

Abstract
X-linked hydrocephalus, MASA syndrome and certain forms of X-linked spastic paraplegia and agenesis of corpus callosum are now known to be due to mutations in the gene for the neural cell adhesion molecule L1 (19, 30). As a result, these syndromes have recently been reclassified as CRASH syndrome, an acronym for Corpus callosum hypoplasia, Retardation, Adducted thumbs, Spasticity and Hydrocephalus (8). A comparison of existing case reports with molecular genetic analysis reveals a striking correlation between the type of mutation in the L1CAM gene and the severity of the disease. Mutations that produce truncations in the extracellular domain of the L1 protein are more likely to produce severe hydrocephalus, grave mental retardation or early death than point mutations in the extracellular domain or mutations affecting only the cytoplasmic domain of the protein. While less severe than extracellular truncations, point mutations in the extracellular domain do produce more severe neurologic problems than mutations in just the cytoplasmic domain.
Keywords: X-linked hydrocephalus, MASA syndrome, L1CAM
Introduction
L1 is a member of the immunoglobulin (Ig) superfamily of cell adhesion molecules (CAMs), and plays an important role in axon outgrowth, fasciculation and neural migration (15, 32). It is a transmembrane glycoprotein of approximately 200 kDa with six Ig-like domains followed by five fibronectin type III domains, a single-pass transmembrane region and a cytoplasmic domain (Fig. 1). Neural L1 is located on the surface of long axons and on growth cones. Schwann cells and a few other cell types, generally of neural crest origin, also express L1. The gene for human L1CAM is located in the Xq28 region of the X chromosome, and its cDNA has a very high degree of homology to mouse L1, including a completely conserved cytoplasmic domain (13).
Fig. 1.
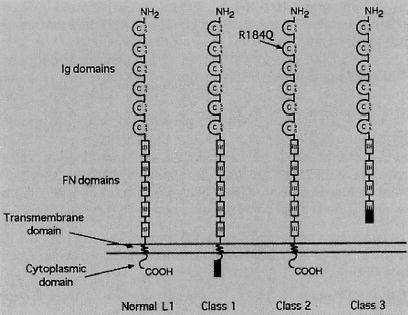
Structure of L1. Normal L1 has 6 Ig domains, 5 fibronectin (FN) type III domains, a short transmembrane region and a 114 amino acid cytoplasmic domain. Class 1 mutations disrupt only the cytoplasmic domain. Class 2 mutations, such as an arginine (R) to glutamine (Q) conversion (37), alter the structure of the extracellular domain. Class 3 mutations result in a stop codon that causes a truncation in the extracellular domain.
The first report of a mutation in the L1CAM gene associated with a neurologic disorder (X-linked hydrocephalus [XLH]) (24) appeared in 1992. Since then, 80 human L1CAM gene mutations have been discovered in patients with either X-linked hydrocephalus (McKusick 307000), MASA syndrome (McKusick 303350), X-linked spastic paraplegia syndrome (SPG1) (McKusick 312900) and X-linked agenesis of the corpus callosum (ACC) (3, 21, 30) X-linked hydrocephalus was originally described by Bickers and Adams in 1949 and attributed to aqueductal stenosis. The syndrome was later characterized by hydrocephalus, mental deficiency, spastic paraplegia and flexion deformity of the thumbs (2). In 1974, Bianchine and Lewis reported a novel X-linked disorder characterized by mental retardation, aphasia, shuffling gale, and adducted thumbs (1). They proposed the acronym “MASA” to name the syndrome. Based on the extensive genetic studies of Kenwrick and associates (17–19, 24) and Willems and colleagues (4, 9, 29, 30), it is clear that these syndromes should be considered as a single clinical entity with varying severity, all resulting from mutations in the L1CAM gene. Willems and his associates have proposed a new terminology to encompass this disorder. “CRASH syndrome” is an acronym for Corpus callosum hypoplasia, Retardation, Adducted thumbs, Spastic paraplegia and Hydrocephalus (8).
While the linkage between mutations in the L1CAM gene and CRASH syndrome is now well established, it is still unclear whether different types of mutations influence the severity of the disease. L1 is known to have both cell signaling and adhesive functions. L1 also binds to several different partners. Therefore, it is likely that mutations that result in null expression of L1 might have a different phenotype than mutations that change only a single amino acid out of the approximately 1400 in an otherwise intact protein. We have reviewed clinical descriptions of 129 individuals from 34 families in which mutations in the L1CAM gene are known from molecular studies and have correlated the severity of the disease with the type of L1CAM mutation.
Methods
Presently 75 different mutations have been detected in 80 families, including three mutations found in two or three unrelated families (as of 1–30–97). These are listed on the L1 World Wide Web site maintained by Willems and associates (http://hgins.uia.ac.be/dnalab/11.html). Thirty-four families having known mutations in L1CAM (4, 7–10, 12, 16–20, 24, 25, 28–30) have been described in the clinical literature (all languages), detailing a total of 129 individuals. Sufficient information is available to analyze severity of hydrocephalus, survival after the first year, degree of mental retardation, and presence or absence of adducted thumbs.
Classification schemes for clinical signs
Hydrocephalus was categorized into three basic groups: severe, moderate or absent. Severe hydrocephalus was defined as ventricular enlargement that required ventriculo-peritoneal shunting (VP shunt), CSF drainage, hydrocephalus that was diagnosed in utero, or described as progressive hydrocephalus or having aqueductal stenosis. Patients whose CT scan or MR images revealed ventricular dilatation with normocephaly or slight macrocephaly with no shunting procedure were classified as having moderate hydrocephalus. Patients with normal head circumference who were not described as being hydrocephalic or who showed no ventricular dilatation by imaging studies were defined as “absent”. Many of the patients in this later category have no published CT or MRI images so it is possible that some might have enlarged ventricles. A total of 129 patients were analyzed.
Mental retardation was analyzed for 79 patients that survived for more than one year and had sufficient description to make categorization possible. Mental retardation was classified into two groups; grave (IQ below 50) and modest (IQ above 50). These descriptors were chosen to avoid confusion with the more standard terms of mild, moderate, severe and profound which we were not able to use due to the nature of the clinical descriptions. When IQ was not specifically mentioned, it was estimated based on the clinical description.
To define survivorship, patients were divided into two groups based on whether they died within the first year or not. Five individuals in which the pregnancies were intentionally terminated were excluded but fetal deaths or spontaneous abortions were included as death within one year. A total of 124 patients were analyzed.
The presence or absence of adducted thumbs was analyzed in 111 of the 129 patients. Eighteen patients were excluded because of a lack of description.
Agenesis of the corpus callosum is commonly found in CRASH syndrome and the corticospinal tract is also often diminished or absent. However, not enough pathological and radiological studies have been performed at this time to permit meaningful correlations with L1CAM mutations. For example, no pathological studies of the medulla have been published on patients with class 1 mutations, apparently because they are very likely to survive beyond 1 year of age. Spastic paraplegia is noted for all CRASH patients but it is rarely described in a way that would permit categorization as severe or modest. For that reason we did not correlate this with the different classes of L1CAM mutations.
Classification of mutations
L1CAM mutations can be arranged into three groups based on how they would affect the expression or structure of the protein (Fig. 1). The first class of mutation includes missense mutations, nonsense mutations, frame shifts, duplications and deletions that affect only the cytoplasmic domain of L1 at the C-terminal end of the protein. Thirty-four patients from five families (18, 23, 29, 30) with different mutations were analyzed in this class. The second class consists of missense point mutations in the extracellular domain. Sixty-nine patients from 18 families in this category have published descriptions (2, 3, 12, 18, 19, 21, 25, 27, 29). The third class of mutation includes nonsense or frame shift mutations that produce a premature stop codon in the extracellular domain of L1. This would result in the L1 failing to remain associated with the cell surface due to loss of its transmembrane domain. Twenty-six patients from 11 families with different mutations were analyzed in this group (4, 12, 18, 19, 24, 28).
Statistics
Categorical modeling methods (11) were used to determine if observed differences between classes were significant, using SAS/STAT, release 6.08 (SAS Institute, Inc.). The analysis determines a chi-sq. value which tests the equality of proportions simultaneously for all classes. Patients for which a particular sign was “not described” were excluded from the analysis. Single degree of freedom tests were used to compare the classes after the initial analysis confirmed there were significant differences at the p = 0.0167 level.
Results
The basic findings are summarized in Figure 2. While individuals with varying degrees of severity for different symptoms can be found in each of the mutation classes, clear differences appear between the three classes. Class 3 mutations that lead to a truncation in the extracellular domain are much more likely to produce a severe form of CRASH syndrome than point mutations of the extracellular domain (class 2) or mutations of the cytoplasmic domain (class 1). Class 2 mutations also are more likely to have severe consequences than class 1 mutations.
Fig. 2.
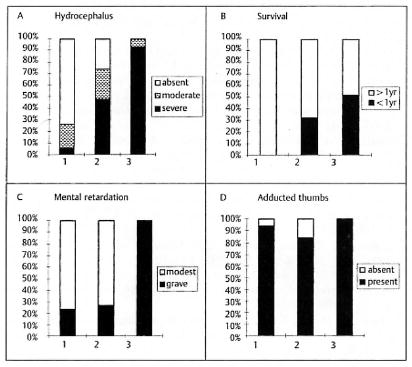
Correlation of class of L1CAM mutation with clinical signs. The percentage of patients with varying degrees of hydrocephalus, survival, mental retardation and presence of adducted thumb are show. Class 1 (1). class 2 (2) and class 3 (3) mutations are defined in the text and illustrated in Figure 1.
Hydrocephalus showed a significant (p < 0.0001) variation between the three classes. Among 34 patients in class 1, 6% showed severe hydrocephalus, 21 % had moderate hydrocephalus, 73% did not have hydrocephalus. Of 69 patients in class 2, 48 % showed severe hydrocephalus, 26 % moderate hydrocephalus and 26 % had no hydrocephalus. Twenty-four patients (92 %) with class 3 mutations showed severe hydrocephalus, and two patients had moderate hydrocephalus. Pairwise comparison confirmed class 1 was significantly different from class 2 (p < 0.0001) and class 2 was significantly different from class 3 (p < 0.0001).
The likelihood of survival showed a similar pattern. In class 1, all patients survived more than a year. For patients in class 2, 32 % died within the first year of life while 50 % of class 3 patients died within the first year. Class 1 showed significantly better survival than both class 2 and class 3 (p < 0.0001), however class 2 was not different from class 3 at the p = 0.01 level (p = 0.0268).
Mental retardation showed significant variation between the three classes. Of 34 patients in class 1, 24 % showed grave mental retardation, while 76% had modest retardation. In class 2 there were 37 patients and 27 % showed grave mental retardation while all 8 patients in class 3 showed grave mental retardation. Statistical analysis showed that class 3 was significantly different from class 1 and 2 (p < 0.0001) but that class 1 and class 2 were not different.
Adducted thumbs showed much less differences between the three groups. In classes 1, 2 and 3, 94, 84 and 100% of the patients presented with adducted thumbs, respectively.
Discussion
L1 is a cell adhesion molecule that promotes cell migration and axon growth. Part of this activity is accounted for by L1 binding to ligands on adjacent cells. Known ligands include L1 itself, two other Ig superfamily adhesion molecules TAG-1/axonin-1 and F3/F11, a proteoglycan called phosphacan, and an integrin, αvβ3 (15). L1-L1 binding is mediated by the 2nd Ig domain (36) while the other binding partners are likely to bind with other regions of L1. L1 also promotes axon growth by acting as a signaling molecule, altering the activities of kinases such as the casein kinase II (34), p90rsk (33) and FGFr (6). Finally the L1 cytoplasmic domain binds the cytoskeleton via ankyrin (5). The multifunctional aspect of L1 most likely accounts for the variation in severity associated with different L1CAM mutations.
Class 3 mutations would be predicted to be the most severe since they would result in a loss of all types of L1-mediated adhesion, whether L1 was binding to L1 or another partner such as axonin-1, phosphacan or an integrin. Mutations of this class would also lose signals generated by L1-associated kinases as well as interactions with the cytoskeleton via ankyrin. This could be a devastating loss where L1 plays a crucial role in axon growth, perhaps at important choice points such as the corticospinal decussation at the pyramids. It is possible that class 3 mutations could result in the production of a secreted form of L1 that might have some deleterious consequence, but at this time there is no data to support this form of L1 secretion in CRASH syndrome.
Class 2 mutations could represent a more subtle form of disruption. A point mutation might disrupt one form of L1 adhesion, such as L1-L1 binding but preserve others, such as L1-integrin binding (37). Since L1-L1 binding clearly generates signals that alter intracellular, second messengers, point mutations in the extracellular domain could alter this signaling capacity but perhaps in a quantitative rather than a qualitative manner.
Class 1 mutations would be expected to disrupt L1-mediated signaling and cytoskeleton interactions but not necessarily L1-mediated adhesion. It has been shown that even if over 110 amino acids from the cytoplasmic domain of L1 are deleted, L1 can still be expressed on the cell surface and mediate adhesion that is indistinguishable from full-length L1 (31).
In the early literature, X-linked hydrocephalus was thought to be due to stenosis of the aqueduct of Sylvius. Subsequently it has been hypothesized that the reduction of aqueduct caliber was produced secondarily by the compression from the enlarged lateral ventricles caused by communicating hydrocephalus (22, 35). The degree and progression of ventricle dilatation in CRASH syndrome is highly variable and ranges from high pressure progressive hydrocephalus to arrested hydrocephalus or no ventricular dilatation. Our analysis shows that loss of cell surface expression of L1 correlates with a very high incidence of severe hydrocephalus. It is still unclear what accounts for this but it is likely to be due to a loss of adhesion rather than just a loss of signaling since mutations of only the cytoplasmic domain are much less likely to cause severe hydrocephalus. The very low survival of class 3 patients and their more grave retardation is also likely due to a loss of L1-mediated adhesion.
The poor survival of patients with class 3 mutations, along with the high incidence of severe hydrocephalus and grave mental retardation in class 3, raises the question as to whether these three signs are interdependent. A patient-by-patient analysis indicates that 96 % patients with moderate or no hydrocephalus survive the first year but only 44% of patients with severe hydrocephalus survive the first year, indicating a strong association between severe hydrocephalus and early mortality. This difference was highly significant (p < 0.0001). The linkage between severe hydrocephalus and severe mental retardation is also strong (p < 0.0001) when compared with no or moderate hydrocephalus. However, 21% of the patients with moderate or no hydrocephalus also have grave mental retardation, suggesting that some additional factor, perhaps failure in the establishment of proper neural connections, causes grave mental retardation in these patients. This analysis does not permit us to conclude that severe hydrocephalus causes poor survival or severe mental retardation but indicates there is a common causal factor.
Adducted thumbs is a specific finding commonly associated with CRASH syndrome (26). The adduction appears to result from loss of innervation of the extensor pollicis longus (EPL) and not loss of input to the appropriate spinal motor neuron pool (14). The fact that nearly all class 1 patients had this sign suggests the adducted thumbs are caused by dysfunction of the cytoplasmic domain, either some signaling function or an essential interaction with the cytoskeleton.
The inter- and intrafamilial variability in phenotype in families with L1CAM mutations has been thought to be wide (10) and our analysis of interfamilial variability is consistent with this. However the intrafamilial variability has been overestimated. There are three families (25, 29) with substantial intrafamilial variability. But many other families (2, 12, 18, 19, 24, 30) show a fairly consistent phenotype. This raises the possibility that intrafamilial variation may depend on genetic background or some other non-L1-associated factor.
Conclusions
CRASH syndrome is a complex clinical entity that shows variation in severity between different affected families and even within given families. A careful review of the literature shows, however, that a strong correlation exists between the type of mutation in the L1CAM gene and tire severity of the disease. This information should help focus future laboratory studies on the molecular causes of different aspects of CRASH syndrome as well as help clinicians make informed decisions and recommendations to families that carry mutations in the L1CAM gene.
Acknowledgments
The authors thank M. L. Hlavin and K. Long for helpful comments on the manuscript. This work was supported by grants from MINDS and NEI to V. Lemmon.
Footnotes
Vance Lemmon, Ph. D. Dept. of Neurosciences, Case Western Reserve University, 2109 Adelbert Rd. Cleveland, OH, 44106-4975 USA
References
- 1.Bianchine JW, Lewis RC. The MASA syndrome: a new heritable mental retardation syndrome. Clin. Genet. 1974;5:298–306. doi: 10.1111/j.1399-0004.1974.tb01697.x. [DOI] [PubMed] [Google Scholar]
- 2.Bickers DS, Adams RD. Hereditary stenosis of the aqueduct of Sylvius as a cause of congenital hydrocephalus. Brain. 1949;72:246–262. doi: 10.1093/brain/72.2.246. [DOI] [PubMed] [Google Scholar]
- 3.Boyd E, Schwartz C, Schroer R, May M, Shapiro S, Arena et al J. Agenesis of the corpus callosum associated with MASA syndrome. Clin Dsymorphology. 1933;2:332–341. [PubMed] [Google Scholar]
- 4.Coucke P, Vits L, Van Camp G, Serville F, Lyonnet S, Kenwrick et al S. Identification of a 5′ splice site mutation in intron 4 of the L1Cam gene in an X-linked hydrocephalus family. Hum. Mol. Genet. 1994;3:671–673. doi: 10.1093/hmg/3.4.671. [DOI] [PubMed] [Google Scholar]
- 5.Davis JQ, Bennett V. Ankyrin binding activity shared by the neurofascin/L1/NrCAM family of nervous system cell adhesion molecules. J. Biol. Chem. 1994;209:27163–27166. [PubMed] [Google Scholar]
- 6.Doherty P, Walsh F. CAM-FGF receptor interactions: A model for axonal growth. Mol. Cell. Neurosci. 1996;8:99–111. doi: 10.1006/mcne.1996.0049. [DOI] [PubMed] [Google Scholar]
- 7.Forrest S, Rosenthal A, Balnaves M. Mutation detection in X-linked hydrocephalus. Am. J. Hum. Genet. 1994;55:A219. [Google Scholar]
- 8.Fransen E, Lemmon V, Van Camp G, Vits L, Coucke P, Willems PJ. CRASH syndrome: a clinical spectrum of corpus callosum hypoplasia, retardation, adducted thumbs, spastic paraparesis and hydrocephalus due to mutations in one single gene, L1. Eur J Human Genetics. 1995;3:273–284. doi: 10.1159/000472311. [DOI] [PubMed] [Google Scholar]
- 9.Fransen E, Schrander-Stumpel C, Vits L, Coucke P, Van Camp G, Willems PJ. X-linked hydrocephalus and MASA syndrome present in one family are due to a single missense mutation in exon 28 of the L1CAM gene. Hum. Molec. Genet. 1994;3:2255–2256. doi: 10.1093/hmg/3.12.2255. [DOI] [PubMed] [Google Scholar]
- 10.Fransen E, Vits L, Van Camp G, Willems P. The clinical spectrum of mutations in L1, a neuronal cell adhesion molecule. Am. J. Med. Gene. 1996;64:73–77. doi: 10.1002/(SICI)1096-8628(19960712)64:1<73::AID-AJMG11>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 11.Grizzle JE, Starmer CF, Koch GG. Analysis of categorical data by linear models. Biometrics. 1969;25:489–504. [PubMed] [Google Scholar]
- 12.Gu SM, Orth U, Veske A, Enders H, Klunder K, Schlosser et al M. Five novel mutations in the L1CAM gene in families with X linked hydrocephalus. J. Med. Genet. 1906;33:103–106. doi: 10.1136/jmg.33.2.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hlavin ML, Lemmon V. Molecular structure and functional testing of human L1CAM: An interspecies comparison. Genomics. 1991;11:416–423. doi: 10.1016/0888-7543(91)90150-d. [DOI] [PubMed] [Google Scholar]
- 14.Holtzman RNN, Garcia L, Koenigsberger R. Hydrocephalus and congenital clasped thumbs: a case report with electromyographic evaluation. Dev. Med. Child. Neurol. 1976;18:521–527. doi: 10.1111/j.1469-8749.1976.tb03692.x. [DOI] [PubMed] [Google Scholar]
- 15.Hortsch M. The L1 family of neural cell adhesion molecules: Old proteins performing new tricks. Neuron. 1996;17:587–593. doi: 10.1016/s0896-6273(00)80192-0. [DOI] [PubMed] [Google Scholar]
- 16.Izumoto S, Yamasaki M, Arita N, Hiraga S, Ohnishi T, Fujitani et al K. A new imitation of the L1CAM gene in an X-linked hydrocephalus family. Child’s Nerv. Syst. 1996;12:742–747. doi: 10.1007/BF00261591. [DOI] [PubMed] [Google Scholar]
- 17.Jouet M, Kenwrick S. Gene analysis of L1 neural cell adhesion molecule in prenatal diagnosis of hydrocephalus. Lancet. 1995;345:161–162. doi: 10.1016/s0140-6736(95)90170-1. [DOI] [PubMed] [Google Scholar]
- 18.Jouet M, Moncla A, Paterson J, Mckeown C, Fryer A, Carpenter et al N. New domains of neural cell-adhesion molecule L1 implicated in x-linked hydrocephalus and MASA syndrome. Am. J. Hum. Genet. 1995;56:1304–1314. [PMC free article] [PubMed] [Google Scholar]
- 19.Jouet M, Rosenthal A, Armstrong G, MacFarlane J, Stevenson R, Paterson et al J. X-linked spastic paraplegia (SPG1), MASA syndrome and X-linked hydrocephalus result from mutations in the L1 gene. Nat. Genetics. 1994;7:402–407. doi: 10.1038/ng0794-402. [DOI] [PubMed] [Google Scholar]
- 20.Jouet M, Rosenthal A, MacFarlane J, Kenwrick S. A missense mutation confirms the L1 defect in X-linked hydrocephalus (HSAS). Nat. Genetics. 1993;4:331. doi: 10.1038/ng0893-331. [DOI] [PubMed] [Google Scholar]
- 21.Kaplan P. X linked recessive inheritance of agenesis of the corpus callosum. J. Med. Genet. 1983;20:122–124. doi: 10.1136/jmg.20.2.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Landrieu P, Ninane J, Ferriere G, Lyon G. Aqueductal stenosis in X-linked hydrocephalus: a secondary phenomenon? Dev. Med. Child. Neurol. 1979;21:637–652. doi: 10.1111/j.1469-8749.1979.tb01678.x. [DOI] [PubMed] [Google Scholar]
- 23.Macias V, Day D, King T, Wilson G. Clasped-thumb mental retardation (MASA) syndrome: confirmation of linkage to Xq28. Am. J. Med. Genet. 1992;43:408–414. doi: 10.1002/ajmg.1320430162. [DOI] [PubMed] [Google Scholar]
- 24.Rosenthal A, Jouet M, Kenwrick S. Aberrant splicing of neural cell adhesion molecule L1 messenger RNA in a family with X-linked hydrocephalus. Nat. Genet. 1992;2:107–112. doi: 10.1038/ng1092-107. [DOI] [PubMed] [Google Scholar]
- 25.Ruiz JC, Cuppens H, Legius E, Fryns JP, Glover T, Marynen et al P. Mutations in L1-CAM in two families with X-linked complicated spastic paraplegia, MASA syndrome, and HSAS. J. Med. Genet. 1995;32:549–552. doi: 10.1136/jmg.32.7.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schrander-Stumpel, C: Clinical and genetic aspects of the X-linked hydrocephalus/MASA spectrum. Amsterdam, Universitaire Pers Maastricht (1995).
- 27.Straussberg R, Blatt I, Brand N, Kessler D, Bat-Miriam Katznelson M, Goodman RM. X-linked mental retardation with bilateral clasped thumbs: report of another affected family. Clin. Gene. 1991;40:337–341. doi: 10.1111/j.1399-0004.1991.tb03105.x. [DOI] [PubMed] [Google Scholar]
- 28.Takechi T, Tohyama J, Kurashige T, Maruta K, Uyemura K, Ohi et al T. A deletion of five nucleotides in the L1CAM gene in a Japanese family with X-linked hydrocephalus. Hum. Genet. 1996;97:353–356. doi: 10.1007/BF02185770. [DOI] [PubMed] [Google Scholar]
- 29.Van Camp G, Vits L, Coucke P, Lyonnet S, Schrander-Stumpel C, Darby et al J. A duplication in the L1CAM gene associated with X-linked hydrocephalus. Nat. Genetics. 1993;4:421–425. doi: 10.1038/ng0893-421. [DOI] [PubMed] [Google Scholar]
- 30.Vits L, Van Camp G, Coucke P, Fransen E, DeBoulle K, Reyniers et al E. MASA syndrome is due to mutations in the neural cell adhesion gene L1CAM. Nat. Genetics. 1994;7:408–413. doi: 10.1038/ng0794-408. [DOI] [PubMed] [Google Scholar]
- 31.Wong EV, Cheng G, Payne HR, Lemmon V. The cytoplasmic domain of the cell adhesion molecule L1 is not required for homophilic adhesion. Neurosci. Lett. 1995;200:155–158. doi: 10.1016/0304-3940(95)12100-i. [DOI] [PubMed] [Google Scholar]
- 32.Wong EV, Kenwrick S, Willems P, Lemmon V. Mutations in the cell adhesion molecule L1 cause mental retardation. Trends Neurosci. 1995;18:168–172. doi: 10.1016/0166-2236(95)93896-6. [DOI] [PubMed] [Google Scholar]
- 33.Wong EV, Schaefer A, Landreth G, Lemmon V. Involvement of p90rsk in neurite outgrowth mediated by the cell adhesion molecule L1. J. Biol. Chem. 1996;271:18217–18223. doi: 10.1074/jbc.271.30.18217. [DOI] [PubMed] [Google Scholar]
- 34.Wong EV, Schaefer AW, Landreth G, Lemmon V. Casein kinase II phosphorylates the neural cell adhesion molecule L1. J. Neurochem. 1995;66:779–786. doi: 10.1046/j.1471-4159.1996.66020779.x. [DOI] [PubMed] [Google Scholar]
- 35.Yamasaki M, Arita N, Hiraga S, Izumoto S, Morimoto K, Nakatani et al S. A clinical and neuroradiological study of X-linked hydrocephalus in Japan. J. Neurosurg. 1995;83:50–55. doi: 10.3171/jns.1995.83.1.0050. [DOI] [PubMed] [Google Scholar]
- 36.Zhao X, Siu CH. Colocalization of the homophilic binding site and the neuritogenic activity of the cell adhesion molecule L1 to its second Ig-like domain. J. Biol. Chem. 1995;270:29413–29421. doi: 10.1074/jbc.270.49.29413. [DOI] [PubMed] [Google Scholar]
- 37.Zhao X, Siu CH. Differential effects of two hydrocephalus/MASA syndrome-related mutations on the homophilic binding and neuritogenic activities of the cell adhesion molecule L1. J. Biol. Chem. 1996;271:6563–6566. doi: 10.1074/jbc.271.12.6563. [DOI] [PubMed] [Google Scholar]