

Abstract
Mesenchymal stem cell-derived extracellular vesicles (MSC-EVs) show potential as neuroregenerative therapies. Incorporating bioactive compounds such as neuropeptides that enhance brain-derived neurotrophic factor (BDNF) expression may amplify their therapeutic potential. We developed a clinical-scale method for loading neuropeptides into MSC-EVs, while preserving their structural integrity and therapeutic functionality. Through scalable 3D bioprocessing, we produced high-purity MSC-EVs and evaluated loading methods for encapsulating neuropeptides and full-length BDNF. EVs were characterized using electron microscopy, nanoparticle tracking analysis, and 3D STORM microscopy. The cellular uptake, distribution, and biological effects of neuropeptide-loaded MSC-EVs were tested in vitro and in vivo. Passive incubation was the optimal loading method for maintaining EV integrity while achieving effective neuropeptide encapsulation. Active loading methods destabilized the EV membrane despite higher encapsulation efficiency. Neuropeptide-loaded MSC-EVs crossed the blood–brain barrier (BBB) and significantly enhanced BDNF expression, neurogenesis, and neuroprotection in vitro, ex vivo, and in vivo. Compared with HEK293-derived extracellular vesicles (HEK-EVs), MSC-EVs demonstrated superior regenerative effects. In a photothrombotic stroke model, intranasal administration of neuropeptide-loaded MSC-EVs reduced infarct size, improved neuronal survival, and activated neuroprotective pathways mediated by Cyclic AMP Response Element-Binding protein (CREB) phosphorylation. We established a clinically scalable approach for producing neuropeptide-loaded MSC-EVs with potential as next-generation, targeted neuroregenerative therapies for treating stroke and other neurological disorders. Importantly, the EVs used in this study were produced under clinically applicable conditions and characterized according to the Minimal Information for Studies of Extracellular Vesicles (MISEV) 2023 guidelines.
Graphical abstract

Supplementary Information
The online version contains supplementary material available at 10.1186/s12951-025-03654-x.
Keywords: Mesenchymal stem cell-derived extracellular vesicles (MSC-EVs), Neuropeptide post loading, Brain-derived neurotrophic factor (BDNF), Intranasal drug delivery, Stroke recovery, Neuroregeneration, STORM super-resolution imaging, MISEV 2023 guidelines
Introduction
Mesenchymal stem cell-derived extracellular vesicles (MSC-EVs) have high potential in neuroregenerative medicine, particularly in neurogenesis and neuroprotection. The application of MSC-EVs, especially when loaded with bioactive compounds, offers a novel approach to enhance therapeutic efficacy for brain disorders. Neuropeptides, which enhance brain-derived neurotrophic factor (BDNF) expression, may augment the therapeutic benefits of MSC-EVs [1], and extracellular vesicles (EVs) can significantly increase the circulation time of neuropeptides and target their delivery to the brain.
Efforts have recently been made to enhance the efficacy of EVs by loading them with various peptides and bioactive compounds. However, the clinical applications of these EVs remain challenging. Optimal methods for loading bioactive compounds onto EVs are lacking because both preloading and post-loading methods have unique advantages and limitations [2, 3]. In clinical application, preloading methods involve genetic engineering of source cells, which can impact their characteristics and regulatory issues, whereas post-loading methods have a lower loading efficiency and can damage the EV membrane and affect their functionality.
Therefore, this study was conducted to explore methods to enhance the clinical applicability of MSC-EVs by loading neuropeptides without altering their EV characteristics, thereby increasing their potential for clinical use. We developed an EV bioprocessing platform designed using a cell non-adhesive microwell-patterned array for scalable production of MSC-EVs [4]. MSC-EV production on a scalable three-dimensional (3D)-bioprocessing method is feasible as well as reduced donor/batch variations and improved outcomes in animal models of ischemic stroke and wound injury [5, 6]. In this study, we compared the therapeutic effects of MSC-EVs and HEK293-derived extracellular vesicles (HEK-EVs) with or without neuropeptide loading. We first evaluated the loading efficiency of full-length BDNF using both passive and active loading strategies. Although active loading resulted in higher encapsulation efficiency, it caused destabilization of the EV membrane and disrupted the expression of EV potency markers. To identify biologically effective candidates, we utilized a PS-SPCL (Positional Scanning–Synthetic Peptide Combinatorial Library) screening strategy and selected ultra-short peptides (2–3 amino acids in length) that significantly upregulate BDNF expression in neuronal cells [7]. Therefore, short neuropeptides consisting of 2–3 amino acids were subsequently encapsulated only through passive loading, which minimized alterations in EV characteristics. Following loading, the physicochemical properties and therapeutic potential of the engineered EVs were comprehensively assessed [8].
We examined the changes in morphology, size, surface marker expression, and cargo composition of EVs before and after neuropeptide loading using a range of imaging and biochemical techniques. Furthermore, we evaluated the cellular uptake and biodistribution of neuropeptide-loaded EVs, as well as their impact on BDNF expression, neurogenesis, and neuroprotective effects through both in vitro and in vivo experiments.
Materials and methods
All studies involving human subjects were approved by the Institutional Review Board of the Samsung Medical Center. Wharton’s Jelly (WJ) was provided to healthy volunteers. All volunteers or their guardians provided written informed consent for participation in the study.
Preparation and isolation of clinical-scale MSC-EVs. Detailed methods are described elsewhere [6] and are provided in the Supplementary data. Schematics of the processes of EV production, isolation, and quality control are shown in Supplementary Fig. S1.
Characterization of EVs: Following the guidelines recommended by the International Society for Extracellular Vesicles (Minimal Information for Studies of Extracellular Vesicles), EVs isolated from WJ-MSC culture medium were characterized in terms of their morphology, size distribution, surface markers, purity, potency markers, efficacy, stability, and safety [9, 10].
1) Nanoparticle tracking analysis (NTA)
The particle concentration and size distribution of EVs were determined by NTA using NanoSight NS300 and Particle Metrix Zeta View® Nanoparticle Tracking Analysis (Particle Metrix, Munich, Germany) according to the manufacturer’s instructions. The samples were diluted from 1:100 to 1:10,000 in ultrapure phosphate-buffered saline (PBS). All samples were measured in duplicate using the same instrument settings. The samples were measured for size and concentration in scatter mode (488 nm laser), and the resulting videos were analyzed with Zeta View® software 8.05.11 (Particle Metrix).
2) Zeta potential characterization
The particle surface charges of the generated nanopowders, including free, cerium-substituted, and drug-loaded nanoparticles, were measured using a Zetasizer (Nano ZS, Malvern Instruments, Malvern, UK). The ZP of JAr EVs was measured three times at 25 °C under the following settings: sensitivity of 85, shutter value of 70, and frame rate of 30 frames/s. ZetaView software was used to collect and analyze the data.
3) Transmission Electron Microscopy (TEM)/Cryo-Electron Microscopy (Cryo-EM)
First, a droplet of concentrated EVs (2 μL) from each sample was placed onto Formvar carbon electron microscopy grids and incubated for 10 min at 22℃ to allow non-specific particle binding. The grids were washed with Milli-Q water. After washing, the grids were transferred to a drop of uranyl acetate solution (pH 7) for approximately 3 min. Excess fluid was removed using filter paper, and the grids were air-dried. The samples were imaged using a Jeol 1010 electron microscope (JEM-1400PLUS; Jeol, Ltd., Tokyo, Japan).
4) SDS-PAGE, Western Blotting, and Protein Concentration Measurement
Protein concentrations were determined using Protein Assay Dye Reagent Concentrate (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s instructions. For total protein analysis, 16 μL of each fraction was mixed with 4 μL of a fivefold-concentrated reducing sample buffer, boiled for 10 min, and loaded on a 10% gradient gel for SDS-PAGE. The proteins were stained with Bio-Safe Coomassie G-250 and detected using a Bio-Rad ChemiDoc imager. A Precision Plus Protein Dual Color Standards (Bio-Rad) was used as a molecular weight standard. For western blotting, proteins were separated by 10% SDS-PAGE and transferred onto nitrocellulose membranes (Bio-Rad, 1,620,112). The blots were blocked for 1 h in Tris-buffered saline (TBS) with 0.1% Tween-20 and 5% nonfat milk powder and incubated overnight with antigen-specific antibodies in TBS containing 0.1% Tween-20 and 3% bovine serum albumin powder. CD9 was detected using anti-rabbit CD9 (D801A, Cell Signaling Technology, Danvers, MA, USA, 1:1000); CD63 with anti-rabbit CD63 (E1W3, Cell Signaling Technology, 1:1000); CD81 with rabbit anti-mouse CD81 (D5O2Q, Cell Signaling Technology, 1:1000); Hsp70 with anti-rabbit Hsp70 (D69, Cell Signaling Technology, 1:1000); and GM130 with anti-rabbit GM130 (D6B1, 1:1000; AF18, Santa Cruz Biotechnology, Dallas, TX, USA, 1:1000). The primary antibodies were labeled with secondary antibodies for 2 h at 22℃ (CSB-PA644737/CSB-PA489724, CUSABIO, Houston, TX, USA; 1:2000). Horseradish peroxidase was detected using the ECL Prime Western Blotting System (RPN2232; Cytiva, Marlborough, MA, USA). Relative signal intensities were determined using the ImageJ software (NIH, Bethesda, MD, USA). All antibodies revealed specific protein signals, as determined by their appropriate molecular weights.
Discovery and functional selection of BDNF-enhancing peptides using a PS-SPCL library
To identify peptides that enhance BDNF expression, we utilized the Positional Scanning–Synthetic Peptide Combinatorial Library (PS-SPCL), a peptide library technology capable of efficiently screening peptide sequences with specific biological activity. The trimer package stock used in this study was provided by the Peptide Library Screening Facility at POSTECH (Pohang University of Science and Technology) [7]. The PS-SPCL approach involves a library of tripeptides, where the first amino acid position is varied (excluding cysteine) from alanine to tyrosine, while the second and third positions are fixed. This configuration enables systematic identification of the most effective amino acid at each position in terms of BDNF induction. Based on the screening, we initially selected 10 candidate neuropeptides and evaluated their ability to upregulate BDNF expression and promote neural regeneration. Among them, NP-8 was identified as the most potent candidate (Fig. 2) [11]. Accordingly, NP-8 was used for all subsequent EV loading and functional efficacy experiments to ensure consistency and reproducibility.
Fig. 2.

Loading of multiple cargo classes into MSC-EVs through active and passive peptide loading methods and NTA results. (A) Positional scanning synthetic peptide combinatorial library (PS-SPCL) is a synthetic peptide library used to efficiently identify peptide sequences that can bind to specific proteins or targets. (B) Amino acid sequences of loading peptides and full length-BDNF and their different drug loading strategies. (C) Conditions of drug loading, cargo encapsulation efficiency (%), and EV recovery (%) under different drug loading strategies. (D and E) Quantification and encapsulation efficiency of drugs in 1 billion EVs (Protein: 2 ug). (F) Size and distribution of drug-loaded EVs measured using NTA. Data are presented as mean ± SD (n = 3) (ns: not significant, *p < 0.05 and ****p < 0.0001)
EV-based drug delivery systems
Neuropeptides were loaded into MSC-EVs using various methods, including passive and active loading [12, 13].
1) Passive incubation
Neuropeptides were passively loaded into MSC-EVs by simple co-incubation (at 22 °C for > 18 h), allowing diffusion to facilitate loading.
2) Electroporation
Neuropeptides were diluted to a concentration of 1 μg/μL with ultrapure water to avoid aggregation at later stages. The neuropeptide was mixed with EVs diluted with 0.1-µm filtered (Nalgene Rapid-Flow; Thermo Fisher Scientific, Waltham, MA, USA) 0.1 M PBS and incubated at 22 °C for 16 h. Prepared electroporation Neon—R buffer was added to the EVs in a 1:1 ratio, and 400 µL of the sample was electroporated in 0.4-cm cuvettes by exponential pulse using an electroporation system (V750, W20, P10, Neon NxT Electroporation system, Thermo Fisher Scientific). The samples were incubated at 37 °C for 30 min, and EVs were isolated by size exclusion chromatography (SEC) [14, 15].
3) Lipofectamine and protein delivery reagents
PULsin nanoparticles were formed using PULsin protein transfection reagent (PolyPlus Transfection, Illkirch-Graffenstaden, France) following the manufacturer’s instructions. To promote complexation between PULsin nanoparticles and anionic EVs, a PULsin/protein ratio four-fold higher than the suggested PULsin/protein ratio (16 µL/1 µg) was used. A mixture of 1 × 109 EVs and 100 ng of BDNF was incubated at 37 °C for 4 h. Lipofectamine nanoparticles were formed using Lipofectamine 2000 transfection reagent (Thermo Fisher Scientific) following the manufacturer’s instructions. A mixture of 1 billion EVs and 100 ng BDNF was incubated at 22℃ for 20 min [16].
4) Removal of unloaded peptide drugs
EVs were isolated by SEC using 70-nm 500 µL qEV columns (qEVoriginal; IZON Science Ltd., Christchurch, New Zealand) collecting the 4th and 5th milliliters of the elute. The elute was concentrated to a final volume of 200–300 µL using 30-kDa spin filters (Amicon Ultra; Millipore, Billerica, MA, USA) through centrifugation at 4000 RCF [17, 18].
5) Peptide quantification after EV loading
To quantify fluorescently labeled neuropeptides, a standard curve was first established by measuring fluorescence across a range of known concentrations. Following EV loading, the fluorescence intensity of the peptide-loaded EVs was measured and used to calculate the absolute concentration of encapsulated peptides based on the standard curve. Neuropeptides were analyzed using fluorescence spectroscopy, and concentrations were determined using a standard curve beginning at 30 μg, with serial 10% dilutions down to 0.1 μg and the blank samples containing ultrapure water and PBS, using the Glomax multi detection system (Promega, Madison, WI, USA). The samples were portioned at 100 µL and evaluated using spectroscopy at excitation and emission wavelengths of 510 and 570 nm, respectively. Effective loading was calculated as a function of μg peptide per billion EV [19].
6) BDNF quantification after EV loading
To measure the amount of BDNF protein encapsulated within EVs, a commercial BDNF ELISA kit (R&D Systems, Cat# DY248) was used according to the manufacturer’s instructions.
7) Drug loading capacity and encapsulation efficiency of peptides
Encapsulation efficiency and loading efficiency were calculated according to Eqs. (1) and (2), respectively.
 |
1 |
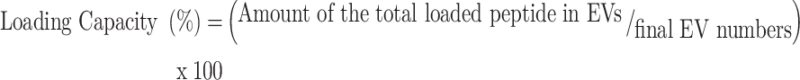 |
2 |
8) EV recovery
The final purified peptide-loaded EV solution was analyzed for EVs recovery using nanoparticle tracking analysis (NTA). EV recovery was calculated according to Eqs. (3), respectively.
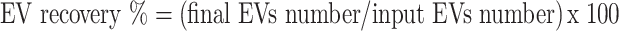 |
3 |
Characterization of exosomal stability following drug loading
1) Quantification of endogenous intraluminal miRNA expression in EVs using RT-qPCR
Total RNA, including small RNAs, was extracted using the (Qiagen miRNeasy micro kit, #217,084) according to the manufacturer’s protocol. For cDNA synthesis, 10 ng of total RNA was reverse transcribed using the TaqMan MicroRNA Reverse Transcription Kit (Cat# 4,366,596, Applied Biosystems) with miRNA-specific stem-loop primers provided in the TaqMan MicroRNA Assays. The probe sets for qPCR were purchased as pre-designed TaqMan MicroRNA Assays from Applied Biosystems (miR-27a-3p: Cat# 478384_mir, miR-92a-3p: #477827_mir, miR-132-3p:_#477,900, miR-181b-5p:_mir, 478583_mir, miR-210-3p: #477970_mir). Quantitative PCR was performed using the TaqMan Universal Master Mix II, no UNG (Cat# 4,440,040, Applied Biosystems) on a Step-One-Plus Real-Time PCR System (Applied Biosystems) under the following conditions: 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 60 s.All reactions were performed in triplicate. Expression levels were normalized to U6 snRNA, and relative quantification was calculated using the 2^–ΔΔCt method.
2) Quantitative ELISA analysis of endogenous surface markers on EV membranes
Exosomal surface markers were quantified by ELISA after lysing 2 × 10⁹ EV particles using RIPA buffer (Cell signaling, #9806). Human CD73 ELISA Kit (abcam. ab254513), Human CD81 ELISA Kit (mybiosouce, #MBS7227291), Human Syntenin ELISA Kit (ancam, ab315038), Human CD63 ELISA Kit (mybiosouce, #MBS289511).
EV labeling
EVs were labeled with PKH26 or PKH67 fluorescent dyes (Sigma-Aldrich, USA) according to the manufacturer’s instructions. The labeled EVs were then purified using ExoQuick-TC™ (System Biosciences, USA) by adding 167 µL ExoQuick solution, incubating at 4 °C for 16 h, and centrifuging at 13,000 rpm for 30 min to pellet the EVs. The EV pellet was resuspended in PBS for further use.
STORM microscopy
We performed stochastic optical reconstruction microscopy (STORM) to visualize each EV at the nanometer scale [20] for high-resolution visualization of the loaded EVs. This technique allows for precise analysis of the localization of peptides within EVs (peptides loaded into the lumen of EVs vs. those bound to the EV surface vs. free in solution), structural integrity, changes in the size/morphology of EVs, and validation of loading efficiency. A representative STORM movie is provided in the Supplementary Data 5 Movie.
1) Sample preparation for EV imaging
A glass-bottomed confocal dish was briefly immersed into 1 M aqueous potassium hydroxide (KOH) (6584–4405; DAEJUNG) solution and it was sonicated for 15 min to remove any contaminants. Then, it was then rinsed three times with distilled water and exposed to UV light for 15 min after suctioning off the remaining water. Following an additional wash with distilled water, the cleaned confocal dish was coated with poly-L-lysine (PLL) (25,988–63-0; Sigma-Aldrich) at room temperature (RT) for 30 min to enhance the adhesion of EVs. After coating, the PLL-coated dishes were washed with distilled water once. The EVs were diluted in 1:2,000 in PBS and then incubated on the PLL-coated dish for 24 h at RT. For EV staining with Cell Mask Deep Red plasma membrane stains (C10046; Thermo Fisher Scientific), EVs on the PLL-coated dish were first incubated with blocking buffer (3% [w/v] bovine serum albumin (BSA) [CNB102-0100; Cell Nest] in PBS) for 30 min at RT. After blocking, the sample was incubated in the Cell Mask Deep Red solution diluted at a ratio of 1:100,000 in PBS. Subsequently, the samples were fixed with 4% paraformaldehyde (PFA) (1574; Electron Microscopy Sciences) in PBS for 10 min at RT. After fixation, the samples were rinsed once with PBS briefly and immediately imaged in STORM imaging buffer. For EV staining with Nile Red membrane dye (415,711,000; Acros Organics), the EVs were diluted in 1:50,000 in PBS and incubated on the PLL-coated dish for 1 h at RT. After washing once briefly with PBS, the samples were stained with 10 nM Nile Red solution in PBS for 30 min and imaged using STORM.
2) STORM imaging
For STORM imaging, the samples were immersed in a buffer solution optimized for STORM imaging, which contained mercaptoethylamine (30,070; Sigma-Aldrich), 5% (w/v) glucose, and oxygen-scavenging enzymes (0.5 mg/mL glucose oxidase [G2133; Sigma-Aldrich] and 38 μg/mL catalase [C3515; Sigma-Aldrich] in PBS at pH 8.5). All STORM imaging was conducted using our custom-built STORM setup with an inverted microscope and 1.49 NA 100 × oil immersion objective lens (CFI SR HP Apo TIRF; Nikon). (Go et al., Structure, 2021; Chung et al., Scientific Reports, 2021) A 647 nm laser (OBIS; Coherent) was employed for imaging Cell Mask-labeled EVs, while a 561 nm laser (OBIS; Coherent) was used for imaging TAMRA-labeled EVs and Nile red-stained EVs. The lasers were directed through the back port of the microscope body, and the angle of incidence was aligned to achieve total internal reflection fluorescence illumination. The CRISP Autofocus system (ASI) was used to stabilize the focus by detecting the separated IR beam reflected at the sample-liquid interface. The emitted fluorescence was filtered using bandpass emission filters (LF408/488/561/635-B; Semrock) and images were recorded using an electron-multiplying charge-coupled device (EMCCD) camera (iXon Ultra 888; Andor). Under these conditions, we could observe the fast photoswitching of TAMRA and Nile red dye molecules, enabling single-molecule localization and hence super-resolution imaging of EVs (Supplementary 5 Movie). Additionally, a cylindrical lens with a focal length of 500 mm.
(LJ1144RM-A; Thorlabs) was used to introduce astigmatism for 3D STORM imaging (Center for Polymers and Composite Materials, Hanyang University, Korea). This cylindrical lens created a distortion in one axis, resulting in elliptical rather than circular point spread functions (PSFs). This distortion enabled the differentiation of axial positions of fluorescent molecules. By employing the observed astigmatism, we reconstructed 3D spatial information by analyzing the altered PSFs from the collected data. By measuring the width of the PSF in the x and y directions, respectively, caused by the cylindrical lens, we could accurately determine the z-coordinate of each molecule, leading to high-resolution 3D imaging. All of 30,000 frames of images were recorded at a frame rate of 70 Hz. To reconstruct the STORM images, the PSFs in each frame were fitted with Gaussian functions to determine the centroid positions. These collected centroids were subjected to drift correction and rendered using various parameters for the final STORM image (Chung et al., 2021; Go et al., 2021; [20].
3) Identification using DBSCAN analysis
EVs can be clearly distinguished from the background signal in STORM images owing to their confined structure and densely concentrated localizations (Jung et al., Frontiers in Cell and Developmental Biology, 2020; Lim et al., Biosensors and Bioelectonics, 2024). We identified and analyzed ~ 1000 EVs from 10 STORM images using DBSCAN analysis, and the size and localization number of each identified EVs were scatter plotted to show their heterogeneity. For DBSCAN analysis, we used the “skeleran.cluster dbscan” algorithm in Python (3.12.0). The main parameters in DBSCAN are eps (ε) and min_samples. The parameter “eps” defines the maximum distance between two data points for them to be regarded as neighbors, indicating the radius of the neighborhood around a data point. The parameter “min_samples” defines the minimum number of data points needed within a neighborhood for a data point to be classified as a core point. By setting the parameters of eps and min_samples appropriately for each STORM images, each EV was successufully identified by being differentiated from the background signal.
Treatment groups
To evaluate MSC- or HEK-EVs loaded neuropeptides (NP) or vesicle controls, six treatment groups were compared in both in vitro and in vivo analyses: MSC-EV-NP, MSC-EV, HEK-EV-NP, HEK-EV, NP, and control(PBS). For in vitro assays, EVs were applied at a concentration of 5 × 10⁸ particles/mL, NP at 500 ng/mL. For in vivo experiments, EVs were administered intranasally at a dose of 6 × 10⁸ particles per treatment, and NP was delivered at 500 ng per mouse.
In vitro studies
1) Cellular uptake assay
To assess the cellular uptake of engineered EVs and peptides, SH-SY5Y cells were seeded onto coverslips and incubated overnight for attachment. The cells were then treated with PKH67- or PKH26-labeled MSC-EVs or MSC-EV-NPs (NP8 or NP9) for 6 h at 37 °C in a humidified CO₂ incubator. To prevent spectral overlap, PKH67 was used with tetramethylrhodamine isothiocyanate (TRITC)-labeled peptides and PKH26 with fluorescein isothiocyanate (FITC)-labeled peptides. TRITC- or FITC-conjugated peptides (NP8, NP9) were passively loaded onto EVs prior to dye labeling or administered alone as controls. After incubation, cells were washed three times with PBS, fixed with 4% paraformaldehyde (PFA) for 15 min at room temperature, and mounted using Fluoroshield™ mounting medium containing DAPI (H-1200–10, VECTASHIELD® Antifade Mounting Medium). Images were acquired using a Zeiss LSM 780 confocal laser scanning microscope(Carl Zeiss, Germany), and colocalization of EV and peptide signals was analyzed using merged fluorescence channels to assess intracellular delivery efficiency. The fluorescence intensity of PKH26-labeled MSC-EVs and fluorescently tagged peptides within cells was quantified using Image J.
2) Neuritogenesis and neurogenesis Assay
SH-SY5Y human neuroblastoma cells were cultured in a 1:1 mixture of Dulbecco’s modified Eagle medium (DMEM; Biowest) and F-12 medium (Biowest), supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells were maintained at 37 °C in a humidified atmosphere containing 5% CO₂. All reagents for cell culture were obtained from HyClone (Logan, UT, USA). To induce neuritogenesis, SH-SY5Y cells were treated with 10 μM retinoic acid (RA) for 9 d, and morphological changes such as neurite elongation and branching were monitored. For neurotrophic stimulation, BDNF and NGF were supplemented at a final concentration of 100 ng/mL each, while NP was treated at 500 ng/mL. MSC-EV and MSC-EV-NP were applied at 5 × 10⁸ particles/mL. ReN VM cells (EMD Millipore) were cultured in ReNcell NSC Maintenance Medium supplemented with 20 ng/mL each of basic fibroblast growth factor (bFGF) and epidermal growth factor (EGF), on laminin-coated plates. Cells were maintained at 37 °C with 5% CO₂. For induction of neurogenesis, bFGF and EGF were withdrawn, and the cells were treated with NGF (100 ng/mL), NP (500 ng/mL), and MSC-EV or MSC-EV-NP (5 × 10⁸ particles/mL). Quantitative analysis of neurite outgrowth was performed using the NeuroCyto plugin to measure neurite length from fluorescence images of neurons stained with TUJ1. The number of neurite tips and total cell count were assessed using ImageJ.
3) Oxygen–glucose deprivation
Oxygen glucose deprivation (OGD) was evaluated on days 10–14 for SH-SY5Y cells, on fully confluent monolayers of cells, and on 14 d-old organotypic brain slice cultures. Following prior culture in glucose-containing medium, the cell culture medium was changed to Dulbecco’s modified Eagle medium without glucose, glutamine, and phenol red (catalog #A1443001, Gibco, Grand Island, NY, USA; Invitrogen, Carlsbad, CA, USA; Thermo Fisher Scientific), and flushed with a gas mixture of 95% N2 and 5% CO2 for 10 min before placing the cells or organotypic slices into a humidified hypoxia chamber (Hypoxie Glove Box HGB-090–1, Toepffer Lab Systems, Adelberg, Germany; 95% N2, 5% CO2, 1% O2, 45% humidity, 37 °C) for the 48 h intervals. OGD was terminated by the return of cells/organotypic slices to glucose-containing medium.
4) Live and dead assay
SH-SY5Y cells were seeded at a density of 5 × 104 cells per well in 96-well plates and cultured in 100 μL DMEM/F12 medium(Biowest) supplemented with 10% FBS, for 10–14 d. The cells were then treated with EVs, neuropeptides, or the vehicle control, followed by OGD stress for 6 h. Cell viability was determined after a 24 h re-oxygenation period using a CCK-8 kit (catalog #CK04, Dojindo, Kumamoto, Japan). Briefly, to each well of a 96-well plate, 10 μL CCK-8 reagent was added to the medium, and the cells were incubated for 2 h at 37 °C. The spectrophotometric absorbance of each well was determined using a Glomax Multi Detection System (Promega) at a wavelength of 450 nm.
5) In vitro BBB studies
We constructed a blood–brain barrier (BBB) model using the trans-well system, where HBEC-5i (brain endothelial cells, ATCC® CRL-2299™, Manassas, VA, USA) were cultured on a membrane insert and astrocytes were co-cultured on the opposite side of the membrane. The BBB model used two cell types to mimic the vascular wall. Initially, astrocytes were seeded at the bottom to form a monolayer, which was then inverted, and HBECs were layered on the inner surface of the trans-well. This setup allows for measurement of Trans-Endothelial Electrical Resistance (TEER) and permeability. During construction of the model, we assessed barrier integrity by measuring TEER. TEER was measured to evaluate the integrity and tightness of the endothelial barrier, with higher TEER values indicating tighter barriers. The integrity and tightness of the endothelial barrier formed a robust barrier after 4 d of culture.
In vivo studies
Animal experimental procedure
All animal experimental procedures were approved by the Institutional Animal Care and Use Committee (Laboratory Animal Research Center, AAALAC International-approved facility) of Samsung Medical Center (Seoul, Korea). C57BL/6 J mice were purchased from Orient Bio Inc. (Seongnam, Korea) and housed at a temperature of 22 ± 1 °C and a relative humidity of 55 ± 10%, under a 12 h light/dark cycle, with ad libitum access to food and water. Animals were anesthetized using a ketamine:xylazine mixture (5:1 ratio) prepared by combining ketamine (Yuhan Ketamine 50Inj.) and xylazine (Rompun Inj.). The anesthetic dose was calculated based on the animal’s body weight, and the mixture was administered intraperitoneally (i.p.). Depth of anesthesia was confirmed by the absence of reflex responses to toe pinching. Once fully anesthetized, the animal was positioned on a surgical platform, and the thoracic cavity was opened to expose the heart. A 21-gauge needle was inserted into the left ventricle, and an incision was made in the right atrium to allow for drainage. Perfusion was performed with 50 mL each of PBS and 4% PFA in PBS. After perfusion, the skull was carefully opened using surgical scissors and forceps. The brain was gently removed and post-fixed in 4% PFA at 4 °C for 24 h. Subsequently, the brain was transferred to a 30% sucrose solution for cryoprotection until it sank, indicating full infiltration.
1) Rose bengal photothrombosis mouse stroke model
Male C57BL/6 mice (7–8 weeks old) were obtained from Orient Bio (Seongnam-si, Korea). To induce photothrombosis, Rose Bengal, a photosensitive dye dissolved in PBS, was injected through the tail vein at a dosage of 0.5 mg/100 μL. To aggregate the dye, an area of 0.6 mm in diameter in the right cerebral cortex of the sensory-motor cortex was locally illuminated with green light (535 ± 25 nm, 36 mV) using an LED lamp (ZEISS, CL 6000 LED) for 3 min through a cranial window. To evaluate the neuroprotective effect of MSC-EV-NPs, two administrations were performed immediately after Rose Bengal photothrombosis modeling, and one additional administration was performed 24 h later via intranasal delivery of MSC-EV-NPs, MSC-EVs, HEK-EV-NPs, peptide only, or vehicle control (each group, n = 6). For intranasal administration, 1 × 10⁹ particles (in 8 μL total volume) were gently applied dropwise to the nostrils using a micropipette under light isoflurane anesthesia. Mice were euthanized 72 h after modeling.
2) Magnetic resonance imaging
Data were acquired on a 7.0 T PharmaScan magnetic resonance imaging scanner (Bruker, Billerica, MA, USA) with a 16-cm bore size and high-performance gradient system (570 mT/m, 130 µs rise-time). The coil used was a volume resonator (diameter = 72 mm) for transmission and mouse brain 2 × 2 phased-array coil for reception. Imaging was performed 72 h after the photothrombosis modeling using the following scanning parameters: all mice were anesthetized with 2% isoflurane while acquiring magnetic resonance images. A T2-weighted spin-echo sequence was used to acquire the images. The parameters were as follows: repetition time/echo time = 3000/60 ms, number of averages = 6, echo train length = 4; in-plane resolution = 100 × 100 μm2; slice thickness = 0.5 mm.
3) Organotypic brain slice cultures
The brains of P5–8 neonatal wild-type C57BL/6 mice were removed following animal handling protocols. The hippocampus and neocortex were dissected, and 350 μm-thick sagittal sections were cut from hippocampal and cortical tissues using a Leica VT1000S Vibrating Blade Microtome (Wetzlar, Germany). Intact sections were carefully selected under a Zeiss Stem 305 dissection microscope (Oberkochen, Germany) in anatomical medium (minimum essential medium, catalog #32,360–026) containing 1% penicillin/streptomycin and 10 mM Tris–HCl, pH 7.2 (Gibco; Invitrogen; Thermo Fisher Scientific), with the medium changed twice per week after initial seeding. The slices were incubated in 4℃ atomical medium for 30 min, and then two sections were plated on polytetrafluoroethylene membrane inserts (0.4 μm, 30 mm in diameter, Merck-Millipore, PICMORG50). The inserts were then incubated for 3 weeks in slice culture medium containing 50% HEPES-buffered minimum essential medium, 25% heat-inactivated horse serum (Merck), 25% Hank’s balanced salt solution (Gibco), and 1 mM L-glutamine (Gibco). At 6 h prior to the OGD hypoxic model experiment, the cells were pretreated with MSC-EV-NPs, MSC-EVs, peptide only, or vehicle control.
4) Fluorescent brain distribution imaging
C57BL/6 male mice were randomly assigned to experimental groups. For near-infrared fluorescence imaging, MSC-EVs were labeled using the ExoGlow-Vivo EV Labeling Kit (System Biosciences, SBI) according to the manufacturer’s protocol. A total of 6 × 10⁸ labeled EVs were intranasal(IN) administered, and whole-brain fluorescence imaging was performed 30 min post-administration using the IVIS Spectrum imaging system (PerkinElmer, MA, USA). Fluorescence signals were captured using excitation/emission wavelengths of 745/820 nm. Data were analyzed with Living Image® software (v4.7.2). For histological tracking studies, MSC-EV-NPs and MSC-EVs were labeled with PKH26 (red) or PKH67 (green) fluorescent dyes (Sigma-Aldrich, USA). Free peptide (FITC- or TRITC-conjugated) or vehicle control was also administered intranasally. At 6 h post-administration, mouse brains were collected and fixed in 4% paraformaldehyde (PFA) overnight at 4 °C, then cryoprotected in 30% sucrose solution. Brains were embedded in OCT compound (Tissue-Tek, Sakura), stored at − 80 °C, and sectioned coronally (20 μm) using a cryostat (Leica CM1950, Leica Biosystems). Sections were mounted using Fluoroshield™ mounting medium containing DAPI (ImmunoBioScience AR-6501–01), and imaged using a Zeiss LSM 780 confocal laser scanning microscope (Carl Zeiss, Germany). Images were collected under identical acquisition parameters and processed using ZEN software.
Molecular biological analysis
1) Western blotting
Total proteins from stroke mice were extracted from the lesional cortex using ice-cold RIPA buffer. Protein lysates (30 μg) were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto nitrocellulose membranes (Bio-Rad, Hercules, CA, USA). After blocking with 5% skimmed milk for 1 h at 22℃. the membranes were incubated with primary antibodies, including rabbit anti-BDNF (1:1000; AB1779SP; Sigma-Aldrich, St. Louis, MO, USA) and mouse anti-β-actin (1:1000; sc-47778; Santa Cruz Biotechnology, Dallas, TX, USA) at 4 °C overnight. After four 10 min washes in TBS with 0.1% Tween 20, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies for 2 h at 22℃ (1:2000; CSB-PA644737/CSB-PA489724; CUSABIO, Houston, TX, USA). The membranes were washed again with TBS with 0.1% Tween 20, and bands were detected using the ECL™ Prime Western Blotting System (RPN2232; Cytiva, Marlborough, MA, USA). The signal was quantified using the ImageJ (ver. 1.54d software, NIH, Bethesda, MD, USA).
2) Measurement of BDNF levels via enzyme-linked immunosorbent assay (ELISA)
Samples were extracted from the brain tissue using RIPA buffer, and assay was carried out using a Human Free BDNF Quantikine ELISA Kit (DBD00; R&D Systems, Minneapolis, MN, USA). The concentration of BDNF in brain tissues was measured according to the manufacturer’s instructions. All kits included standard proteins; therefore, the amounts of protein and EV counts were determined based on the standard curve from each kit.
3) Reverse transcription-quantitative polymerase chain reaction(RT-qPCR)
Brain sections of mouse sacrificed after I.N. administration were homogenizedand total RNA was extracted according to the manufacturer’s instructions (MN740955.50, Macherey–Nagel). 1ug of RNA was reversely transcribed to cDNA using a rever transcription kit(AccuPower® CycleScript™ RT PreMix, Bioneer). RT-qPCR was performed with CFX Opus 96 Real-time PCR system to amplify samples in triplicate. The cycle time(Ct) vaule of the CREB gene was normalized with GAPDH from the same sample. The relative transcripts were calculated using the 2 − ΔΔCt method. The sequences of the CREB primer were as follow: Forward: 5’—CCCACTGTAACCTTAGTGCAG—3’, Reverse: 5’- CCCCTGGTGCATCAGAAGAT—3’and GAPDH primer were as follow: Forward: 5’—TGATGACATCAAGAAGGTGGTGA AG—3’, Reverse: 5’—TCCTTGGAGGCCATGTAGGCCAT—3’.
4) MTT assay
To assess cell viability, the slices were incubated with 0.5 mg/mL MTT solution (Sigma-Aldrich, M2128) at 37 °C for 1 h. After incubation, the resulting formazan crystals were dissolved by adding 100 µL of dimethyl sulfoxide (DMSO; Sigma-Aldrich, D8418) and mixed thoroughly to ensure complete dissolution. The optical density (OD) of the dissolved formazan solution was measured at 570 nm using a microplate reader to quantify cell viability.
Statistical analyses
Statistical analyses were performed using the SPSS (version 24.0; SPSS, Inc., Chicago, IL, USA) and GraphPad Prism 9 (GraphPad, Inc., San Diego, CA, USA) software. The normality of the data was evaluated using the D’Agostino–Pearson test. One-way and two-way analyses of variance with Tukey’s multiple comparison test were used to analyze the three groups. Student’s t-test and Wilcoxon–Mann–Whitney U test were used for paired and unpaired analyses of the two groups, respectively. Statistical analysis results are indicated in the figure legends. Results are expressed as the mean ± standard error. Statistical significance was defined as p < 0.05.
Results
EV characterization
The number of MSC-EVs obtained from the 3D culture system was approximately 8155 ± 28 particles per cell. MSC-EVs had a typical round shape, as observed by transmission electron microscopy (TEM) (Fig. 1A). The mean particle diameter was 116.5 ± 1.4 nm, and the mode particle diameter was 113.1 ± 25.5 nm (Fig. 1B). The size and distribution of MSC-EVs were analyzed using NTA (NanoSight NS300 and ZetaView) (Fig. 1B). The total protein/particle ratio was 4.71 μg/1010 particles. In addition to MSC-EVs and HEK-EVs were included as a loading control for drug encapsulation and used in parallel experiments.
Fig. 1.

Characterization of EVs and STORM imaging. (A) Naïve EVs imaged using electron TEM. (B) Histogram representing the size distributions and concentrations of EVs using NanoSight and ZetaView. (C) EV-positive markers, including CD63, CD9, CD81, Syntenin-1, and Alix (western blotting); EV-negative markers, including GM130, Lamin A/C, calreticulin, calnexin, and CYCS (western blotting). (D and E) (Left) Representative 2D STORM images of Nile red-labeled EVs derived from MSCs (D) and HEK293 cells (E). Quantitative analysis showed the distribution of full-width half-maximum (FWHM) and localization number per vesicle. (F and G) 3D STORM images of MSC-EVs (F) and HEK-EVs (G), with middle panels showing x–y cross-sections and right panels depicting cross-sectional localization profiles (white dashed boxes), indicating hollow spherical structures. Scale bars: 100 nm
Specific contaminating proteins, including GM130, lamin A/C (nuclear), calreticulin (ER), calnexin (ER), and CYCS (mitochondria), were identified using western blotting but were not detected in the extracted MSC-EVs. We examined the expression levels of CD markers such as CD9, CD63, and CD81, as well as markers associated with the endosomal sorting complex required for transport (ESCRT) system, including Syntenin-1 and Alix, by western blotting. EVs had a higher subpopulation of CD63 + cells than of CD9 + or CD81 + cells. The exosome markers Syntenin-1 and Alix, which are associated with the ESCRT system, also exhibited high expression levels and purity (Fig. 1C).
To analyze the membrane structure of EVs, STORM imaging was performed after labeling neutral and amphipathic lipids on the EV surface with Nile red. 2D STORM imaging of MSC- and HEK- EVs revealed ring-like distributions of fluorescent spots along the vesicle membrane. MSC-EVs exhibited a broader full width at half maximum (FWHM) of 110.86 nm and a higher average number of localized fluorescent signals per vesicle, with 221 localizations, compared to HEK-EVs, which showed an FWHM of 104.86 nm and 192 localizations (Fig. 1D and E). 3D STORM imaging of Nile red-labeled EVs further revealed hollow, spherical vesicle structures with bilaterally symmetric fluorescence signals along the membrane. The peak-to-peak distance between opposing membrane signals was approximately 84 nm, reflecting consistent membrane spacing characteristic of intact vesicles (Fig. 1F and G).
Validation of drug loading of full-length BDNF protein and short sequence neuropeptides within MSC-EVs and HEK-EVs, and assessment of EV stability"
Short sequence peptides were selected using PS-SPCL technology to identify those that enhanced BDNF function (Fig. 2A). We identified four type peptides with BDNF-enhancing properties; their nucleotide sequences and drug-loading methods are shown in Fig. 2B.
To load full-length BDNF protein into MSC-EVs and HEK-EVs, various drug-loading methods were employed, including passive incubation, active loading via electroporation, and attempts to induce fusion between liposomes and EVs using Lipofectamine and a protein delivery reagent. For the loading of neuropeptides (NPs) into MSC-EVs and HEK-EVs, only passive incubation was employed. Active loading methods, such as electroporation, were excluded due to concerns regarding the preservation of the intrinsic properties and stability of EVs after drug loading. The short-sequence peptides possess physicochemical characteristics and molecular size that allow them to diffuse across the EV lipid bilayer and be encapsulated within the vesicles (Fig. 2C). NTA was conducted after drug loading for each method and cargo loading to determine the EV recovery rate and changes in EV size (Fig. 2C and F). To assess the encapsulation efficiency of each intended cargo, FITC-labeled neuropeptides were analyzed by fluorescence spectroscopy, while EV-encapsulated BDNF protein was quantified using ELISA. (Fig. 2D and E). BDNF protein exhibited low encapsulation efficiency (8.6–19.5 ng per 1 × 10⁹ EV particles), except under active loading conditions. As a large protein molecule, BDNF showed poor loading efficiency via methods other than active loading. In contrast, neuropeptides achieved high encapsulation efficiency (> 500 ng per 1 × 10⁹ EV particles) in both MSC-EVs and HEK-EVs. This may be attributed to their extremely small size typically only 2 to 3 amino acids in length which allows them to diffuse across lipid membranes. Furthermore, their inherent lipophilicity is presumed to facilitate passive transmembrane loading. Passive incubation was effective for loading short neuropeptides without significantly altering the EV characteristics, whereas active loading methods, although efficient, caused EV membrane destabilization. The active loading method (electroporation) showed high efficiency in encapsulating BDNF protein; however, it resulted in damage to both CD and potency markers (Supplementary Fig. S2). Furthermore, cryo-electron microscopy (cryo-EM) analysis demonstrated swelling and disruption of the EV lipid bilayer, consistent with the observed loss of surface and cargo markers, suggesting compromised structural integrity and limited suitability for drug encapsulation (Fig. 5D).
Fig. 5.

Characterization of EV properties following peptide loading. (A) Expression levels of five MSC-EV specific miRNAs across MSC-EVs, HEK-EV-NPs, and MSC-EV-NPs were quantified using RT-qPCR. (B) CD63 levels, an EV-specific marker, were measured in EVs using ELISA. (C and D) Electron microscopy images of EVs after peptide loading, obtained using of TEM and cryo-EM. Data are expressed as the mean ± SD (n = 5). One-way analysis of variance followed by Tukey’s multiple comparisons test was performed (**p < 0.01, ***p < 0.001, ****p < 0.0001 vs. control; ###p < 0.001, ####p < 0.001 for comparisons between MSC-EV and MSC-EV-NP)
Quantitative and qualitative results of drug loading with neuropeptides into EVs
In subsequent experiments, NPs were loaded only through passive loading to enhance the stability of MSC-EVs (Fig. 3A). Based on in vitro efficacy screening (Fig. 6), neuropeptide 8 (NP8) was selected as the representative cargo. All peptide-loading experiments shown in Figs. 3, 4, 5 were performed using NP8, and the resulting EVs are hereafter referred to as MSC-EV-NPs or HEK-EV-NPs. NPs were loaded into MSC-EVs and HEK-EVs at various concentrations, with no significant differences in the drug-loading results between the two types of EVs. To quantitatively evaluate the amount of encapsulated peptide, FITC-labeled neuropeptides were loaded into EVs and subsequently measured. For accurate concentration assessment, a standard curve was established using the fluorescently labeled peptides (Fig. 3B). Notably, the amount of NPs that could be encapsulated within 1 × 10⁹ EVs was limited, and it was difficult to encapsulate more than 580 ng of peptide. When loading was performed at various input concentrations, a substantial amount of unencapsulated residual peptide was observed at higher concentrations, resulting in reduced calculated loading efficiency (Fig. 3C and D). However, when NPs were loaded at an appropriate concentration (e.g., 500 ng input), the loading efficiency exceeded 82% (Fig. 3 D, 500 ng loading condition). Based on these results, we selected 450 ng as the optimal peptide loading dose for 1 × 10⁹ MSC-EVs. This condition was used for repeated experiments and subsequent efficacy evaluation both in vivo and in vitro. To evaluate whether the peptide encapsulation process affected the physicochemical properties of the EVs, we measured the zeta potential and particle size after peptide loading. No significant changes were observed, indicating that the surface charge and colloidal stability of the EVs were preserved throughout the loading process (Fig. 3E and F).
Fig. 3.

Loading of neuropeptides into extracellular vesicles (EVs) via passive loading into MSC-EVs. (A) Schematic of drug loading into EVs and removal of unencapsulated drug. (B) Quantified using a standard curve with fluorescent peptides. Fluorescently labeled neuropeptides were used to establish a standard calibration curve based on serial dilutions, enabling quantification of absolute peptide concentrations. (C and D) Following encapsulation into EVs, fluorescence intensity measurements were conducted, and the amount of peptide loaded into EVs was calculated by referencing the established standard curve. Encapsulation efficiency % and loading peptide capacity (concentration). Peptide loading capacity was evaluated through fluorescent quantification of FITC-labeled peptide cargo. (E and F) Zeta potential measurements can provide information on the stability and surface properties of EVs. Data are presented as mean ± SD (n = 3)
Fig. 6.

Cellular uptake and functional effects of neuropeptide-loaded MSC-EVs in SH-SY5Y cells. (A) Representative immunofluorescence images of SH-SY5Y cells treated for 24 h with FITC-labeled neuropeptides and PKH26-labeled EVs (5 × 108 particles). EVs were derived from HEK293 or MSCs and labeled with PKH26 to assess targeting efficiency. (B) Comparison of naive MSC-EVs and neuropeptide-loaded MSC-EV-NPs. Co-localization of PKH67-labeled EVs and TRITC-labeled peptides was observed, and the degree of overlap was quantified. (C) Representative images and quantitative analysis of SH-SY5Y neurite outgrowth and neuronal differentiation following NP8-MSC-EV treatment, assessed by TUJ1 (βIII-tubulin) immunostaining. Scale bar: 100 µm. (D) Western blot analysis of molecular mechanisms underlying the neurogenic effects of neuropeptides and MSC-EVs. (E) MSC-EV carriers significantly enhanced intracellular uptake of peptides and increased p-CREB expression compared to free peptide treatment. Data are presented as mean ± SD (n = 5 fields of view per group). Statistical analysis was performed using one-way ANOVA followed by Tukey’s multiple comparisons test. (**p < 0.01, ***p < 0.001, ****p < 0.0001 vs. control.)
Fig. 4.

Neuropeptide Topology Analysis by STORM Microscopy: Super-resolution STORM imaging was used to visualize the spatial distribution and intravesicular topology of neuropeptides following encapsulation, enabling high-resolution assessment of their localization within EVs. (A) Representative 3D stochastic optical reconstruction microscopy (STORM) images of TAMRA-labeled EVs and the x–y-cross-section images, Scale bars: 100 nm. (B) (Left) Size measurement of single EV in terms of full-width half-maximum (FWHM) of the localization distribution in x and y directions. (Right) Distribution of the size (FWHM) and localization number of EVs measured from STORM images. (C) Comparison of mean size and localization number of EVs labeled with Nile red and TAMRA identified by DBSCAN. Data are presented as mean ± SD (n = 3, ***p < 0.001)
In addition, STORM imaging demonstrated successful intraluminal localization of NPs within the EVs (Fig. 4A and B). Specifically, STORM images of TAMRA-labeled-NP-loaded EVs were smaller than those of Nile red-stained EVs, suggesting that NPs were inside the lipid membrane of EVs. In contrast to Nile red-stained EVs, TAMRA-labeled-NP-loaded EVs did not show a hollow structure in the x–y cross-sectional image, suggesting that NPs were loaded inside the EV lumen (Fig. 4).
Changes in EV characteristics and properties following peptide drug loading
Loading EVs with peptide drugs can alter their characteristics, making it crucial to measure these changes to ensure the efficacy and safety of EV-based drug delivery systems. To verify that the modified EVs retained their desired properties and effectively loaded therapeutic peptides, their size, morphology, surface charge, membrane integrity, cargo composition, cellular uptake, biological activity, stability, and release kinetics were measured. To evaluate the impact of the drug-loading procedure on EV composition, we assessed whether key surface markers (CD63) and representative endogenous cargo molecules (a set of five miRNAs identified as markers within MSC- EVs) were altered or lost following peptide encapsulation. The characteristics of EVs did not change after NP loading in either HEK-EVs or MSC-EVs (Fig. 5A and B). MSC-EVs exhibited higher expression levels of CD63 and five miRNAs associated with stem cell regenerative efficacy compared to HEK-EVs, as measured by ELISA and quantitative reverse transcription-polymerase chain reaction (qRT-PCR), respectively. However, the expression levels of CD63 and the five miRNAs were not affected by peptide loading. In addition, the size and morphological characteristics, as assessed by NTA, TEM, and cryo-EM, remained unchanged following NP loading (Fig. 5C and D).
In contrast, structural disruption observed in BDNF-loaded EVs, as shown in Fig. 5D. This alteration likely results primarily from the electroporation process, which is known to perturb lipid vesicle membranes. Such disruption may impair EV-mediated cellular interactions, as vesicle uptake and intracellular trafficking often rely on membrane-associated proteins and conserved surface features.
Cell uptake studies of peptide-loaded extracellular vesicles
A cellular uptake assay using FITC- or TRITC-labeled peptides demonstrated that peptide-loaded EVs were effectively internalized by SH-SY5Y cells. In contrast to unmodified MSC-EVs, which showed no detectable peptide signal, NP8-loaded EVs exhibited clear intracellular fluorescence. Notably, the co-localization of EV markers (PKH26 or PKH67) with TRITC-tagged peptides confirmed that the peptides were incorporated into EVs and delivered as EV cargo (Fig. 6B). The results were consistent for HEK-EV-NPs and NP9-loaded MSC-EVs (Fig. 6A). Co-localization of EV labeling and fluorescent peptides confirmed the incorporation of neuropeptides into EVs. Notably, FITC/TRITC-labeled neuropeptides were predominantly localized within the cytoplasm of SH-SY5Y cells. This cytoplasmic localization was observed not only for peptide NP-8 but also for NP-9 (Fig. 6B). However, given the observed increase in cyclic AMP response element-binding protein (CREB) phosphorylation at the protein level, it is likely that a portion of the peptides delivered via EVs may have modulated the expression of nuclear proteins after cytoplasmic delivery in neurons (Fig. 6E). CREB is a key transcription factor that regulates various genes in neurons, notably promoting BDNF expression. It exerts this function by binding to the cAMP response element (CRE) in the promoters of target genes, thereby enhancing transcription of genes such as those involved in BDNF–TrkB signaling pathways (Fig. 6D). These findings suggest that neuropeptides may enhance CREB phosphorylation either directly or indirectly, and further demonstrate that EV-mediated delivery improves their intracellular uptake and biological efficacy compared to free peptide administration (Fig. 6E).
Evaluation of the biological activity of neuropeptide-loaded MSC-derived EVs
Biological responses, including signaling pathways activated by the peptide, in target cells and tissues were evaluated to assess the therapeutic effect of the loaded EVs. Specifically, NPs enhancing BDNF function were assessed for their effect on BDNF expression in neurons using western blotting, and their efficacy in promoting neurogenesis was evaluated (Fig. 6C and D). NP8 and NP9 notably increased BDNF expression and activated the tropomyosin receptor kinase B (TrkB) receptor (Fig. 6D). To ensure targeted and consistent evaluation of therapeutic efficacy, we selected a single neuropeptide with the highest neuroregenerative capacity for EV loading. Neuritogenesis assays conducted in SH-SY5Y neuroblastoma cells revealed that NP-8 exhibited the most robust induction of neurite outgrowth, validating its suitability as the lead cargo candidate for subsequent encapsulation and delivery experiments (Fig. 6D). The NP8 peptide showed the strongest neurotrophic effects in SH-SY5Y neurons compared to the other NPs (Fig. 6C and D). Compared with NPs delivered without a carrier, MSC-EV-NPs showed enhanced cellular uptake and subsequently elevated BDNF expression in neurons (Fig. 6E). Notably, MSC-EV-NPs at normal (1X) and double (2X) concentrations exhibited a dose-dependent enhancement of p-CREB activation, and both doses showed higher efficacy compared to equivalent NP treatments. These results suggest that NP8, when encapsulated in MSC-EVs, promotes CREB phosphorylation more effectively than free peptide administration.
MSC-EVs loaded with BDNF-enhancing neuropeptides promote neuroregeneration and neuroprotection
We investigated whether encapsulation of the BDNF-enhancing peptide NP-8 into MSC-derived extracellular vesicles (MSC-EVs) enhances their ability to promote neuritogenesis and neurogenesis. In SH-SY5Y cells, MSC-EVs loaded with NP-8 (MSC-EV-NPs) significantly enhanced axonal outgrowth, increased the number of neurite tips, and promoted neuronal cell proliferation compared to unmodified MSC-EVs (p < 0.005; Fig. 7A and B). Furthermore, when applied to differentiated neural progenitor cells derived from ReNcell VM, MSC-EV-NPs at both standard (1X) and double (2X) concentrations induced a dose-dependent increase in neurite length and neuronal proliferation. Both doses outperformed treatment with NP or NGF alone (Fig. 7C and D). In addition, MSC-EV-NPs demonstrated superior neuroregenerative effects compared to NGF or BDNF treatment alone, indicating a synergistic potentiation conferred by EV-mediated delivery of neuropeptides. In a hypoxia-induced injury model, MSC-EV-NPs protected neurons from apoptosis and significantly enhanced cell survival (Fig. 7E and F). Compared to HEK-EV-NPs, MSC-EV-NPs exhibited superior effects on neurite outgrowth and neuronal proliferation, underscoring the intrinsic neuroregenerative capacity of MSC-derived EVs (Supplementary Fig. S3).
Fig. 7.

Neuronal Regeneration and Protection Induced by MSC-EVs Loaded with BDNF-Enhancing Neuropeptides (MSC-EV-NPs). (A and B) SH-SY5Y cells were pre-differentiated with retinoic acid (RA, 10 μM) for 9 days and then treated with either solvent (control), NGF, BDNF, MSC-EVs, or MSC-EV-NPs for 24 h. Quantification was based on measurements from 200 neurites. (A) Representative fluorescence images. (B) Quantification graphs. Scale bar: 100 µm. (C and D) Differentiated ReNcell VM cells were treated with NGF, naïve MSC-EVs, MSC-EV-NPs, or NPs for 24 h, followed by analysis of neurogenesis. (C) Representative fluorescence images. (D) Quantification graphs. Scale bar: 300 µm. (E) Live/dead assay showing neuronal survival and protective effects after pretreatment with MSC-EV-NPs followed by oxygen–glucose deprivation (OGD). Scale bar: 20 µm. Data are presented as mean ± SD (n = 5 fields of view per group). Statistical analysis was performed using one-way ANOVA followed by Tukey’s multiple comparisons test. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 vs. control; ###p < 0.001, ####p < 0.0001 for comparisons between MSC-EVs and MSC-EV-NPs.)
These in vitro findings were extended by ex vivo studies, which demonstrated additional neuroprotective effects. Mouse brain slices were cultured for 2 weeks to create a hypoxic model. Brain slices exposed to a hypoxic environment for > 12 h were used for cell viability analyses. Neuronal tissue viability, as measured by the MTT assay, indicated neuroprotective effects following treatment with MSC-EV-NPs (Fig. 8B and C). Elevated BDNF expression was further confirmed in ex vivo organotypic brain slice cultures treated with neuropeptide-loaded EVs (Fig. 8D). Compared to treatment with MSC-EVs alone, MSC-EV-NPs significantly enhanced neuronal regeneration, as indicated by increased axon length, number of neurite terminals, and neuronal proliferation, and also exhibited neuroprotective effects.
Fig. 8.

Temporal analysis of neuronal apoptosis in the ex vivo brain. (A and B) Quantitative data of MTT colorimetric assays after exposure of hippocampal fragments to OGD after MSC-EV-NP pretreatment. (C) CCK assay and BDNF expression (D). Data are expressed as the mean ± SD (n = 5). One-way analysis of variance followed by Tukey’s multiple comparisons test was performed (**p < 0.01, ***p < 0.001, ****p < 0.0001 vs. OGD(+); ###p < 0.001, ####p < 0.001 for comparisons between MSC-EV and MSC-EV-NP)
Brain targeting of neuropeptide-loaded MSC-EVs
IN administration is increasingly recognized as a promising strategy for the noninvasive delivery of therapeutic agents to the central nervous system (CNS), bypassing the BBB and enabling rapid drug absorption through the nasal mucosa. In our study, MSC-EVs delivered via the IN route effectively localized to multiple brain regions, consistent with previous findings that IN delivery results in higher brain accumulation and prolonged retention of extracellular vesicles [21].
To assess the biodistribution of EVs following intranasal administration, MSC-EV-NPs were labeled with ExoGlow™ and subjected to in vivo fluorescence imaging. No signal was detected in the brains of PBS-treated mice(Control), whereas strong near-infrared fluorescence was observed in the brains of mice treated with MSC-EV-NPs within 30 min, confirming effective brain accumulation (Fig. 9A and B). Biodistribution analysis revealed additional fluorescence signals in peripheral organs, with the lungs showing relatively higher intensity among non-brain tissues (Supplementary Fig. S4).
Fig. 9.

Brain delivery and distribution of fluorescence-labeled EVs after intranasal administration. (A) Representative IVIS images of mouse brains 30 min after IN administration of Control and ExoGlow-labeled MSC-EV-NPs. (B) Quantification of fluorescence signals in panel A. Data are expressed as the mean ± SD. Unpaired two-tailed t-test was performed (n = 3, ***p < 0.001). (C and D) Distribution of EVs in mouse brain tissue by immunohistochemistry (IHC). Representative images of localization by IHC in coronal Sects. 6 h after intranasal administration. (E) Quantification of FITC-positive cells in various brain regions. (F) Expression level of BDNF for each brain region was measured using ELISA. Data are expressed as the mean ± SD (n = 5). One-way analysis of variance followed by Tukey’s multiple comparisons test was performed (**p < 0.01, ***p < 0.001, ****p < 0.0001 vs. control; ##p < 0.01, ###p < 0.001, ####p < 0.001 for comparisons between MSC-EV and MSC-EV-NP)
To verify the targeted delivery of peptides and peptide-loaded EVs in vivo, FITC-labeled NPs or PKH26-labeled MSC-EV-NPs were administered intranasally, and their biodistribution in the brain was assessed. When intranasally administered, fluorescently tagged MSC-EV-NPs reached the olfactory bulb, frontal cortex, and hippocampus, but not when the peptide was administered alone (Fig. 9C and D). The number of FITC-positive cells significantly increased after treatment with MSC-EV-NPs compared to after treatment with NPs alone (Fig. 9E). To confirm the increase in BDNF expression following treatment with MSC-EV-NPs, in vivo analysis was conducted in the olfactory bulb, cortex, and hippocampus (Fig. 9F). BDNF expression levels were higher in the MSC-EV-NP group compared to the NP treatment group, particularly in the olfactory bulb, after intranasal administration. MSC-EV-NPs and MSC-EVs demonstrated comparable brain distribution following intranasal administration. Although low-molecular-weight compounds such as NPs hold promise for brain delivery, their therapeutic efficacy is often limited by inherent instability and low permeability. Encapsulation within MSC-EVs significantly improves their brain-targeting efficiency, underscoring the potential of MSC-EVs as a stable and effective delivery platform.
Neuroprotective effects of MSC-EV-NPs in an animal model of stroke
Finally, the effects of MSC-EV-NPs were evaluated in a mouse model of stroke because BDNF plays a crucial role in neuroprotection and recovery after acute ischemic stroke. In a photothrombotic stroke model, intranasal administration of MSC-EV-NPs enhanced BDNF expression and significantly (p < 0.0001) reduced infarct areas compared to controls (Fig. 10 B and C). Elevated BDNF expression levels were consistently observed across multiple brain regions following MSC-EV-NP administration. In MSC-EV-NP treated tissues, a concurrent increase in CREB expression and BDNF protein level was observed, suggesting a potential link between CREB activation and BDNF upregulation (Fig. 10 D and E,F). These neuroprotective effects were mediated by the phosphorylation of CREB, a key transcription factor in BDNF signaling. Notably, NPs alone failed to confer neuroprotective or regenerative effects, likely due to poor brain permeability. However, when delivered via MSC-EVs as a drug delivery system, NPs exhibited enhanced brain targeting and therapeutic efficacy, resulting in improved neuronal protection and tissue recovery in ischemic regions. Together, these findings highlight the potential of MSC-EV-NPs as a neuroprotective delivery platform that facilitates BDNF-associated recovery mechanisms.
Fig. 10.

Neuroprotective effect of neuropeptide loaded MSC-EVs in a mouse model of stroke. (A) Experimental design and timeline for animal model. (B) Representative photographs of stained coronal brain sections evaluated using magnetic resonance imaging and whole-brain at 3 d after modeling. The lack of formazan production (white tissue) indicates an infarct area with a white line. (C) Infarct volume was quantified by ImageJ software. (D and E) BDNF expression levels in the whole mouse brain. (F) Expression of CREB was measured using RT-qPCR 24 h after intranasal administration. Data are expressed as the mean ± SD (n = 5). One-way analysis of variance followed by Tukey’s multiple comparisons test was performed (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 vs. control; ##p < 0.01, ###p < 0.001 for comparisons between MSC-EV and MSC-EV-NP)
Discussion
EVs loaded with neuropeptides demonstrated consistent quality and purity. Short amino acid-based neuropeptides were efficiently loaded into MSC-EVs via passive incubation based on diffusion, preserving their structural integrity and functional activity. Additionally, MSC-EVs loaded with neuropeptides showed significantly enhanced therapeutic effects compared to neuropeptides or MSC-EVs alone. In vitro and ex vivo findings indicated that neuropeptide-loaded MSC-EVs significantly enhanced BDNF expression and promoted neurogenesis and neuroprotection against hypoxic damage. These effects were more pronounced in MSC-EVs than those in HEK-EVs, highlighting their intrinsic regenerative capacity. An in vitro BBB model confirmed the ability of peptide-loaded EVs to traverse the barrier. Complementary in vivo experiments demonstrated that intranasally administered neuropeptide-loaded MSC-EVs bypassed the BBB via the olfactory nerve pathway, enabling targeted delivery to key brain regions implicated in stroke recovery. This approach resulted in a marked reduction in infarct volume and a significant upregulation of BDNF expression, underscoring the neuroprotective potential of the EV-based delivery system. Mechanistically, these effects were mediated through phosphorylation of the CREB, thereby enhancing BDNF gene transcription and protein synthesis. Various peptides have been applied to EVs, including ischemic myocardium-targeting peptides, the neuroprotective peptide NR2B9c, and cell-penetrating peptides [22–24]. In this study, BDNF peptides were tested to enhance BDNF expression, promoting neurogenesis and providing neuroprotection against hypoxic damage. BDNF is a multifaceted neurotrophin that strongly contributes to neuronal survival, plasticity, mood regulation, cognitive abilities, and new neuron generation in the brain. BDNF, with a molecular weight of 27 kDa, was administered directly into the brain via intracerebroventricular administration. In contrast, neuropeptides can substitute for neurotrophic factors. Neuro peptide (NP8) drugs can cross the BBB because of their small size. However, neuropeptides have a short half-life in the body because they are rapidly degraded by proteases and immune-related mechanisms. This effect can be mitigated by encapsulating neuropeptides within EVs, which helps protect them from degradation and immune responses. In addition, both BDNF and BDNF peptides have low target cell delivery rates. To date, no preclinical and clinical studies have demonstrated the successful therapeutic effects of direct BDNF or BDNF peptide administration [1, 12, 15].
Our study demonstrated that neuropeptides can be controllably loaded into EVs using passive incubation. When neuropeptides were loaded at an appropriate concentration, the loading efficiency exceeded 80%. The selection of the optimal neuropeptide-loading dose influenced not only the number of neuropeptides encapsulated within the EVs but also the amount of residual neuropeptides that remained unloaded. The methods used to load drugs into EVs can vary depending on the characteristics of the cargo [8]. In this study, we demonstrated that neuropeptides were efficiently loaded into MSC-EVs using passive incubation methods, which preserved both their integrity and functional properties. Zeta potential measurements and STORM imaging confirmed that the neuropeptides were successfully loaded within the EV lumen. In contrast, active loading methods caused destabilization of the EV membrane [25].
Our results indicate that while delivering large BDNF protein molecules to the brain as therapeutic agents is challenging, short neuropeptides that perform similar functions can easily be encapsulated in EVs, even using a passive incubation method, which can serve as drug delivery vehicles to prevent proteases from targeting neuronal cells in the brain. Various nanocarriers have been developed; however, issues such as instability in vivo, particle heterogeneity during large-scale production, and side effects in human trials have led to a continued demand for biocompatible nanocarriers [26]. Among the numerous available nanocarriers, MSC-EVs were recently recognized for their numerous advantages. MSC-EVs cross the BBB and deliver neuropeptides directly to the central nervous system. Furthermore, although this study employed intranasal administration to bypass the BBB, evidence of BBB permeability is essential for future studies aiming at direct brain-targeted therapies. To demonstrate this, an in vitro BBB bilayer model was established, and it was confirmed that MSC-derived EVs possess inherent BBB permeability (Supplementary Fig. S5).
Additionally, MSC-EVs contain stem cell-derived bioactive substances, including growth factors, cytokines, and miRNAs, which confer intrinsic neuroprotective and regenerative properties [27]. The combined (“add-on”) effect of MSC-EVs, which include neuroprotective cargo (such as miRNAs), and encapsulated neuropeptides synergistically enhances neuroprotection and neurogenesis, as shown by superior neuroregenerative effects compared to loading them into HEK-EVs. The benefits of encapsulation of neuropeptides with MSC-EVs are as follows. First, our results showed that small-sized NPs can encapsulate EVs from various cell sources, protecting the neuropeptides from degradation and enhancing their stability and bioavailability (Protection). Second, MSC-EVs can either traverse the BBB or bypass it via intranasal administration, enabling targeted delivery to specific neural cells and thereby improving the precision of neuropeptide delivery (Targeted Delivery). We demonstrated that neuropeptides could be successfully delivered to the brain. Finally, our findings suggest that the synergy between MSC-EVs and neuropeptides enhances their therapeutic efficacy, promoting neural repair and regeneration (Enhanced Efficacy).
The strengths of this study include: (a) the clinical scale MSC-EV therapeutic used in this study was produced and isolated at a GMP facility, (b) all processes were performed according to the current guidelines recommended by the ISEV (International Society for Extracellular Vesicles), known as MISEV (Minimal Information for Studies of Extracellular Vesicles 2018 and 2023). [9, 10], and (c) the characteristics of EVs before and after neuropeptide loading, including the EV markers and cargoes, and STORM imaging findings, were measured.
However, this study had some limitations.
First, although our in vitro and ex vivo models of neuronal cells and BBB systems offer important preliminary data, they do not fully recapitulate the complexity of in vivo systems and human clinical settings.
Second, although the passive loading method showed acceptable EV stability and loading efficacy, long-term studies are needed because bioactive compounds may not be stably incorporated and may be released prematurely. Moreover, passive loading is inherently limited to lipophilic cargos that can diffuse across the EV lipid bilayer, and it is only effective for small and short peptides. As a result, the encapsulation of large biomolecules such as full-length BDNF necessitates the use of active loading strategies, such as electroporation. However, these methods can induce undesirable alterations in EV properties, including disruption of the lipid bilayer structure (as demonstrated by Cryo-EM imaging) and modification of MSC-EV potency marker expression, such as changes in CD marker profiles. These structural and molecular changes may compromise essential EV functions, including tissue homing ability, cellular uptake via endocytosis, and the preservation of endogenous luminal cargo (e.g., miRNAs, proteins). Moreover, active loading approaches inherently restrict the selection of cargo due to limitations imposed by molecular size, physicochemical characteristics, and structural stability, thereby narrowing the range of therapeutically viable molecules that can be encapsulated.
Third, although we demonstrated the role of BDNF and CREB phosphorylation in mediating neuroprotection and provided supporting experimental evidence, further mechanistic analysis is required. Additional research should investigate other signaling and molecular pathways involved in neurogenesis.
Finally, although we compared different loading methods, a comparative study of the methods used to remove unincorporated neuropeptides is needed, as these methods can impact not only loading efficacy but also therapeutic efficacy. Addressing these limitations is crucial for advancing the clinical application of neuropeptide-loaded MSC-EVs for the treatment of acute ischemic stroke and other neurological disorders.
Overall, our findings provide strong evidence for the therapeutic efficacy of neuropeptide-loaded MSC-EVs as a neuroregenerative medicine with high clinical applicability. Encapsulating neuropeptides into clinical-grade MSC-EVs represents a cutting-edge approach to enhance neuroprotection and neurogenesis by leveraging the unique advantages of MSC-EVs, such as their ability to cross the BBB, biocompatibility, and intrinsic regenerative properties, to overcome the limitations of neuropeptide therapy. The enhanced and consistent therapeutic effects, along with the effective delivery of bioactive compounds to the brain, make this approach highly promising for developing treatments for stroke and various neurological conditions.
Supplementary Information
Acknowledgements
This work was supported by the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea under Grant No. RS-2022-KH129441 and K-Brain Project of the National Research Foundation (NRF) funded by the Korean government (MSIT) under Grant No. RS-2024-00399320.
Abbreviations
- MSC-EVs:
Extracellular vesicles
- EVs:
Mesenchymal stem cell-derived extracellular vesicles
- HEK-EVs:
HEK293-derived extracellular vesicles
- NPs:
Neuro peptides
- BBB:
Blood-brain barrier
- BDNF:
Brain-derived neurotrophic factor
- TrkB:
Tropomyosin receptor kinase B
- CREB:
Cyclic AMP response element-binding protein
- CRE:
CAMP response element
- NGF:
Nerve growth factor
- PBS:
Phosphate-buffered saline
- TBS:
Tris-buffered saline
- FBS:
Fetal bovine serum
- PFA:
Paraformaldehyde
- FITC:
Fluorescein isothicyanate
- TRITC:
Tetramethylrhodamine isothicyanate
- OGD:
Oxygen glucose deprivation
- IN:
Intranasal
- STORM:
Stochastic optical reconstruction microscopy
- qRT-PCR:
Quantitative reverse transcription-polymerase chain reaction
- ELISA:
Enzyme-linked immunosorbent assay
- SEC:
Size exclusion chromatography
- NTA:
Nanoparticle tracking analysis
- TEM:
Transmission electron microscopy
- Cryo-EM:
Cryo-electron microscopy
- TEER:
Trans-endothelial electrical resistance
Author contributions
J.E.K. and O.Y.B. designed the study, analyzed all samples, interpreted the data, and wrote the manuscript. J.E.K. and Y.E.J. acquired and analyzed the data. E.H.K. and S.W.Y. revised the study for important intellectual content. J.E.K. and O.Y.B. reviewed the report and provided scientific advice. O.Y.B. designed and funded the study, analyzed all samples, interpreted the data, wrote, and approved the submission of the manuscript.
Funding
S&E Bio Inc. provided support for this study in the form of salaries to J.E.K., E.H.K. The funders had no role in the study design, data collection and analysis, decision to publish, or manuscript preparation. The specific roles of the authors are described in the author contributions statement.
Data availability
No datasets were generated or analysed during the current study.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Ji-Eun Kim, Ye Eun Ji have contributed equally to this work.
References
- 1.Nagahara AH, Tuszynski MH. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat Rev Drug Discov. 2011;10:209–19. 10.1038/nrd3366. [DOI] [PubMed] [Google Scholar]
- 2.Le Saux S, et al. Interest of extracellular vesicles in regards to lipid nanoparticle based systems for intracellular protein delivery. Adv Drug Deliv Rev. 2021;176: 113837. 10.1016/j.addr.2021.113837. [DOI] [PubMed] [Google Scholar]
- 3.Yang C, et al. Extracellular vesicles and their engineering strategies, delivery systems, and biomedical applications. J Control Release. 2024;365:1089–123. 10.1016/j.jconrel.2023.11.057. [DOI] [PubMed] [Google Scholar]
- 4.Cha JM, et al. Efficient scalable production of therapeutic microvesicles derived from human mesenchymal stem cells. Sci Rep. 2018;8:1171. 10.1038/s41598-018-19211-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim J, et al. Clinical-scale mesenchymal stem cell-derived extracellular vesicle therapy for wound healing. Int J Mol Sci. 2023. 10.3390/ijms24054273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Son JP, et al. Mesenchymal stem cell-extracellular vesicle therapy for stroke: scalable production and imaging biomarker studies. Stem Cells Transl Med. 2023;12:459–73. 10.1093/stcltm/szad034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim G, Kim H, Jung W, Park D, Shin M. Novel peptides for enhancing BDNF expression and pharmaceutical compositions for prevention or treatment of Alzheimer’s or Parkinson’s disease containing the same. Korean Patent KR10–2011–0050134A. Published May 13, 2011.
- 8.Kimiz-Gebologlu I, Oncel SS. Exosomes: Large-scale production, isolation, drug loading efficiency, and biodistribution and uptake. J Control Release. 2022;347:533–43. 10.1016/j.jconrel.2022.05.027. [DOI] [PubMed] [Google Scholar]
- 9.Thery C, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7: 1535750. 10.1080/20013078.2018.1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Welsh JA, et al. Minimal information for studies of extracellular vesicles (MISEV2023): from basic to advanced approaches. J Extracell Vesicles. 2024;13: e12404. 10.1002/jev2.12404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schneider EL, Craik CS. Positional scanning synthetic combinatorial libraries for substrate profiling. Methods Mol Biol. 2009;539:59–78. 10.1007/978-1-60327-003-8_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Castilla PEM, et al. Extracellular vesicles as a drug delivery system: A systematic review of preclinical studies. Adv Drug Deliv Rev. 2021;175:113801. 10.1016/j.addr.2021.05.011. [DOI] [PubMed] [Google Scholar]
- 13.Koh HB, et al. Exosome-based drug delivery: translation from bench to clinic. Pharmaceutics. 2023;15:2042. 10.3390/pharmaceutics15082042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Z, et al. Encapsulation and assessment of therapeutic cargo in engineered exosomes: a systematic review. J Nanobiotechnol. 2024;22:18. 10.1186/s12951-023-02259-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Man F, et al. Engineered small extracellular vesicles as a versatile platform to efficiently load ferulic acid via an “esterase-responsive active loading” strategy. Front Bioeng Biotechnol. 2022;10:1043130. 10.3389/fbioe.2022.1043130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang L, et al. Research advances of engineered exosomes as drug delivery carrier. ACS Omega. 2023;8:43374–87. 10.1021/acsomega.3c04479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zeng H, et al. Current strategies for exosome cargo loading and targeting delivery. Cells. 2023;12:1416. 10.3390/cells12101416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang H, et al. Exosomes as smart drug delivery vehicles for cancer immunotherapy. Front Immunol. 2023;13:1093607. 10.3389/fimmu.2022.1093607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jeyaram A, et al. Enhanced loading of functional miRNA cargo via pH gradient modification of extracellular vesicles. Mol Ther. 2020;28:975–85. 10.1016/j.ymthe.2019.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods. 2006;3:793–5. 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Betzer O, Perets N, Angel A, Motiei M, Sadan T, Yadid G, Offen D, Popovtzer R. In vivo neuroimaging of exosomes using gold nanoparticles. ACS Nano. 2017;11:10883–93. 10.1021/acsnano.7b04495. [DOI] [PubMed] [Google Scholar]
- 22.Haroon K, et al. Engineered exosomes mediated targeted delivery of neuro protective peptide NR2B9c for the treatment of traumatic brain injury. Int J Pharm. 2024;649: 123656. 10.1016/j.ijpharm.2023.123656. [DOI] [PubMed] [Google Scholar]
- 23.Nebogatova J, et al. A method for using cell-penetrating peptides for loading plasmid DNA into secreted extracellular vesicles. Biomolecules. 2023. 10.3390/biom13121751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yin T, et al. Exosome-based WTAP siRNA delivery ameliorates myocardial ischemia-reperfusion injury. Eur J Pharm Biopharm. 2024;197: 114218. 10.1016/j.ejpb.2024.114218. [DOI] [PubMed] [Google Scholar]
- 25.Armstrong JP, Holme MN, Stevens MM. Re-engineering extracellular vesicles as smart nanoscale therapeutics. ACS Nano. 2017;11:69–83. 10.1021/acsnano.6b07607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tenchov R, et al. Exosomes—nature’s lipid nanoparticles, a rising star in drug delivery and diagnostics. ACS Nano. 2022;16:17802–46. 10.1021/acsnano.2c08774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang ZG, Buller B, Chopp M. Exosomes – beyond stem cells for restorative therapy in stroke and neurological injury. Nat Rev Neurol. 2019;15:193–203. 10.1038/s41582-018-0126-4. [DOI] [PubMed] [Google Scholar]
- 28.Chung, J., et al., 2021. Super-resolution imaging of platelet-activation process and its quantitative analysis. Scientific Reports. 11, 10511. 10.1038/s41598-021-89799-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Go, S., et al., 2021. Super-resolution imaging reveals cytoskeleton-dependent organelle rearrangement within platelets at intermediate stages of maturation. Structure. 29, 810–822.e3. 10.1016/j.str.2021.06.001 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No datasets were generated or analysed during the current study.