

Abstract
The binding of p53 protein to DNA is stimulated by its interaction with covalent as well as noncovalent modifiers. We report the identification of a factor from HeLa nuclear extracts that activates p53 DNA binding. This factor was purified to homogeneity and identified as the high mobility group protein, HMG-1. HMG-1 belongs to a family of highly conserved chromatin-associated nucleoproteins that bend DNA and facilitate the binding of various transcription factors to their cognate DNA sequences. We demonstrate that recombinant His-tagged HMG-1 enhances p53 DNA binding in vitro and also that HMG-1 and p53 can interact directly in vitro. Unexpectedly, HMG-1 also stimulates DNA binding by p53Δ30, a carboxy-terminally deleted form of the protein that is considered to be constitutively active, suggesting that HMG-1 stimulates p53 by a mechanism that is distinct from other known activators of p53. Finally, using transient transfection assays we show that HMG-1 can increase p53 and p53Δ30-mediated transactivation in vivo. HMG-1 promotes the assembly of higher order p53 nucleoprotein structures, and these data, along with the fact that HMG-1 is capable of bending DNA, suggest that HMG-1 may activate p53 DNA binding by a novel mechanism involving a structural change in the target DNA.
Keywords: p53 protein, HMG-1, DNA-binding activation, transcription
The DNA-binding activity of p53 is central to its biological function as a tumor suppressor (for review, see Gottlieb and Oren 1996; Ko and Prives 1996; Levine 1997). In keeping with its global role as a cell cycle checkpoint factor, p53 can induce either cell cycle arrest or apoptosis, thus ensuring genetic stability. Several genes have now been identified to function downstream of p53 in the DNA damage response pathway: These include p21/Waf1/Cip1 (El-Diery et al. 1993), GADD45 (Kastan et al. 1992), cyclin G (Okamoto and Beach 1994; Zauberman et al. 1995), bax (Miyashita and Reed 1995), mdm-2 (Barak et al. 1993; Wu et al. 1993), IGF-BP3 (Buckbinder et al. 1995), and many others. Each of these genes has been shown to contain one or more p53-responsive sites in its promoter region that conform to the consensus binding site identified by El-Diery et al. (1992) and to be directly activated by p53 following genotoxic insult.
The region in p53 that contributes to sequence-specific DNA binding spans the central portion of the protein, extending approximately from amino acid 100 to 300 (Cho et al. 1994 and references therein). Nearly all tumor-derived mutations map within this region, and most of these mutants exhibit defective DNA-binding abilities (Vogelstein and Kinzler 1992). A better understanding of the precise functioning of this domain and its regulation has therefore been the focus of several laboratories. Given the importance of p53 DNA binding, it is likely that this activity is subject to careful and possibly multilevel regulation to correctly orchestrate p53 function at the correct times in the cell cycle. It is therefore not surprising that in the context of the full-length protein, the activity of the DNA-binding domain is modulated both by other domains of the protein as well as by other cellular proteins/factors. Support for this comes from observations implicating the basic carboxyl terminus of p53 (residues 363–393) in regulating p53 DNA binding. Manipulation of this region by phosphorylation (Hupp et al. 1992), binding by monoclonal antibody pAb 421 (Hupp et al. 1992, 1995; Halazonetis et al. 1993), or interaction with single-stranded DNA (Jayaraman and Prives 1995) activates sequence-specific DNA binding. Deletion of this region results in a p53 molecule that is considered to be constitutively active for DNA binding (Hupp et al. 1992; Jayaraman et al. 1997a). Furthermore, addition in trans of the entire p53 carboxyl terminus (residue 311–393) (Jayaraman and Prives 1995), or short peptides spanning the critical basic residues, results in stimulation of p53 DNA binding (Hupp et al. 1995; Shaw et al. 1996; Jayaraman et al. 1997a). Finally, we recently purified the redox/repair protein Ref-1 from HeLa nuclear extracts and showed that it is a potent activator of full-length but not carboxy-terminally deleted p53 DNA binding both in vitro and in vivo (Jayaraman et al. 1997a). Taken together, the data imply that the carboxyl terminus of p53 plays a role in negative regulation of sequence-specific DNA binding by the central portion of the protein. These observations have led to the suggestion that there may exist in cells at least two forms of p53—a latent (inefficient DNA-binding) species and an active (efficient DNA-binding) one—and that conversion from the former to the latter occurs upon reception of the appropriate signal (Halazonetis et al. 1993; Hupp and Lane 1994).
In this paper we report the purification and identification from HeLa nuclear extracts of another regulator of p53 DNA binding, high mobility group protein 1 (HMG-1). Our data suggest that HMG-1 enhances p53 DNA binding and transactivation functions by a mechanism that is at least in part distinct from relief of repression by the p53 carboxyl terminus.
Results
Purification of a heat-stable activator of p53 from HeLa nuclear extracts
We purified previously a p53 stimulatory activity, Ref-1, a redox/DNA repair factor, from HeLa nuclear extracts and characterized its interactions with p53 (Jayaraman et al. 1997a). Ref-1 was purified from the 500 mm salt eluate of a phosphocellulose P-11 column. When we tested other fractions from the P-11 column for their ability to affect p53 DNA binding using the electrophoretic mobility shift assay (EMSA), we found that the P-11.85 fraction (0.85 m KCl eluate) markedly stimulated DNA binding by p53 (Fig. 1A, cf. lanes 1 and 2). The stimulated complex was supershifted by two different p53-specific monoclonal antibodies, pAb 1801 and pAb 421 (lanes 10,11), indicating that it contained p53. Competition assays using an excess of unlabeled oligonucleotides containing either wild-type or mutant p53 binding sites showed that the stimulation was sequence specific (cf. lanes 6 and 7 with lanes 8 and 9). Two lines of evidence argued against the possibility that this stimulatory activity was also Ref-1. First, we were unable to detect any Ref-1 protein in the 0.85 m eluate with anti-Ref-1 polyclonal antibodies (data not shown). Second, unlike Ref-1, the activity in this fraction was heat stable: It still retained considerable p53 activating function after incubation at 70°C for 15 min (lane 3). Accordingly, we decided to isolate and identify the putative p53 activator in the HeLa P-11.85 fraction.
Figure 1.

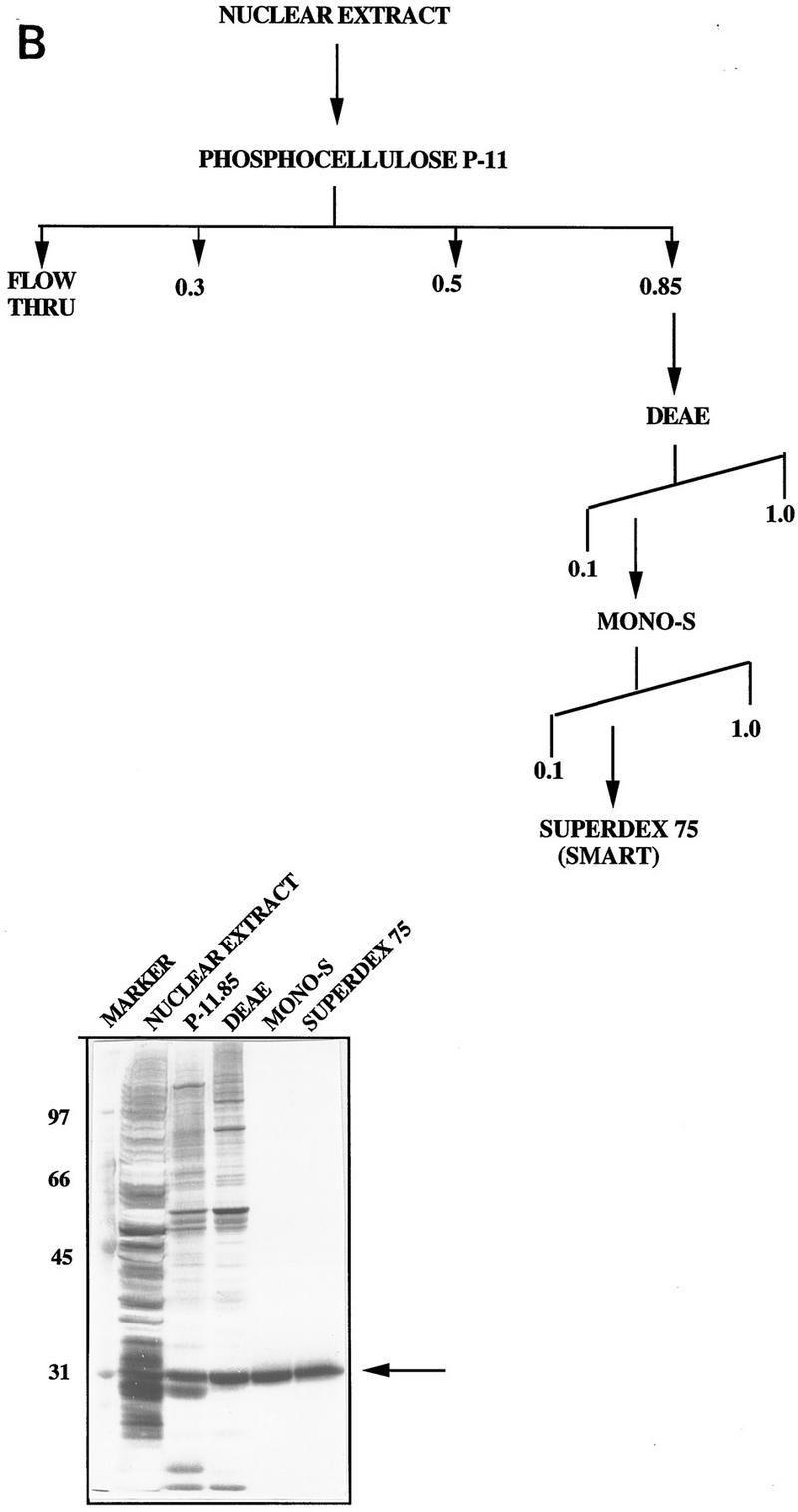
(A) HeLa nuclear fraction P-11.85 stimulates p53 DNA binding. p53 protein (90 ng) was bound to 32P-labeled GADD45 oligonucleotides either alone (lane 1) or in the presence of the P-11.85 fraction and p53 monoclonal antibodies pAb 421 and pAb 1801 or competitor DNA as indicated and analyzed by EMSA and autoradiography. Lanes 3 and 5 contain the P-11.85 fraction heat-treated for 5 min at 70°C. Reaction mixtures in lanes 4 and 5 contain no p53 protein. Wild-type (wt) or mutant (mt) consensus site-containing oligonucleotides were added in 10-fold (lanes 6,8) or 20-fold (lanes 7,9) molar excess over probe. The arrow indicates the p53–DNA complex; the bracket indicates p53–DNA complexes supershifted by antibodies. Not shown is free unbound probe at bottom of gel. (B) Purification of the p53 activator protein from HeLa nuclear fraction P-11.85. (Top) Protocol for purification of p53 DNA-binding stimulatory activity from HeLa nuclear extract. (Bottom) Aliquots of peak fraction from each step in A were analyzed by SDS-PAGE and silver staining. The arrow indicates purified protein.
We initially tested the properties of the p53-stimulating factor on a variety of FPLC columns to derive the final purification scheme (Fig. 1B, top). Fractions from each chromatographic step that showed stimulatory activity were pooled, dialyzed, and loaded onto the next column. We were successful in purifying the stimulatory factor to relative homogeneity such that the peak fraction from the final Superdex-75 column consisted of a single predominant band on a silver stained gel (Fig. 1B, bottom). The relative molecular mass of this protein was estimated to be ∼29 kD, based on its electrophoretic mobility.
The stimulatory activity in P-11.85 is HMG-1
To identify this novel p53 activator, the ∼29-kD protein band was isolated and subjected to proteolytic digestion. Automated protein sequencing of one of the resulting polypeptides revealed a 13-amino-acid sequence that corresponded to a region (residues 150–162) within a previously characterized protein, HMG-1. Confirmation of the identity of the p53 stimulatory factor as HMG-1 was based on several independent lines of evidence. First, the electrophoretic mobility on SDS–polyacrylamide gels of the protein we had isolated was similar to that reported for HMG-1 (for review, see Bustin and Reeves 1996). Second, the peak fraction from the Superdex-75 column was specifically recognized by an affinity purified anti-HMG-1 polyclonal antibody (Fig. 2A). Third, HMG-1 from a different source and purified by a different procedure was also capable of activating p53 (Fig. 2B). The stimulation of p53 by HMG-1, purified from calf thymus by acid extraction (Romani et al. 1979; Hamada and Bustin 1985), is virtually indistinguishable from that by HMG-1 that we purified from HeLa nuclear extracts (cf. lanes 1 and 2). The magnitude of stimulation of p53 DNA binding produced by HMG-1 varied with different preparations of the protein and was between 10-fold and 50-fold. Additionally, HMG-2, a protein highly homologous to HMG-1 and a member of the HMG-1 box-containing class of proteins, was also capable of eliciting a similar stimulation (data not shown). The stimulation was sequence specific as tested by challenging with an excess of wild-type or mutant p53 binding site-containing unlabeled oligonucleotides (cf. lanes 3 and 4). The effect was specific to HMG-1 (and HMG-2) and was not seen with nonspecific proteins such as bovine serum albumin (BSA) or fetal bovine serum (FBS) (data not shown). Although p53-specific monoclonal antibodies could supershift the stimulated complexes, similar addition of a polyclonal anti-HMG-1 antibody neither supershifted the complexes nor disrupted the stimulation (Fig. 2B, lanes 8,9). Furthermore, the lack of a change in electrophoretic mobility between the unstimulated and stimulated complexes suggested that the interaction between p53 and HMG-1 was either transient or not stable enough to withstand the conditions of the EMSA. Calf thymus-purified HMG-1 also displayed heat stability even after heating at 90°C (lane 5).
Figure 2.
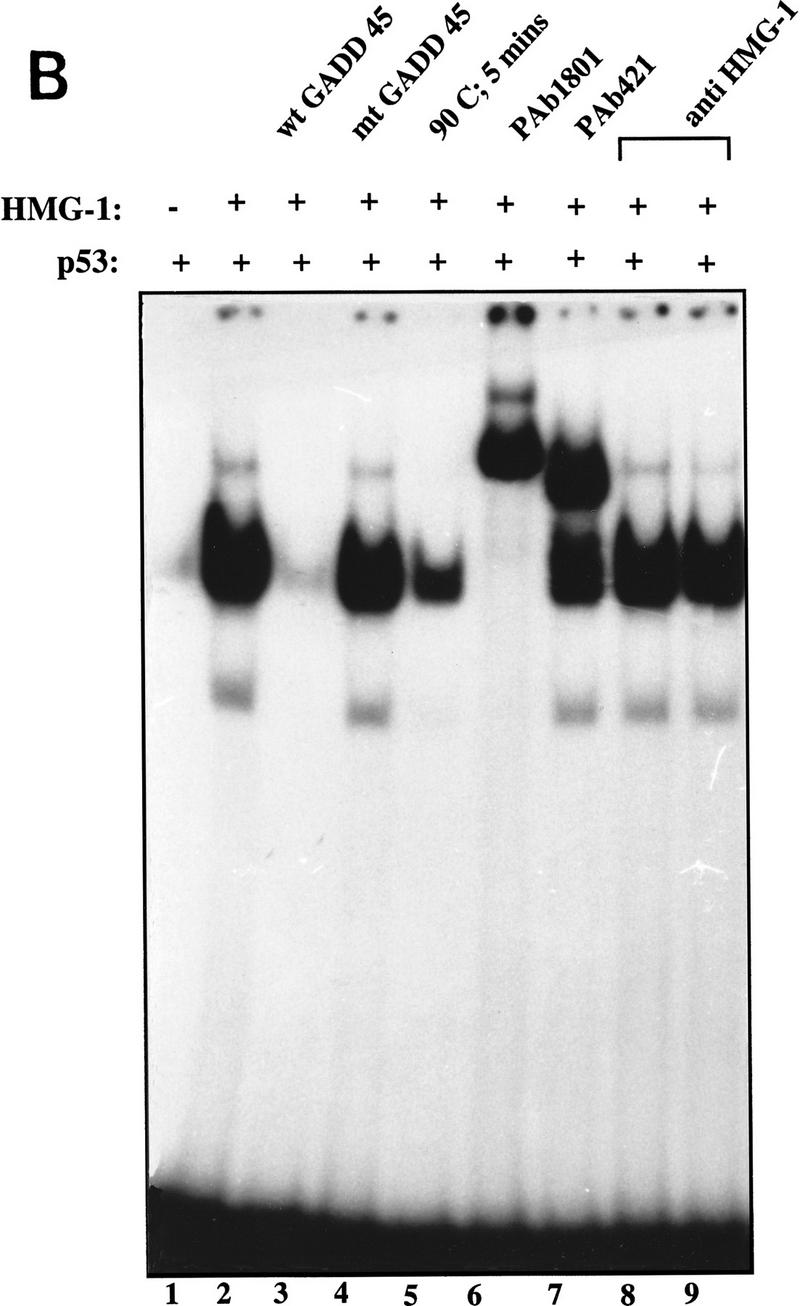


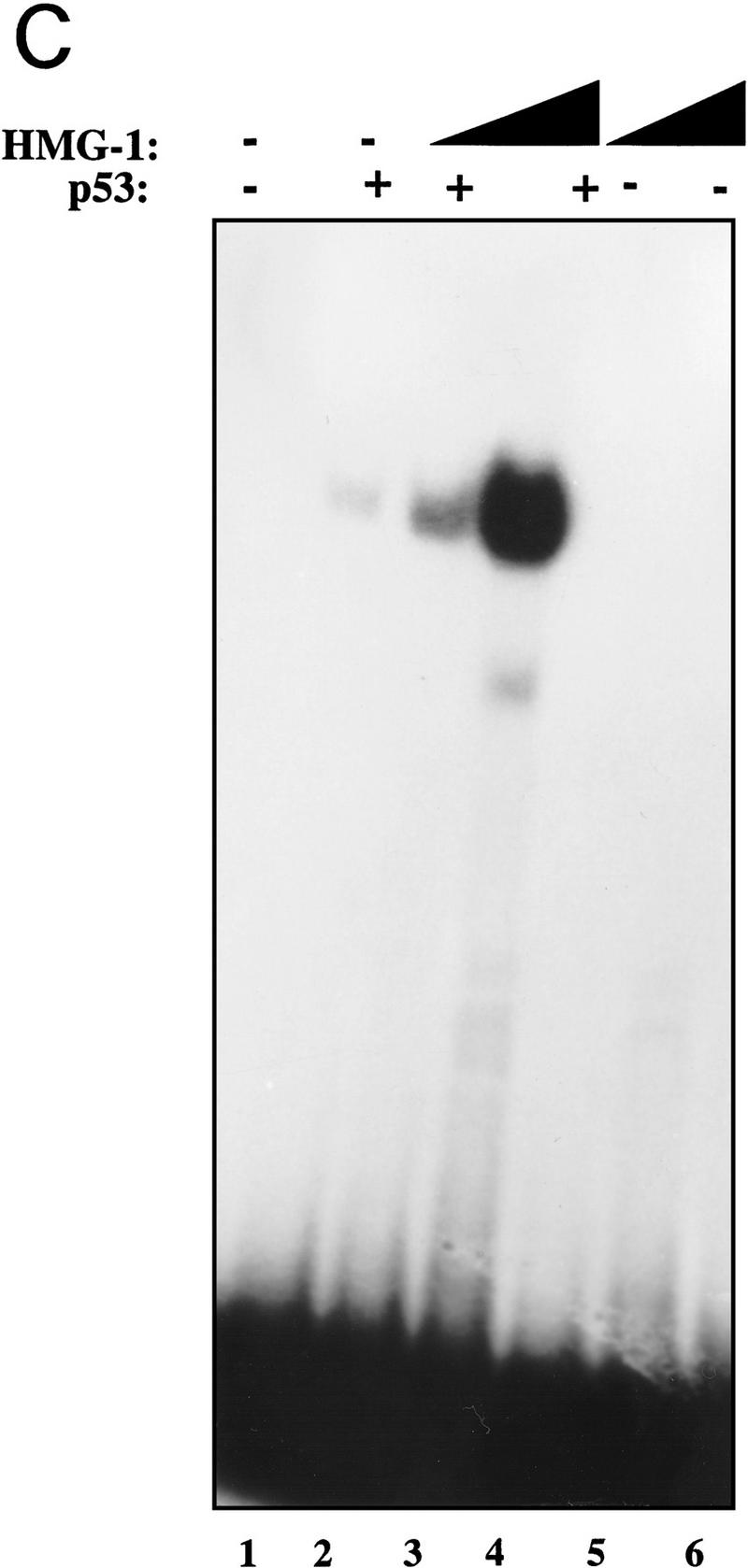
Identification of HMG-1 as the p53 stimulating protein from HeLa P-11.85 fraction. (A) HMG-1 purified from calf thymus or HeLa P-11.85 Superdex-75 peak fraction or immunopurified p53 protein was subjected to SDS-PAGE and transferred to nitrocellulose, and the resulting blots were probed with affinity-purified polyclonal anti-HMG-1 antibody. (B) DNA binding by p53 (90 ng) in the absence (lane 1) or presence of calf thymus-purified HMG-1 (100 ng) (lanes 2–8) was analyzed by EMSA as in Fig. 1. Antibodies and 20-fold excess competitor DNA were added as indicated. Lane 8 contains mixtures in which antibody and proteins were added simultaneously, whereas in lane 9 HMG-1 was preincubated with anti-HMG-1 antibody for 20 min prior to addition to reaction mixture. (C) Recombinant HMG-1 enhances DNA binding by p53. DNA-binding mixtures contained recombinant His–HMG-1 (50 or 100 ng), either in the absence (lanes 5,6) or presence of p53 (90 ng) (lanes 2–4). Lane 1 contains probe alone. (D) DNA-binding reaction mixtures were as above but in the absence of poly[d(I-C)]. Binding mixtures contained either 8 (lanes 2–4,12,13) or 4 ng (lanes 7–9,10,11) of p53 in the absence (lanes 2,7,10,11) or presence (lanes 3,4, 8,9,12,13) of His–HMG-1 (60 or 120 ng). HMG-1 (60 ng) was present in mixtures shown in lanes 12 and 13. Monoclonal antibody pAb 421 was added as indicated. Only the DNA–protein complexes are shown as in Fig 1A.
Final verification of the identity of the p53-activating factor came from testing recombinant HMG-1 purified from bacteria (see Materials and Methods for details). As can be seen from Figure 2C, His-tagged recombinant HMG-1 protein was fully capable of activating DNA binding by p53 (cf. lanes 3 and 4 with lane 2). HMG-1 by itself showed no binding to the probe at these levels (lanes 5 and 6). Careful titration of HMG-1 followed by quantitation of the resulting p53–DNA complexes revealed that ∼50 ng of purified His–HMG-1 was required for eliciting a 2- to 3-fold stimulation of ∼90 ng of p53 protein, indicating that HMG-1 exerted its effects in the stoichiometric range (data not shown). At higher concentrations (in the microgram range), HMG-1 was itself capable of binding nonspecifically to DNA (data not shown). Such nonspecific DNA binding by HMG-1 has been described previously (for review, see Bustin and Reeves 1996). This observation raised the possibility that the stimulation of p53 at lower concentrations of HMG-1 may be a consequence of HMG-1 protein binding to and sequestering the nonspecific poly[d(I-C)] present in the reaction mixtures, thereby freeing the specific probe for increased binding by p53. As seen in Figure 2D, HMG-1 was able to stimulate p53 DNA binding significantly in the absence of any nonspecific carrier DNA (cf. lanes 3 and 4 with lane 2 and lanes 8 and 9 with lane 7). The effect of adding the carboxy-terminal stimulatory antibody pAb 421 in conjunction with HMG-1 was also examined. At the levels of pAb 421 used, HMG-1 was able to stimulate DNA binding over and above that seen with pAb 421 alone (cf. lanes 10 and 11 with lanes 12 and 13) suggesting that HMG-1 might enhance p53 DNA binding by a mechanism independent from that involving the carboxyl terminus of p53.
HMG-1 can stimulate a carboxy-terminally truncated p53 (p53Δ30)
As discussed in the introductory section, many activators of full-length p53 work by relieving repression by the carboxy-terminal 30 amino acids and are incapable of stimulating DNA binding by p53 lacking this autoinhibitory region (p53Δ30). Ref-1 activates full-length p53 but not p53Δ30 (Jayaraman et al. 1997a). We were therefore interested in seeing if HMG-1 would similarly fail to activate p53Δ30. Remarkably, we observed a dramatic stimulation of p53Δ30 upon addition of increasing amounts of HMG-1 to two different levels of p53Δ30 tested (Fig. 3A). Additionally, both the “core” DNA-binding domain of p53 as well p53Δ96 (p53 lacking the amino-terminal 96 amino acids) were stimulated by HMG-1 (Fig. 3B). These results are particularly interesting because they imply the existance of a level of regulation of p53 DNA binding that is distinct from relief of repression by its carboxy-terminal domain and redox.
Figure 3.
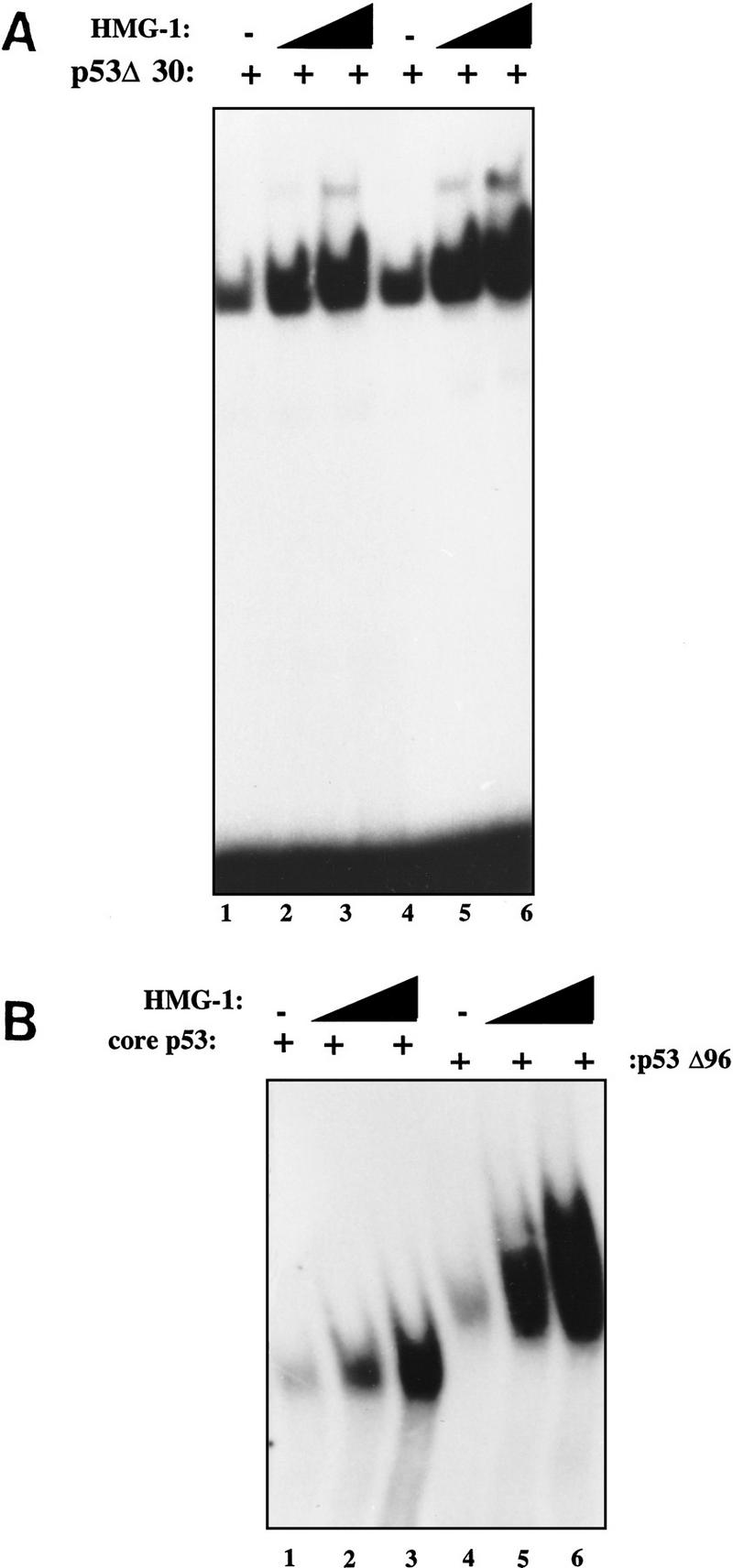
HMG-1 can stimulate DNA binding by p53Δ30 as well as by core p53. (A) Ten nanograms (lanes 1–3) or 20 ng (lanes 4–6) of p53Δ30 was bound to 32P-labeled GADD45 oligonucleotides and analyzed by EMSA in the absence (lanes 1,4) or presence (lanes 2,3,5,6) of HMG-1 (50 or 100 ng). (B) DNA-binding mixtures contained either core domain p53 (lanes 1–3) or p53Δ96 (lanes 4–6) in the presence (lanes 2,3,5,6) or absence (lanes 1,4) of HMG-1 (50 or 100 ng). Shown are p53–DNA complexes only as in Fig. 1A.
HMG-1 promotes cooperative p53 interactions on DNA
To further understand the mechanistic basis of the stimulatory effect of HMG-1, we examined its effect on p53 binding to a longer DNA fragment. p53 exhibits striking cooperativity when bound to its consensus site in the context of a relatively long strand of DNA (L. Jayaraman and C. Prives, unpubl.; see below). We therefore decided to examine the effect of HMG-1 on this characteristic feature of p53. Figure 4A (left panel) shows the effect of increasing amounts of HMG-1 on p53 binding to the GADD45 consensus site in the context of a 120-bp-long probe. Addition of HMG-1 resulted not only in a stimulation of DNA binding by p53 but also in a marked shift in the mobility of the p53–DNA complexes (cf. lane 2 with lanes 3–6), whereas HMG-1 itself did not show any binding to the probe (lanes 7–9). One possibility was the formation of a stable complex between p53 and HMG-1 in the context of a longer DNA molecule, resulting in a retardation of mobility of the protein–DNA complexes. However, addition of anti-HMG-1 polyclonal antibodies to the reaction mixture did not result in a further shift in mobility (data not shown; Fig. 4B), indicating that HMG-1 was not a component of the supershifted complex. If the higher mobility complexes consisted entirely of p53 bound to DNA, it was possible that HMG-1 was, in fact, enhancing cooperative binding by p53. To verify whether this was indeed the case, we directly compared the mobility shift produced in the presence of HMG-1 with that produced on addition of increasing amounts of p53 protein alone to a given amount of probe. As can be seen from Figure 4A (right panel), the pattern of complexes produced in both cases is very similar. We also examined the effect of HMG-1 on p53Δ30 binding to the longer DNA probe (Fig. 4B). As seen with full-length p53, HMG-1 promoted the formation of higher order p53Δ30–DNA complexes (lanes 3–5), and addition of anti-HMG-1 antibody did not cause a supershift of these complexes (lanes 6–8). The most likely interpretation of these data is that bending of DNA by HMG-1 promotes cooperative interactions between p53 tetramers.
Figure 4.

HMG-1 promotes co-operative p53 interactions on DNA. (A, Left) Reaction mixtures contained increasing amounts of HMG-1 (50, 75, 100, or 150 ng) in the absence (lanes 7–9) or presence (lanes 3–6) of full-length p53 (50 ng) using a 120-bp 32P-labeled DNA fragment containing the GADD45 p53 binding site. Lane 1 contains no protein. Mixtures in lane 2 contained 50 ng of p53 alone. (Right) Reaction mixtures contain increasing amounts (50, 100, 200, or 400 ng) of p53 (lanes 2–5) or increasing amounts of HMG-1 (50 or 125 ng) with 50 ng of p53 (lanes 6,7). Lane 1 contains no protein. (B) Reaction mixtures contained p53Δ30 (10 ng), alone (lane 2) or with 75 (lanes 3,6), 100 (lanes 4,7), or 150 (lanes 5,8) ng of HMG-1 with or without excess anti-HMG-1 antibody as indicated. Mixtures in lanes 9–11 contained HMG-1 protein at the above concentrations without p53.
p53 binds directly to HMG-1 in vitro
As shown above, an association between p53 and HMG-1 could not be demonstrated by EMSA. Nevertheless our results suggested that the two proteins might interact specifically, however transiently. This was tested by performing a Far Western experiment in which purified HMG-1 protein and, as positive and negative controls, two versions of human MDM2 (HDM2), wild-type HDM2 and dl166 HDM2, which cannot bind p53 (Chen et al. 1993; Shieh et al. 1997), were resolved by SDS-PAGE, and the polypeptides were transferred to nitrocellulose (Fig. 5, see left panel for Ponceau S stain of transferred proteins). When the ability of purified p53 to recognize the immobilized proteins was examined, p53 clearly recognized both HMG-1 and full-length HDM2 but not the truncated HDM2 (dl166) protein that cannot bind to p53 (Fig. 5, right panel). Attesting further to the specificity of the interaction between p53 and HMG-1 were additional Far Western experiments in which, whereas p53 consistently bound to HMG-1 protein, it failed to interact with equivalent quantities of immobilized bovine serum albumin, myosin, β-galactosidase, and phosphorylase B (data not shown). These experiments indicate that the affinity of p53 for HDM2 is greater than for HMG-1, although the relative difference in the interaction of p53 with these two proteins has not been quantitated. That HMG-1 binds specifically to p53 protein may help to elucidate its stimulatory effect on p53 DNA binding.
Figure 5.

p53 binds HMG-1 in vitro. Calf-thymus-purified HMG-1 (0.25, 0.5, 1.0, and 2.0 μg), truncated dl166 HDM2 (1 μg) or full-length HDM2 (1 μg) as indicated, and molecular mass marker polypeptides were resolved by SDS-PAGE and transferred to nitrocellulose. (Right) Filter was incubated with human p53 protein (1 μg in 5 ml) followed by detection with pAb 421 as described in Materials and Methods. (Left) Ponceau S staining of filter. The lower amounts of HMG-1 were below the level of detection by Ponceau S. Arrows indicate position of HMG-1 protein.
Full-length p53 as well as p53Δ30 can be stimulated by HMG-1 in vivo
Our results with full-length and carboxy-terminally deleted p53 in vitro prompted us to test the effect of HMG-1 on the activity of both these proteins in vivo. To do this, HMG-1 cDNA was placed under the control of a cytomegalovirus (CMV) promoter, and this construct was then used to test the ability of either full-length p53 or p53Δ30 to activate transcription from a promoter containing a p53-responsive site, using transient transfection assays. Although HMG-1 is an abundant nuclear protein (Singh and Dixon 1990, and references therein), we reasoned that the large amount of p53 produced upon transfection might be capable of responding to increased HMG-1 levels. p53–null H1299 cells were transfected with plasmids expressing p53 or p53Δ30 and HMG-1 either separately or together, along with a luciferase reporter construct driven by a portion of the cyclin G promoter (Jayaraman et al. 1997a). Our results indicate that there was dramatically better activation of the cyclin G promoter upon cotransfection of both HMG-1 and p53 than was obtained by transfecting p53 alone (Fig. 6A). In addition, transfection experiments with the Bax promoter showed that HMG-1 and p53 can cooperate in activating this promoter as well (data not shown). Importantly, and consistent with our in vitro results, HMG-1 was also capable of significantly activating carboxy-terminally deleted p53Δ30 (Fig. 6B). This is the first demonstration of a protein exerting a stimulatory effect on p53Δ30 in cells. The cyclin G promoter itself was not affected by HMG-1, implying that HMG-1 was directly targeting p53 (or p53Δ30). These data suggest a role for HMG-1 in p53 function in vivo and strengthen our hypothesis that HMG-1 activates p53 by a novel and hitherto unidentified mechanism.
Figure 6.


HMG-1 stimulates transactivation by wild-type p53 as well as p53Δ30 in transfected cells. (A,B) H1299 cells were transiently transfected with wild-type p53 (300 ng) (A) or p53Δ30 (35 ng) (B) or HMG-1 expression plasmid (2.5 μg) or both p53 and HMG-1 expression plasmids as indicated. In all cases, total DNA transfected was normalized with equivalent amounts of control parental vectors. The reporter construct used was cyclin G–luc (1 μg). (Y-axis) Luciferase activity is represented as fold transactivation relative to samples transfected with reporter constructs and parental control plasmids. The fold activation represents an average of triplicate samples.
Discussion
The multifunctional nature of HMG proteins
HMG-1 belongs to a class of nonhistone chromosomal nuclear proteins that are highly conserved among mammals (Bustin and Reeves 1996). They have been shown to enhance the sequence-specific DNA binding of a variety of proteins such as the progesterone receptor (Oñate et al. 1994), the estrogen receptor (Verrier et al. 1997), HOX proteins (Zappavigna et al. 1996), and the POU domain-containing proteins Oct1, Oct2, and Oct6 (Zwilling et al. 1995) and to increase specific transcription by RNA polymerase II and III in vitro (Tremethick and Molloy 1986, 1988; Singh and Dixon 1990; Ge and Roeder 1994; Ogawa et al. 1995). HMG-1/2 proteins contain three domains, two highly basic amino-terminal domains A and B (termed HMG-1 boxes) and an acidic carboxy-terminal domain. These proteins exhibit interesting interactions with DNA: They recognize and bind a variety of DNA structures including single-stranded DNA, palindromes, B-Z DNA junctions, four-way junctions, cruciforms, and stem loops (for review, see Bustin and Reeves 1996). DNA binding by HMG-1/2 is therefore non-sequence specific but is DNA conformation dependent (Bianchi et al. 1989). Additionally, and perhaps most relevant to the experiments described here, HMG-1 can bend DNA (Paull et al. 1993; Pil et al. 1993; Oñate et al. 1994; for review, see Grosschedl et al. 1994). More recently it was shown that V(D)J cleavage by Rag1 and Rag2 proteins is stimulated by HMG-1/2 proteins (Agarwal and Schatz 1997; Sawchuck et al. 1997; van Gent et al. 1997).
How does HMG-1 activate p53?
Previous reports on regulation of p53 interactions with DNA have implicated its carboxyl terminus in negatively regulating DNA binding by the central core domain of p53. Supporting this was the observation that carboxy-terminally deleted p53 appeared to be constitutively active for DNA binding (Hupp et al. 1992; Jayaraman et al. 1997a). Our observation that HMG-1 stimulates both full-length p53 and p53Δ30 in vitro and in vivo suggests a regulatory mechanism distinct from relief of carboxy-terminal repression. Moreover, the antibody pAb 421 (Fig. 2D) and the redox/repair protein Ref-1 (N. Moorthy, L. Jayaraman and C. Prives, unpubl.) appear to function additively with HMG-1 in enhancing DNA binding by p53. Because alleviation of hindrance by the p53 carboxyl terminus is an unlikely mode of action of HMG-1 on p53, we must consider alternate mechanisms. There is now increasing evidence implicating the involvement of the HMG-1 box in mediating the assembly of higher order nucleoprotein complexes based on their protein–protein and protein–DNA complex forming abilities (Paull et al. 1993; Zappavigna et al. 1996; van Gent et al. 1997; Verrier et al. 1997). One could similarly ask whether HMG-1/2 proteins enhance p53 DNA binding through a direct association with p53 or solely by dint of their interactions with DNA (or both).
Our experiments show that HMG-1 does not alter the electrophoretic mobility of p53 complexed with oligonucleotides and that HMG-1-stimulated p53–DNA complexes cannot be supershifted by an affinity-purifed polyclonal antibody directed against HMG-1. Nevertheless, we were able to demonstrate a direct interaction between p53 and HMG-1 by an alternative approach. What could be the reasons for the apparent failure to detect HMG-1 in the stimulated complex? Either binding to p53 or to DNA may mask the HMG-1 antibody epitope, or p53 and HMG-1 may associate to form a ternary complex with DNA, but this interaction is transient or too unstable to withstand the conditions of the gel shift assay, resulting in the dissociation of HMG-1 prior to entry into the gel. Although others too have failed to demonstrate an interaction between HMG-1/2 and other DNA-binding proteins using EMSA, in some cases direct interactions of HMG-1/2 with DNA-binding proteins have been described (Zwilling et al. 1995; Zappavigna et al. 1996; Agarwal and Schatz 1997). It remains to be determined what regions of p53 and HMG-1 are responsible for their interaction with each other.
The role of DNA bending in p53 binding
The ability of HMG-1 proteins to bend DNA contributes significantly to their ability to stimulate transcription factors. For instance, transcriptional stimulation of the T-cell receptor enhancer element by Lef-1 protein is dependent on both specific DNA bending as well as protein–DNA interactions by this HMG-1 box containing protein (Giese et al. 1992). Similarly, the RNA polymerase I transcription factor UBF mediates looping of a 180-bp promoter region into a single turn around itself to stimulate RNA polymerase I transcription of ribosomal RNA (Bazett-Jones et al. 1994). HMG-1 stimulation of progesterone receptor (PR) binding seems to involve a structural change in target DNA as a direct consequence of HMG-1’s ability to flex DNA (Oñate et al. 1994). Finally, the stimulatory effects of HMG-1/2 proteins on V(D)J recombination is likely to require the DNA-binding property of these HMG proteins (Agarwal and Schatz 1997; Sawchuck et al. 1997; van Gent et al. 1997).
There are three lines of evidence supporting the likelihood that the DNA-bending activity of HMG-1 is responsible for its stimulation of p53. First, our own results strongly support DNA bending as the mechanism by which HMG-1 increases p53 activity. (1) HMG-1 stimulation of p53Δ30 is unique because no activator of p53 DNA binding has been shown to augment the binding of this carboxy-terminally truncated form of p53 to DNA. Additionally, when added together PAb 421 and HMG-1 stimulate p53 DNA binding more than either does alone (Fig. 2D). This suggests a form of activation different from previously described modes. Given that HMG-1 is a well-characterized DNA-bending protein, it is reasonable to assume that this property is related to its effect on p53. (2) The p53 core crystal structure (Cho et al. 1994) and a systematic analysis of p53 binding to DNA substructures within the consensus site by Wang et al. (1995) has shown that not only does p53 bind DNA as a tetramer but also that co-operative interactions between monomers greatly facilitates binding. Consistent with this, we found that HMG-1 dramatically stimulated p53 co-operative interactions on a longer DNA fragment. With long fragments, at low concentrations of p53, one p53 tetramer makes appropriate contacts with the two half-sites that make up the p53 consensus site. We propose that in the presence of HMG-1, bending of the DNA between the two half-sites opens up more space such that two (or more) p53 tetramers can now contact two half-sites, and such loading of two p53 tetramers on the same DNA strand would cause the observed “supershift” in mobility shown in Figure 4B. Conversely, when increasing amounts of p53 alone are added to a given amount of DNA, excess p53 protein similarly causes multiple p53 tetramers to bind the same DNA strand. The failure to demonstrate these higher order forms with shorter DNA molecules may be owing to spatial constraints forbidding two tetramers on adjacent half-sites. It is conceivable that the increased specific affinity of p53 for DNA prebent by HMG-1 may result in displacement of HMG-1, hence our failure to detect a ternary complex. (3) Stimulation of p53 DNA binding by HMG-1 was two to threefold greater when p53 half sites were separated by 10, 15, or 30 bp than when they were adjacent (data not shown). The best explanation for these data is that flexure of DNA by HMG-1 provides an opportunity for more stable contacts between p53 molecules on DNA.
The second line of evidence supporting DNA bending as the mode by which HMG-1 stimulates p53 is derived from the observation that p53 itself can bend the DNA to which it binds. Balagurumoorthy et al. (1995) have shown that DNA binding by the core DNA-binding domain of p53 results in bending of the target DNA, an observation strengthened by evidence that the stability of the p53–DNA complex is greatly enhanced by the DNA-bending angle (Nagaich et al. 1997a). DNA bending may help relieve steric hindrance between the subunits of p53 tetramers on DNA (Nagaich et al. 1997b). Proteins such as HMG-1 that bend DNA efficiently, thus providing prebent consensus site DNA to p53, would therefore serve to increase DNA binding by p53. Alternatively, at high concentrations of p53, bending of DNA would also stabilize interactions between p53 tetramers as shown in Figure 4A (right panel, lanes 2–5).
The final supporting evidence is derived from the fact that many HMG-1 box-containing proteins recognize DNA containing CA/TG dinucleotides (Landsman and Bustin 1993). Although HMG-1/2 proteins do not show sequence specificity in their interactions with DNA, the fact that CA/TG dinucleotides induce a local deformation in DNA is consistent with the observation that these proteins can bind bent DNA and bend it still further. Interestingly, the core and critical sequence within the p53 cognate site is 5′-CATG 3′. Thus, it is likely that HMG-1/2 proteins can recognize a p53-binding site and, although they do not form a stable interaction with this DNA, they can induce a bend thus making it a better binding target for p53.
Consequences of p53–HMG-1 interaction in vivo
Our data showing that HMG-1 enhances p53 DNA binding in vitro was supported by its marked stimulatory effect on transactivation by p53 in vivo. It was particularly satisfying to demonstrate that the carboxy-terminally deleted form of p53, p53Δ30, could also be stimulated by HMG-1 in cells. This provided strong confirmatory support for the notion that the effect of HMG-1 on p53 is unique and separate from the now rather large class of agents that serve to relieve negative regulation by the carboxyl terminus. Although HMG-1 proteins are rather abundant in cells, our data show that they can be limiting when p53, a potential target, is overexpressed such as in transient transfection assays. Future experiments will hopefully provide evidence linking p53 and HMG-1 in a more physiological setting. However, it is noteworthy that HMG-1 can also specifically recognize and bind to cisplatin DNA adducts (Huges et al. 1992; Pil and Lippard 1992). p53 too can recognize and bind to sites in DNA that contain certain kinds of DNA lesions such as insertion/deletion mismatches (Lee et al. 1995), and thus it is predicted to also interact with bulky adducts formed by treatment with chemotherapeutic drugs such as cisplatin. Furthermore, p53 can potentially associate with a number of protein components of the excision repair machinery (for review, see Ko and Prives 1996). Localization of p53 at the site of DNA lesion, either through its inherent ability to recognize damaged DNA or by interaction with various repair-related proteins, might bring p53 and HMG-1 into proximity, with the resulting recruitment by p53 of HMG-1 to p53-binding sites in its target promoters.
Materials and methods
Purification of p53 proteins
Flu peptide-tagged human full-length p53, p53Δ30 and p53Δ96 were constructed as detailed in Jayaraman et al. (1997a,b). Sf21 cells were infected with human p53 or p53Δ30 recombinant virus, harvested 48 hr postinfection, and p53 protein was immunopurified essentially as described in Jayaraman et al. (1997b). Elution was performed using either the flu peptide YPYDVPDYA (for latent p53) or the 421 peptide KKGQSTSRHKK (for active p53), which were purchased from the Cold Spring Harbor protein chemistry facility. Oxidized p53 was prepared similarly except that the flu peptide was used for the elution step and extraction, elution, and dialysis were done in buffers lacking DTT.
DNA binding
EMSA was performed as described previously (Jayaraman and Prives 1995). The oligonucleotide probe containing the GADD45 site is as follows: 5′-AATTCTCGAGCAGAACATGTCTAAGCATGCTGGGCTCGAG-3′. The probe was labeled by the Klenow fragment of Escherichia coli DNA polymerase. The 120-bp GADD45 p53-binding site was prepared and labeled as detailed in Jayaraman and Prives (1995). Reaction mixtures contained 8 μl of 5× EMSA buffer (100 mm HEPES at pH 7.9, 125 mm KCl, 0.5 mm EDTA, 50% glycerol, 10 mm MgCl), 2 μl of 40 mm spermidine, 2 μl of 10 mm DTT, 2 μl of 0.5% NP-40, 2 μl of 60 mg/ml double-stranded poly [d(I-C)], 4 μl of BSA (1 mg/ml), 32P-labeled probe DNA (3 ng), proteins and antibodies as indicated, and water in a total volume of 40 μl. Reaction mixtures were incubated at room temperature for 30 min; 20 μl of each reaction mixture was then loaded onto a native 4% polyacrylamide gel containing 0.5× Tris-borate/EDTA (TBE) buffer, 1 mm EDTA, and 0.05% NP-40 and electrophoresed in 0.5% TBE at 4°C at 200–250 V for 2 hr. DNA–protein complexes were quantified by PhosphorImaging using ImageQuant software.
Purification of HMG-1 from HeLa cells
Nuclear extracts (7 mg/ml), prepared from ∼50 liters of HeLa cells according to the procedure of Dignam et al. (1983) and dialyzed into BC100 buffer (100 mm KCl, 20 mm Tris at pH 8.0, 0.2 mm EDTA, and 20% glycerol), were aliquoted, frozen, and stored at −80°C. Protein purification was performed at 4°C, and samples were kept on ice. All chromatographic steps were performed by FPLC, except for the Superdex-75 fractionation that was performed on the SMART fractionation system (Pharmacia). HeLa nuclear extract (300 mg) was loaded onto a 20-ml phosphocellulose P-11 column and washed with two column volumes of BC100. The bound protein was eluted in stepwise fashion with BC100 buffer containing 0.3, 0.5, and 0.85 m KCl. In this and all subsequent chromatographic steps, protein concentration was monitored by continuous UV absorption at 280 nm. The p53 stimulatory fractions from the P-11.85 eluate (as assayed by gel retardation analysis) were pooled, dialyzed against BC100 buffer, and loaded onto a 10-ml DEAE column equilibrated in the same buffer. The stimulatory activity was eluted using a linear gradient of 0.1–1 m KCl in BC100 and dialyzed against BC100 buffer and further fractionated by Mono-S chromatography. The stimulatory activity was once again eluted using a linear gradient of 0.1–1 m KCl in BC100 buffer, and peak fractions were pooled and dialyzed before gel-filtration chromatography on a Superdex-75 column. The protein composition of different column fractions was analyzed on 10% SDS–polyacrylamide gels and silver-stained for visualization of bands. The protein concentration of pooled fractions from individual columns was estimated using the method Bio-Rad protein determination kit.
Antibodies
Polyclonal anti HMG-1 antibodies that were used in the initial identification of HMG-1 from HeLa cells were affinity-purified on Sepharose columns containing HMG-1 protein and have been described previously (Bustin 1989). A polyclonal antiserum (Cocalico Biologicals, Inc.) generated against bacterially expressed HMG-1 protein was used in subsequent experiments.
Construction, expression, and purification of recombinant HMG-1
Recombinant His-tagged HMG-1 was constructed as follows: Primers complimentary to the 5′ and 3′ ends of human HMG-1 were used in PCR amplification of HMG-1 cDNA obtained by reverse transcription from total HeLa cell poly(A) mRNA. A single PCR product of appropriate size was obtained and cloned into the pRSET vector resulting in the second residue of HMG-1 affixed to six histidines present immediately downstream of the start codon in the vector. The ligated product was first transformed into DH5α and subsequently into BL21 for expression. Expression of His-tagged HMG-1 was induced by addition of 1 mm IPTG for 2 hr, and the protein was purified by binding to Ni–agarose beads and eluting with imidazole. Substantial quantities of relatively pure HMG-1 protein were obtained, the identity of which was confirmed by reactivity with the polyclonal anti-HMG-1 antibody (data not shown).
Far Western analysis
Polypeptides were separated by SDS-PAGE and transferred to nitrocellulose. After incubation in denaturation buffer (6 m guanidine–HCL in PBS) twice for 5 min at 4°C, filters were then incubated 6 × 10 min in serial dilutions (1:1) of denaturation buffer in PBS + 1 mm DTT. The membrane was blocked in PBS containing 0.5% Tween 20 and 5.0% nonfat dry milk (NFDM) for 45 min at 20°C. After washing twice in PBS/Tween 20/0.25% NFDM, the blot was incubated with p53 (0.2 μg/ml) in PBS/Tween 20/0.25% NFDM containing 1 mm DTT and 0.5 mm PMSF for 2 hr. The filter was washed 4 × 10 min in PBS/Tween 20/0.25% NFDM, the second wash containing 0.0001% glutaraldehyde. Blots were then probed using pAb 421 as primary antibody and goat anti-mouse IgG as secondary antibody and detected using ECL (Amersham).
Transfection assays
H1299 cells (American Type Culture Collection) were maintained at 37°C in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% FBS. p53 and p53Δ30 cDNAs were under the control of the cytomegalovirus (CMV) promoters (Jayaraman et al. 1997a). HMG-1 cDNA was obtained by PCR amplification using appropriate 5′- and 3′-end primers and cloned under the control of a CMV promoter. The reporter construct used was pGL–cyclin G–LUC, containing 1.48 kb of the 5′ region of the rat cyclin G gene (Jayaraman et al. 1997a). Prior to transfection, cells were seeded at 0.6 × 106 cells per 60-mm-diameter dish for luciferase assays. Cells were transfected by the calcium phosphate method, and the precipitate was left on the cells for 5 hr, after which the cells were plated in fresh DMEM supplemented with 10% FBS. Luciferase assays were performed 18–24 hr later. When appropriate, DNA of the parental vector was included to keep the total amount of transfected DNA constant in each sample. Luciferase assays were essentially as described by Friedlander et al. (1996). Each transfection experiment was performed in triplicate cultures.
Acknowledgments
We are grateful for the expert technical assistance of E. Freulich and S. Landau. R. Tacke is thanked for assistance and advice in generating recombinant HMG-1 protein. S.-Y. Shieh is thanked for providing advice on the Far Western protocol. L.J. would like to thank the members of the Prives laboratory for helpful and interesting discussions. This work was supported by NIH grants CA58316 to C.P. and GM37971 to J.L.M.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL prives@cubsps.bio.columbia.edu; FAX (212) 865-8246.
References
- Agarwal A, Schatz DG. RAG1 and RAG2 form a stable postcleavage synaptic complex with DNA containing signal ends in V(D)J recombination. Cell. 1997;89:43–53. doi: 10.1016/s0092-8674(00)80181-6. [DOI] [PubMed] [Google Scholar]
- Balagurumoorthy P, Sakamoto H, Lewis MS, Zambrano N, Clore GM, Groneborn AM, Appella E, Harrington RE. Four p53 DNA-binding domain peptides bind natural p53 response elements and bend the DNA. Proc Natl Acad Sci. 1995;92:8591–8595. doi: 10.1073/pnas.92.19.8591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak Y, Juven T, Haffner R, Oren M. mdm2 expression is induced by wild type p53 activity. EMBO J. 1993;12:461–468. doi: 10.1002/j.1460-2075.1993.tb05678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxevanis AD, Landsman D. The HMG-1 box protein family: Classification and functional relationships. Nucleic Acids Res. 1995;23:1604–1613. doi: 10.1093/nar/23.9.1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazett-Jones DP, LeBlanc B, Herfort M, Moss T. Short-range DNA looping by the Xenopus HMG-box transcription factor, xUBF. Science. 1994;264:1134–1137. doi: 10.1126/science.8178172. [DOI] [PubMed] [Google Scholar]
- Bianchi ME, Beltrame M, Paonessa G. Specific recombination of cruciform DNA by nuclear protein HMG-1. Science. 1989;243:1056–1059. doi: 10.1126/science.2922595. [DOI] [PubMed] [Google Scholar]
- Buckbinder L, Talbott R, Velasco-Miguel S, Takenaka I, Faha B, Seizinger BR, Kley N. Induction of the growth inhibitor IGF-binding protein 3 by p53. Nature. 1995;377:646–649. doi: 10.1038/377646a0. [DOI] [PubMed] [Google Scholar]
- Bustin M. Preparation and application of immunological probes for nucleosomes. Methods Enzymol. 1989;170:214–251. doi: 10.1016/0076-6879(89)70049-5. [DOI] [PubMed] [Google Scholar]
- Bustin M, Reeves R. High mobility group proteins. Prog Nucleic Acids Res Mol Biol. 1996;54:35–100. doi: 10.1016/s0079-6603(08)60360-8. [DOI] [PubMed] [Google Scholar]
- Chen J, Marechal V, Levine AJ. Mapping of the p53 and mdm-2 interaction domains. Mol Cell Biol. 1993;13:4107–4114. doi: 10.1128/mcb.13.7.4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Gorina S, Jeffery PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science. 1994;265:346–355. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- Dignam JD, Martin PL, Shastry BS, Roeder RG. Eukaryotic gene transcription with purified components. Methods Enzymol. 1983;101:582–598. doi: 10.1016/0076-6879(83)01039-3. [DOI] [PubMed] [Google Scholar]
- El-Diery WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nature Genet. 1992;1:45–49. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- El-Diery WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- Friedlander P, Haupt Y, Prives C, Oren M. A mutant p53 that discriminates between p53 responsive genes cannot induce apoptosis. Mol Cell Biol. 1996;16:4961–4971. doi: 10.1128/mcb.16.9.4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese K, Amsterdam A, Grosschedl R. DNA-binding properties of the HMG domain of the lymphoid-specific transcriptional regulator Lef-1. Genes & Dev. 1991;5:2567–2578. doi: 10.1101/gad.5.12b.2567. [DOI] [PubMed] [Google Scholar]
- Ge H, Roeder RG. The high mobility group protein HMG-1 can reversibly inhibit class II gene transcription by interaction with the TATA-binding protein. J Biol Chem. 1994;269:17136–17140. [PubMed] [Google Scholar]
- Gottlieb MT, Oren M. p53 in growth control and neoplasia. Biochim Biophys Acta. 1996;1287:77–102. doi: 10.1016/0304-419x(95)00019-c. [DOI] [PubMed] [Google Scholar]
- Grosschedl R, Giese K, Pagel J. HMG domain proteins: Architectural elements in the assembly of nucleoprotein structures. Trends Genet. 1994;10:94–100. doi: 10.1016/0168-9525(94)90232-1. [DOI] [PubMed] [Google Scholar]
- Halazonetis TD, Davis LJ, Kandil AN. Wild-type p53 adopts a “mutant”-like conformation when bound to DNA. EMBO J. 1993;12:1021–1028. doi: 10.1002/j.1460-2075.1993.tb05743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada H, Bustin M. Chromosomal proteins HMG-1 and -2 distinguish between S1 sensitive sites in super-coiled DNA. Biochemistry. 1985;24:1428–1433. doi: 10.1021/bi00327a022. [DOI] [PubMed] [Google Scholar]
- Huges EN, Engelsberg BN, Billings PC. Purification of a nuclear protein that binds cis-platinum damaged DNA. J Biol Chem. 1992;267:13520–13527. [PubMed] [Google Scholar]
- Hupp TR, Lane DP. Allosteric activation of latent p53 tetramers. Curr Biol. 1994;4:865–875. doi: 10.1016/s0960-9822(00)00195-0. [DOI] [PubMed] [Google Scholar]
- Hupp TR, Meek DW, Midgley CA, Lane DP. Regulation of the specific DNA binding function of p53. Cell. 1992;71:875–886. doi: 10.1016/0092-8674(92)90562-q. [DOI] [PubMed] [Google Scholar]
- Hupp TR, Sparks A, Lane DP. Small peptides activate the latent sequence specific DNA binding function of p53. Cell. 1995;83:237–245. doi: 10.1016/0092-8674(95)90165-5. [DOI] [PubMed] [Google Scholar]
- Jayaraman L, Prives C. Activation of p53 sequence-specific DNA binding by short single strands of DNA requires the p53 C-terminus. Cell. 1995;81:1021–1029. doi: 10.1016/s0092-8674(05)80007-8. [DOI] [PubMed] [Google Scholar]
- Jayaraman L, Murthy KGK, Zhu C, Curran T, Xanthoudakis S, Prives C. Identification of redox/ repair protein Ref-1 as a potent activator of p53. Genes & Dev. 1997a;11:558–570. doi: 10.1101/gad.11.5.558. [DOI] [PubMed] [Google Scholar]
- Jayaraman L, Freulich E, Prives C. Functional dissection of the p53 tumor suppressor protein. Methods Enzymol. 1997b;283:245–256. doi: 10.1016/s0076-6879(97)83020-0. [DOI] [PubMed] [Google Scholar]
- Kastan MB, Zhan Q, El-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace Jr AJ. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in Ataxia-Telangiectasia. Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- Ko LJ, Prives C. p53: Puzzle and paradigm. Genes & Dev. 1996;10:1054–1072. doi: 10.1101/gad.10.9.1054. [DOI] [PubMed] [Google Scholar]
- Landsman D, Bustin M. A signature for the HMG-1 box DNA-binding proteins. Bioessays. 1993;15:539–546. doi: 10.1002/bies.950150807. [DOI] [PubMed] [Google Scholar]
- Lee S, Elenbaas B, Levine A, Griffith J. p53 and its 14 kDa C-terminal domain recognize primary DNA damage in the form of insertion/deletion mismatches. Cell. 1995;81:1013–1020. doi: 10.1016/s0092-8674(05)80006-6. [DOI] [PubMed] [Google Scholar]
- Levine AJ. p53, the cellular gatekeeper for growth and development. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–299. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- Nagaich AK, Appella E, Harrington RE. DNA bending is essential for the site-specific recognition of DNA response elements by the DNA binding domain of the tumor suppressor protein p53. J Biol Chem. 1997a;272:14842–148949. doi: 10.1074/jbc.272.23.14842. [DOI] [PubMed] [Google Scholar]
- Nagaich AK, Zhurkin VB, Sakamoto H, Gorin AA, Clore GM, Gronenborn AM, Appellam E, Harrington RE. Architectural accommodation in the complex of four p53 DNA binding domain peptides with the p21/waf1/cip1 DNA response element. J Biol Chem. 1997b;272:14830–14841. doi: 10.1074/jbc.272.23.14830. [DOI] [PubMed] [Google Scholar]
- Ogawa Y, Aizawa S, Shirakawa H, Yoshida M. Stimulation of transcription accompanying relaxation of chromatin structure in cells overexpressing high mobility group 1 protein. J Biol Chem. 1995;270:9272–9280. doi: 10.1074/jbc.270.16.9272. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Beach D. Cyclin G is a transcriptional target of the p53 tumor suppressor protein. EMBO J. 1994;13:4816–4822. doi: 10.1002/j.1460-2075.1994.tb06807.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oñate SA, Prendergast P, Wagner JP, Nissen M, Reeves R, Pettijohn DE, Edwards DP. The DNA-bending protein HMG-1 enhances progesterone receptor binding to its target DNA sequences. Mol Cell Biol. 1994;14:3376–3391. doi: 10.1128/mcb.14.5.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull T, Haykinson M, Johnson RC. The non-specific DNA-binding and bending proteins HMG-1 and HMG-2 promote the assembly of complex nucleoprotein structures. Genes & Dev. 1993;7:1521–1534. doi: 10.1101/gad.7.8.1521. [DOI] [PubMed] [Google Scholar]
- Pil PM, Lippard SJ. Specific binding of chromosomal protein HMG-1 to DNA damaged by the anticancer drug cisplatin. Science. 1992;256:234–237. doi: 10.1126/science.1566071. [DOI] [PubMed] [Google Scholar]
- Pil PM, Chow CS, Lippard SJ. High mobility group protein 1 mediates DNA bending as determined by ring-closures. Proc Natl Acad Sci. 1993;90:9465–9469. doi: 10.1073/pnas.90.20.9465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romani M, Rodman TC, Vidali G, Bustin M. Serological analysis of the species specificity of the high mobility group proteins. J Biol Chem. 1979;254:2918–2922. [PubMed] [Google Scholar]
- Sawchuck DJ, Weis-Garcia F, Malik S, Besmer E, Bustin M, Nussenzweig MC, Cortes P. V(D)J recombination: Modulation of RAG1 and RAG2 cleavage activity on 12/23 substrates by whole cell extract and DNA-bending proteins. J Exp Med. 1997;185:2025–2032. doi: 10.1084/jem.185.11.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw P, Freeman J, Bovey R, Iggo R. Regulation of specific DNA binding by p53: Evidence for a role for O-glycosylation and charged residues at the carboxy-terminus. Oncogene. 1996;12:921–930. [PubMed] [Google Scholar]
- Shieh S-Y, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by mdm2. Cell. 1997;91:325–334. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- Singh J, Dixon GH. High mobility group proteins 1 and 2 function as general class II transcription factors. Biochemistry. 1990;29:6295–6302. doi: 10.1021/bi00478a026. [DOI] [PubMed] [Google Scholar]
- Tremethick DJ, Molloy PL. High mobility group proteins 1 and 2 stimulate transcription in vitro by RNA polymerase II and III. J Biol Chem. 1986;261:6986–6992. [PubMed] [Google Scholar]
- ————— Effects of high mobility group proteins 1 and 2 on initiation and elongation of specific transcription by RNA polymerase II in vitro. Nucleic Acids Res. 1988;16:11105–11122. doi: 10.1093/nar/16.23.11107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gent DC, Hiom K, Paull TT, Gellert M. Stimulation of V(D)J cleavage by high mobility group proteins. EMBO J. 1997;16:2665–2670. doi: 10.1093/emboj/16.10.2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verrier CS, Roodi N, Yee CJ, Bailey LR, Jensen RA, Bustin M, Parl FF. High-mobility group (HMG) protein and TATA-binding protein-associated factor TAF(II)30 affect estrogen receptor-mediated transcriptional activation. Mol Endochrinol. 1997;11:1009–1019. doi: 10.1210/mend.11.8.9962. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Kinzler KW. p53 function and dysfunction. Cell. 1992;70:523–526. doi: 10.1016/0092-8674(92)90421-8. [DOI] [PubMed] [Google Scholar]
- Wang Y, Schwedes JF, Parks D, Mann K, Tegtmeyer P. Interaction of p53 with its consensus DNA-binding site. Mol Cell Biol. 1995;15:2157–2165. doi: 10.1128/mcb.15.4.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes & Dev. 1993;7:1126–1132. doi: 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]
- Zappavigna V, Falciola L, Citterich M, Mavilio F, Bianchi ME. HMG-1 interacts with HOX proteins and enhances their DNA binding and transcriptional activation. EMBO J. 1996;15:4981–4991. [PMC free article] [PubMed] [Google Scholar]
- Zauberman A, Lubp A, Oren M. Identification of p53 target genes through immune selection of genomic DNA: The cyclin G gene contains two distinct p53 binding sites. Oncogene. 1995;10:2361–2366. [PubMed] [Google Scholar]
- Zwilling S, Konig H, Wirth T. High mobility group protein 2 functionally interacts with the POU domains of octomer transcription factors. EMBO J. 1995;14:1198–1208. doi: 10.1002/j.1460-2075.1995.tb07103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]