

Abstract
mRNA vaccines for infectious diseases prevent diseases by stimulating the body to produce specific immune responses through mRNA molecules encoding pathogenic proteins. Compared to traditional vaccines (e.g., inactivated, live attenuated, subunit, recombinant protein and viral vectors), mRNA vaccines offer several advantages including high safety, potent efficacy, scalable large-scale production, and cost-effectiveness. mRNA vaccines have demonstrated significant potential in combating infectious diseases since their inception. In particular, during the 2019 Coronavirus Disease (COVID-19) pandemic, the mRNA vaccines delivered with lipid nanoparticles (LNPs) have been developed by BioNTech and Moderna, their exceptional protective efficacy and extensive clinical application further proved the rapid responsiveness of mRNA vaccines in addressing emerging infectious diseases. This success has brought mRNA vaccines back into the spotlight of the scientific community. This article reviews the molecular biological basis, delivery systems, and immune mechanisms of mRNA vaccines, as well as the progress of research and clinical trials related to mRNA vaccines targeting the COVID-19 virus, influenza virus, rabies virus, Zika virus, human immunodeficiency virus, and Mycobacterium tuberculosis (M.tb), while also discussing the current challenges faced in the application of mRNA vaccines. These discussions provide a theoretical foundation and practical guidance for the future development of mRNA vaccines targeting bacterial infectious diseases such as tuberculosis (TB).
Supplementary Information
The online version contains supplementary material available at 10.1186/s40001-025-03060-x.
Keywords: MRNA vaccines, MRNA vaccine components, Delivery systems, Immune mechanisms, Clinical trials
Introduction
Vaccines have played a critical role in global public health as a powerful weapon against infectious diseases caused by bacteria or viruses. They have evolved through three generations: the first generation of traditional vaccines (including attenuated live vaccines, inactivated vaccines, and toxoid vaccines), the second generation of genetically engineered vaccines (mainly subunit vaccines, synthetic peptide vaccines, and recombinant genetically engineered live vaccines), and the third generation of vaccines (mainly including DNA vaccines, recombinant viral vector vaccines, and mRNA vaccines) [1]. Vaccine research has increasingly demonstrated its important role in disease prevention and treatment [1].
Traditional vaccines have demonstrated a notable preventive effect against infectious diseases, such as smallpox and poliomyelitis. However, it is necessary to take into consideration their associated risks, and research, development, and production cycles, as well as the limitations in aspects, such as scalability and adaptability, to new pathogens [2]. Second-generation recombinant vaccines have also played a crucial role in reducing epidemics such as hepatitis B. However, they usually require a suitable expression system and the combined use of effective adjuvants to be effective [3]. The mRNA vaccine in the third generation of vaccines has played a crucial role in ending the COVID-19 pandemic. Compared to traditional vaccines (e.g., inactivated, live attenuated, subunit, recombinant protein, and viral vectors), mRNA vaccines offer many advantages, including rapid design and synthesis, the ability to adapt the sequence based on viral mutations or the structure of emerging pathogens, and rapid large-scale production with high cost-effectiveness. Importantly, mRNA vaccines are non-infectious and do not integrate into the host’s genome, effectively avoiding the risk of potential mutations and ensuring safety.
In 1961, mRNA was identified as an intermediary that carries genetic information to ribosomes for protein synthesis. With the development of in vitro transcription (IVT) technology for RNA production, synthetic mRNAs can be engineered to resemble mature and processed mRNA molecules and scientists have begun to use mRNAs to express desired proteins [4]. Initially, the use of mRNA to express proteins in vivo was limited by its instability and the genetically inflammatory nature of unmodified nucleotides. However, with the rapid advances in delivery technology and the development of nucleoside modification, sequence/codon optimization, and design of the UTR techniques to address these issues, mRNA technology has rapidly expanded in biomedical applications [5]. The major milestone in the development of mRNA vaccines was the widespread clinical use of two rapidly developed and licensed COVID-19 mRNA vaccines by BioNtech/Pfizer and Moderna during the COVID-19 pandemic in 2021, which showed significant preventive effects. The mRNA vaccines using lipid nanoparticles (LNPs) as delivery materials overcome the inherent limitations of bare mRNA molecules [6].
Although mRNA vaccines are still being administered, adverse reactions to them are constantly being reported [7] and they still have the following limitations: (1) Storage and transportation requirements: The strict ultra-low-temperature cold-chain requirements increase costs and affect accessibility, posing challenges to regions with weak infrastructure [8]. (2) Short-term adverse reactions: Including local pain and systemic symptoms, such as fatigue and fever. The probability of severe allergic reactions is extremely low, but emergency treatment is required [9]. (3) Long-term safety: The application of new nanotechnology lacks long-term follow-up data. Although there is no evidence showing an impact on fertility or the development of chronic diseases [10]. (4) Immune tolerance differences: There is heterogeneity in the response of individuals to mRNA vaccines. For instance, the antibody production in the elderly or those with weakened immunity is relatively weak. Therefore, adjutants (such as TLR agonists) or multivalent vaccines need to be developed to enhance the response [11]. (5) Scalability: The production process of mRNA vaccines involves multiple steps, including mRNA synthesis, purification, and formulation with delivery vehicles (such as lipid nanoparticles). Each step requires strict quality control, and any disruption in the supply chain of raw materials or equipment can affect production capacity.
Currently, research on mRNA vaccines for various diseases has grown exponentially, and mRNA vaccines for some infectious diseases have entered clinical trials. mRNA vaccine has brought new hope for controlling these diseases [5]. Tuberculosis (TB) is currently an infectious disease with high morbidity and mortality, and the Bacillus Calmette–Guérin (BCG) vaccine remains the only licensed vaccine for human use in the prevention of tuberculosis. Newborns vaccinated with BCG can reduce their risk of Mycobacterium tuberculosis (M.tb) infection up to the age of 10 years [12]. However, the protective efficacy of BCG vaccination in adults varies widely, ranging from 0 to 80% [13]. Therefore, there is an urgent need to develop a TB vaccine with high protective efficacy. The emerging mRNA vaccine technology undoubtedly offers new hope for TB vaccine research. Therefore, this paper will review the current progress of mRNA vaccine research in terms of structure, type, immunogenicity regulation, delivery methods, mechanism of inducing immune response, and mRNA vaccines for various diseases that have entered into clinical trials, as well as put forward the current challenges and look forward to the possible future research directions, providing a theoretical basis for future mRNA vaccine research.
mRNA vaccine structural elements
mRNA structure and its optimization
Currently, mRNA IVT technology has reached a relatively advanced stage, with the most common methods using phage T3, T7, or SP6 RNA polymerase and linearized DNA for mRNA synthesis [14]. The conventional mRNA has a specific structure, including: 5’-end cap structure (5’ cap), 5’-end untranslated region (UTR), protein-encoding open reading frame (ORF), 3’-end translated region, and the polyadenylated poly(A) tail [15]. To promote the structural integrity and translational capability of mRNA vaccines, it is important to consider proper sequence design as an effective strategy to improve their stability. This involves enhancing the 5’ end (e.g., through chemical modification of the 5’ cap and incorporation of a shorter 5’ UTR), refining the ORF region (e.g., by codon optimization and preventing the formation of excessively strong stem-loop structures within the ORF), and improving the 3’ end (e.g., by introducing an AU-rich sequence into the 3’ UTR, selecting the optimal poly(A) tail length), and implementing nucleotide modifications.
5’ cap
The chemical modification at the 5’-end of the mRNA molecule, known as the 5’-cap structure, which is formed gradually during the transcription and processing of mRNA is not derived directly from the coding sequence of the DNA template. The core 7-methylguanosine (m7G) nucleotide represents a non-template-dependent addition linked to the first nucleotide encoded by the template via a distinctive 5’–5’ triphosphate bond. It plays a multitude of critical roles in molecular biology. For instance, during cytoplasmic mRNA translation process, it facilitates the recognition and binding of ribosome to mRNA, thereby participating in the initiation of protein translation and enabling the onset of protein synthesis. Additionally, the 5’ cap collaborates with the 3’-end poly(A) tail, poly(A)-binding proteins, RNA helicase, and other initiation factors (such as eukaryotic initiation factor 4E (eIF4E)) to enhance translation efficiency [5, 16]. At the same time, the 5’ cap structure protects the mRNA from degradation by the 5’–3’ exonuclease. In addition, the 5’ cap converts 5’-triphosphate (5’-PPP) into m⁷GpppN through capping enzymes, such as Vaccinia Capping Enzyme, or co-transcriptional capping enzymes, such as CleanCap. This process involves the coverage of free phosphate groups, thereby preventing RIG-I-mediated immune activation [19], reducing immunogenicity, and enhancing RNA stability.
The role of the mRNA 5’ cap has contributed to the development of mRNA vaccines and their use in disease prevention and treatment. There are two main methods of capping during mRNA transcription in vitro: enzymatic capping (post-transcriptional) and co-transcriptional capping, but they do not occur simultaneously during IVT. Enzymatic capping uses RNA 5’-triphosphatase (RTPase) and guanylyl transferase (GTase) to form a 5’-cap structure. RTPase hydrolyzes the 5’ γ-phosphate of mRNA to form β-phosphate, and then β-phosphate transfers guanosine mono-phosphorus (GMP) to the 5’-diphosphate RNA under the action of GTase to form the GpppNp-RNA structure. Guanine-N7 methyltransferase (N7MTase) methylates the guanosine moiety of the cap to form the Cap0 structure (m7GpppNp). The Cap0 structure is essential for the efficient translation of mRNAs as it serves as the basic form which protein factors bind and drive related functions. However, the Cap0 structure mRNA is perceived by the host as exogenous RNA, thereby triggering an inflammatory response by the host’s natural immune system [17]. The formation of the Cap1 structure (m7GpppN1mp) by 2’-O-methylation of the first nucleotide in the mRNA in the presence of 2’-O-methyltransferase (2’-O-MTase) effectively reduces the immunogenicity of mRNAs in cells [18]. In addition, it assists cellular sensors and the type I interferon (IFN) signaling pathways in distinguishing the Cap1 structure from other exogenous RNAs, thereby preventing activation of the innate immune response [19]. Moderna’s COVID-19 vaccine, known as mRNA-1273, uses this approach to cap the mRNA [20]. Further methylation at the 2’-hydroxyl position of the second nucleotide at the 5’-end of the mRNA results in the formation of the Cap2 structure (m7GpppN1mpN2mp) [21]. Recent studies have shown that while the Cap1 structure does indeed significantly reduce the immunogenicity of mRNAs, cellular mRNAs with only Cap1 type can still activate inflammatory antiviral mechanisms, while Cap2 can provide crucial additional protection [22]. Although Cap 2 is present in 10–15% of eukaryotic cells, its role is not fully understood. Enzymatic capping is characterized by transcription followed by capping, with a capping rate of almost 100% [26]. However, the cost of various enzymes is high, the production process involves many steps with complicated quality control, and the enzymatic (post-transcriptional) capping is also time-intensive.
The second method is co-transcriptional capping, which uses capping analogs (such as the first generation of mCap, the second generation of ARCA, and the third generation of Cap1 cap structural analogs) to achieve co-transcriptional capping of mRNA [21, 23]. Currently, the CleanCap® capping analog products developed by TriLink Company can co-transcribe with the target mRNA to form the Cap1 structure with a capping rate of more than 90%, effectively solving the problems of low efficiency (a capping rate of co-transcriptional capping is less than 70%) and high enzyme cost (Enzymatic capping) in the traditional capping methods [24]. BioNTech’s COVID-19 mRNA vaccines (BNT162b1 and BNT162b2) used CleanCap® capping analogs for vaccine capping [25]. Co-transcriptional capping is currently the most widely used method in IVT. It is characterized by one-step cap addition, simple operation and high success rate of capping, which can rapidly increase the yield of mRNA vaccines. However, the current cost of synthesizing cap analog is relatively high. Newly introduced Cap2 cap structural analogs, such as 3’-O-Me-m7G(5’)ppp(5’)(2’OMeG)p(2’OMEG), can be synthesized and capped more efficiently, with lower immunogenicity and higher protein translation efficiency [26].
UTR
UTRs (5’UTR and 3’UTR) are non-coding regions at the 5’ and 3’ ends of mRNA molecules. They can affect mRNA translation, half-life, and mRNA localization in cells. The 5’UTR is mainly derived from genes, such as globin, heat shock protein 70 (Hsp70), and hydroxy-steroid dehydrogenase (3β-HSD), prevalently [27, 28]. Its main role is to engage in the translation of downstream ORFs [29, 30]. The 3’ UTR is mainly derived from genes encoding the hemoglobin subunits α (HBA) and β (HBB), as well as the genes for albumin (Alb), heat shock protein 70 (Hsp70), tyrosine hydroxylase (TH), and collagen α1(COL1A1) [31, 32]. Their main function is to maintain mRNA stability and improve translational efficiency [33, 34]. UTRs are optimized to enhance mRNA stability and improve the translation efficiency. Therefore, when designing mRNA, it is crucial to avoid placing the upstream open reading frames and start codons (AUG) within the 5’UTR, which prevents expression repression and mRNA degradation. In addition, the 5’UTR should not be too long so as not to a secondary structure that makes it difficult to bind to the ribosome [35]. Meanwhile, the Kozak sequence was added to the 5’UTR region to enhance the start codon recognition ability and improve the translation efficiency [36]. The AU-rich elements (AREs) in the 3’UTR are cis-acting regions that that interacts with ARE-binding proteins to regulate mRNA stability and steady-state expression [44]. The number and arrangement of AUUUA repetitions or UUAUUUAUU (U- rich) or AU-rich (AUAUAUA) in the AREs have a big effect on how the poly(A) tail is cut off and how the RNA breaks down afterward. GU-rich sequences (GREs) interact with the CELF1 protein in human cells, facilitating mRNA degradation. Consequently, the proper modification of AREs and GREs can preserve mRNA stability [37–39].
ORF
The ORF is a crucial segment in mRNA that encodes proteins and influences mRNA stability and translational efficiency. Optimizing the ORF primarily involves substituting uncommon codons in the foreign mRNA with the prevalent codons of the host cell, thereby reducing the presence of rare codons and enhancing translation efficiency. Certain research indicates that CureVac (a biopharmaceutical company headquartered in Germany) substitutes A or U in the third nucleotide of the CVnCoV codon with C or G to enhance the GC content of the ORF, thereby improving the stability and translation efficiency of the mRNA [40]. Not all proteins necessitate outstanding translation efficiency, some require reduced efficiency to achieve proper folding [41]. Consequently, selecting a suitable optimization approach to equilibrate the translation rate and precision is essential.
Poly(A) tail
The poly(A) tail is an extended sequence of poly-adenosine. The length of the poly(A) tail influences mRNA stability and translation efficiency [42]. It inhibits mRNA degradation and establishes a closed loop with eIF4E and the 5’ cap to augment mRNA stability [43]. However, this tail is unstable and can be lost or cut off. Currently, there are two methods to polyadenylate mRNA: the first is the traditional enzymatic polyadenylation, which is unable to change the length of the tail [16], and the second is to design specific poly(A) sequences on DNA templates, which are then transcribed into poly(A) tail whose length can be changed [44]. A truncated poly(A) tail is incapable of promoting mRNA translation. Consequently, the ideal length of the poly(A) tail sequence enhances the translation efficiency and stability of mRNA [45, 46]. In a recent study, the poly(A) tail sequences were separated and connected by spacer sequences in the middle which could significantly reduce the recombination events during plasmid DNA amplification, maintain the tail length, and at the same time did not affect the translation efficiency and half-life of the mRNA transcribed in vitro [47]. Using branched poly(A) tails and the multiorgan protection sequence, these two key technological innovations in the development of linearly modified mRNA (modRNA) vaccines have significantly enhanced the stability, expression efficiency, and tissue targeting capabilities of traditional mRNA vaccines [55, 56].
Types of mRNA vaccines
Non-replicating mRNA vaccines
Non-replicating mRNA vaccines only encode the target antigen and do not contain replicase genes. Their diminutive size (about 2–3 kb), uncomplicated structure, and high specificity of the immune response make them distinctive. Currently, researchers have established two categories of non-replicating mRNA vaccines: the unmodified (natural) mRNA vaccination and the modified mRNA vaccine (Fig. 1A and B). The last one uses 1-methylpseudouridine or pseudouridine instead of uridine nucleoside, which might make mRNA work better at translation and make cells more stable [48]. Compared to self-amplifying mRNA, constrained expression of non-replicating mRNA requires higher doses to achieve the desired vaccination effect, or a comprehensive optimization process to elicit an effective immune response at reduced doses [49, 50].
Fig. 1.
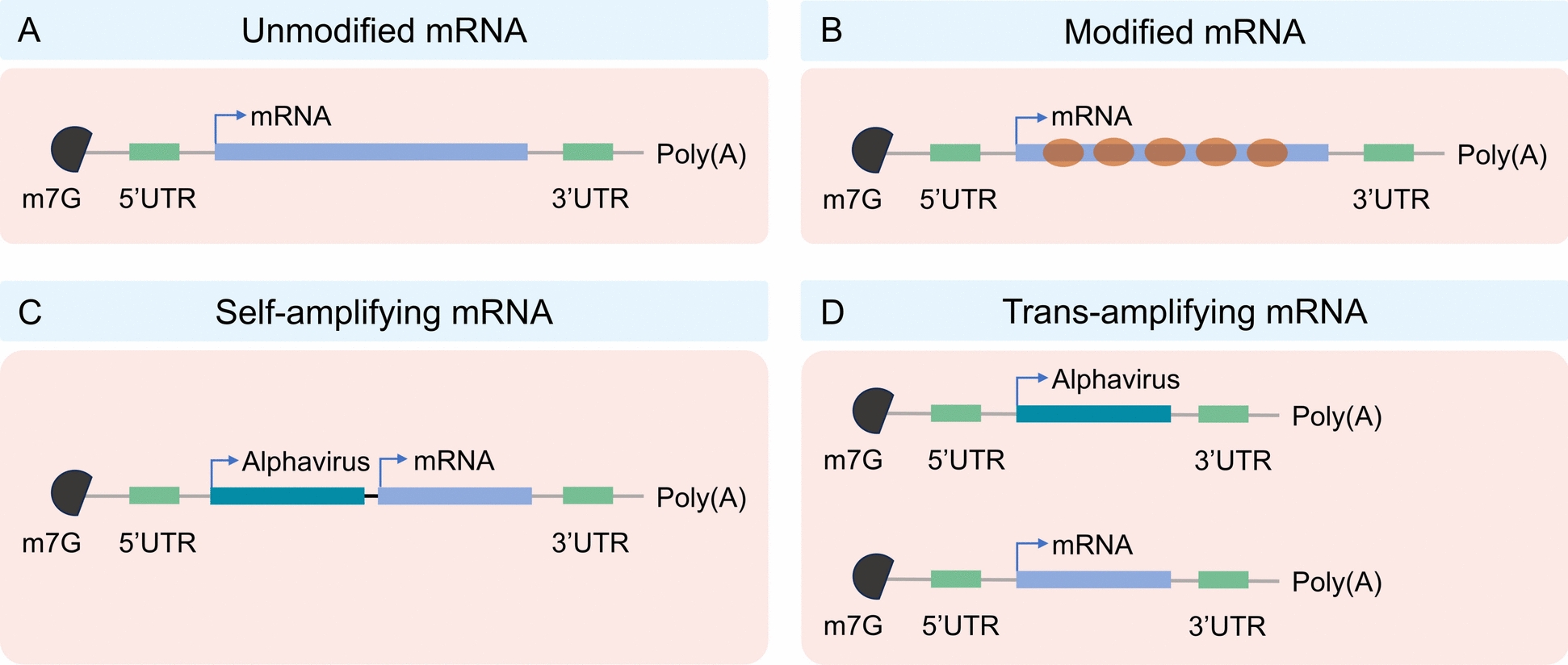
Types of mRNA vaccines
Self-amplifying RNA (saRNA) vaccines
Non-replicating mRNA vaccines are extremely simple to transfect in vitro, nonetheless, they necessitate high dosages or numerous vaccinations to provoke a sufficient immune response. As an alternative technology addressing these concerns, saRNA vaccines mimic alphaviruses by delivering genetic information encoding the target antigen and other genes (e.g., viral RNA-dependent RNA polymerase), and are self-amplifying, autonomously replicating within host cells to significantly enhance target protein expression [51] (Fig. 1C). This technology is expected to trigger a strong immune response at low doses and prolong protection [52–55]. Moreover, saRNA can diminish the reliance on delivery materials (e.g., LNP), hence reducing expenses (Less LNP, lower dose) and adverse consequences (e.g., LNP toxicity, unmodified mRNA may be recognized by RIG-1/MDA5 to elicit an interferon response) [65]. These benefits could facilitate the vaccination of a greater number of individuals in a reduced timeframe during a pandemic [56]. In 2021, British scientists executed the inaugural clinical study of the novel saRNA COVID-19 vaccine (COVAC1). The findings indicated that 87% of vaccinated individuals elicited a protective immunological response at a modest dosage, with no safety concerns reported [57]. Recently, CSL and Arcturus Therapeutics collaborated to produce the novel coronavirus vaccine ARCT-154, the first commercially authorized saRNA vaccine for adult COVID-19, suitable for both primary and booster vaccinations in adults [58]. The saRNA vaccine has emerged as a leader in the next generation of RNA vaccines.
However, saRNA/replicons also have their limitations. The body may generate T cell responses against the replicase, and repeated injections may lead to the suppression of its expression. This severely limits the potential of saRNA vaccines for multiple booster immunizations and is one of the main obstacles in their clinical application. Furthermore, since the replicase cannot recognize many modified bases, replicons have long been limited to using natural uridine, which is a compatibility issue for base modification [59]. However, two recent reports showed that saRNA can be modified with other nucleotides, such as 5-hydroxymethylcytidine, 5-methylcytidine (5-mC), and 5-methyluridine [60, 61]. In comparison to conventional mRNA, saRNA possesses a more extensive molecular structure and exhibits a reduced yield during in vitro synthesis. To overcome this technical limitation, Beissert et al. created an enhanced saRNA, known as trans-amplified RNA (taRNA), in 2020 (Fig. 1D). This is a novel trans-amplified RNA system characterized by excellent translation efficiency and minimal cellular translation interference, representing a significant advancement in RNA technology. This method makes the replicase complex work better by working together with two vectors: one that contains the viral immunogen and the other that contains the alphavirus replicon. Experimental findings confirm the use of taRNA in influenza hemagglutinin antigen-encoding RNA, showing that a minimal dose of 50 ng can significantly enhance the expression of the viral immunogen, thereby inducing an immunological protective response [62]. It is noteworthy that taRNA can solely encode the replicase, bypassing the length limitation of the target gene, thereby diminishing the risk of recombinant viral particles and enhancing safety [63]. This taRNA vaccination technology is adept at addressing rapidly evolving pathogens and new infectious illnesses [62].
Circular RNA (circRNA)
circRNAs are generated through back-splicing, wherein their 3’ and 5’ ends become covalently linked, forming a closed circular structure devoid of free termini. This unique topology constitutes the fundamental basis for circRNAs stability. Cellular exoribonucleases primarily recognize and cleave the free ends (5’ or 3’) of linear RNAs [64, 65]. Owing to the absence of free ends, circRNAs exhibit significant resistance to degradation by these enzymes. Experimental evidence confirms that circRNAs possess marked resistance to RNase R, a potent 3’ → 5’ exoribonuclease that efficiently degrades linear RNAs [66, 67].
This covalently closed circular architecture confers exceptional stability, enabling sustained and prolonged expression of antigenic proteins in vivo and offering a transformative solution to the transient expression profile inherent to linear mRNA therapeutics. Consequently, circRNA technology lays a solid foundation for developing next-generation RNA vaccines and therapeutics with enhanced efficacy (potentiating stronger immune responses), improved dosing convenience (potentially requiring lower doses and fewer administrations), and broader applicability (e.g., in oncology and therapeutic protein delivery).
Delivery systems for mRNA vaccines
Nucleases readily destroy mRNA, which is too large and negatively charged to pass through cell membranes. Research indicates that the absorption rate of naked mRNA molecules by cells is below 1 in 100,000 [5]. Administering mRNA vaccines using designated vectors is necessary to achieve optimal efficacy. The vector of choice must successfully penetrate the target cell membrane, internalize within the cell, avoid the endosome, release the mRNA into the cytoplasm, and aid in the translation process. At the same time, mRNA vaccine delivery vectors can change both the amount and quality of antigen protein production, which in turn changes how the host immune system responds [68]. This discussion will address several delivery systems, including lipid vector derivatives, polymers and protamine.
Lipid carrier derivatives
Lipid nanoparticles (LNPs)
The primary purpose of LNP, a lipid-based nanoparticle, is to deliver mRNA to the cytoplasm. By joining with the lipid bilayer of the early endosome, these nanoparticle carriers can move mRNA into the cell quickly and efficiently. This procedure not only liberates the mRNA into the cytoplasm but also safeguards it against degradation by ribonucleases (RNases) in systemic circulation [69]. LNP is mostly made up of four main parts: ionizable lipids (40–50%), cholesterol (38–45%), auxiliary phospholipids (10–12%), and polyethylene glycol (PEG) lipids (1–2%) [70]. These components collaborate to encapsulate and safeguard unprotected mRNA [71]. Ionizable amino lipids facilitate the release of mRNA from the endosome by interacting with the endosomal membrane [72]. PEG can make lipid nanoparticles stay in circulation longer, stop mRNA from joining with plasma proteins, and speed up the clearance process. Phospholipids and cholesterol contribute to the stabilization of lipid nanoparticle structure [73].
But endosomal escape, which is necessary for RNA expression, also directly triggers inflammation by causing endosomal membrane damage, However, inflammation can be controlled by inhibiting galectins (large hole detectors) or using biodegradable lipids, which create smaller holes that are reparable by the endosomal sorting complex required for transport (ESCRT) pathway [74]. In addition, the amine headgroups in ionizable lipids drive LNP immunogenicity by binding to Toll-like receptor 4 and CD1d and by promoting lipid-raft formation. Immunogenic LNPs favor a type-1 T helper cell-biased immune response marked by increase in the immunoglobulins IgG2c and IgG1 and in the pro-inflammatory cytokines tumor necrosis factor, interferon γ, and interleukins IL-6 and IL-2 [75].
The delivery efficiency of LNP primarily relies on cationic or ionizable lipids, such as DLin-DMA, DLin-KC2-DMA [76], and DLin-MC3-DMA [77], which have been meticulously designed and synthesized. Additionally, C12-200 [78] and cKK-E12 [79] have been identified through high-throughput screening in combinatorial libraries. The next generation of ionizable lipids includes derivatives, such as DLin-MC3-DMA L319 [80], C12-200 and cKK-E12 [81–83], ALC-0315, and SM-102, which are used in COVID-19 vaccines [84], TT3 and its biodegradable derivative FTT5 [84, 85], vitamin-derived lipids ssPalmE [86] and VcLNP [87], A9 [88], L5 [89], A18 Lipid [90], ATX Lipid [91] and LP01 [92]. Most of these lipids have the capability to be biodegraded.
DLin-MC3-DMA has been approved for clinical treatment of RNA by ionizing cationic lipid [93]. Moderna used DLin-MC3-DMA ionizable lipids to make mRNA vaccines against the Zika virus and influenza. They then conducted both preclinical and clinical trials on these vaccines [94, 95]. Subsequent studies revealed the breakdown of the dialkyl tail of the dilauryl lipid in the ionizable lipid, suggesting that repeated administration of the drug could potentially increase its toxicity over time [96]. Consequently, Moderna has created a novel ionizable lipid, Lipid H, SM-102. The novel lipid features an enlarged branching structure at the tail end, and the incorporation of an ester bond improves biodegradability [89, 97]. Additionally, BioNTech has used the lipid nanoparticle form of its COVID-19 mRNA vaccine along with the ionizable lipid ALC-0315, which has a chemical structure similar to SM-102 and has also shown good results [96]. G-LNPs, which are lipid-modified poly-(guanine-lipoic acid) polymers, are a new type of lipid nanoparticle for mRNA vaccines that has been discovered recently. G-LNPs can greatly improve the translation efficiency of mRNA that is encapsulated and reduce inflammation after vaccination by blocking reactive oxygen species that slow down translation and cause inflammatory reactions. Consequently, this markedly enhances the translation efficiency of the administered mRNA and diminishes inflammation post-vaccination. In vivo studies show that G-LNPs deliver mRNA effectively, triggering a strong immune response against tumors. This suggests that G-LNPs-based mRNA vaccines will be a powerful and flexible way to treat cancer [98].
LNPs have two benefits for mRNA vaccine vector: first, it protects mRNA from being broken down by endosomal enzymes [99], which improves delivery efficiency [100]. Second, it makes it easier for mRNA to be delivered for expression through a variety of biological processes, showing great biocompatibility. Therefore, experts view LNP as a promising mRNA delivery vector due to its excellent biocompatibility and high delivery efficiency. Currently, mRNA vaccines and therapeutics targeting COVID-19 and influenza extensively utilize LNP [101, 102].
Xu et al. developed the AGILE platform, integrating deep learning and combinatorial chemistry, to accelerate the development of ionizable lipids. Through virtual screening and experimental validation, two new lipids, H9 and R6, were discovered, which exhibited excellent performance in muscles and macrophages respectively, demonstrating the great potential of the combination of deep learning and combinatorial chemistry, and providing a new tool for personalized delivery of mRNA therapy. By optimizing the lipid structure and formulation, AGILE significantly improved the efficiency and specificity of mRNA delivery, opening up new avenues for vaccines and gene therapy [103]. In addition, Witten J et al. [104] also developed a deep learning (DL)-based method, LiON (Lipid Optimization using Neural Networks), to accelerate the design of novel ionizable lipids and enhance their delivery efficiency in different tissues. Through model prediction and experimental validation, two highly efficient lipid structures, FO-32 and FO-35, were discovered. They show great potential in muscle injection vaccines, nasal vaccines, and lung gene therapy. Particularly, the whole lung transfection ability of FO-32 and FO-35 is of great significance in the treatment of obstructive lung diseases such as cystic fibrosis. Vander Straeten A et al. successfully developed an automated microneedle vaccine printer (MVP) device, which can efficiently produce microneedle patches (MNPs) containing LNP mRNA vaccines. The MNPs can be stably stored at room temperature for 6 months and still induce a strong immune response [105].
The exposure of the mRNA to the water at the core of an LNP makes the mRNA very vulnerable, and the instability of LNP mRNA has become a key factor affecting the quality of the formulation. Compared with the LNP mRNA system, the siRNA-LNP drug Onpattro™ has a significantly longer shelf life, suggesting that the current stability bottleneck is not the LNP but the mRNA. Lyophilization, as an effective preservation method, avoids the protein denaturation and loss of biological activity that may occur under high temperatures in traditional drying methods. Therefore, it is considered particularly suitable for the long-term preservation of biological macromolecular products, such as mRNA vaccines and enzyme preparations [106]. Munoz-Moreno R et al. compared the stability of freeze-dried formulations (stored at 5 °C) and freeze formulations (stored at − 70 °C). The experimental data showed that in the 24-month stability test, the EC50 value (the in vitro expression of half-effective concentration) of the freeze-dried formulation was close to that of the freeze formulation (the freeze-dried version was only slightly higher), indicating that the freeze-drying process had a negligible impact on the structural integrity of mRNA. The immunogenicity test revealed that the level of gE-specific IgG antibodies induced by the freeze-dried vaccine was not significantly different from that of the freeze formulation, and was significantly higher than that of the control group [107].
In the phase I clinical study, the cytomegalovirus vaccine developed by Moderna (mRNA-1647) was in the form of a freeze-dried liquid formulation. However, in the subsequent phase II study, the formulation of this vaccine changed to a freeze-dried form. According to relevant reports, the freeze-dried formulation can be stably stored for up to 18 months under refrigeration conditions, and it can be clearly observed that the freeze-dried formulation has significant advantages in improving the stability of the formulation. Additionally, BioNTech/Pfizer is also conducting a phase III clinical trial to evaluate the freeze-dried version of its BNT162b2 vaccine. Recently, Reckitt Benckiser successfully developed the RH109 lyophilized mRNA vaccine [108], which has entered the clinical phase. The company developed three freeze-dried vaccines targeting the wild-type, Delta, and Omicron SARS-CoV-2 variants. These vaccines demonstrated efficacy in inducing high IgG titers and neutralization responses alongside long-term stability during storage at both 4 °C and 25 °C. This stability profile suggests potential to address the storage and transportation challenges associated with current mRNA vaccines. However, the specific stabilizing excipients used in the formulation and the detailed lyophilization protocols were not disclosed by the company.
Freeze-drying technology has become an important tool in modern vaccine research and global distribution by enhancing vaccine stability, simplifying storage conditions, ensuring efficacy and reducing costs. With the rapid development of nucleic acid vaccines such as mRNA, further optimization of the freeze-drying process (such as rapid freeze-drying and the application of new protective agents) will continue to drive the innovation of vaccine technology.
Nanostructured lipid carrier (NLC)
The use of NLC facilitated the successful development of the world’s first saRNA vaccine, ARCT-154, for adult COVID-19. This vector can maintain the vaccine in a freeze-dried state, with a shelf life of up to 21 months under refrigeration and 8 months at ambient temperature. Furthermore, the utilization of this vaccine has markedly enhanced its thermal stability. It can keep being immunogenic for at least 6 months at room temperature or under normal refrigeration after being lyophilized, which solves a big technical problem in making RNA vaccines. This saRNA vaccine also works well to make the SARS-CoV-2 spike protein antigen, which causes a strong humoral and cell-specific immune response in mice without the need for nucleoside modification. For the ARCT-154 saRNA vaccine, the lowest dose needed is only 5 μg [72]. For comparison, the Moderna mRNA vaccine needs 100 μg and the BioNTech/Pfizer mRNA vaccine needs 30 μg. In conclusion, NLC delivery vehicles demonstrate the capability to enhance RNA technology and facilitate the broader application of RNA vaccines for the ongoing COVID-19 pandemic and future infectious disease pandemics [109].
Polymers
Due to their excellent physical stability, polymer carriers find extensive use in mRNA distribution. Two primary categories of polymer delivery vehicles are polyethylenimine (PEI) and poly-(β-amino ester) (PBAE). Currently, PEI serves as the primary delivery method for mRNA vaccines [110–112]. PEI exhibits early cytotoxicity attributable to its elevated molecular weight, thereby constraining its applicability. Research indicates that the amalgamation of PEI with PEG or cyclodextrin can diminish the positive surface charge and mitigate cytotoxicity [113]. A 2007 study showed that PBAE may effectively transfect mRNA in serum-free protein systems [114]. However, a study in 2007 revealed its potential for mRNA delivery, particularly in a serum protein-free system where PBAE achieved more efficient mRNA transfection in vitro [115]. This discovery has facilitated a broader range of studies and uses of PBAE. Researchers Kaczmarek et al. made a polymer-based delivery system in 2016 called a PBAE and lipid–PEG complex. This system showed a lot of stability in serum and good mRNA delivery. Following intravenous administration, the mouse lungs effectively identified the target mRNA product [116].
Protamine
Drug delivery, diagnostics, and vaccine development use peptide- or protein-based biomaterials because of their biocompatibility and availability [117, 118]. Positively charged cationic peptides readily transport negatively charged mRNA through electrostatic interactions. Cationic peptides facilitate the delivery of negatively charged mRNA via electrostatic interactions, wherein the positive charge originates from lysine and arginine residues, enabling the mRNA to adhere firmly to the cationic peptide [119]. Protamine, a cationic peptide, establishes a stable compound with mRNA via electrostatic interactions, thereby safeguarding it from RNase-mediated destruction [120]. However, the tight binding between protamine [41] and mRNA may inhibit the translation process when using only protamine-formulated RNA [121]. However, CureVac’s RNActive technology can circumvent this issue, the RNActive® vaccine technology effectively addresses the issue of protamine–mRNA complex inhibiting translation through a dual-component system. Its core technical principle includes: exposed optimized mRNA and protamine–mRNA complex. Specifically: the exposed mRNA achieves a 4–fivefold enhancement in expression through sequence optimization (UTR modification, codon optimization, etc.), while the protamine complex mainly fulfills the immune adjuvant function mediated by TLR7/8. The optimized mRNA is combined with fish protamine at a specific ratio to form a dual-component vaccine system. The naked mRNA enters the cytoplasm through scavenger receptors for efficient antigen expression; the protamine complex enters the endosome through endocytosis and activates innate immunity through TLR7/8. This innovative design not only overcomes the inhibitory effect of the simple protamine complex on translation but also realizes self-adjunctive activity [122]. It has shown superior protective efficacy to traditional mRNA vaccines in preclinical models of influenza vaccines and tumor vaccines [123, 124].
Mechanisms of immune response induced by mRNA vaccines
Once mRNA vaccines enter cells, they serve as pathogen-associated molecular patterns (PAMPs) that are recognized by the pattern recognition receptors (PRRs) of the host cell [125]. PRRs bind to specific ligands, triggering type I interferon and NF-κB signaling pathways, which in turn mediate innate and adaptive immune responses. The most notable PRRs that recognize exogenous RNA include TLRs located on endosomes and retinoic acid-inducible gene I-like receptors (RLRs) found in the cytoplasm. The TLR family consists of TLR3, TLR7, and TLR8. TLR3 is capable of recognizing and binding double-stranded RNA (dsRNA) that is more than 45 base pairs in length, as well as single-stranded RNA (ssRNA) with secondary structure or dsRNA derived from viral replication intermediates. This process regulates the activation of the IFN-I pathway and the secretion of cytokines and chemokines [126]. TLR7 can bind both dsRNA and ssRNA, and its activation process can enhance antigen presentation, promote cytokine secretion, and stimulate B cell responses [127]. In comparison to TLR3 and TLR7, TLR8 is limited to ssRNA [128]. RLRs consist of RIG-I and Melanoma differentiation-related gene 5 (MDA5) [129]. RIG-I primarily detects the 5’-terminal triphosphate (5’-ppp) of dsRNA and stimulates the production of IFN-I [130–132]. MDA5 is a cytosolic RNA sensor that can recognize long dsRNA produced during RNA virus replication and RNA transcription in vitro [133]. However, research has demonstrated that the dsRNA-activated protein kinase receptor (PKR) and oligoadenylate synthetase (OAS) pathway are activated, leading to the inhibition of mRNA translation [134] and the promotion of RNA degradation [135]. Therefore, it is crucial to purify mRNA.
Purified mRNA, which contains only ssRNA, is endogenously taken up by the delivery system (Fig. 2). It is then released to endosomes and translated to protein by ribosomes. The translated proteins activate the immune system in two main ways: First, they are proteasome proteins that turn into peptides, which then combine with major histocompatibility complex (MHC) class I molecules on the cell surface antigen. These molecules bind with T cell receptors (TCRs), thereby activating CD8+T cells to kill infected cells by secreting perforin and granzyme. Second, exogenous antigens can be internalized by cells, processed into peptide fragments within cellular compartments, and presented via MHC class II molecules on the cell surface to CD4+T cells. CD4+T cells can either activate cellular immune response through secretion of inflammatory cytokines or activate humoral immune response through mutual activation of B cells [136].
Fig. 2.
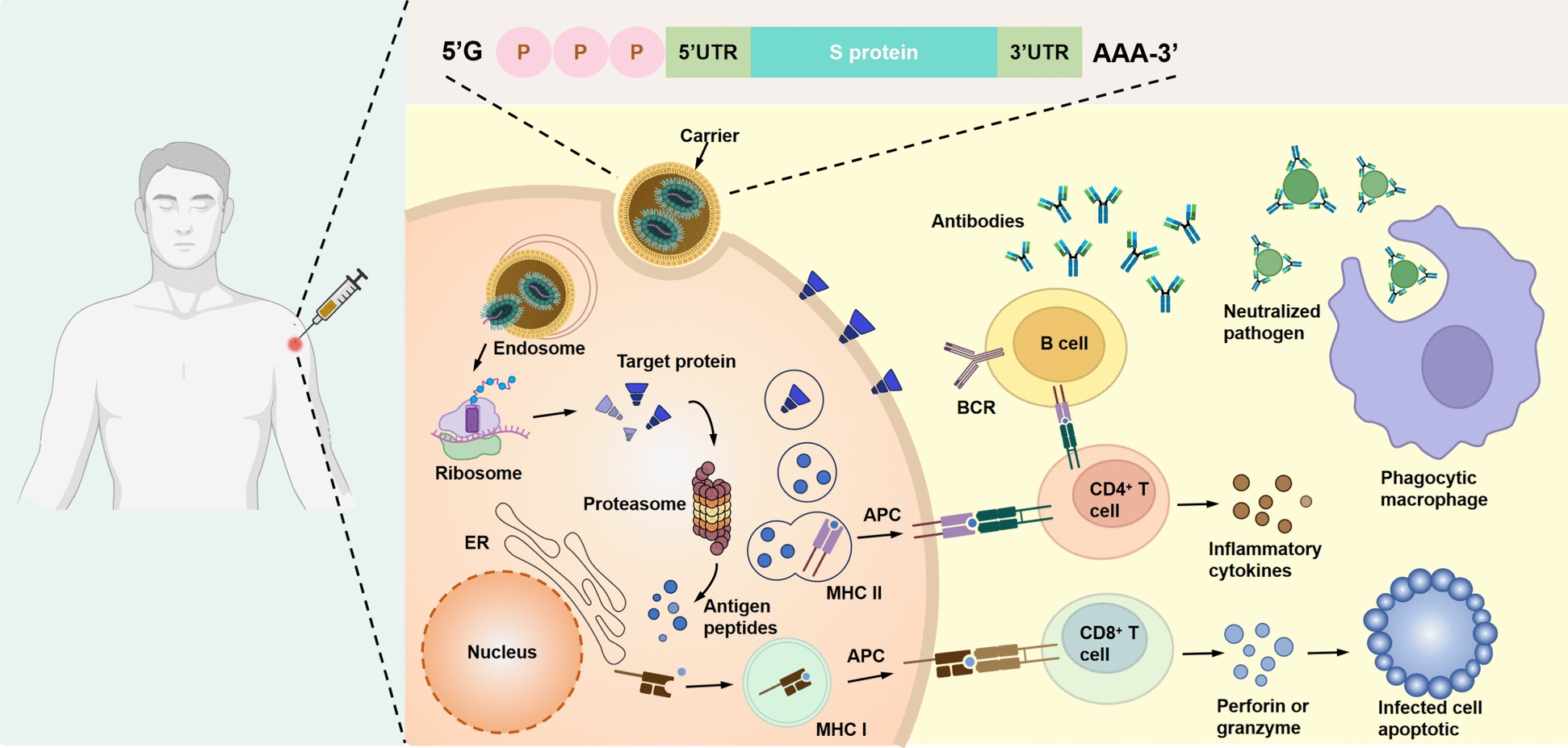
Mechanism of cellular and humoral immune responses induced by mRNA vaccines
Research and clinical trials on mRNA vaccines for infectious diseases
COVID-19
At the conclusion of 2019, the emergence of the novel coronavirus precipitated widespread infection and mortality, presenting a significant threat to the global community. In response to this problem, the mRNA-1273 vaccine developed by Moderna and the BNT162b2 mRNA vaccine created by BioNTech in partnership with Pfizer received emergency authorization from the US FDA in December 2020. Nonetheless, SARS-CoV-2 persists in mutating, and the infection incidence remains elevated. Research aimed at the development of efficacious pharmaceuticals and vaccines continues to advance swiftly. Currently, multiple mRNA vaccines have commenced clinical trials (Table 1).
Table 1.
Clinical application of COVID-19 mRNA vaccine
| Type of candidate vaccine | Types of RNA | Trial characteristics | Developers | Phase | Number |
|---|---|---|---|---|---|
| mRNA-1273 (Spikevax) | mRNA | 2 doses; Day 0 + 28; IM | Moderna + National Institute of Allergy and Infectious Diseases (NIAID) | Phase 4 | NCT04760132 |
| BNT162b2 (3 LNP-mRNAs), also known as"Comirnaty" | mRNA | 2 doses; Day 0 + 21; IM | Pfizer/BioNTech + Fosun Pharma | Phase 4 | NCT04674189 |
| CVnCoV Vaccine | mRNA | 2 doses; Day 0 + 28; IM | CureVac AG | Phase 3 | NCT04760132 |
| ARCT-021 | saRNA | NR; NR; IM | Arcturus Therapeutics | Phase 2 | NCT04668339 |
| LNP-nCoVsaRNA | saRNA | 2 doses; NR; IM | Imperial College London | Phase 1 | ISRCTN17072692 |
| SARS-CoV-2 mRNA vaccine (ARCoV) | mRNA | 2 doses; Day 0 + 14 or Day 0 + 28; IM | Academy of Military Science (AMS), Walvax Biotechnology and Suzhou Abogen Biosciences | Phase 3 | NCT04847102 |
| ChulaCov19 mRNA vaccine | mRNA | 2 doses; Day 0 + 21; IM | Chulalongkorn University | Phase 2 | NCT05605470 |
| PTX-COVID19-B, mRNA vaccine | mRNA | 2 doses; Day 0 + 28; IM | Providence Therapeutics | Phase 3 | NCT05534035 |
| CoV2 SAM (LNP) vaccine. A self-amplifying mRNA (SAM) lipid nanoparticle (LNP) platform + Spike antigen | saRNA | 2 doses; Day 0 + 30; IM | GlaxoSmithKline | Phase 1 | NCT04758962 |
| mRNA-1273.351 | mRNA | 3 doses; Day 0 or Day 0 + 28 or Day 56; IM | Moderna + National Institute of Allergy and Infectious Diseases (NIAID) | Phase 4 | EUCTR2021-000930-32 |
| MRT5500, an mRNA vaccine candidate | mRNA | 2 doses; Day 0 + 21; IM | Sanofi Pasteur and Translate Bio | Phase 2 | NCT04798027 |
| DS-5670a, coronavirus-modified uridine RNA vaccine (SARS-CoV-2) | mRNA | 2 doses; NR; IM | Daiichi Sankyo Co., Ltd | Phase 2/3 | JPRN-jRCT2071210106 |
| HDT-301: Self-replicating mRNA vaccine formulated as a lipid nanoparticle | saRNA | 2 doses; Day 0 + 28; IM | SENAI CIMATEC | Phase 2/3 | NCT05542693 |
| mRNA-1283 | mRNA | 2 doses; Day 0 + 28; IM | ModernaTX, Inc | Phase 1 | NCT04813796 |
| EXG-5003; a temperature-sensitive self-replicating RNA vaccine expressing the receptor binding domain of the SARS-CoV-2 spike protein | saRNA | 1 dose; Day 0; ID | Elixirgen Therapeutics, Inc | Phase 1/2 | NCT04863131 |
| mRNA COVID-19 vaccine (SW-BIC-213) | mRNA | 2 doses; TBD; IM | Shanghai East Hospital and Stemirna Therapeutics | Phase 3 | NCT05580159 |
| LNP-nCOV saRNA-02 vaccine; Self-amplifying RNA (saRNA) encapsulated in lipid nanoparticles (LNP) | saRNA | 2 doses; Day 0 + 28; IM | MRC/UVRI and LSHTM Uganda Research Unit | Phase 1 | NCT04934111 |
| mRNA-1273.211. A multivalent booster candidate combining mRNA-1273 plus mRNA-1273.351 | mRNA | 1 dose; Day 0; IM | ModernaTX, Inc | Phase 2/3 | NCT04927065 |
| ARCT-154 mRNA Vaccine | saRNA | 2 doses; Day 0 + 28; IM | Arcturus Therapeutics, Inc | Phase 3 | ISRCTN15779782 |
| ARCT-165 mRNA Vaccine | saRNA | 2 doses; Day 0 + 29; IM | Arcturus Therapeutics, Inc | Phase 1/2 | NCT05037097 |
| ARCT-021 mRNA Vaccine | saRNA | 2 doses; Day 0 + 30; IM | Arcturus Therapeutics, Inc | Phase 1/2 | NCT05037097 |
| HDT-301 vaccine | saRNA | 1–2 doses; Day 0 ± 56; IM | HDT Bio | Phase 1 | NCT05132907 |
| VLPCOV-01, self-amplifying RNA vaccine against the coronavirus | saRNA | 2 doses; NR; IM | VLP Therapeutics Japan GK | Phase 1 | jRCT2071210067 |
| EG-COVID vaccine | mRNA | 3 doses; Day 0 + 21 + 42; IM | EyeGene Inc | Phase 1/2 | NCT05188469 |
| Coronavirus mRNA vaccine (LVRNA009) | mRNA | 2 doses; Day 0 + 28; IM | AIM Vaccine and Liverna Therapeutics | Phase 3 | NCT05428592 |
| mRNA-1273.529—Booster | mRNA | 1 dose; Day 0; IM | ModernaTX, Inc | Phase 2/3 | NCT05249829 |
| CV2CoV, mRNA vaccine | mRNA | 1 dose; Day 0; IM | CureVac AG | Phase 1 | NCT05260437 |
| mRNA vaccine (MIPSCo-mRNA-RBD-1) | mRNA | 1 dose; Day 0; IM | University of Melbourne | Phase 1 | NCT05272605 |
| A Lyophilized COVID-19 mRNA Vaccine | mRNA | 1 dose; Day 0; IM | Jiangsu Rec-Biotechnology Co., Ltd | Phase 1 | NCT05351450 |
| COVID-19 mRNA Vaccine (SYS6006) | mRNA | 2 doses; Day 0 + 21; IM | CSPC ZhongQi Pharmaceutical Technology Co., Ltd | Phase 2 | NCT05439824 |
| mRNA GEMCOVAC-19 (COVID-19 vaccine) | mRNA | 2 doses; Day 0 + 28; IM | Gennova Biopharmaceuticals Limited | Phase 2/3 | CTRI/2022/04/041880 |
| Lyophilized COVID-19 mRNA Vaccine | mRNA | 1 dose; Day 0; IM | Wuhan Recogen Biotechnology Co., Ltd | Phase 1 | NCT05366296 |
| A self-amplifying RNA (saRNA) boost vaccines (AAHI-SC2 and AAHI-SC3) | saRNA | 1 dose; Day 0; IM | ImmunityBio, Inc | Phase 1/2 | NCT05370040 |
| RQ3013: SARS-CoV-2 mRNA Chimera Vaccine | mRNA | 1 dose; Day 0; IM | Walvax Biotechnology; Shanghai RNACure Biopharma | Phase 1 | NCT05396573 |
| mRNA-1273.214 (Booster) | mRNA | 2 doses; Day 0 + 55; IM | ModernaTX | Phase 3 | NCT05436834 |
| mRNA-1073; (COVID-19/Influenza) Vaccine | mRNA | 2 dose; Day 0; IM | ModernaTX | Phase 1/2 | NCT05375838 |
| RVM-V001 | mRNA | 1 dose; Day 0; IM | RVAC Medicines | Phase 1 | NCT05420077 |
| ABO1009-DP (COVID-19 Omicron) mRNA Vaccine; ABO1020 vaccine | mRNA | 1 dose; Day 0; IM | Suzhou Abogen Biosciences Co., Ltd | Phase 1 | NCT05433194 |
| Self-Amplifying Messenger Ribonucleic Acid (samRNA) Vaccines | saRNA | 2 doses; Day 0 + 28; IM | Gritstone bio, Inc | Phase 1 | NCT05435027 |
| Investigational CV0501 mRNA COVID-19 Vaccine | mRNA | 1 dose; Day 0; IM | GlaxoSmithKline | Phase 1 | NCT05477186 |
| GLB-COV2-043, an mRNA booster vaccine candidate | mRNA | 1 dose; Day 0; IM | GreenLight Biosciences, Inc | Phase 1/2 | NCT05602961 |
| mRNA-based COVID-19 vaccine (COReNAPCIN) | mRNA | 1 dose; Day 0; IM | ReNAP Technology | Phase 1 | IRCT20230131057293N1 |
| JCXH-221, an mRNA-based | mRNA | 1 dose; Day 0; IM | Immorna Biotherapeutics, Inc | Phase 1/2 | NCT05743335 |
*The data are sourced from https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines (25 December 2024)
Moderna’s mRNA-1273 encodes the spike protein of SARS-CoV-2, encapsulated in LNP. It commenced phase I clinical trials in under 10 weeks following the publication of the initial SARS-CoV-2 genome sequencing, an unparalleled occurrence in the pharmaceutical business [137]. A phase I clinical investigation (ClinicalTrials.gov: NCT04283461) involved 45 healthy adults aged 18 to 55 years who received varying doses of the vaccine (50, 100, and 250 μg). Regardless of age, this study revealed a dose-dependent antibody response among subjects following two vaccine doses. Over fifty percent of the subjects encountered systemic adverse reactions, particularly in the high-dose group (250 μg), with the prevalence of severe adverse events reaching 21%. Despite the decline in neutralizing and binding antibody levels over time, they persisted at a comparatively elevated level [138]. A phase II clinical trial (ClinicalTrials.gov: NCT04405076) showed that after the second dose of the vaccine, the 100 μg dose caused higher levels of binding and neutralizing antibodies. During the study, no major side effects were reported. The results validate the safety and immunogenicity of 50 μg and 100 μg mRNA-1273 in a two-dose immunization protocol [139]. Over 30,000 volunteers participated in the phase III clinical investigation (ClinicalTrials.gov: NCT04470427). Among the participants, 2.2% had prior SARS-CoV-2 infections before the experiment commenced. The findings indicated that significant adverse effects were infrequent following two doses of the vaccination. Furthermore, the vaccination demonstrated 94.1% efficacy in preventing COVID-19 infection [140]. In February 2021, an extensive phase IV clinical trial (ClinicalTrials.gov: NCT04760132) was initiated to more accurately assess the vaccine’s efficacy and safety.
The BNT162b2 vaccine is made up of an LNP-formulated nucleoside-modified mRNA that codes for the full trans-membrane S protein in its pre-fusion conformation (S-pp). A phase I/II study (ClinicalTrials.gov: NCT04380701) with healthy people ages from 19 to 55 showed that the BNT162b2 vaccine worked to produce both antibody and T cell responses after the first dose, with booster doses ranging from 1 to 30 μg. When compared to the titer in plasma from people who had a booster shot seven days later, the geometric mean titer (GMT) of SARS-CoV-2 serum 50% neutralization ranged from 0.3-fold (1 μg dose group) to 3.3-fold (30 μg dose group). The study’s results indicated that immunization with BNT162b2 at a tolerable dosage can elicit an adaptive mixed humoral and cellular immune responses, which collectively leads to protection against COVID-19 [141]. The outcomes of the phase III clinical trial (ClinicalTrials.gov: NCT04368728), which included over 30,000 participants, demonstrated the significant efficacy of the BNT162b2 vaccine. The vaccine demonstrated 52% efficacy following the initial dose and 95% efficacy after the administration of two doses in preventing mild to moderate SARS-CoV-2 infection. This research indicated that the occurrence of severe adverse events post-vaccination was minimal and not substantially different from the placebo cohort, demonstrating the vaccine’s safety [142]. The phase IV study (ClinicalTrials.gov: NCT04844489) commenced in February 2021. This is a longitudinal study to assess the efficacy and longevity of the BNT162b2 mRNA vaccine and the safety of SARS-CoV-2 inoculation in the general populace.
The global advancement of COVID-19 mRNA vaccines is vigorously encouraged, with numerous vaccine candidates undergoing different phases of clinical trials. CanSino Biologics Inc. is undertaking phase I and II clinical trials (ClinicalTrials.gov: NCT05373485 and NCT05373472) of a COVID-19 mRNA vaccine (LNP) to assess its safety and immunogenicity in adults aged 18 and older. A total of 300 participants participated in these two studies, categorized into low-dose (30 μg) and high-dose (50 μg) groups, and vaccinated on days 0 and 21, respectively. Nevertheless, the outcomes of these experiments remain unpublished. CureVac has started a phase II/III trial (NCT04652102) to test the CVnCoV SARS-CoV-2 mRNA vaccine (LNP) candidate to see how well it works and how safe it is. The trial enrolled 39,693 individuals, with the experimental group administered 12 μg of the CVnCoV vaccination intramuscularly on days 1 and 29. The findings indicated that the vaccination demonstrated efficacy of 70.7% and 77.2% against mild-to-severe novel coronavirus pneumonia in individuals aged 18 to 60. This affirmed that the two-dose CVnCoV vaccination regimen is efficacious in preventing symptomatic COVID-19 in adults and possesses a dependable safety profile [143]. SinoMicro has commenced phase I/II clinical research to assess the safety, immunogenicity, and immunological durability of the COVID-19 mRNA vaccine (LNP) in healthy individuals aged 18 and above. The trial enlisted 480 participants, categorized into a high-dose group (45 μg) and a low-dose group (25 μg), who got intramuscular injections every 29 days. The clinical trial has concluded, but its results remain unpublished. The outcomes of these clinical trials are essential for assessing the efficacy and safety of mRNA vaccines in preventing COVID-19 and will furnish significant scientific data to bolster the global response to the pandemic.
Influenza
Among the most extensively researched vaccinations are mRNA vaccines targeting influenza viruses. Owing to the elevated mutation rate of influenza viruses, vaccine research and development must proceed in tandem with the evolving viral strains; hence, mRNA vaccines are particularly advantageous. For instance, during the lethal H7N9 influenza virus strain epidemic in China in 2013, researchers successfully cloned the HA gene into the pDNA template of the self-amplifying mRNA vaccine, leading to the successful production of the saRNA vaccine within 8 days of the HA gene sequence’s release [144] (Supplementary Table 1).
In 2017, Moderna executed a pioneering trial to assess the safety and immunogenicity of mRNA vaccines VAL-506440 and VAL-339851 against the potentially pandemic avian influenza viruses H10N8 and H7N9 for the first time (ClinicalTrials.gov: NCT03076385, NCT03345043). VAL-506440 and VAL-339851 mRNA vaccines consisted of chemically modified mRNAs encoding the full-length, membrane-bound form of the hemagglutinin (HA) glycoprotein from the H10N8 influenza strain (A/Jiangxi-Donghu/346/2013) or the H7N9 influenza strain (A/Anhui/1/2013) [155], an LNP delivery system was used [104]. A single facility in Germany conducted two randomized, placebo-controlled, double-blind Phase I clinical trials for H10N8 and in the United States for H7N9. The H10N8 vaccine research comprised 201 participants aged from 18 to 64 years, whereas the H7N9 vaccine study involved 156 participants aged from 18 to 49 years. Participants were administered two doses of either the vaccination or a placebo, spaced three weeks apart. The H10N8 vaccine trial assessed intramuscular doses ranging from 25 to 400 μg and intradermal doses of 25 and 50 μg. All the people who were given the 100 μg dose showed hemagglutination inhibition (HAI) levels of 1:40 or higher, and 87.0% of them showed microneutralization (MN) levels of 1:20 or higher. Zero. The 25 μg intradermal dosage elicited HAI titers exceeding 1:40 in 64.7% of subjects. The H7N9 vaccine trial assessed intramuscular dose levels of 10, 25, and 50 μg. This study showed that injecting 10, 25, or 50 μg of the drug into the muscle caused HAI titers of 1:40 or more in 36.0%, 96.3%, and 89.7% of the subjects, respectively. The 10 μg and 25 μg doses attained 100% MN titers in all subjects, whereas the 50 μg dose group reached 96.6%. Both H10N8 and H7N9 mRNA intramuscular vaccinations had favorable safety profiles and immunogenicity, with no major side events linked to the vaccines described. These experiments represent the inaugural mRNA vaccines targeting the H10N8 and H7N9 influenza viruses. They are not only well tolerated but also elicit a robust humoral immune response, offering a novel method for the prevention of future pandemic influenza [145].
Rabies
Rabies is a critical, acute zoonotic infectious illness responsible for around 59,000 fatalities annually. The rabies virus (RABV), consisting of structural proteins and negative-stranded RNA, is the causal agent of the disease. Glycoproteins are the principal component of RABV and serve as its primary antigen, eliciting a particular immunological response in the host. Rabies is highly virulent and exhibits a dismal prognosis once it manifests. At present, vaccination is the most efficacious method for preventing and managing the dissemination of the rabies virus (Supplementary Table 1).
Recently, CureVac initiated a proof-of-concept human clinical trial to investigate an mRNA-based rabies vaccine. CV7201 is an unmodified, yet sequence-optimized mRNA molecule encoding a rabies virus glycoprotein in which the core delivery system is the protamine complex (ClinicalTrials.gov: NCT02241135). This research was done for the first time in healthy adults, recruiting participants aged 18 to 40 years, all of whom had not received prior rabies vaccinations. The participants were delivered three doses of the CV7201 vaccine, with dosages varying from 80 to 640 μg, via either intradermal or intramuscular injection using standard syringes with needles and three different types of needle-free devices. In the latter group of vaccinated individuals, certain volunteers received a booster dosage one year afterward. The majority of participants reported mild to moderate injection-site reactions post-vaccination, while 78% exhibited systemic adverse effects, including fever, exhaustion, and pain. The trial results showed that the needle-injected vaccine failed to elicit a sufficient antibody response at various dosages and administration modes. Still, the mRNA vaccine candidate worked well to make a strong immune response against viral antigens when given through a needle-free device. The vaccination showed satisfactory safety and received adequate tolerance [146]. CureVac initiated a phase I clinical investigation (ClinicalTrials.gov: NCT03713086) to assess the safety, reactogenicity, and immunogenicity of a low-dose unmodified mRNA rabies vaccine, CV7202. This research involved 55 healthy adults aged from 18 to 40 years who received intramuscular injections of 1 μg, 2 μg, or 5 μg of CV7202 on days 1 and 29, respectively. The results showed that both the 1 μg and 2 μg doses of the CV7202 vaccine were well tolerated and caused a rabies-neutralizing antibody response in all of the people who were vaccinated, which was in line with WHO guidelines. Nonetheless, the 5 μg dose of the vaccine exhibited suboptimal reactogenicity [147].
Zika virus
The Zika virus (ZIKV), identified in 1947, is a member of the flavivirus family and transmitted via mosquitoes [148]. Recent research indicates that ZIKV is linked to congenital anomalies and neurological conditions, including microcephaly in infants and Guillain–Barré syndrome in adults [149]. Researchers led by Norbert Pardi and others have shown that nucleoside-modified mRNA-LNP vaccines targeting the pre-membrane and envelope (prM-E) glycoprotein of the 2013 ZIKV outbreak strain can produce a strong and long-lasting neutralizing antibody response after a single low-dose intradermal immunization in both mice and nonhuman primates. This shows that nucleoside-modified mRNA-LNP vaccines can quickly build up long-lasting immunity and are a good way to control ZIKV around the world [150]. The modified mRNA vaccine that Justin M. Richner et al. created effectively protects against ZIKV-related illness and can be customized to make a person less likely to get dengue virus (DENV) in future [94]. Phase I clinical trials conducted by Moderna to assess the safety and immunogenicity of mRNA-1325 (ClinicalTrials.gov: NCT03014089) and mRNA-1893 (ClinicalTrials.gov: NCT04064905) against Zika virus indicated that mRNA-1893 showed superior efficacy compared to mRNA-1325 [151]. Previous animal model studies demonstrated that the neutralizing antibody titer of mRNA-1893 was approximately 20 times greater than that of mRNA-1325 [152]. A phase II trial concerning mRNA-1893 is now in progress.
Human immunodeficiency virus (HIV)
HIV is a persistent virus that can integrate into the host cell’s DNA, remaining for the lifespan of the infected cell. HIV infection poses a significant threat to world health, with almost 2 million new adult infections annually. Consequently, the advancement of secure and efficacious HIV preventive strategies is essential to mitigating the global AIDS epidemic. Researchers have already shown that adding nucleosides to the HIV-1 envelope protein (Env) 1086 C and changing it could make the mRNA-LNP vaccine work better against cells in the serum of rabbits and rhesus monkeys after they were vaccinated [153]. Moreover, mRNA vaccines that co-express membrane-anchored HIV-1 Env and simian immunodeficiency virus (SIV) Gag proteins to generate virus-like particles (VLPs) can elicit the generation of broadly neutralizing antibodies and diminish the risk of infection in rhesus monkeys [154]. A lot of research has shown that NanoVac formulations work better than LNP vectors at lowering the amount of HIV-1 virus in the body [155]. New research shows that using germline-targeted priming-boost immunogens along with mRNA-LNP technology can create progenitors of broadly neutralizing antibodies BG18 against the V3-glycan region of HIV-1. This is an innovative way to make HIV vaccines more effective [156]. HIV mRNA vaccines have initiated numerous clinical trials (Supplementary Table 1). The results of the first phase of iHIVARNA-01’s clinical study, which was started by Judit Pich Martínez (ClinicalTrials.gov: NCT02413645), showed that iHIVARNA was safe, well tolerated, and able to cause a moderate HIV-specific T cell response [157]. The phase II clinical trial (ClinicalTrials.gov: NCT02888756) proved that iHIVARNA-01, a possible vaccine that could be used along with or instead of cART for people with chronic HIV infection, was safe [158]. The US National Institute of Allergy and Infectious Diseases finished a phase I clinical trial (ClinicalTrials.gov: NCT05217641) with healthy, uninfected adults to test the safety and immunogenicity of an HIV trimer mRNA vaccine. The results have not been made public yet. The International AIDS Vaccine Initiative paid for a separate phase I trial that looked at the safety and immunogenicity of the eOD-GT8 60-polymer mRNA vaccine (mRNA-1644) and the Core-g28v2 60-polymer mRNA vaccine (mRNA-1644v2-Core) (ClinicalTrials.gov: NCT05217641). The results have not been made public yet.
Tuberculosis
TB is an infectious disease caused by M.tb. Currently, the BCG vaccine remains the only approved anti-TB vaccine and has shown significant protective effects on infants and young children [159]. However, its effectiveness in protecting against pulmonary TB in adults is limited [160–162].
In 2004, the Tascon RE team at the National Institute for Medical Research in London, United Kingdom, conducted the first experimental study of an mRNA vaccine against TB. This study involved using nude IVT mRNA encoding the immunodominant antigen MPT83 to immunize mice (four times with an interval of 3 weeks). The researchers successfully induced a protective immune response against M.tb challenge in a short period of time [163]. In 2010, the Coelho-Castelo AA team of the University of Sao Paulo, Brazil, extracted the mRNA of Hsp65 protein from Mycobacterium leprae. They inoculated mice with a single dose of 10 μg of mRNA-Hsp65 through the nasal route. Two weeks after immunization, the mice were euthanized. The final experimental results demonstrated that this method could induce protective immune responses in the mouse TB model and reduce the bacterial load and inflammatory response in the lungs of mice [164]. In 2022, the Coler RN team at the Seattle Children’s Research Institute in the United States, along with Seattle HDT Bio biotechnology companies, reported on the use of the replication rep RNA platform encoding ID91 TB vaccine candidate antigens in preprints on the BioRxiv platform. They utilized a first-generation unstructured NLC lipid carrier to coat ID91 and develop a rep RNA TB vaccine. In a mouse model, this vaccine demonstrated medium immunogenicity and protective effects. These findings represent the first evidence that the repRNA platform is a viable system for TB vaccination [165]. In addition, immunoinformatics methods can also be utilized in the development of TB mRNA vaccines. This includes epitope screening, construction of novel vaccines, and immune simulation, all of which will contribute to the further advancement of TB mRNA vaccines [166–168]. Currently, BioNTech is actively developing an mRNA vaccine for TB, and its research and development pipeline includes several vaccine candidates. Associated research has advanced to the phase I clinical trial stage (ClinicalTrials.gov: NCT05537038 and NCT05547464). The primary objective of these trials is to assess the safety and immunogenicity of the vaccine, with no particular data on protective effectiveness disclosed to date.
Conclusion, challenges, and future prospects
The successful development of a COVID-19 vaccine has showcased the significant potential of mRNA vaccine technology for disease prevention and control, surpassing traditional vaccinations. This achievement motivates global scientists to diligently create new mRNA vaccines for both infectious and non-infectious disorders, signaling a significant transformation in disease prevention and management. We anticipate a substantial rise in mRNA vaccine candidates and further advancements in clinical trials in the forthcoming years. The distinctive benefit of mRNA vaccine technology is in its capacity to encode several antigens, facilitating the advancement of a novel class of infectious disease vaccines. This enables the prevention of various infectious illnesses with a single vaccine, significantly enhancing the practicality and efficacy of vaccination.
Notwithstanding the numerous benefits of mRNA vaccines, they encounter some obstacles throughout the research and development phase. First, the production process of mRNA vaccines faces three major technical challenges, which are the disadvantages of its application, such as the complex multi-step in vitro synthesis system, the dual threshold of equipment and technology, and the cost challenge of scale-up production. The production process involves enzymatic reactions, such as in vitro transcription of linearized plasmid DNA, 5’ capping, and polyadenylation tail modification, as well as purification steps to remove impurities [49]. This process must establish a strict aseptic reaction system and precise molecular biology control standards. In addition, specific equipment, such as bioreactors, chromatography systems, and nano-liposome encapsulation devices, is required during the production process, and each production batch needs to undergo integrity testing, which increases the technical complexity. Moreover, from laboratory scale to commercial production, the reaction efficiency may decline, and the cumulative yield loss in the purification steps can reach 40%, resulting in a significantly higher unit dose cost than recombinant protein vaccines [169].
Second, mRNA vaccines synthesize in the cytoplasm and do not interact with human DNA, but their potentially robust immunogenic response may elicit a significant type I interferon and pro-inflammatory cytokine response, heightening the risk of inflammation and autoimmune disorders, particularly in the elderly, children, pregnant women, and individuals with chronic conditions such as autoimmune diseases. Therefore, preclinical and clinical trials must thoroughly evaluate the safety of vaccinations across various groups. Furthermore, various factors (such as pH value, temperature, and ionic concentration) can affect the stability of mRNA, making it difficult to maintain a stable state. Additionally, multiple factors—including pH, temperature, and ionic concentration—contribute to the limited stability of mRNA. It is worth noting that current research is breaking through process bottlenecks through the following directions: developing freeze-dried formulations, optimizing cap analog structures, and developing continuous flow production technologies, which are expected to improve the accessibility of mRNA vaccines in future.
Regarding the safety of mRNA vaccines and the known adverse immune reactions, Gurudeeban Selvaraj et al. reported [170] that the main reason for allergic reactions caused by mRNA vaccines lies in the presence of allergenic components in the vaccines, such as glycoproteins (the amino acid residue sequence 437–508 of polyphenolase) or stabilizers (such as sucrose and polyethylene glycol). These molecules can promote or stimulate the reaction between allergen-specific antibodies and IgE in the body, thereby triggering allergic inflammatory responses [171]. Allergens may also further lead to the release of inflammatory mediators (including histamine, prostaglandins, leukotrienes, and proteases) and pro-inflammatory cytokines, thereby causing allergic reactions, which include nausea, vomiting, red skin, rash, laryngeal edema, wheezing, tachycardia, hypotension, and cardiovascular failure. Although significant reductions in the immunostimulatory effect of mRNA and enhancement of the immunogenicity of LNP were achieved through synthetic modification [172], when mRNA at a dose exceeding a certain level or certain ionizable lipids were administered in the form of LNP, it might trigger immune activation and cytotoxicity, thereby causing allergic reactions in individuals vaccinated with the vaccine. These reactions are mainly related to the lipid content and payload of LNPs. To address the immune stimulation caused by LNP mRNA, major clinical studies have focused on the use of the glucocorticoid receptor (GR) agonist dexamethasone [173] as well as corticosteroids to reduce the immune stimulation of mRNA [174]. Furthermore, an increasing number of clinical trials have confirmed the effectiveness and safety of mRNA vaccines in treating infectious diseases, providing valuable references for the development of vaccines for various diseases.
In conclusion, we should take advantage of the COVID-19 pandemic’s impetus to conduct in-depth research on the properties and potential degradation mechanisms of mRNA vaccines in order to develop safe, stable, and effective mRNA vaccines against other bacterial infectious diseases (such as tuberculosis) and make ground-breaking advances in human and public health.
Supplementary Information
Acknowledgements
Not applicable.
Author contributions
Y.L. and X.W. jointly conceptualized this scoping review. Z.Z. and J.D. and D.Z. extracted the data from the articles. Z.Z. and D.Z. cross-checked the extracted data. Z.Z. prepared the initial manuscript, Y.L., X.W. and R.H. edited the initial manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
The funders had no role in the design of the study, in the collection, analyses, or interpretation of data; in the writing of the article; or in the decision to publish the results.
Availability of data and materials
No datasets were generated or analysed during the current study.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Zhen Zhang, Jingli Du and Danyang Zhang contributed equally to this study.
Contributor Information
Rui Han, Email: shhr950822@163.com.
Xueqiong Wu, Email: xueqiongwu@139.com.
Yan Liang, Email: amy5919@sina.com.
References
- 1.Plotkin SA. Vaccines: the fourth century. Clin Vaccine Immunol. 2009;16(12):1709–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gote V, et al. A comprehensive review of mRNA vaccines. Int J Mol Sci. 2023. 10.3390/ijms24032700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shan P, et al. A new nano adjuvant of PF3 used for an enhanced hepatitis B vaccine. Front Bioeng Biotechnol. 2022;10: 903424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brenner S, Jacob F, Meselson M. An unstable intermediate carrying information from genes to ribosomes for protein synthesis. Nature. 1961;190:576–81. [DOI] [PubMed] [Google Scholar]
- 5.Sahin U, Karikó K, Türeci Ö. mRNA-based therapeutics–developing a new class of drugs. Nat Rev Drug Discov. 2014;13(10):759–80. [DOI] [PubMed] [Google Scholar]
- 6.Mohanty P, et al. Emerging perspectives on RNA virus-mediated infections: from pathogenesis to therapeutic interventions. World J Virol. 2023;12(5):242–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang L, et al. Recent advances in lipid nanoparticles for delivery of mRNA. Pharmaceutics. 2022. 10.3390/pharmaceutics14122682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pardi N, Krammer F. mRNA vaccines for infectious diseases - advances, challenges and opportunities. Nat Rev Drug Discov. 2024;23(11):838–61. [DOI] [PubMed] [Google Scholar]
- 9.Kubota T, et al. Case report: isolated, unilateral oculomotor palsy with anti-GQ1b antibody following COVID-19 vaccination. F1000Res. 2021;10: 1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang G, et al. mRNA vaccines in disease prevention and treatment. Signal Transduct Target Ther. 2023;8(1):365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen J, Chen J, Xu Q. Current developments and challenges of mRNA vaccines. Annu Rev Biomed Eng. 2022;24:85–109. [DOI] [PubMed] [Google Scholar]
- 12.Whitlow E, Mustafa AS, Hanif SNM. An overview of the development of new vaccines for tuberculosis. Vaccines (Basel). 2020. 10.3390/vaccines8040586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schrager LK, et al. The status of tuberculosis vaccine development. Lancet Infect Dis. 2020;20(3):e28–37. [DOI] [PubMed] [Google Scholar]
- 14.Karmacharya P, Patil BR, Kim JO. Recent advancements in lipid-mRNA nanoparticles as a treatment option for cancer immunotherapy. J Pharm Investig. 2022;52(4):415–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamamoto A, et al. Current prospects for mRNA gene delivery. Eur J Pharm Biopharm. 2009;71(3):484–9. [DOI] [PubMed] [Google Scholar]
- 16.Weissman D. mRNA transcript therapy. Expert Rev Vaccines. 2015;14(2):265–81. [DOI] [PubMed] [Google Scholar]
- 17.To KKW, Cho WCS. An overview of rational design of mRNA-based therapeutics and vaccines. Expert Opin Drug Discov. 2021;16(11):1307–17. [DOI] [PubMed] [Google Scholar]
- 18.Ramanathan A, Robb GB, Chan SH. mRNA capping: biological functions and applications. Nucleic Acids Res. 2016;44(16):7511–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang J, et al. Quantifying the RNA cap epitranscriptome reveals novel caps in cellular and viral RNA. Nucleic Acids Res. 2019;47(20): e130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Corbett KS, et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature. 2020;586(7830):567–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kwon H, et al. Emergence of synthetic mRNA: In vitro synthesis of mRNA and its applications in regenerative medicine. Biomaterials. 2018;156:172–93. [DOI] [PubMed] [Google Scholar]
- 22.Despic V, Jaffrey SR. mRNA ageing shapes the Cap2 methylome in mammalian mRNA. Nature. 2023;614(7947):358–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pardi N, Hogan MJ, Weissman D. Recent advances in mRNA vaccine technology. Curr Opin Immunol. 2020;65:14–20. [DOI] [PubMed] [Google Scholar]
- 24.Henderson JM, et al. Cap 1 messenger RNA synthesis with co-transcriptional cleancap(®) analog by in vitro transcription. Curr Protoc. 2021;1(2): e39. [DOI] [PubMed] [Google Scholar]
- 25.Sahin U, et al. Publisher correction: COVID-19 vaccine BNT162b1 elicits human antibody and T(H)1 T cell responses. Nature. 2021;590(7844):E17. [DOI] [PubMed] [Google Scholar]
- 26.胡勇, et al., 一种Cap2结构5′帽子类似物及其制备方法和应用. p. 15.
- 27.Carralot JP, et al. Polarization of immunity induced by direct injection of naked sequence-stabilized mRNA vaccines. Cell Mol Life Sci. 2004;61(18):2418–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weng Y, et al. The challenge and prospect of mRNA therapeutics landscape. Biotechnol Adv. 2020;40: 107534. [DOI] [PubMed] [Google Scholar]
- 29.Balzer Le S, et al. Dual UTR-a novel 5’ untranslated region design for synthetic biology applications. Synth Biol (Oxf). 2020;5(1): ysaa006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leppek K, Das R, Barna M. Functional 5’ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat Rev Mol Cell Biol. 2018;19(3):158–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karikó K, et al. Increased erythropoiesis in mice injected with submicrogram quantities of pseudouridine-containing mRNA encoding erythropoietin. Mol Ther. 2012;20(5):948–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sepac A, et al. Comparison of cardiomyogenic potential among human ESC and iPSC lines. Cell Transplant. 2012;21(11):2523–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mayr C. Regulation by 3’-untranslated regions. Annu Rev Genet. 2017;51:171–94. [DOI] [PubMed] [Google Scholar]
- 34.Matoulkova E, et al. The role of the 3’ untranslated region in post-transcriptional regulation of protein expression in mammalian cells. RNA Biol. 2012;9(5):563–76. [DOI] [PubMed] [Google Scholar]
- 35.Pelletier J, Sonenberg N. Insertion mutagenesis to increase secondary structure within the 5’ noncoding region of a eukaryotic mRNA reduces translational efficiency. Cell. 1985;40(3):515–26. [DOI] [PubMed] [Google Scholar]
- 36.Zarghampoor F, et al. Improved translation efficiency of therapeutic mRNA. Gene. 2019;707:231–8. [DOI] [PubMed] [Google Scholar]
- 37.Mayr C. What are 3’ UTRs doing? Cold Spring Harb Perspect Biol. 2019. 10.1101/cshperspect.a034728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fotin-Mleczek M, et al. Messenger RNA-based vaccines with dual activity induce balanced TLR-7 dependent adaptive immune responses and provide antitumor activity. J Immunother. 2011;34(1):1–15. [DOI] [PubMed] [Google Scholar]
- 39.Eberhardt W, et al. Modulation of mRNA stability as a novel therapeutic approach. Pharmacol Ther. 2007;114(1):56–73. [DOI] [PubMed] [Google Scholar]
- 40.Brüssow H. mRNA vaccines against COVID-19: a showcase for the importance of microbial biotechnology. Microb Biotechnol. 2022;15(1):135–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schlake T, et al. Developing mRNA-vaccine technologies. RNA Biol. 2012;9(11):1319–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gallie DR. The cap and poly(A) tail function synergistically to regulate mRNA translational efficiency. Genes Dev. 1991;5(11):2108–16. [DOI] [PubMed] [Google Scholar]
- 43.Lima SA, et al. Short poly(A) tails are a conserved feature of highly expressed genes. Nat Struct Mol Biol. 2017;24(12):1057–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holtkamp S, et al. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood. 2006;108(13):4009–17. [DOI] [PubMed] [Google Scholar]
- 45.Tang TTL, Passmore LA. Recognition of poly(A) RNA through its intrinsic helical structure. Cold Spring Harb Symp Quant Biol. 2019;84:21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu S, Kim VN. A tale of non-canonical tails: gene regulation by post-transcriptional RNA tailing. Nat Rev Mol Cell Biol. 2020;21(9):542–56. [DOI] [PubMed] [Google Scholar]
- 47.Trepotec Z, et al. Segmented poly(A) tails significantly reduce recombination of plasmid DNA without affecting mRNA translation efficiency or half-life. RNA. 2019;25(4):507–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karikó K, et al. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol Ther. 2008;16(11):1833–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rosa SS, et al. mRNA vaccines manufacturing: challenges and bottlenecks. Vaccine. 2021;39(16):2190–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sandbrink JB, Shattock RJ. RNA vaccines: a suitable platform for tackling emerging pandemics? Front Immunol. 2020;11: 608460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lundstrom K. Self-replicating RNA viruses for RNA therapeutics. Molecules. 2018. 10.3390/molecules23123310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vogel AB, et al. Self-amplifying RNA vaccines give equivalent protection against influenza to mRNA vaccines but at much lower doses. Mol Ther. 2018;26(2):446–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Geall AJ, et al. Nonviral delivery of self-amplifying RNA vaccines. Proc Natl Acad Sci U S A. 2012;109(36):14604–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brito LA, et al. A cationic nanoemulsion for the delivery of next-generation RNA vaccines. Mol Ther. 2014;22(12):2118–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bloom K, van den Berg F, Arbuthnot P. Self-amplifying RNA vaccines for infectious diseases. Gene Ther. 2021;28(3–4):117–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Blakney AK, Ip S, Geall AJ. An update on self-amplifying mRNA vaccine development. Vaccines. 2021;9(2): 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pollock KM, et al. Safety and immunogenicity of a self-amplifying RNA vaccine against COVID-19: covac1, a phase I, dose-ranging trial. EClinicalMedicine. 2022;44: 101262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Oda Y, et al. Immunogenicity and safety of a booster dose of a self-amplifying RNA COVID-19 vaccine (ARCT-154) versus BNT162b2 mRNA COVID-19 vaccine: a double-blind, multicentre, randomised, controlled, phase 3, non-inferiority trial. Lancet Infect Dis. 2024;24(4):351–60. [DOI] [PubMed] [Google Scholar]
- 59.Silva-Pilipich N, et al. Self-amplifying RNA: a second revolution of mRNA vaccines against COVID-19. Vaccines. 2024. 10.3390/vaccines12030318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Komori M, et al. Incorporation of 5 methylcytidine alleviates innate immune response to self-amplifying RNA vaccine. bioRxiv, 2023.
- 61.McGee JE, et al. Complete substitution with modified nucleotides in self-amplifying RNA suppresses the interferon response and increases potency. Nat Biotechnol. 2025;43(5):720–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Beissert T, et al. A trans-amplifying RNA vaccine strategy for induction of potent protective immunity. Mol Ther. 2020;28(1):119–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Perenkov AD, et al. In vitro transcribed RNA-based platform vaccines: past, present, and future. Vaccines. 2023. 10.3390/vaccines11101600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yin X, Li H, Zhou Y. Circular RNAs in viral infection and antiviral treatment. Cells. 2024. 10.3390/cells13232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang M, et al. Circular RNAs: a novel type of non-coding RNA and their potential implications in antiviral immunity. Int J Biol Sci. 2017;13(12):1497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu L, et al. Circular RNAs: isolation, characterization and their potential role in diseases. RNA Biol. 2017;14(12):1715–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu R, et al. CircRNA: regulatory factors and potential therapeutic targets in inflammatory dermatoses. J Cell Mol Med. 2022;26(16):4389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Maruggi G, et al. mRNA as a transformative technology for vaccine development to control infectious diseases. Mol Ther. 2019;27(4):757–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hou X, et al. Lipid nanoparticles for mRNA delivery. Nat Rev Mater. 2021;6(12):1078–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Buck J, et al. Lipid-based DNA therapeutics: hallmarks of non-viral gene delivery. ACS Nano. 2019;13(4):3754–82. [DOI] [PubMed] [Google Scholar]
- 71.Kim J, et al. Self-assembled mRNA vaccines. Adv Drug Deliv Rev. 2021;170:83–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ramishetti S, et al. A combinatorial library of lipid nanoparticles for RNA delivery to leukocytes. Adv Mater. 2020;32(12): e1906128. [DOI] [PubMed] [Google Scholar]
- 73.Samaridou E, Heyes J, Lutwyche P. Lipid nanoparticles for nucleic acid delivery: current perspectives. Adv Drug Deliv Rev. 2020;154–155:37–63. [DOI] [PubMed] [Google Scholar]
- 74.Omo-Lamai S, et al. Lipid nanoparticle-associated inflammation is triggered by sensing of endosomal damage: engineering endosomal escape without side effects. bioRxiv, 2024.
- 75.Chaudhary N, et al. Amine headgroups in ionizable lipids drive immune responses to lipid nanoparticles by binding to the receptors TLR4 and CD1d. Nat Biomed Eng. 2024;8(11):1483–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Semple SC, et al. Rational design of cationic lipids for siRNA delivery. Nat Biotechnol. 2010;28(2):172–6. [DOI] [PubMed] [Google Scholar]
- 77.Jayaraman M, et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew Chem Int Ed Engl. 2012;51(34):8529–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Akinc A, et al. A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nat Biotechnol. 2008;26(5):561–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dong Y, et al. Lipopeptide nanoparticles for potent and selective siRNA delivery in rodents and nonhuman primates. Proc Natl Acad Sci U S A. 2014;111(11):3955–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Maier MA, et al. Biodegradable lipids enabling rapidly eliminated lipid nanoparticles for systemic delivery of RNAi therapeutics. Mol Ther. 2013;21(8):1570–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Whitehead KA, et al. Degradable lipid nanoparticles with predictable in vivo siRNA delivery activity. Nat Commun. 2014;5: 4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fenton OS, et al. Synthesis and biological evaluation of ionizable lipid materials for the in vivo delivery of messenger RNA to B lymphocytes. Adv Mater. 2017. 10.1002/adma.201606944. [DOI] [PubMed] [Google Scholar]
- 83.Fenton OS, et al. Bioinspired alkenyl amino alcohol ionizable lipid materials for highly potent in vivo mRNA delivery. Adv Mater. 2016;28(15):2939–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Crommelin DJA, et al. Addressing the cold reality of mRNA vaccine stability. J Pharm Sci. 2021;110(3):997–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang X, et al. Functionalized lipid-like nanoparticles for in vivo mRNA delivery and base editing. Sci Adv. 2020. 10.1126/sciadv.abc2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li B, et al. An orthogonal array optimization of lipid-like nanoparticles for mRNA delivery in vivo. Nano Lett. 2015;15(12):8099–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hou X, et al. Vitamin lipid nanoparticles enable adoptive macrophage transfer for the treatment of multidrug-resistant bacterial sepsis. Nat Nanotechnol. 2020;15(1):41–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Conway A, et al. Non-viral delivery of Zinc Finger Nuclease mRNA enables highly efficient in vivo genome editing of multiple therapeutic gene targets. Mol Ther. 2019;27(4):866–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sabnis S, et al. A novel amino lipid series for mRNA delivery: improved endosomal escape and sustained pharmacology and safety in non-human primates. Mol Ther. 2018;26(6):1509–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Miao L, et al. Delivery of mRNA vaccines with heterocyclic lipids increases anti-tumor efficacy by STING-mediated immune cell activation. Nat Biotechnol. 2019;37(10):1174–85. [DOI] [PubMed] [Google Scholar]
- 91.Ramaswamy S, et al. Systemic delivery of factor IX messenger RNA for protein replacement therapy. Proc Natl Acad Sci U S A. 2017;114(10):E1941-e1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Finn JD, et al. A single administration of CRISPR/Cas9 lipid nanoparticles achieves robust and persistent in vivo genome editing. Cell Rep. 2018;22(9):2227–35. [DOI] [PubMed] [Google Scholar]
- 93.Ferraresso F, et al. Comparison of DLin-MC3-DMA and ALC-0315 for siRNA delivery to hepatocytes and hepatic stellate cells. Mol Pharm. 2022;19(7):2175–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Richner JM, et al. Modified mRNA vaccines protect against Zika virus infection. Cell. 2017;168(6):1114-1125.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen S, et al. Influence of particle size on the in vivo potency of lipid nanoparticle formulations of siRNA. J Control Release. 2016;235:236–44. [DOI] [PubMed] [Google Scholar]
- 96.Buschmann MD, et al. Nanomaterial delivery systems for mRNA vaccines. Vaccines (Basel). 2021. 10.3390/vaccines9010065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hassett KJ, et al. Optimization of lipid nanoparticles for intramuscular administration of mRNA vaccines. Mol Ther Nucleic Acids. 2019;15:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yang K, et al. Biodegradable lipid-modified poly(guanidine thioctic acid)s: a fortifier of lipid nanoparticles to promote the efficacy and safety of mRNA cancer vaccines. J Am Chem Soc. 2024. 10.1021/jacs.3c14010. [DOI] [PubMed] [Google Scholar]
- 99.Lou G, et al. Delivery of self-amplifying mRNA vaccines by cationic lipid nanoparticles: the impact of cationic lipid selection. J Control Release. 2020;325:370–9. [DOI] [PubMed] [Google Scholar]
- 100.Tang B, Qian Y, Fang G. Development of lipid-polymer hybrid nanoparticles for improving oral absorption of Enoxaparin. Pharmaceutics. 2020. 10.3390/pharmaceutics12070607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pardi N, et al. mRNA vaccines - a new era in vaccinology. Nat Rev Drug Discov. 2018;17(4):261–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gómez-Aguado I, et al. Nanomedicines to deliver mRNA: state of the art and future perspectives. Nanomaterials. 2020. 10.3390/nano10020364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Xu Y, et al. AGILE platform: a deep learning powered approach to accelerate LNP development for mRNA delivery. Nat Commun. 2024;15(1):6305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Witten J, et al. Artificial intelligence-guided design of lipid nanoparticles for pulmonary gene therapy. Nat Biotechnol. 2024. [DOI] [PMC free article] [PubMed]
- 105.Vander Straeten A, et al. A microneedle vaccine printer for thermostable COVID-19 mRNA vaccines. Nat Biotechnol. 2024;42(3):510–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Meng Y, et al. Cell-free expression of a therapeutic protein Serratiopeptidase. Molecules. 2023. 10.3390/molecules28073132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Munoz-Moreno R, et al. A highly stable lyophilized mRNA vaccine for Herpes zoster provides potent cellular and humoral responses. NPJ Vaccines. 2025;10(1):49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ai L, et al. Lyophilized mRNA-lipid nanoparticle vaccines with long-term stability and high antigenicity against SARS-CoV-2. Cell Discov. 2023;9(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Voigt EA, et al. A self-amplifying RNA vaccine against COVID-19 with long-term room-temperature stability. NPJ Vaccines. 2022;7(1):136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zeng C, et al. Formulation and delivery technologies for mRNA vaccines. Curr Top Microbiol Immunol. 2022;440:71–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chahal JS, et al. Dendrimer-RNA nanoparticles generate protective immunity against lethal Ebola, H1N1 influenza, and Toxoplasma gondii challenges with a single dose. Proc Natl Acad Sci U S A. 2016;113(29):E4133–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Li W, et al. West Nile virus infectious replicon particles generated using a packaging-restricted cell line is a safe reporter system. Sci Rep. 2017;7(1):3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lv P, et al. Modified-epsilon-polylysine-grafted-PEI-β-cyclodextrin supramolecular carrier for gene delivery. Carbohydr Polym. 2017;168:103–11. [DOI] [PubMed] [Google Scholar]
- 114.Wilson DR, et al. Continuous microfluidic assembly of biodegradable poly(beta-amino ester)/DNA nanoparticles for enhanced gene delivery. J Biomed Mater Res A. 2017;105(6):1813–25. [DOI] [PubMed] [Google Scholar]
- 115.Zugates GT, et al. Rapid optimization of gene delivery by parallel end-modification of poly(β-amino ester)s. Mol Ther. 2007;15(7):1306–12. [DOI] [PubMed] [Google Scholar]
- 116.Kaczmarek JC, et al. Polymer-lipid nanoparticles for systemic delivery of mRNA to the lungs. Angew Chem Int Ed Engl. 2016;55(44):13808–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Maassen SJ, van der Schoot P, Cornelissen J. Experimental and theoretical determination of the pH inside the confinement of a virus-like particle. Small. 2018;14(36): e1802081. [DOI] [PubMed] [Google Scholar]
- 118.Thaxton CS, et al. Templated spherical high density lipoprotein nanoparticles. J Am Chem Soc. 2009;131(4):1384–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Qiu Y, et al. Effective mRNA pulmonary delivery by dry powder formulation of PEGylated synthetic KL4 peptide. J Control Release. 2019;314:102–15. [DOI] [PubMed] [Google Scholar]
- 120.Hoerr I, et al. In vivo application of RNA leads to induction of specific cytotoxic T lymphocytes and antibodies. Eur J Immunol. 2000;30(1):1–7. [DOI] [PubMed] [Google Scholar]
- 121.Scheel B, et al. Immunostimulating capacities of stabilized RNA molecules. Eur J Immunol. 2004;34(2):537–47. [DOI] [PubMed] [Google Scholar]
- 122.Kallen KJ, et al. A novel, disruptive vaccination technology: self-adjuvanted RNActive(®) vaccines. Hum Vaccin Immunother. 2013;9(10):2263–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Schnee M, et al. An mRNA vaccine encoding rabies virus glycoprotein induces protection against lethal infection in mice and correlates of protection in adult and newborn pigs. PLoS Negl Trop Dis. 2016;10(6): e0004746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Papachristofilou A, et al. Phase Ib evaluation of a self-adjuvanted protamine formulated mRNA-based active cancer immunotherapy, BI1361849 (CV9202), combined with local radiation treatment in patients with stage IV non-small cell lung cancer. J Immunother Cancer. 2019;7(1):38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Liehl P, et al. Host-cell sensors for Plasmodium activate innate immunity against liver-stage infection. Nat Med. 2014;20(1):47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bergougnan C, et al. Physical and immunological barrier of human primary nasal epithelial cells from non-allergic and allergic donors. World Allergy Organ J. 2020;13(3): 100109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hua Z, Hou B. TLR signaling in B-cell development and activation. Cell Mol Immunol. 2013;10(2):103–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ablasser A, et al. Selection of molecular structure and delivery of RNA oligonucleotides to activate TLR7 versus TLR8 and to induce high amounts of IL-12p70 in primary human monocytes. J Immunol. 2009;182(11):6824–33. [DOI] [PubMed] [Google Scholar]
- 129.Kato H, Oh SW, Fujita T. RIG-I-like receptors and type I interferonopathies. J Interferon Cytokine Res. 2017;37(5):207–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rehwinkel J, et al. RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell. 2010;140(3):397–408. [DOI] [PubMed] [Google Scholar]
- 131.Goubau D, et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5’-diphosphates. Nature. 2014;514(7522):372–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Schlee M. Master sensors of pathogenic RNA - RIG-I like receptors. Immunobiology. 2013;218(11):1322–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Feng Q, et al. MDA5 detects the double-stranded RNA replicative form in picornavirus-infected cells. Cell Rep. 2012;2(5):1187–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Linares-Fernández S, et al. Tailoring mRNA vaccine to balance innate/adaptive immune response. Trends Mol Med. 2020;26(3):311–23. [DOI] [PubMed] [Google Scholar]
- 135.Pulit-Penaloza JA, Scherbik SV, Brinton MA. Activation of Oas1a gene expression by type I IFN requires both STAT1 and STAT2 while only STAT2 is required for Oas1b activation. Virology. 2012;425(2):71–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Fang E, et al. Advances in COVID-19 mRNA vaccine development. Signal Transduct Target Ther. 2022;7(1):94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Abdelzaher HM, et al. RNA vaccines against infectious diseases: vital progress with room for improvement. Vaccines. 2021. 10.3390/vaccines9111211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jackson LA, et al. An mRNA vaccine against SARS-CoV-2 - preliminary report. N Engl J Med. 2020;383(20):1920–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chu L, et al. A preliminary report of a randomized controlled phase 2 trial of the safety and immunogenicity of mRNA-1273 SARS-CoV-2 vaccine. Vaccine. 2021;39(20):2791–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Baden LR, et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N Engl J Med. 2021;384(5):403–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sahin U, et al. BNT162b2 induces SARS-CoV-2-neutralising antibodies and T cells in humans. medRxiv, 2020: p. 2020.12.09.20245175.
- 142.Polack FP, et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N Engl J Med. 2020;383(27):2603–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kremsner PG, et al. Efficacy and safety of the CVnCoV SARS-CoV-2 mRNA vaccine candidate in ten countries in Europe and Latin America (HERALD): a randomised, observer-blinded, placebo-controlled, phase 2b/3 trial. Lancet Infect Dis. 2022;22(3):329–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hekele A, et al. Rapidly produced SAM(®) vaccine against H7N9 influenza is immunogenic in mice. Emerg Microbes Infect. 2013;2(8): e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Feldman RA, et al. mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine. 2019;37(25):3326–34. [DOI] [PubMed] [Google Scholar]
- 146.Alberer M, et al. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: an open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet. 2017;390(10101):1511–20. [DOI] [PubMed] [Google Scholar]
- 147.Aldrich C, et al. Proof-of-concept of a low-dose unmodified mRNA-based rabies vaccine formulated with lipid nanoparticles in human volunteers: a phase 1 trial. Vaccine. 2021;39(8):1310–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Dick GW, Kitchen SF, Haddow AJ. Zika virus. I. isolations and serological specificity. Trans R Soc Trop Med Hyg. 1952;46(5):509–20. [DOI] [PubMed] [Google Scholar]
- 149.Pierson TC, Graham BS. Zika virus: immunity and vaccine development. Cell. 2016;167(3):625–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Pardi N, et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature. 2017;543(7644):248–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Essink B, et al. The safety and immunogenicity of two Zika virus mRNA vaccine candidates in healthy flavivirus baseline seropositive and seronegative adults: the results of two randomised, placebo-controlled, dose-ranging, phase 1 clinical trials. Lancet Infect Dis. 2023;23(5):621–33. [DOI] [PubMed] [Google Scholar]
- 152.Bollman B, et al. An optimized messenger RNA vaccine candidate protects non-human primates from Zika virus infection. NPJ Vaccines. 2023;8(1):58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Pardi N, et al. Characterization of HIV-1 nucleoside-modified mRNA vaccines in rabbits and rhesus macaques. Mol Ther Nucleic Acids. 2019;15:36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zhang P, et al. A multiclade env-gag VLP mRNA vaccine elicits tier-2 HIV-1-neutralizing antibodies and reduces the risk of heterologous SHIV infection in macaques. Nat Med. 2021;27(12):2234–45. [DOI] [PubMed] [Google Scholar]
- 155.Xu Y, et al. Short carbon nanotube-based delivery of mRNA for HIV-1 vaccines. Biomolecules. 2023. 10.3390/biom13071088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Xie Z, et al. mRNA-LNP HIV-1 trimer boosters elicit precursors to broad neutralizing antibodies. Science. 2024;384(6697): eadk0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Leal L, et al. Phase I clinical trial of an intranodally administered mRNA-based therapeutic vaccine against HIV-1 infection. AIDS. 2018;32(17):2533–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.de Jong W, et al. iHIVARNA phase IIa, a randomized, placebo-controlled, double-blinded trial to evaluate the safety and immunogenicity of iHIVARNA-01 in chronically HIV-infected patients under stable combined antiretroviral therapy. Trials. 2019;20(1):361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Adesanya OA, et al. Bacillus Calmette-guerin (BCG): the adroit vaccine. AIMS Microbiol. 2021;7(1):96–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Colditz GA. Efficacy of BCG vaccine in the prevention of tuberculosis. Meta-analysis of the published literature. JAMA. 1994;271(9):698–702. [PubMed] [Google Scholar]
- 161.Fine PE. Variation in protection by BCG: implications of and for heterologous immunity. Lancet. 1995;346(8986):1339–45. [DOI] [PubMed] [Google Scholar]
- 162.Singh AK, Netea MG, Bishai WR. BCG turns 100: its nontraditional uses against viruses, cancer, and immunologic diseases. J Clin Invest. 2021. 10.1172/JCI148291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Xue T, et al. RNA encoding the MPT83 antigen induces protective immune responses against Mycobacterium tuberculosis infection. Infect Immun. 2004;72(11):6324–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lorenzi JC, et al. Intranasal vaccination with messenger RNA as a new approach in gene therapy: use against tuberculosis. BMC Biotechnol. 2010;10:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Larsen SE, et al. An RNA-based vaccine platform for use against Mycobacterium tuberculosis. Vaccines. 2023. 10.3390/vaccines11010130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Sharma R, et al. An immunoinformatics approach to design a multi-epitope vaccine against Mycobacterium tuberculosis exploiting secreted exosome proteins. Sci Rep. 2021;11(1): 13836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Al Tbeishat H. Novel in silico mRNA vaccine design exploiting proteins of M. tuberculosis that modulates host immune responses by inducing epigenetic modifications. Sci Rep. 2022;12(1): 4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Shahrear S, Islam A. Modeling of MT. P495, an mRNA-based vaccine against the phosphate-binding protein PstS1 of Mycobacterium tuberculosis. Mol Divers. 2023;27(4):1613–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Khalid K, Poh CL. The promising potential of reverse vaccinology-based next-generation vaccine development over conventional vaccines against antibiotic-resistant bacteria. Vaccines. 2023. 10.3390/vaccines11071264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Selvaraj G, et al. Are the allergic reactions of COVID-19 vaccines caused by mRNA constructs or nanocarriers? Immunological insights. Interdiscip Sci. 2021;13(2):344–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Aalberse RC. Structural biology of allergens. J Allergy Clin Immunol. 2000;106(2):228–38. [DOI] [PubMed] [Google Scholar]
- 172.Kauffman KJ, et al. Efficacy and immunogenicity of unmodified and pseudouridine-modified mRNA delivered systemically with lipid nanoparticles in vivo. Biomaterials. 2016;109:78–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Abrams MT, et al. Evaluation of efficacy, biodistribution, and inflammation for a potent siRNA nanoparticle: effect of dexamethasone co-treatment. Mol Ther. 2010;18(1):171–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Chen S, et al. Dexamethasone prodrugs as potent suppressors of the immunostimulatory effects of lipid nanoparticle formulations of nucleic acids. J Control Release. 2018;286:46–54. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No datasets were generated or analysed during the current study.