Fig. 10H). In contrast, at 3 h of co-exposure, neither MeHg nor AG490 affected Nqo1 and Sod2 mRNA expression (Sup. Figure 5B, C).
In cells pre-treated with C188-9 and co-exposed with MeHg, a significant interaction was observed in the expression of Hmox1 mRNA at 24 h of co-exposure (FIntC(2,18) = 4.623, p = 0.024) (Fig. 10K). The highest C188-9 concentration (30 µM) enhanced MeHg-induced Hmox1 gene expression (post hoc p < 0.001 compared to MeHg, and p < 0.001 compared to 30 µM C188-9 alone). Similar effects were observed at 3 h, where MeHg induced Hmox1 gene expression (FMeHg(1,30) = 28.946, p < 0.001) (Sup. Figure 5 F), and C188-9 further upregulated it (FC188(2,30) = 21.500, p < 0.001) (Sup. Figure 5 F). Additionally, we measured Slc7a11 mRNA expression at 24 h. While no effect of MeHg was detected, C188-9 upregulated Slc7a11 mRNA expression (FC188(2,18) = 8.375, p = 0.003) (Fig. 10L). In contrast, at 3 h, MeHg did not significantly affect Nqo1 and Sod2 gene expression (Sup. Figure 5G, H). However, C188-9 induced Nqo1 mRNA expression at 3 h (FC188(2,30) = 21.702, p < 0.001) (Sup. Figure 5G). Finally, Nfe2l2 gene expression, which encodes NRF2, was not affected by MeHg, AG490, or C188-9 (Sup. Figure 5D, I).
Altogether, these data suggest that STAT3 inhibitors may mediate activation of antioxidant defense.
STAT3 Inhibition Did not Prevent MeHg-induced Interleukin 6 Release in C8-D1A Astrocytic Cells
Due to the crucial role of STAT3 in neuroinflammation [49, 50] we measured the expression of some inflammatory cytokines. Although it has been reported that MeHg induces Tnf gene expression [51], we did not detect any significant effect. Neither AG490 nor C188-9 affected Tnf mRNA expression (Sup. Figure 5E, J).
MeHg significantly induced Il-6 mRNA expression in cells pre-treated 1 h with AG490 and co-exposed with MeHg for 3 h (FMeHg(1,30) = 20.547, p < 0.001) (Fig. 11A). Conversely, the highest concentration of AG490 (100 µM) decreased Il-6 mRNA expression (FAG(2,30) = 4.318, p = 0.022; post hoc p = 0.045 compared to non-exposed cells) (Fig. 11A). However, this decrease in the mRNA expression did not correlate with decreased IL-6 release into the medium after 16 h of co-exposure (Fig. 11B). Notably, MeHg significantly increased IL-6 release in these cells (FMeHg(1,20) = 20.692, p < 0.001) (Fig. 11B).
Fig. 11.

STAT3 inhibition did not prevent MeHg IL-6 release in C8-D1A astrocytic cells. Cells were pretreated with 0, 10 or 100 µM AG490 (A, B) or with 0, 3, or 30 µM C188-9 (C, D), followed by co-treatment with 0 or 10 µM MeHg. Il-6 mRNA expression (A, C) was measured by qPCR at 3 h after adding MeHg. IL-6 release into the medium (B, D) was measured at 16 h after adding MeHg Data are presented as mean ± SD. Statistical significance was determined using two-way ANOVA followed by Bonferroni’s post-hoc analysis. When data did not meet the assumptions of normality, a logarithmic or square root transformation was employed before conducting the two-way ANOVA. p < 0.05 was considered statistically significant. * denotes a significant difference
In cells pre-treated with C188-9 and co-exposed to MeHg, the toxic compound significantly induced Il-6 gene expression (FMeHg(1,30) = 10.702, p = 0.003) (Fig. 11C), but no effects of C188-9 were observed on it. In these cells, MeHg significantly increased IL-6 release in the medium (FMeHg(1,20) = 11.957, p = 0.002) (Fig. 11D). However, after 16 h, C188-9 induced a significant increase in IL-6 release (FC188(1,20) = 5.789, p = 0.026) (Fig. 11D).
While MeHg consistently enhances IL-6 expression and release, the role of STAT3 modulating IL-6 appears to be more complex.
N-acetylcysteine Protected C8-D1A Astrocytic Cells from MeHg-induced Oxidative Stress, Partially Inhibiting STAT3 Phosphorylation
In addition to classical kinase activation, STAT3 is also regulated by oxidative stress [52]. Therefore, MeHg may directly mediate STAT3 activation by binding to STAT3 cysteines, or indirectly by increasing ROS levels. To better understand how MeHg induced STAT3 activation, we inhibited MeHg-induced oxidative stress using two common antioxidants, NAC and Trolox [53].
First, we studied the effects of NAC in cell survival. MeHg significantly induced cytotoxicity (FMeHg(1,30) = 5.113, p = 0.031) (Fig. 12A) and decreased cell viability (FMeHg(1,30) = 5.036, p = 0.032) (Fig. 12B) at 24 h. However, NAC did not affect cell survival under these conditions.
Fig. 12.
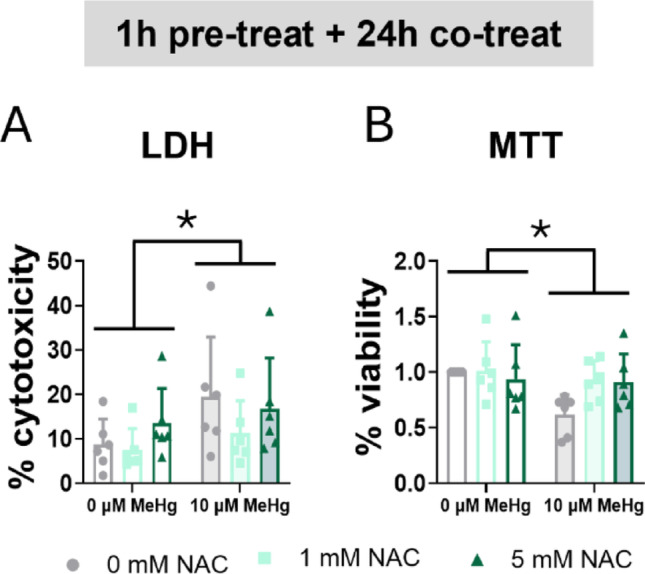
N-acetylcysteine failed to counteract MeHg-induced cytotoxicity. Cells were pretreated with 0, 1, or 5 mM NAC, followed by co-treatment with 0 or 10 µM MeHg. (A) Cytotoxicity was measured using LDH assay after 24 h of MeHg addition. (B) Cell viability was assessed by MTT assay after 24 h of MeHg addition. Data are presented as mean ± SD. Statistical significance was determined using two-way ANOVA followed by Bonferroni’s post-hoc analysis. When data did not meet the assumptions of normality, a logarithmic or square root transformation was employed before conducting the two-way ANOVA. p < 0.05 was considered statistically significant. * denotes a significant difference
Second, we measured the antioxidant effect of NAC on MeHg-induced ROS levels. MeHg increased ROS production, reaching significance at 3 h (FMeHg(1,12) = 4.789, p = 0.049) (Fig. 13C), whereas NAC significantly decreased ROS production at 30 min (FNAC(2,12) = 290.735, p < 0.001) (Fig. 13A), 1 h (FNAC(2,12) = 229.490, p < 0.001) (Fig. 13B), 3 h (FNAC(2,12) = 90.950, p < 0.001) (Fig. 13C), 6 h (FNAC(2,12) = 34.811, p < 0.001) (Fig. 13D), and at 24 h (FNAC(2,12) = 12.364, p = 0.001) (Fig. 13E).
Fig. 13.

N-acetylcysteine reduced MeHg-induced ROS production. Cells were pretreated with 0, 1, or 5 mM NAC, followed by co-treatment with 0 or 10 µM MeHg. ROS levels were measured using CM-H2DCFDA probe at 30 min (A), 1 h (B), 3 h (C), 6 h (D), and 24 h (E) after MeHg addition. Data are presented as mean ± SD. Statistical significance was determined using two-way ANOVA followed by Bonferroni’s post-hoc analysis. When data did not meet the assumptions of normality, a logarithmic or square root transformation was employed before conducting the two-way ANOVA. p < 0.05 was considered statistically significant. * denotes a significant difference
Finaly, we assessed whether NAC reversed MeHg-induced STAT3 phosphorylation. A significant interaction was found in the phosphorylated STAT3 to total STAT3 ratio (FIntN(2,36) = 3.457, p = 0.042) (Fig. 14A, D). Whereas MeHg significantly increased the phosphorylated STAT3 to total STAT3 ratio (post hoc p = 0048 compared to non-exposed cells), NAC treatment partially blocked this induction (Fig. 14A, D).
Fig. 14.

MeHg-induced STAT3 phosphorylation was partially mediated by ROS. Cells were pretreated with 0, 1, or 5 mM NAC, followed by co-treatment with 0 or 10 µM MeHg. (A) phosphorylated STAT3 to total STAT3 ratio, (B) HO-1, and (C) Nrf2 protein levels were measured by western blot 24 h after MeHg addition. (D) shows representative densitometry images. Data are presented as mean ± SD. Statistical significance was determined using two-way ANOVA followed by Bonferroni’s post-hoc analysis. When data did not meet the assumptions of normality, a logarithmic or square root transformation was employed before conducting the two-way ANOVA. p < 0.05 was considered statistically significant. * denotes a significant difference
To confirm if the antioxidant effects of NAC were mediated by antioxidant defense induction, we measured HO-1 protein levels at 24 h of co-exposure. A significant interaction between MeHg and NAC was found (FIntN(2,36) = 3.614, p = 0.037) (Fig. 14B, D). MeHg significantly increased HO-1 protein levels (post hoc p < 0.001 compared to non-exposed cells), which were restored in cells treated with NAC (for 1 mM NAC post hoc p < 0.001 compared to MeHg and for 5 mM NAC post hoc p < 0.001 compared to MeHg). In contrast, no effects of MeHg nor NAC were detected in Nrf2 protein levels at 24 h (Fig. 14C, D).
To further understand the effects of ROS on MeHg-induced STAT3 regulation, we also used Trolox as an antioxidant. Similarly, at 24 h, MeHg significantly induced cytotoxicity (FMeHg(1,30) = 23.942, p < 0.001) (Sup. Figure 6 A) and decreased cell viability (FMeHg(1,30) = 50.225, p < 0.001) (Sup. Figure 6B). As observed in NAC-treated cells, Trolox did not restore cell survival (Sup. Figure 6 A, B). MeHg significantly elevated ROS levels at 3 h (FMeHg(1,12) = 8.747, p = 0.012) (Sup. Figure 7 C), 6 h (FMeHg(1,12) = 10.650, p = 0.007) (Sup. Figure 7D), and 24 h (FMeHg(1,12) = 19.067, p < 0.001) (Sup. Figure 7E). Unlike NAC, Trolox’s effects on ROS production were less evident; only the low concentration of Trolox (25 µM) significantly decreased ROS levels at 30 min (FTx(2,12) = 11.427, p = 0.002; post hoc p = 0.001 compared to non-exposed cells) (Sup. Figure 7 A), 1 h (FTx(2,12) = 11.211, p = 0.002; post hoc p = 0.001 compared to non-exposed cells) (Sup. Figure 7B), and 3 h (FTx(2,12) = 5.050, p = 0.026; post hoc p = 0.027 compared to non-exposed cells) (Sup. Figure 7 C).
Neither MeHg nor Trolox had any significant effect at 24 h on phosphorylated STAT3 to total STAT3 ratio (Sup. Figure 8 A, D) or Nrf2 protein levels (Sup. Figure 8 C, D). Whereas MeHg significantly increased HO-1 protein expression (FMeHg(1,36) = 185.363, p < 0.001), Trolox failed to modulate its expression (Sup. Figure 8 C, D). Our data suggests that under our conditions, Trolox was less efficient than NAC, counteracting MeHg-induced oxidative stress.
Discussion
The signaling pathways involved in the deleterious effects of MeHg toxicity are not fully characterized. This novel study explores the role of the STAT3 signaling pathway in regulating oxidative stress induced by MeHg. Here, we demonstrate that acute/short term (≤ 24 h) MeHg induced STAT3 activation. Additionally, we show that inhibiting the JAK2/STAT3 pathway exacerbated acute MeHg-induced oxidative stress, leading to enhanced activation of antioxidant defenses in C8-D1A astrocytic cells. Furthermore, MeHg-induced STAT3 phosphorylation is partially mediated by the increase in ROS production.
MeHg has been shown in numerous studies to modify protein phosphorylation in a concentration- and cell-type-dependent manner, resulting in the dysregulation of signaling pathways [54–56]. However, very little is known about the effects of MeHg on the STAT3 signaling pathway. Tan et al. found that ERK/MAPKs and STAT3 signaling pathways may be related to the hormesis of MeHg-HSA in N9 microglial cells [20], while Jebbett and colleagues found that specific levels of MeHg enhanced CNTF-evoked STAT3 phosphorylation, target-gene expression, and glial differentiation in mouse cortical neural progenitor cells [18]. For the first time, the role of STAT3 in MeHg toxicity was implicated in hypothalamic neuronal cells, where MeHg induced STAT3 phosphorylation in a time- and concentration-dependent manner, exacerbating oxidative stress [21]. To understand whether the observed STAT3 functions in hypothalamic neuronal cells are specific to neuronal cells or represent a conserved mechanism in other cell types, we aimed to investigate the role of STAT3 in MeHg-induced toxicity in an astrocytic cell model.
Consistent with previous findings, MeHg induced a time- and concentration-dependent increase in ROS production in C8-D1A astrocytic cells. Our results align with numerous studies reporting that MeHg increases ROS production in astrocytic cells, leading to cellular death [11, 29, 57–60]. It is known that MeHg activates Nrf2 signaling pathway, upregulating antioxidant genes such as HO-1, NQO1, and SLC7A11 in various cell types, including astrocytes, microglia, and neuronal cells [11, 29, 40, 42, 58]. MeHg increased Nrf2 protein levels in a concentration- and time- dependent manner in C8-D1A astrocytic cells. Following MeHg exposure, we also observed an upregulation of antioxidant genes such as Hmox1 and Nqo1, further confirming activation of Nrf2 signaling pathway by MeHg. Interestingly, MeHg also induced a concentration- and time-dependent increase in STAT3 phosphorylation, and in its downstream target genes, Stat3 and Socs3, suggesting that MeHg mediates the activation of STAT3 signaling pathway. Research on the effects of MeHg on STAT3 is limited. Although some studies have suggested that MeHg induced STAT3 phosphorylation in vitro in N9 microglial, SH-SY5Y neuroblastoma, mouse cortical neuronal progenitor, and in GT1-7 hypothalamic neuronal cells [18, 20, 21], and in vivo in the brain of Wistar rats and the hypothalami of C57BL/6 mice [19, 24], its role in MeHg toxicity is not fully understood.
In this study, we found that pharmacological inhibition of STAT3 led to a significant decrease in cell viability in MeHg-exposed cells. This effect was particularly pronounced when STAT3 was inhibited using 30 µM C188-9, which targets its SH2 domain and prevents its dimerization and nuclear translocation [46]. Interestingly, C188-9 exhibited a biphasic pattern depending on the concentration and treatment duration. At 3 h, the lower concentration used (3 µM C188-9) suggested a transient, potentially protective effect against cell death. However, this initial efficacy was not sustained. In contrast, the higher concentration of C188-9 increased LDH release at 6 and 24 h, indicating cell membrane damage. This was confirmed by decreased MTT reduction at 24 h, demonstrating a clear cytotoxic effect over time.
STAT3 has been shown to play a neuroprotective role against glutamate-induced excitotoxicity in primary cultures of dorsal root ganglion neurons and glioblastoma cells [61, 62]. Activated STAT3 promotes cell survival and proliferation through the upregulation of Cyclin D1, PIM-1, and BCL-2 [63–65]. Interestingly, several studies have suggested that MeHg interferes with cell cycle progression, leading to cell death [66–68]. MeHg exposure regulated Cyclin D1 in a concentration-dependent manner, with distinct patterns observed in different brain regions. Higher MeHg concentrations increased Cyclin D1, whereas lower concentrations decreased it [69]. Notably, MeHg reduced Cyclin D1 expression in hippocampus, while in cerebellum no effects were observed [66]. The effects of MeHg on Cyclin D1 may involve the regulation of NF-κ B and STAT3 signaling pathways. We previously reported that in GT1-7 hypothalamic cells, MeHg-induced mortality correlated with the decrease of Ccnd1 and Pim1 mRNA after inhibition of JAK2/STAT3 [21].
The inhibition of STAT3 in GT1-7 hypothalamic neuronal cells exacerbated oxidative stress and increased antioxidant genes, suggesting a possible role in antioxidant defense [21]. Notably, when STAT3 activity was inhibited by AG490, which targets its upstream kinase JAK2, ROS production was exacerbated. These findings align with a previous study indicating that disruption of astrocyte STAT3 signaling decreases mitochondrial function and increases oxidative stress in vitro [70]. Surprisingly, this effect was not observed when STAT3 activity was prevented by blocking SH2 domain dimerization using C188-9, which partially ameliorated MeHg-induced superoxide production. The reasons for these discrepancies are unclear. Studies have shown that STAT3 could be localized in the mitochondria [71–73]. STAT3 mitochondrial localization has been linked to phosphorylation of the serine 727 residue of STAT3 [71, 72]. Blocking the STAT3 SH2 domain is linked with a decrease in STAT3 phosphorylation at the serine 727 residue and impaired mitochondrial function [74]. In PC12 cells, a serine dominant negative mutant of STAT3 reduced nerve growth factor-induced ROS production [75], suggesting a role for serine-phosphorylated STAT3 in mitochondrial ROS production. Therefore, the absence of C188-9 effects on MeHg-induced ROS production observed in our cells, may be mediated by inhibition of serine phosphorylation of STAT3 and a failure in translocation into the mitochondria. Further experiments measuring serine phosphorylation and STAT3 mitochondrial localization may help to clarify these results.
During oxidative stress conditions, Nrf2 translocates into the cell nucleus, forms a dimer with sMaf, and binds to antioxidant response elements (AREs) located in the regulatory regions of genes responsible for cellular defense [41]. Numerous studies have revealed an indirect relationship between STAT3 and Nrf2, wherein decreased STAT3 phosphorylation results in increased Nrf2 expression [76]. The relationship between STAT3 and Nrf2 is also supported by computational analysis of the connection between Nrf2 and STAT1 and STAT3 [77]. As expected, HO-1 levels were increased in MeHg-exposed cells compared to the control, suggesting the activation of the Nrf2 signaling pathway. This finding is consistent with the established role of Nrf2 in mediating cellular resistance and protecting against toxicity [78]. Furthermore, activation of the Nrf2/HO-1 signaling pathway has been shown to inhibit excessive oxidative stress and inflammation [23, 79]. In this study, inhibition of STAT3 pathway enhanced the MeHg-induced expression of antioxidant enzymes, especially HO-1. Direct inhibition of STAT3 with C188-9 further enhanced MeHg-induced Hmox1 gene expression, suggesting a role for STAT3 in regulating antioxidant stress defense.
Another key antioxidant defense mechanism against MeHg is GSH. In vivo models have shown that developmental exposure to MeHg decreases GSH brain levels [39, 80]. Similar results have been observed in primary astrocytic cultures [11, 81, 82]. Although MeHg-induced GSH depletion is an expected event, surprisingly, in our cell model, MeHg did not affect total or reduced GSH levels. Similar to our findings, astrocytoma cells have shown resistance to MeHg-induced GSH depletion [83]. Altogether, these results suggest that different cell types may present different vulnerabilities to MeHg. More interestingly, the effects of STAT3 inhibition on GSH levels were observed. AG490 induced GSH depletion, and this effect was exacerbated in cells exposed to MeHg. Therefore, adequate GSH levels are needed to ensure protection against MeHg. GSH depletion leads to enhanced MeHg toxicity [84]. Similar effects of AG490 inhibitor have been shown in other disease models where oxidative stress plays a pathological role. In myocardial ischemia and infarction models, the protective effects of GSH-inducing substances were abolished by AG490 treatment [85–88], suggesting a role for JAK2/STAT3 in modulating GSH levels. Interestingly, direct inhibition of STAT3 via blocking its SH2 domain using the C188-9 inhibitor did not elicit any effect on GSH levels, suggesting that GSH may be regulated by JAK2 rather than by STAT3. It is important to note that the AG490 inhibitor mediates inhibition of STAT3 phosphorylation by targeting its upstream kinase, JAK2. It is known that JAK2 also regulates phosphorylation of other STAT members, such as STAT1, STAT2, or STAT5 [89–91]. Therefore, we cannot rule out that the AG490-induced GSH decrease may be mediated by other STATs.
Although the potential role of inflammation in MeHg-induced neurodegeneration remains to be elucidated, studies suggest that MeHg triggers neuroinflammatory markers [92–94]. In vivo studies have demonstrated that MeHg exposure increased IL-6 serum levels in rodents [95–97]. Consistently, in vitro studies using glial cells have shown that MeHg induces IL-6 at high concentrations [98–102]. In our study, MeHg induced Il-6 gene expression and release in C8-D1A astrocytic cells, supporting the published data. Studies in different disease models have shown that AG490 treatment counteracts IL-6 induction [103–106], suggesting a role for the JAK2/STAT3 pathway in the regulation of IL-6 expression. AG490 inhibited MeHg-induced Il-6 gene expression, but it did not affect IL-6 release. Contrary to the AG490, C188-9 did not affect IL-6 gene expression in C8-D1A astrocytic cells. Similar findings have been reported in kidney and fibroblast cells, where the C188-9 failed to modulate IL-6 expression [107, 108]. Although we did not observe changes in gene expression, the C188-9 increased IL-6 release. The differences observed between both inhibitors suggest that other pathways may be implicated in MeHg-induced IL-6 regulation. Additionally, Wruck et al. (2011) propose an interaction between Nrf2 and IL-6, suggesting that Nrf2 may play a role in inflammatory responses by binding to antioxidant response elements in the promoter region of the Il-6 gene and activating its transcription [109]. Therefore, a link between antioxidant and anti-inflammatory mechanisms may exist. Further research is needed to investigate the implication of inflammatory genes such as the IL-6 gene in the defense response against MeHg toxicity.
The nuclear factor kappa B (NF-κB) signaling pathway induces the expression of pro-inflammatory cytokines, such as IL-6 [110, 111]. Notably, STAT3 and NF-κB exhibit crosstalk. They can interact directly or indirectly, regulating biological functions, such as inflammatory processes [112, 113]. STAT3 and NF-κB co-regulate the expression of several genes, including Socs3, Bcl2, and Il-6 [112, 114]. This crosstalk is further supported by the physical interaction between STAT3 and p65/p50 NF-κB subunits [115]. Similarly, Nrf2 also interacts with NF-κB, suggesting crosstalk [116]. Both pathways are activated upon exposure to different stressors, such as lipopolysaccharide or cigarette smoke [117, 118]. Several mechanisms regulated the intricate interplay between NF-κB and Nrf2. NF-κB represses Nrf2 transcription through competition for the transcription co-activator CREB (CBP) or by recruiting histone deacetylase 3 to the antioxidant response elements (ARE) sequences [119]. In summary, these interconnected pathways form a sophisticated regulatory core that coordinates both inflammatory and antioxidant responses.
Intracellular ROS are involved in MeHg toxicity [57], playing a pivotal role in the activation of antioxidant defense mechanisms, such as Nrf2 pathway [41, 120]. Similar to Nrf2, STAT3 is phosphorylated in response to oxidative stress [121–123]. NAC, a derivative of the naturally occurring amino acid L-cysteine, and Trolox, a water-soluble vitamin E analog, are two common antioxidants that may mediate protection against MeHg neurotoxicity [53]. Once MeHg reaches the cell, it could induce ROS production, which in turn may activate STAT3. Alternatively, MeHg may directly bind to cysteines present in STAT3, leading to aberrant activation of signaling pathway. NAC and Trolox decreased MeHg-Induced ROS levels in C8-D1A cells; however, Trolox was less efficient than NAC. Consistent with the changes in ROS levels, NAC decreased HO-1 protein expression, and ameliorated MeHg-induced STAT3 phosphorylation. Interestingly, Trolox failed to modulate the expression of these proteins. Trolox’s inability to counteract MeHg-induced HO-1 protein or Hmox1 mRNA has been previously described in HEK293, HeLa cells and cerebellar primary astrocytic cultures [13, 59]. The differences observed between Trolox and NAC could be explained by the chelating effects of NAC. NAC binds MeHg through its thiol groups, facilitating the reduction of MeHg within the cell by inhibiting the entry of the NAC-MeHg complexes and by promoting MeHg efflux [59, 124]. Interestingly, NAC only partially reversed MeHg-induced phosphorylated to total STAT3 ratio, suggesting that MeHg-induced STAT3 activation is only partly mediated by the increase in ROS production. Therefore, MeHg may mediate STAT3 activation by directly binding to it or indirectly by inactivating the enzymatic activity of upstream inhibitors such as PTP1B. Although NAC was effective in partially reversing MeHg-induced STAT3 phosphorylation and modulating HO-1 expression, these effects may primarily reflect its capacity to chelate MeHg, thereby reducing its bioavailability, rather than a direct antioxidant action. This limitation should be considered when interpreting the redox-related effects observed in this study. Future experiments using mitochondria-targeted ROS scavengers such as Mito-TEMPO will be necessary to clarify these mechanisms.
However, we measured antioxidant effects at 24 h, a time point where the STAT3 phosphorylation had already begun to revert to basal levels. While our aim was to assess the sustained effects of MeHg and antioxidant co-treatment, this timing might not fully capture the STAT3 activation driven by early ROS signaling. Therefore, future studies including earlier time points will be essential to better define the temporal dynamics of ROS-mediated STAT3 activation and its functional consequences.
In this study, we explored the complex relationship between oxidative stress, Nrf2, and JAK2/STAT3 signaling pathways in MeHg neurotoxicity. Our data demonstrated that MeHg induces STAT3 activation, and the dysregulation of this pathway exacerbates MeHg toxicity, suggesting a possible role for STAT3 in mediating its detrimental effects. These effects are similar to those we previously reported in the hypothalamic neuronal GT1-7 cell line, supporting the notion that MeHg-induced STAT3 activation may represent a shared mechanism with neurons. However, whether other CNS cell types, such as microglia, also exhibit this mechanism remains to be explored. Further analysis is needed to achieve a better understanding of these intricate mechanisms.
Conclusions
In conclusion, the data presented in this study demonstrates the neuroprotective role of STAT3 as a defense response in MeHg-induced toxicity. STAT3 Inhibitors decreased cell proliferation, increased ROS production levels, and induced an aberrant activation of antioxidant defense in MeHg-exposed C8-D1A cells, leading to an exacerbation of MeHg cytotoxicity. Future research is warranted to fully understand the protective role of STAT3 and its relation to the Nrf2-signaling pathway, as well as the expression of antioxidant genes.
Supplementary Information
Below is the link to the electronic supplementary material.
Author Contributions
Conceptualization, A.A. and B.F.; Formal analysis, A.A. and B.F.; Funding acquisition, A.B.B. and M.A.; Investigation, A.A., M.W., and B.F.; Methodology, A.A, M.W., A.S., and B.F.; Resources, A.S., J.B.R., A.B.B., and M.A.; Supervision, M.A. and B.F.; Validation, A.S., J.B.R., A.B.B., and M.A.; Visualization, A.S. and J.B.R.; Writing– original draft, A.A., M.A., and B.F.; Writing– review & editing, M.W., A.S., J.B.R., and A.B.B. All authors approved the final manuscript.
Funding
Michael Aschner and Aaron B Bowman were supported in part by a grant from The National Institute of Environmental Health Sciences (NIEHS), grant number R01ES007331.
Data Availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
Declarations
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.W. World Health Organization, Mercury and health, (2017)
- 2.Depew DC, Burgess NM, Campbell LM (2013) Spatial patterns of Methylmercury risks to common loons and piscivorous fish in Canada. Environ Sci Technol 47:13093–13103 [DOI] [PubMed] [Google Scholar]
- 3.Wren CD (1986) A review of metal accumulation and toxicity in wild mammals. I. Mercury. Environ Res 40:210–244 [DOI] [PubMed] [Google Scholar]
- 4.E. Environmental Protection Agency, Basic Information about Mercury, (2023)
- 5.Cariccio VL, Sama A, Bramanti P, Mazzon E (2019) Mercury involvement in neuronal damage and in neurodegenerative diseases. Biol Trace Elem Res 187:341–356 [DOI] [PubMed] [Google Scholar]
- 6.Kerper LE, Ballatori N, Clarkson TW (1992) Methylmercury transport across the blood-brain barrier by an amino acid carrier. Am J Physiol 262:R761–R765 [DOI] [PubMed] [Google Scholar]
- 7.Ajsuvakova OP, Tinkov AA, Aschner M, Rocha JBT, Michalke B, Skalnaya MG, Skalny AV, Butnariu M, Dadar M, Sarac I, Aaseth J, Bjorklund G (2020) Sulfhydryl groups as targets of mercury toxicity. Coordin Chem Rev 417 [DOI] [PMC free article] [PubMed]
- 8.Fernandes Azevedo B, Barros Furieri L, Pecanha FM, Wiggers GA, Frizera Vassallo P, Ronacher Simoes M, Fiorim J, Rossi P, de Batista M, Fioresi L, Rossoni I, Stefanon MJ, Alonso M, Salaices (2012) Valentim vassallo, toxic effects of mercury on the cardiovascular and central nervous systems. J Biomed Biotechnol 2012:949048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.do Nascimento JL, Oliveira KR, Crespo-Lopez ME, Macchi BM, Maues LA, Pinheiro Mda C, Silveira LC, Herculano AM (2008) Methylmercury neurotoxicity & antioxidant defenses. Indian J Med Res 128:373–382 [PubMed] [Google Scholar]
- 10.Liao Y, Peng S, He L, Wang Y, Li Y, Ma D, Wang Y, Sun L, Zheng H, Yang W, Dai F, Zhao J (2021) Methylmercury cytotoxicity and possible mechanisms in human trophoblastic HTR-8/SVneo cells. Ecotoxicol Environ Saf 207:111520 [DOI] [PubMed] [Google Scholar]
- 11.Ni M, Li X, Yin Z, Sidoryk-Wegrzynowicz M, Jiang H, Farina M, Rocha JB, Syversen T, Aschner M (2011) Comparative study on the response of rat primary astrocytes and microglia to Methylmercury toxicity. Glia 59:810–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fujimura M, Usuki F, Nakamura A (2019) Fasudil, a Rho-Associated coiled Coil-Forming protein kinase inhibitor, recovers Methylmercury-Induced axonal degeneration by changing microglial phenotype in rats. Toxicol Sci 168:126–136 [DOI] [PubMed] [Google Scholar]
- 13.Takanezawa Y, Sakai K, Nakamura R, Ohshiro Y, Uraguchi S, Kiyono M (2023) Inhibition of p38 Mitogen-Activated protein kinases attenuates Methylmercury toxicity in SH-SY5Y neuroblastoma cells. Biol Pharm Bull 46:1203–1210 [DOI] [PubMed] [Google Scholar]
- 14.Wang HQ, Man QW, Huo FY, Gao X, Lin H, Li SR, Wang J, Su FC, Cai L, Shi Y, Liu B, Bu LL (2020) STAT3 pathway in cancers: past, present, and future. MedComm 3(2022):e124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wong GL, Manore SG, Doheny DL, Lo HW (2022) STAT family of transcription factors in breast cancer: pathogenesis and therapeutic opportunities and challenges. Semin Cancer Biol 86:84–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gu Y, Mohammad IS, Liu Z (2020) Overview of the STAT-3 signaling pathway in cancer and the development of specific inhibitors. Oncol Lett 19:2585–2594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li J, Yin Z, Huang B, Xu K, Su J (2022) Stat3 signaling pathway: A future therapeutic target for Bone-Related diseases. Front Pharmacol 13:897539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jebbett NJ, Hamilton JW, Rand MD, Eckenstein F (2013) Low level Methylmercury enhances CNTF-evoked STAT3 signaling and glial differentiation in cultured cortical progenitor cells. Neurotoxicology 38:91–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferrer B, Prince LM, Tinkov AA, Santamaria A, Farina M, Rocha JB, Bowman AB, Aschner M (2021) Chronic exposure to Methylmercury enhances the anorexigenic effects of leptin in C57BL/6J male mice. Food Chem Toxicol 147:111924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tan Q, Zhang M, Geng L, Xia Z, Li C, Usman M, Du Y, Wei L, Bi H (2019) Hormesis of methylmercury-human serum albumin conjugate on N9 microglia via erk/mapks and STAT3 signaling pathways. Toxicol Appl Pharmacol 362:59–66 [DOI] [PubMed] [Google Scholar]
- 21.Ferrer B, Suresh H, Santamaria A, Rocha JB, Bowman AB, Aschner M (2021) The antioxidant role of STAT3 in methylmercury-induced toxicity in mouse hypothalamic neuronal GT1-7 cell line. Free Radic Biol Med 171:245–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carocci A, Rovito N, Sinicropi MS, Genchi G (2014) Mercury toxicity and neurodegenerative effects. Rev Environ Contam Toxicol 229:1–18 [DOI] [PubMed] [Google Scholar]
- 23.Minj E, Upadhayay S, Mehan S (2021) Nrf2/HO-1 signaling activator Acetyl-11-keto-beta Boswellic acid (AKBA)-Mediated neuroprotection in Methyl Mercury-Induced experimental model of ALS. Neurochem Res 46:2867–2884 [DOI] [PubMed] [Google Scholar]
- 24.Kumar S, Mehan S, Khan Z, Das Gupta G, Narula AS (2024) Guggulsterone selectively modulates STAT-3, mTOR, and PPAR-Gamma signaling in a Methylmercury-Exposed experimental neurotoxicity: evidence from CSF, blood plasma, and brain samples. Mol Neurobiol [DOI] [PubMed]
- 25.Ceyzeriat K, Abjean L, Carrillo-de Sauvage MA, Ben Haim L, Escartin C (2016) The complex STATes of astrocyte reactivity: how are they controlled by the JAK-STAT3 pathway? Neuroscience 330:205–218 [DOI] [PubMed] [Google Scholar]
- 26.Ni M, Li X, Rocha JB, Farina M, Aschner M (2012) Glia and Methylmercury neurotoxicity. J Toxicol Environ Health A 75:1091–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meydan N, Grunberger T, Dadi H, Shahar M, Arpaia E, Lapidot Z, Leeder JS, Freedman M, Cohen A, Gazit A, Levitzki A, Roifman CM (1996) Inhibition of acute lymphoblastic leukaemia by a Jak-2 inhibitor. Nature 379:645–648 [DOI] [PubMed] [Google Scholar]
- 28.Redell MS, Ruiz MJ, Alonzo TA, Gerbing RB, Tweardy DJ (2011) Stat3 signaling in acute myeloid leukemia: ligand-dependent and -independent activation and induction of apoptosis by a novel small-molecule Stat3 inhibitor. Blood 117:5701–5709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Culbreth M, Zhang Z, Aschner M (2017) Methylmercury augments Nrf2 activity by downregulation of the Src family kinase Fyn. Neurotoxicology 62:200–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dos Santos AA, Lopez-Granero C, Farina M, Rocha JBT, Bowman AB, Aschner M (2018) Oxidative stress, caspase-3 activation and cleavage of ROCK-1 play an essential role in MeHg-induced cell death in primary astroglial cells. Food Chem Toxicol 113:328–336 [DOI] [PubMed] [Google Scholar]
- 31.Shapiro AM, Chan HM (2008) Characterization of demethylation of Methylmercury in cultured astrocytes. Chemosphere 74:112–118 [DOI] [PubMed] [Google Scholar]
- 32.Aschner M (2012) Considerations on Methylmercury (MeHg) treatments in in vitro studies. Neurotoxicology 33:512–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choi BH, Lapham LW, Amin-Zaki L, Saleem T (1978) Abnormal neuronal migration, deranged cerebral cortical organization, and diffuse white matter astrocytosis of human fetal brain: a major effect of Methylmercury poisoning in utero. J Neuropathol Exp Neurol 37:719–733 [DOI] [PubMed] [Google Scholar]
- 34.Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta delta C(T)) method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 35.Farina M, Aschner M, Rocha JB (2011) Oxidative stress in MeHg-induced neurotoxicity. Toxicol Appl Pharmacol 256:405–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ghizoni H, de Souza V, Straliotto MR, de Bem AF, Farina M, Hort MA (2017) Superoxide anion generation and oxidative stress in methylmercury-induced endothelial toxicity in vitro. Toxicol Vitro 38:19–26 [DOI] [PubMed] [Google Scholar]
- 37.Chang JY, Tsai PF (2008) Prevention of methylmercury-induced mitochondrial depolarization, glutathione depletion and cell death by 15-deoxy-delta-12,14-prostaglandin J(2). Neurotoxicology 29:1054–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Farina M, Aschner M (2019) Glutathione antioxidant system and methylmercury-induced neurotoxicity: an intriguing interplay. Biochim Biophys Acta Gen Subj 1863:129285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stringari J, Nunes AK, Franco JL, Bohrer D, Garcia SC, Dafre AL, Milatovic D, Souza DO, Rocha JB, Aschner M, Farina M (2008) Prenatal Methylmercury exposure hampers glutathione antioxidant system ontogenesis and causes long-lasting oxidative stress in the mouse brain. Toxicol Appl Pharmacol 227:147–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Toyama T, Sumi D, Shinkai Y, Yasutake A, Taguchi K, Tong KI, Yamamoto M, Kumagai Y (2007) Cytoprotective role of Nrf2/Keap1 system in Methylmercury toxicity. Biochem Biophys Res Commun 363:645–650 [DOI] [PubMed] [Google Scholar]
- 41.Unoki T, Akiyama M, Kumagai Y, Goncalves F.M., Farina M, da Rocha J.B.T., Aschner M (2018) Molecular pathways associated with Methylmercury-Induced Nrf2 modulation. Front Genet 9:373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang L, Jiang H, Yin Z, Aschner M, Cai J (2009) Methylmercury toxicity and Nrf2-dependent detoxification in astrocytes. Toxicol Sci 107:135–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coronell-Tovar A, Pardo JP, Rodriguez-Romero A, Sosa-Peinado A, Vasquez-Bochm L, Cano-Sanchez P, Alvarez-Anorve LI (2024) Gonzalez-Andrade, protein tyrosine phosphatase 1B (PTP1B) function, structure, and Inhibition strategies to develop antidiabetic drugs. FEBS Lett 598:1811–1838 [DOI] [PubMed] [Google Scholar]
- 44.Kolodziej-Sobczak D, Sobczak L, Laczkowski KZ (2024) Protein tyrosine phosphatase 1B (PTP1B): A comprehensive review of its role in pathogenesis of human diseases. Int J Mol Sci 25 [DOI] [PMC free article] [PubMed]
- 45.Hillmer EJ, Zhang H, Li HS, Watowich SS (2016) STAT3 signaling in immunity. Cytokine Growth Factor Rev 31:1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bharadwaj U, Eckols TK, Xu X, Kasembeli MM, Chen Y, Adachi M, Song Y, Mo Q, Lai SY, Tweardy DJ (2016) Small-molecule Inhibition of STAT3 in radioresistant head and neck squamous cell carcinoma. Oncotarget 7:26307–26330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Myers MP, Andersen JN, Cheng A, Tremblay ML, Horvath CM, Parisien JP, Salmeen A, Barford D, Tonks NK (2001) TYK2 and JAK2 are substrates of protein-tyrosine phosphatase 1B. J Biol Chem 276:47771–47774 [DOI] [PubMed] [Google Scholar]
- 48.Funes SC, Rios M, Fernández-Fierro A, Covián C, Bueno SM, Riedel CA, Mackern-Oberti JP, Kalergis AM (2020) Naturally derived Heme-Oxygenase 1 inducers and their therapeutic application to Immune-Mediated diseases. Front Immunol 11 [DOI] [PMC free article] [PubMed]
- 49.Matsuda T (2023) The physiological and pathophysiological role of IL-6/STAT3-Mediated signal transduction and STAT3 binding partners in therapeutic applications. Biol Pharm Bull 46:364–378 [DOI] [PubMed] [Google Scholar]
- 50.Panda SP, Kesharwani A, Datta S, Prasanth D, Panda SK, Guru A (2024) JAK2/STAT3 as a new potential target to manage neurodegenerative diseases: an interactive review. Eur J Pharmacol 970:176490 [DOI] [PubMed] [Google Scholar]
- 51.Iwai-Shimada M, Takahashi T, Kim MS, Fujimura M, Ito H, Toyama T, Naganuma A, Hwang GW (2016) Methylmercury induces the expression of TNF-alpha selectively in the brain of mice. Sci Rep 6:38294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Butturini E, Carcereri de A, Prati, Mariotto S (2020) Redox regulation of STAT1 and STAT3 signaling. Int J Mol Sci 21 [DOI] [PMC free article] [PubMed]
- 53.Kaur P, Aschner M, Syversen T (2011) Biochemical factors modulating cellular neurotoxicity of Methylmercury. J Toxicol 2011:721987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ke T, Goncalves FM, Goncalves CL, Dos Santos AA, Rocha JBT, Farina M, Skalny A, Tsatsakis A, Bowman AB, Aschner M (2019) Post-translational modifications in MeHg-induced neurotoxicity. Biochim Biophys Acta Mol Basis Dis 1865:2068–2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sarafian T, Verity MA (1990) Altered patterns of protein phosphorylation and synthesis caused by Methyl mercury in cerebellar granule cell culture. J Neurochem 55:922–929 [DOI] [PubMed] [Google Scholar]
- 56.Yagame H, Horigome T, Ichimura T, Uchiyama J, Omata S (1994) Differential effects of Methylmercury on the phosphorylation of protein species in the brain of acutely intoxicated rats. Toxicology 92:101–113 [DOI] [PubMed] [Google Scholar]
- 57.Aschner M, Syversen T, Souza DO, Rocha JB, Farina M (2007) Involvement of glutamate and reactive oxygen species in Methylmercury neurotoxicity. Braz J Med Biol Res 40:285–291 [DOI] [PubMed] [Google Scholar]
- 58.Liu W, Xu Z, Yang T, Deng Y, Xu B, Feng S (2016) Tea polyphenols protect against Methylmercury-Induced cell injury in rat primary cultured astrocytes, involvement of oxidative stress and glutamate uptake/metabolism disorders. Mol Neurobiol 53:2995–3009 [DOI] [PubMed] [Google Scholar]
- 59.Sasaki S, Negishi T, Tsuzuki T, Yukawa K (2023) Methylmercury-induced reactive oxygen species-dependent and independent dysregulation of MAP kinase-related signaling pathway in cultured normal rat cerebellar astrocytes. Toxicology 487:153463 [DOI] [PubMed] [Google Scholar]
- 60.Shanker G, Aschner JL, Syversen T, Aschner M (2004) Free radical formation in cerebral cortical astrocytes in culture induced by Methylmercury. Brain Res Mol Brain Res 128:48–57 [DOI] [PubMed] [Google Scholar]
- 61.Corbetta C, Di Ianni N, Bruzzone MG, Patane M, Pollo B, Cantini G, Cominelli M, Zucca I, Pisati F, Poliani PL, Finocchiaro G, Pellegatta S (2019) Altered function of the glutamate-aspartate transporter GLAST, a potential therapeutic target in glioblastoma. Int J Cancer 144:2539–2554 [DOI] [PubMed] [Google Scholar]
- 62.Wen SY, Li AM, Mi KQ, Wang RZ, Li H, Liu HX, Xing Y (2017) In vitro neuroprotective effects of ciliary neurotrophic factor on dorsal root ganglion neurons with glutamate-induced neurotoxicity. Neural Regen Res 12:1716–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bachmann M, Moroy T (2005) The serine/threonine kinase Pim-1. Int J Biochem Cell Biol 37:726–730 [DOI] [PubMed] [Google Scholar]
- 64.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao YX, Pestell RG, Albanese C, Darnell JE (1999) Stat3 as an oncogene. Cell 98:295–303 [DOI] [PubMed] [Google Scholar]
- 65.Lu L, Dong J, Wang L, Xia Q, Zhang D, Kim H, Yin T, Fan S, Shen Q (2018) Activation of STAT3 and Bcl-2 and reduction of reactive oxygen species (ROS) promote radioresistance in breast cancer and overcome of radioresistance with niclosamide. Oncogene 37:5292–5304 [DOI] [PubMed] [Google Scholar]
- 66.Falluel-Morel A, Sokolowski K, Sisti HM, Zhou X, Shors TJ, Dicicco-Bloom E (2007) Developmental mercury exposure elicits acute hippocampal cell death, reductions in neurogenesis, and severe learning deficits during puberty. J Neurochem 103:1968–1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Malfa GA, Tomasello B, Sinatra F, Villaggio G, Amenta F, Avola R, Renis M (2014) Reactive response evaluation of primary human astrocytes after Methylmercury exposure. J Neurosci Res 92:95–103 [DOI] [PubMed] [Google Scholar]
- 68.Xu M, Yan C, Tian Y, Yuan X, Shen X (2010) Effects of low level of Methylmercury on proliferation of cortical progenitor cells. Brain Res 1359:272–280 [DOI] [PubMed] [Google Scholar]
- 69.Fujimura M, Usuki F (2015) Low concentrations of Methylmercury inhibit neural progenitor cell proliferation associated with up-regulation of glycogen synthase kinase 3beta and subsequent degradation of Cyclin E in rats. Toxicol Appl Pharmacol 288:19–25 [DOI] [PubMed] [Google Scholar]
- 70.Sarafian TA, Montes C, Imura T, Qi JW, Coppola G, Geschwind DH, Sofroniew MV (2010) Disruption of astrocyte STAT3 signaling decreases mitochondrial function and increases oxidative stress. PLoS ONE 5 [DOI] [PMC free article] [PubMed]
- 71.Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE (2009) Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 324:1713–1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, Derecka M, Szczepanek K, Szelag M, Gornicka A, Moh A, Moghaddas S, Chen Q, Bobbili S, Cichy J, Dulak J, Baker DP, Wolfman A, Stuehr D, Hassan MO, Fu XY, Avadhani N, Drake JI, Fawcett P, Lesnefsky EJ, Larner AC (2009) Function of mitochondrial Stat3 in cellular respiration. Science 323:793–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang R, Rincon M (2016) Mitochondrial Stat3, the need for design thinking. Int J Biol Sci 12:532–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Genini D, Brambilla L, Laurini E, Merulla J, Civenni G, Pandit S, D’Antuono R, Perez L, Levy DE, Pricl S, Carbone GM, Catapano CV (2017) Mitochondrial dysfunction induced by a SH2 domain-targeting STAT3 inhibitor leads to metabolic synthetic lethality in cancer cells. Proc Natl Acad Sci U S A 114:E4924–E4933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou LH, Too HP (2011) Mitochondrial localized STAT3 is involved in NGF induced neurite outgrowth. PLoS ONE 6 [DOI] [PMC free article] [PubMed]
- 76.Mohamed MZ, Hafez HM, Mohamed HH, Zenhom NM (2018) STAT3 and Nrf2 pathways modulate the protective effect of verapamil on lung injury of diabetic rats. Endocr Regul 52:192–198 [DOI] [PubMed] [Google Scholar]
- 77.Turei D, Papp D, Fazekas D, Foldvari-Nagy L, Modos D, Lenti K, Csermely P, Korcsmaros T (2013) NRF2-ome: an integrated web resource to discover protein interaction and regulatory networks of NRF2. Oxid Med Cell Longev 2013:737591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ma Q (2013) Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 53:401–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang B, Yin C, Zhou Y, Wang Q, Jiang Y, Bai Y, Qian H, Xing G, Wang S, Li F, Feng Y, Zhang Y, Cai J, Aschner M, Lu R (2019) Curcumin protects against methylmercury-induced cytotoxicity in primary rat astrocytes by activating the Nrf2/ARE pathway independently of PKCdelta. Toxicology 425:152248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Franco JL, Teixeira A, Meotti FC, Ribas CM, Stringari J, Garcia Pomblum SC, Moro AM, Bohrer D, Bairros AV, Dafre AL, Santos AR, Farina M (2006) Cerebellar thiol status and motor deficit after lactational exposure to Methylmercury. Environ Res 102:22–28 [DOI] [PubMed] [Google Scholar]
- 81.Dong L, Yang B, Zhang Y, Wang S, Li F, Xing G, Farina M, Zhang Y, Appiah-Kubi K, Tinkov AA, Aschner M, Shi H, Liu T, Lu R (2022) Ferroptosis contributes to methylmercury-induced cytotoxicity in rat primary astrocytes and Buffalo rat liver cells. Neurotoxicology 90:228–236 [DOI] [PubMed] [Google Scholar]
- 82.Kaur P, Aschner M, Syversen T (2006) Glutathione modulation influences Methyl mercury induced neurotoxicity in primary cell cultures of neurons and astrocytes. Neurotoxicology 27:492–500 [DOI] [PubMed] [Google Scholar]
- 83.Robitaille S, Mailloux RJ, Chan HM (2016) Methylmercury alters glutathione homeostasis by inhibiting glutaredoxin 1 and enhancing glutathione biosynthesis in cultured human Astrocytoma cells. Toxicol Lett 256:1–10 [DOI] [PubMed] [Google Scholar]
- 84.Shanker G, Syversen T, Aschner JL, Aschner M (2005) Modulatory effect of glutathione status and antioxidants on methylmercury-induced free radical formation in primary cultures of cerebral astrocytes. Brain Res Mol Brain Res 137:11–22 [DOI] [PubMed] [Google Scholar]
- 85.Chen H, Chen YJ, Wang X, Yang J, Huang CX (2020) Edaravone attenuates myocyte apoptosis through the JAK2/STAT3 pathway in acute myocardial infarction. Free Radical Res 54:351–359 [DOI] [PubMed] [Google Scholar]
- 86.Li Q, Ding JQ, Xia BY, Liu K, Zheng KL, Wu JJ, Huang C, Yuan XM, You QS (2024) L-theanine alleviates myocardial ischemia/reperfusion injury by suppressing oxidative stress and apoptosis through activation of the JAK2/STAT3 pathway in mice. Mol Med 30 [DOI] [PMC free article] [PubMed]
- 87.Xia BY, Ding JQ, Li Q, Zheng KL, Wu JJ, Huang C, Liu K, You QS, Yuan XM (2024) Loganin protects against myocardial ischemia-reperfusion injury by modulating oxidative stress and cellular apoptosis via activation of JAK2/ STAT3 signaling. Int J Cardiol 395 [DOI] [PubMed]
- 88.Shati AA, El-kott AF (2019) Acylated Ghrelin prevents doxorubicin-induced cardiac intrinsic cell death and fibrosis in rats by restoring IL-6/JAK2/STAT3 signaling pathway and Inhibition of STAT1. N-S Arch Pharmacol 392:1151–1168 [DOI] [PubMed] [Google Scholar]
- 89.de Dios N, Orrillo S, Irizarri M, Theas MS, Boutillon F, Candolfi M, Seilicovich A, Goffin V, Pisera D, Ferraris J (2019) JAK2/STAT5 pathway mediates Prolactin-Induced apoptosis of lactotropes. Neuroendocrinology 108:84–97 [DOI] [PubMed] [Google Scholar]
- 90.Gong L, Manaenko A, Fan R, Huang L, Enkhjargal B, McBride D, Ding Y, Tang J, Xiao X, Zhang JH (2018) Osteopontin attenuates inflammation via JAK2/STAT1 pathway in hyperglycemic rats after intracerebral hemorrhage. Neuropharmacology 138:160–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang K, Wang C, Xiao F, Wang H, Wu Z (2008) JAK2/STAT2/STAT3 are required for myogenic differentiation. J Biol Chem 283:34029–34036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Novo JP, Martins B, Raposo RS, Pereira FC, Oriá RB, Malva JO (2021) and C. Fontes-Ribeiro, cellular and molecular mechanisms mediating Methylmercury neurotoxicity and neuroinflammation. Int J Mol Sci 22 [DOI] [PMC free article] [PubMed]
- 93.Panzenhagen AC, Petry FD, Alves-Teixeira A, Santos L, Carazza-Kessler FG, Gelain DP, Moreira JCF (2024) Biomarkers of Methylmercury neurotoxicity and neurodevelopmental features: A systematic review. Food Chem Toxicol 191 [DOI] [PubMed]
- 94.Shinoda Y, Akiyama M, Toyama T (2023) Potential association between Methylmercury neurotoxicity and inflammation. Biol Pharm Bull 46:1162–1168 [DOI] [PubMed] [Google Scholar]
- 95.Muniroh M, Gumay AR, Indraswari DA, Bahtiar Y, Hardian H, Bakri S, Maharani N, Karlowee V, Koriyama C, Yamamoto M (2020) Activation of MIP-2 and MCP-5 expression in Methylmercury-Exposed mice and their suppression by N-Acetyl-L-Cysteine. Neurotox Res 37:827–834 [DOI] [PubMed] [Google Scholar]
- 96.Ortega HG, Lopez M, Takaki A, Huang QH, Arimura A, Salvaggio JE (1997) Neuroimmunological effects of exposure to Methylmercury forms in the Sprague-Dawley rats. Activation of the hypothalamic-pituitary-adrenal axis and lymphocyte responsiveness. Toxicol Ind Health 13:57–66 [DOI] [PubMed] [Google Scholar]
- 97.Chen X, Ren Q, Wu F, Zhu K, Tao JY, Zhang AH (2024) Exposure to four typical heavy metals induced telomere shortening of peripheral blood mononuclear cells in relevant with declined urinary aMT6s in rats. Ecotox Environ Safe 283 [DOI] [PubMed]
- 98.Chang JY (2007) Methylmercury causes glial IL-6 release. Neurosci Lett 416:217–220 [DOI] [PubMed] [Google Scholar]
- 99.Muniroh M, Khan N, Koriyama C, Akiba S, Vogel CFA, Yamamoto M (2015) Suppression of methylmercury-induced IL-6 and MCP-1 expressions by -acetylcysteine in U-87MG human Astrocytoma cells. Life Sci 134:16–21 [DOI] [PubMed] [Google Scholar]
- 100.Noguchi Y, Shinozaki Y, Fujishita K, Shibata K, Imura Y, Morizawa Y, Gachet C, Koizumi S (2013) Astrocytes protect neurons against Methylmercury via ATP/P2Y Receptor-Mediated pathways in astrocytes. PLoS ONE 8 [DOI] [PMC free article] [PubMed]
- 101.Shinozaki Y, Nomura M, Iwatsuki K, Moriyama Y, Gachet C, Koizumi S (2014) Microglia trigger astrocyte-mediated neuroprotection via purinergic gliotransmission. Sci Rep-Uk 4 [DOI] [PMC free article] [PubMed]
- 102.Yamamoto M, Khan N, Muniroh M, Motomura E, Yanagisawa R, Matsuyama T, Vogel CFA (2017) Activation of interleukin-6 and-8 expressions by Methylmercury in human U937 macrophages involves rela and p50. J Appl Toxicol 37:611–620 [DOI] [PubMed] [Google Scholar]
- 103.Guo J, Wang LY, Wu J, Xu LF, Sun M (2020) The JAK2 inhibitor AG490 regulates the Treg/Th17 balance and alleviates DSS-induced intestinal damage in IBD rats. Clin Exp Pharmacol P 47:1374–1381 [DOI] [PubMed] [Google Scholar]
- 104.Gyurkoyska V, Iyanoyska N (2015) Tyrosine kinase inhibitor tyrphostin AG490 reduces liver injury in LPS-induced shock. Eur J Pharmacol 751:118–126 [DOI] [PubMed] [Google Scholar]
- 105.Wang YY, Lin SY, Chang CY, Wu CC, Chen WY, Liao SL, Chen YF, Wang WY, Chen CJ (2021) Jak2 inhibitor AG490 improved poststroke central and peripheral inflammation and metabolic abnormalities in a rat model of ischemic stroke. Antioxidants-Basel 10 [DOI] [PMC free article] [PubMed]
- 106.Xu HW, Fang XY, Liu XW, Zhang SB, Yi YY, Chang SJ, Chen H (2023) Wang, α-Ketoglutaric acid ameliorates intervertebral disk degeneration by blocking the IL-6/JAK2/STAT3 pathway. Am J Physiol-Cell Ph 325:C1119–C1130 [DOI] [PubMed] [Google Scholar]
- 107.Ono Y, Saito M, Sakamoto K, Maejima Y, Misaka S, Shimomura K, Nakanishi N, Inoue S, Kotani J (2022) C188-9, a specific inhibitor of STAT3 signaling, prevents thermal burn-induced skeletal muscle wasting in mice. Front Pharmacol 13:1031906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang J, Sun Q, Zhang J, Wang H, Liu H (2022) Classical signaling and Trans-Signaling pathways stimulated by megalobrama amblycephala IL-6 and IL-6R. Int J Mol Sci 23 [DOI] [PMC free article] [PubMed]
- 109.Wruck CJ, Streetz K, Pavic G, Götz ME, Tohidnezhad M, Brandenburg LO, Varoga D, Eickelberg O, Herdegen T, Trautwein C, Cha KM, Kan YW, Pufe T (2011) Nrf2 induces Interleukin-6 (IL-6) expression via an antioxidant response element within the IL-6 promoter. J Biol Chem 286:4493–4499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Brasier AR (2010) The nuclear factor-kappaB-interleukin-6 signalling pathway mediating vascular inflammation. Cardiovasc Res 86:211–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liu T, Zhang L, Joo D, Sun SC (2017) NF-kappaB signaling in inflammation. Signal Transduct Target Ther 2:17023– [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Grivennikov SI, Karin M (2010) Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev 21:11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lee H, Herrmann A, Deng JH, Kujawski M, Niu G, Li Z, Forman S, Jove R, Pardoll DM (2009) Yu, persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell 15:283–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dhar K, Rakesh K, Pankajakshan D, Agrawal DK (2013) SOCS3 promotor hypermethylation and STAT3-NF-kappaB interaction downregulate SOCS3 expression in human coronary artery smooth muscle cells. Am J Physiol Heart Circ Physiol 304:H776–H785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Han SS, Yun H, Son DJ, Tompkins VS, Peng L, Chung ST, Kim JS, Park ES, Janz S (2010) NF-kappaB/STAT3/PI3K signaling crosstalk in iMyc E mu B lymphoma. Mol Cancer 9:97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wardyn JD, Ponsford AH, Sanderson CM (2015) Dissecting molecular cross-talk between Nrf2 and NF-kappaB response pathways. Biochem Soc Trans 43:621–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hong H, Lou S, Zheng F, Gao H, Wang N, Tian S, Huang G, Zhao H (2022) Hydnocarpin D attenuates lipopolysaccharide-induced acute lung injury via MAPK/NF-kappaB and Keap1/Nrf2/HO-1 pathway. Phytomedicine 101:154143 [DOI] [PubMed] [Google Scholar]
- 118.Liu Y, Li A, Feng X, Jiang X, Sun X, Huang W, Zhu X, Zhao Z (2018) l-Menthol alleviates cigarette smoke extract induced lung injury in rats by inhibiting oxidative stress and inflammation via nuclear factor kappa B, p38 MAPK and Nrf2 signalling pathways. RSC Adv 8:9353–9363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Liu GH, Qu J, Shen X (2008) NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim Biophys Acta 1783:713–727 [DOI] [PubMed] [Google Scholar]
- 120.Ni MW, Li X, Yin ZB, Jiang HY, Sidoryk-Wegrzynowicz M, Milatovic D, Cai JY, Aschner M (2010) Methylmercury induces acute oxidative stress, altering Nrf2 protein level in primary microglial cells. Toxicol Sci 116:590–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Carballo M, Conde M, El Bekay R, Martín-Nieto J, Camacho MJ, Monteseirín J, Conde J, Bedoya FJ, Sobrino F (1999) Oxidative stress triggers STAT3 tyrosine phosphorylation and nuclear translocation in human lymphocytes. J Biol Chem 274:17580–17586 [DOI] [PubMed] [Google Scholar]
- 122.Liu TS, Castro S, Brasier AR, Jamaluddin M, Garofalo RP, Casola A (2004) Reactive oxygen species mediate virus-induced STAT activation - Role of tyrosine phosphatases. J Biol Chem 279:2461–2469 [DOI] [PubMed] [Google Scholar]
- 123.Simon AR, Rai U, Fanburg BL, Cochran BH (1998) Activation of the JAK-STAT pathway by reactive oxygen species. Am J Physiol-Cell Ph 275:C1640–C1652 [DOI] [PubMed] [Google Scholar]
- 124.Trumpler S, Nowak S, Meermann B, Wiesmuller GA, Buscher W, Sperling M, Karst U (2009) Detoxification of mercury species–an in vitro study with antidotes in human whole blood. Anal Bioanal Chem 395:1929–1935 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of this study are available from the corresponding author upon reasonable request.